

Abstract
Li–Fraumeni syndrome (LFS) is a cancer predisposition syndrome caused by a germline mutation of the TP53 gene on chromosome 17p13.1. It has an autosomal dominant pattern of inheritance with high penetrance. These patients have a very high lifetime cumulative risk of developing multiple malignancies and have a strong family history of early-onset malignancies. The protein p53, encoded by TP53, has a complex set of genome-preserving functions initiated during episodes of cellular stress and DNA damage. In LFS, TP53 gene mutations cause the loss of function of p53, leading to downstream events permissive for development of various malignancies throughout life. The LFS component tumors include soft tissue sarcomas, osteosarcoma, premenopausal breast cancer, brain tumors, and adrenal cortical carcinomas. Multiple types of sarcomas have been reported in association with LFS; this review article will focus on the most frequently encountered pediatric sarcomas associated with TP53 mutations.
Keywords: Li–Fraumeni syndrome, TP53, p53, rhabdomyosarcoma, osteosarcoma
Introduction
Drs. Frederick Li and Joseph F. Fraumeni Jr. were the first to report four families of children with rhabdomyosarcoma (RMS) or other soft tissue sarcomas in infancy, and a strong family history of breast cancer and other malignancies affecting their parents and relatives at a young age.1 Subsequent studies by Birch et al verified this association as a clinical syndrome and named it Li–Fraumeni syndrome (LFS).2 In addition to the originally described tumors, brain tumors, osteosarcoma, leukemia, and adrenal cortical carcinoma were added to the syndrome.2 3 4 5 In 1990, after molecular testing for TP53 germline mutations was developed, it was used to confirm the link between p53 and LFS.6 7 Since then, nearly 250 mutations throughout the TP53 gene have been detected.8 The majority of tumor-associated TP53 mutations occur within the DNA-binding domain, resulting in a defective protein.9
Genetics
TP53 is an important tumor suppressor gene. Located on chromosome 17p13.1, it belongs to a p53 family with more than 40 different isoforms that have a multiplicity of complex cellular functions, including cell-cycle arrest, DNA repair, genomic stability, senescence, apoptosis, stemness, differentiation, autophagy, angiogenesis, autophagy, drug sensitivity, and metabolism.10 Deregulation of p53 is the most commonly inactivated protein in human cancer.11 Due to its critical role in preventing the propagation of DNA mutations, TP53 has been named the “guardian of the genome.”12 During physiologic conditions, p53 protein levels remain low. During times of cellular stress and DNA damage, p53 is stabilized, activated, and subsequently translocated to the nucleus. Once in the nucleus, it acts as a transcription factor regulating the activity of a set of genes involved with cell-cycle arrest, apoptosis, and DNA repair.12 13 14
A mutation in another gene, hCHK2, has also been associated with LFS.15 hCHK2 is a tumor suppressor gene that is activated in response to DNA damage. It encodes for a kinase enzyme that results in cell-cycle arrest by phosphorylating protein products of mutated genes. The mutation was first detected by Bell et al in a family with RMS, brain tumors, and multiple cases of early-onset breast cancer, yet no evidence of TP53 mutation.15 Only a small number of families carry this mutation. LFS patients with TP53 or hCHK2 mutations present a similar range of malignancies and are clinically indistinguishable.
LFS has an autosomal dominant pattern of inheritance with high penetrance. The overall lifetime risk of developing cancer varies slightly by sex, with women having a higher overall risk by age 50 years compared with men (93 vs. 68%). Women also tend to present at an earlier age (female: 29 years vs. male: 40 years). Although most patients have a positive family history of early-onset cancer, the frequency of de novo germline TP53 ranges between 7 and 20%.9 16
Clinical Findings
The classic clinical characterization of LFS by Li and Fraumeni includes an initial patient with a sarcoma, a first-degree relative with cancer before the age of 45 years, and a first- or second-degree relative with cancer before the age of 45 years or a sarcoma at any age.3 Since then, families with similar features, but not meeting the full criteria for LFS, have been identified using two different sets of criteria proposed by Birch et al, Eeles, and Chompret et al.2 17 18 19 The Birch et al criteria do not require the initial presentation of a sarcoma. The Eeles criteria only require two first- or second-degree relatives with LFS component tumors at any age. Following Chompret et al's less strict criteria, patients with LFS component tumors but no family history can be included as having LFS.
The LFS component tumors constitute a long list of sarcomas, carcinomas, brain tumors, and leukemias, far beyond the scope of a single review article. Analysis of the histological typing of tumors associated with TP53 germline mutations proved difficult because, in a considerable fraction of neoplasms, no histopathologic data have been reported. The most commonly identified sarcoma in LFS is osteosarcoma, corresponding to 12.6% of cases, followed by brain tumors (12%) and soft tissue sarcomas (11.6%). Of the soft tissue sarcomas, RMSs are the most frequently identified. Other less frequent sarcomas reported include fibrosarcomas (which is no longer considered a true entity), atypical fibroxanthomas, leiomyosarcomas, orbital liposarcomas, spindle cell sarcomas, and undifferentiated pleomorphic sarcomas.20 This review will focus on the two most common pediatric malignancies associated with TP53 mutations: RMS and osteosarcoma.
Rhabdomyosarcoma
The Intergroup Rhabdomyosarcoma Study Group has defined RMS as a malignant neoplasm showing exclusive evidence of skeletal muscle differentiation by histologic morphology or ancillary techniques such as immunohistochemical stains or molecular genetics.21 22 RMSs represent the largest group of soft tissue sarcomas in children, adolescents, and young adults, and overall constitute 3% of all childhood cancers, with a yearly incidence rate of 4.5 per million in individuals aged less than 20 years in the United States. The majority of these tumors are diagnosed in children and adolescents, with more than half of them occurring before 10 years of age.22 23 24 25
The current World Health Organization classification divides RMSs into four subtypes: embryonal, alveolar, spindle cell/sclerosing, and pleomorphic.25 The first two subtypes are most frequently encountered in the pediatric population. The spindle cell/sclerosing subtype affects children and adults, while the pleomorphic RMS is most rare and tends to be limited to adults. RMS most often occurs in the head and neck region—including orbit and meninges—followed by genitourinary tract, limbs, and the trunk, in decreasing order of frequency.
Embryonal rhabdomyosarcomas (ERMSs) are the most common subtype, accounting for 60% of childhood cases, or 2.6 per million children aged less than 15 years in the United States. Thirty-six percent of cases occur in children less than 5 years of age, while only 18% occur in adolescents, with a moderate male predominance.22 25 Histologically, ERMSs are composed of primitive mesenchymal cells in various stages of early myogenesis. The cells are small, round to ovoid, and poorly differentiated or undifferentiated, with a high nuclear:cytoplasmic ratio and variable cytoplasmic features depending on the degree of cytodifferentiation. Occasional rhabdomyoblasts may progressively acquire more features of differentiation toward skeletal cell formation, forming elongated “strap cells,” which may exhibit cross-striations visible by light microscopy. Bright eosinophilia and multinucleation may also be seen with terminal differentiation, as well as postchemotherapy. These cells tend to occur in a myxoid background, with variable degrees of cellularity.
Botryoid RMSs are a subgroup of ERMS with a unique clinical and pathologic presentation. They arise beneath mucosal surfaces of viscera such as the genitourinary tract (urinary bladder, vagina), biliary tract, pharynx, conjunctiva, or auditory canal. In these sites, they grow as exophytic, edematous, often myxomatous polyps, appearing as grapelike masses.22 25 ERMSs are aneuploid with multiple chromosomal changes, including whole chromosome gains or losses. Polysomy 8 occurs frequently, as do extra copies of chromosomes 2 and 20. Loss of chromosomes 10 and 15 is also common. In most cases, there is loss of heterozygosity (LOH) in 11p15.5, which contains IGF2 and genes encoding several tumor suppressors.26 ERMSs in patients with TP53 germline mutations are more likely to be anaplastic, which is defined by the presence of markedly enlarged atypical cells with hyperchromatic nuclei. On occasion, bizarre multipolar mitotic figures may be encountered27 (Fig. 1A, B).
Fig. 1.
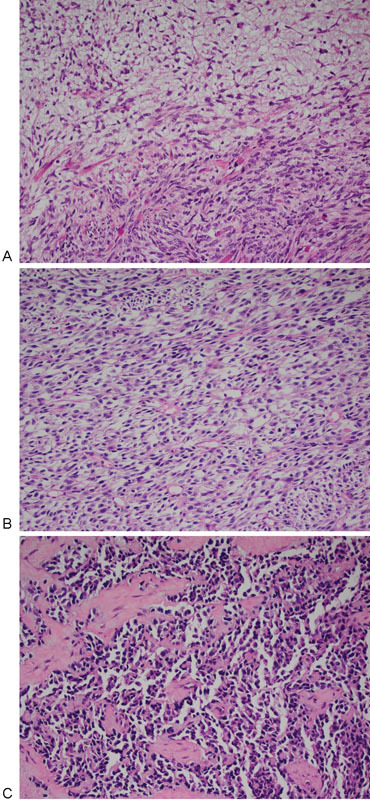
(A) Embryonal rhabdomyosarcoma with areas of high and low cellularity. (B) Embryonal rhabdomyosarcoma with a diffuse arrangement of spindle cells with slight cellular pleomorphism. (C) Alveolar rhabdomyosarcoma with discohesive cells and fibrous septae.
Alveolar RMSs (ARMSs) account for 20% of pediatric RMSs and occur most often in adolescents and young adults, being less common in younger children. These tumors are known for the recurrent translocations t(2;13)(q35;q14) or t(1;13)(p36;q14), resulting in PAX3-FOXO1 and PAX7-FOXO1 fusion genes, respectively. PAX3-FOXO1 fusion is encountered in 55% of cases, while PAX7-FOXO1 is encountered in 22%.25 The resultant fusion proteins result in high expression of potent pro-oncogenic transcription factors that alter the growth and apoptotic pathways and facilitate ineffective myogenic differentiation.25 26 It is noteworthy that 23% of ARMSs are PAX fusion-negative (PFN).
Histologically, ARMSs are highly cellular tumors composed of larger, more rounded undifferentiated cells, admixed with variable numbers of rhabdomyoblasts and multinucleated cells with a peripheral, wreath-like arrangement of their nuclei. The nuclei of ARMS cells tend to be larger and slightly more pleomorphic than those of ERMSs. Nucleoli are present in some cells. The classic histologic pattern is formed by a cohesive arrangement of the cells on thin collagenous septa, and discohesion of the cells detached from the septa. This creates the characteristic alveolar-like pattern of these tumors (Fig. 1C). Cross-striations are not commonly seen in ARMSs. On occasion, these tumors have a paucity of collagenous septa and less discohesion of the tumor cells, forming the solid variant of ARMS.22 In a retrospective review, it was noted that tumors with a solid pattern, lacking any alveolar spaces, were more likely to be PFN (78%, vs. 16% with alveolar pattern).28 In a study of early mutational processes and genomic events in RMSs, the tumors were subdivided into PAX fusion–positive RMS (PFP-RMS) and PFN-RMS.29 It was shown that the initiating common lesions in the majority of PFN-RMS are the combination of LOH of 11p15.5 and point mutations in members of the RAS pathway. A smaller number of PFN-RMS demonstrate LOH of chromosome 17, encompassing two island regions centered around TP53 and NF1.29 Another link between p53 inactivation and RMS was identified by integrated genetic and epigenetic analysis. Seki et al described four distinct tumor subtypes based on methylation patterns, correlating strongly with histological phenotypes, mutation or copy number (CN) profiles, and clinical behavior.30 Their A1 and A2 subtypes correspond to ARMS and PFP-RMS, and their E1 and E2 subtypes to ERMS and PFN-RMS. The E1 and E2 subtypes are characterized by high frequency of CN alterations, allelic imbalances, FGFR4/RAS/AKT mutations, and PTEN epigenetic silencing. Of note was the identification of p53 inactivation (45.5%) in the E2 subtype, along with a higher mutation frequency of the FGFR4/RAS/AKT pathway (81.8% in E2, vs. 54.5% in E1 and 17.9% in A1/A2). The E2 subgroup was associated with a significantly worse overall survival regardless of stage, age, and the site of tumor involvement. Furthermore, TP53 mutations significantly affected the overall survival of patients; however, E1/E2 classification had a greater impact than TP53 mutations on overall survival.30
Osteosarcoma
Conventional osteosarcomas, the most common primary cancer of bone, are high-grade malignancies in which osteoid or bone matrix is produced by the tumor cells. The majority of these tumors arise within the metaphysis of long bones, near to the growth plates. They tend to occur in adolescents and young adults, with most cases developing between 10 and 14 years of age. There is a second peak in frequency, occurring in adults over 60 years of age. The annual incidence is 4.4 per million persons between the age of 0 and 24 years, 1.7 per million between the age of 25 and 59 years, and 4.2 per million after 60 years of age. This sarcoma is slightly more frequent in males (male to female ratio of 1.34:1). Overall, it is considered a rare tumor, since it represents fewer than 1% of all malignancies diagnosed in the United States.31 32 33 Histologically, the tumor cells typically demonstrate significant pleomorphism and variable degrees of hyperchromasia. They tend to be large, pleomorphic cells, ranging from round to fusiform to spindle. They produce variable amounts of osteoid matrix, fibroblastic stroma, and cartilage, and are thus histologically subdivided into osteoblastic, fibroblastic, and chondroblastic subtypes, respectively, depending on the predominant matrix (Fig. 2A–C).
Fig. 2.
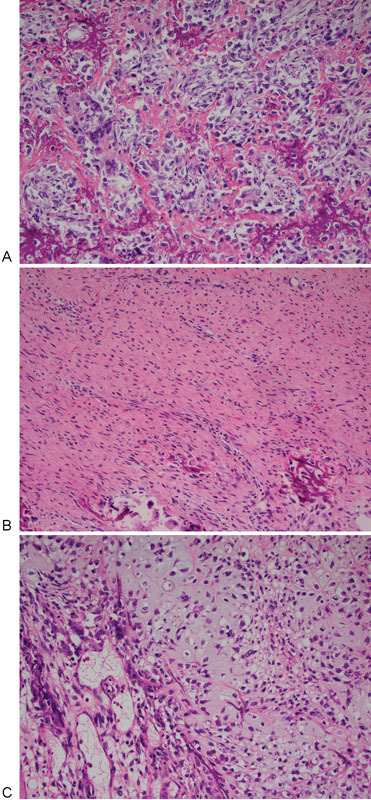
(A) Osteosarcoma with osteoblastic component. (B) Osteosarcoma with fibroblastic component. (C) Osteosarcoma with chondroblastic component.
Osteosarcomas have highly complex and inconsistent karyotypes, with complicated genomic rearrangements involving any chromosome and a large number of numerical and structural chromosomal abnormalities. The most frequent genetic alterations in conventional osteosarcomas are losses of portion of chromosomes 3q, 6q, 10, 13, 17p, and 18q, and gains of portions of chromosomes 1p, 1q, 6p, 8q, and 17p.31 34 35 Although there is a massive amount of information regarding the molecular genetics of osteosarcomas, the genetic etiology of osteosarcomas is still unknown. It is well known that osteosarcoma occurs at higher-than-expected frequencies in individuals with LFS, hereditary retinoblastoma (germline RB1 inactivation), Rothmund–Thomson syndrome (germline RECQL4 inactivation), Bloom syndrome (BLM inactivation), and Werner syndrome (germline WRN inactivation). These syndromes have in common an inactivation of protein products involved in genomic stabilization.35 36 Genomic instability can result in numerical chromosomal instability or structural chromosomal instability. The former leads to CN variations, and the latter to aberrant genomic rearrangements as a result of ineffective DNA damage response mechanisms. Mutations in TP53 are highly associated with genomic instability in osteosarcoma36 and have been identified in 10 to 39% of cases. Sporadic or germline inactivation of the tumor suppressor protein pRB occurs in 50% of osteosarcomas. pRB is essential for preventing cell-cycle progression through G1/S following DNA damage. LOH or deletion of RB1, located in chromosome 13q14.2, has been detected in 19 to 67% of osteosarcomas, and RB1 mutations in 25 to 35% of cases. LOH of the 17p13.1 locus has been identified in 29 to 42% of sporadic osteosarcomas.36
Conclusion
The pediatric sarcomas associated with TP53 germline mutations have numerous genetic alterations permitted by ineffective protection of the integrity of the genome. There is still a great deal to be learned about the intricate mechanisms that result in the many tumors seen in LFS.
References
- 1.Li F P, Fraumeni J F Jr. Soft-tissue sarcomas, breast cancer, and other neoplasms. A familial syndrome? Ann Intern Med. 1969;71(4):747–752. doi: 10.7326/0003-4819-71-4-747. [DOI] [PubMed] [Google Scholar]
- 2.Birch J M, Hartley A L, Marsden H B, Harris M, Swindell R. Excess risk of breast cancer in the mothers of children with soft tissue sarcomas. Br J Cancer. 1984;49(3):325–331. doi: 10.1038/bjc.1984.51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Li F P, Fraumeni J F Jr, Mulvihill J J. et al. A cancer family syndrome in twenty-four kindreds. Cancer Res. 1988;48(18):5358–5362. [PubMed] [Google Scholar]
- 4.Birch J M, Hartley A L, Blair V. et al. Identification of factors associated with high breast cancer risk in the mothers of children with soft tissue sarcoma. J Clin Oncol. 1990;8(4):583–590. doi: 10.1200/JCO.1990.8.4.583. [DOI] [PubMed] [Google Scholar]
- 5.Lustbader E D, Williams W R, Bondy M L, Strom S, Strong L C. Segregation analysis of cancer in families of childhood soft-tissue-sarcoma patients. Am J Hum Genet. 1992;51(2):344–356. [PMC free article] [PubMed] [Google Scholar]
- 6.Malkin D, Li F P, Strong L C. et al. Germ line p53 mutations in a familial syndrome of breast cancer, sarcomas, and other neoplasms. Science. 1990;250(4985):1233–1238. doi: 10.1126/science.1978757. [DOI] [PubMed] [Google Scholar]
- 7.Srivastava S, Zou Z Q, Pirollo K, Blattner W, Chang E H. Germ-line transmission of a mutated p53 gene in a cancer-prone family with Li-Fraumeni syndrome. Nature. 1990;348(6303):747–749. doi: 10.1038/348747a0. [DOI] [PubMed] [Google Scholar]
- 8.Varley J, Haber D A. Familial breast cancer and the hCHK2 1100delC mutation: assessing cancer risk. Breast Cancer Res. 2003;5(3):123–125. doi: 10.1186/bcr582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Pinto E M, Ribeiro R C, Figueiredo B C, Zambetti G P. TP53-associated pediatric malignancies. Genes Cancer. 2011;2(4):485–490. doi: 10.1177/1947601911409745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Pflaum J, Schlosser S, Müller M. p53 family and cellular stress responses in cancer. Front Oncol. 2014;4:285. doi: 10.3389/fonc.2014.00285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Levine A J, Oren M. The first 30 years of p53: growing ever more complex. Nat Rev Cancer. 2009;9(10):749–758. doi: 10.1038/nrc2723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lane D P. Cancer. p53, guardian of the genome. Nature. 1992;358(6381):15–16. doi: 10.1038/358015a0. [DOI] [PubMed] [Google Scholar]
- 13.Vogelstein B, Lane D, Levine A J. Surfing the p53 network. Nature. 2000;408(6810):307–310. doi: 10.1038/35042675. [DOI] [PubMed] [Google Scholar]
- 14.Blattner C, Hay T, Meek D W, Lane D P. Hypophosphorylation of Mdm2 augments p53 stability. Mol Cell Biol. 2002;22(17):6170–6182. doi: 10.1128/MCB.22.17.6170-6182.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bell D W, Varley J M, Szydlo T E. et al. Heterozygous germ line hCHK2 mutations in Li-Fraumeni syndrome. Science. 1999;286(5449):2528–2531. doi: 10.1126/science.286.5449.2528. [DOI] [PubMed] [Google Scholar]
- 16.Gonzalez K D, Noltner K A, Buzin C H. et al. Beyond Li Fraumeni syndrome: clinical characteristics of families with p53 germline mutations. J Clin Oncol. 2009;27(8):1250–1256. doi: 10.1200/JCO.2008.16.6959. [DOI] [PubMed] [Google Scholar]
- 17.Eeles R A. Germline mutations in the TP53 gene. Cancer Surv. 1995;25(25):101–124. [PubMed] [Google Scholar]
- 18.Kamihara J, Rana H Q, Garber J E. Germline TP53 mutations and the changing landscape of Li-Fraumeni syndrome. Hum Mutat. 2014;35(6):654–662. doi: 10.1002/humu.22559. [DOI] [PubMed] [Google Scholar]
- 19.Chompret A, Abel A, Stoppa-Lyonnet D. et al. Sensitivity and predictive value of criteria for p53 germline mutation screening. J Med Genet. 2001;38(1):43–47. doi: 10.1136/jmg.38.1.43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kleihues P, Schäuble B, zur Hausen A, Estève J, Ohgaki H. Tumors associated with p53 germline mutations: a synopsis of 91 families. Am J Pathol. 1997;150(1):1–13. [PMC free article] [PubMed] [Google Scholar]
- 21.Qualman S J, Coffin C M, Newton W A. et al. Intergroup Rhabdomyosarcoma Study: update for pathologists. Pediatr Dev Pathol. 1998;1(6):550–561. doi: 10.1007/s100249900076. [DOI] [PubMed] [Google Scholar]
- 22.Fletcher C DM. China: Churchill Livingstone Elsevier; 2007. Soft tissue tumors; pp. 1527–1592. [Google Scholar]
- 23.Gurney J G, Davis S, Severson R K, Fang J Y, Ross J A, Robison L L. Trends in cancer incidence among children in the U.S. Cancer. 1996;78(3):532–541. doi: 10.1002/(SICI)1097-0142(19960801)78:3<532::AID-CNCR22>3.0.CO;2-Z. [DOI] [PubMed] [Google Scholar]
- 24.Ognjanovic S, Linabery A M, Charbonneau B, Ross J A. Trends in childhood rhabdomyosarcoma incidence and survival in the United States, 1975-2005. Cancer. 2009;115(18):4218–4226. doi: 10.1002/cncr.24465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Parham D, Barr F G, Montgomery E A, Nascimiento A F. Lyon: International Agency for Research on Cancer; 2013. Skeletal-muscle tumours; pp. 123–135. [Google Scholar]
- 26.Parham D M, Barr F G. Classification of rhabdomyosarcoma and its molecular basis. Adv Anat Pathol. 2013;20(6):387–397. doi: 10.1097/PAP.0b013e3182a92d0d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hettmer S, Archer N M, Somers G R. et al. Anaplastic rhabdomyosarcoma in TP53 germline mutation carriers. Cancer. 2014;120(7):1068–1075. doi: 10.1002/cncr.28507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Parham D M, Qualman S J, Teot L. et al. Correlation between histology and PAX/FKHR fusion status in alveolar rhabdomyosarcoma: a report from the Children's Oncology Group. Am J Surg Pathol. 2007;31(6):895–901. doi: 10.1097/01.pas.0000213436.99492.51. [DOI] [PubMed] [Google Scholar]
- 29.Chen L, Shern J F, Wei J S. et al. Clonality and evolutionary history of rhabdomyosarcoma. PLoS Genet. 2015;11(3):e1005075. doi: 10.1371/journal.pgen.1005075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Seki M, Nishimura R, Yoshida K. et al. Integrated genetic and epigenetic analysis defines novel molecular subgroups in rhabdomyosarcoma. Nat Commun. 2015;6:7557. doi: 10.1038/ncomms8557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Unni K K, Inwards C Y. China: Churchill Livingstone Elsevier; 2007. Tumors of the osteoarticular system; pp. 1593–1652. [Google Scholar]
- 32.Mirabello L, Troisi R J, Savage S A. Osteosarcoma incidence and survival rates from 1973 to 2004: data from the Surveillance, Epidemiology, and End Results Program. Cancer. 2009;115(7):1531–1543. doi: 10.1002/cncr.24121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Rosenberg A E, Cleton-Jansen A-M, de Pinieux G, Deyrup A T, Hauben E, Squire J. Lyon: International Agency for Research on Cancer; 2013. Conventional osteosarcoma; pp. 282–288. [Google Scholar]
- 34.Selvarajah S, Yoshimoto M, Ludkovski O. et al. Genomic signatures of chromosomal instability and osteosarcoma progression detected by high resolution array CGH and interphase FISH. Cytogenet Genome Res. 2008;122(1):5–15. doi: 10.1159/000151310. [DOI] [PubMed] [Google Scholar]
- 35.Martin J W, Squire J A, Zielenska M. The genetics of osteosarcoma. Sarcoma. 2012;2012:627254. doi: 10.1155/2012/627254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Mirabello L, Yeager M, Mai P L. et al. Germline TP53 variants and susceptibility to osteosarcoma. J Natl Cancer Inst. 2015;107(7):djv101. doi: 10.1093/jnci/djv101. [DOI] [PMC free article] [PubMed] [Google Scholar]