

Abstract
Multiple endocrine neoplasia (MEN) syndrome is a familial cancer syndrome characterized by neuroendocrine tumors. The syndrome encompasses four major subtypes: MEN1, MEN2A, MEN2B, and MEN4. MEN1 is caused by mutations in the MEN1 gene, MEN2A and MEN2B are caused by mutations in RET, and MEN4 is caused by mutations in CDKNB1. All are inherited in an autosomal dominant pattern, but de novo cases do arise. While all subtypes are associated with neuroendocrine tumors, each has characteristic organ involvement. Identifying patients with the syndrome can aid in proper screening and treatment.
Keywords: multiple endocrine neoplasia, MEN1, MEN2, MEN4, neuroendocrine tumors, familial cancer syndrome
Introduction
Multiple endocrine neoplasia (MEN) syndrome is actually a series of syndromes: MEN1, MEN2A, MEN2B, and MEN4. All are typified by neoplastic proliferations of neuroendocrine cells, but the endocrine organs that are classic for each are different. Unlike many of the hereditary cancer syndromes in which one gene mutation predisposes individuals to different histologic variants of carcinoma throughout the body, MEN is unique in that the tumors that present in patients with the syndrome are all histologically quite similar, but three different genes have been associated with each of the major syndrome subtypes. MEN1 mutations typify MEN1, while RET mutations are seen in MEN2A and MEN2B, and finally, CDKN1B mutations are causal in MEN4.
Neuroendocrine tumors are composed of an organoid pattern, nests, or a solid sheet of tumor cells with classic salt-and-pepper nuclei and abundant granular cytoplasm. Typically, the cells are bland in appearance. Immunohistochemical stains for synaptophysin and chromogranin are frequently employed to identify these tumors, in addition to stains for the particular hormonal byproducts of a given tumor (i.e., calcitonin in the case of medullary thyroid carcinoma). While many neuroendocrine tumors are functional and secrete hormones, some are nonfunctioning.
This review will describe the major genetic findings associated with each of the MEN variants, along with the major neuroendocrine tumors and nonneuroendocrine associations that correspond to each of the syndromes. While MEN1, MEN2, and MEN4 are all inherited in an autosomal dominant fashion, sporadic cases can occur. Familial cases should not pose significant diagnostic dilemmas to the pediatric geneticist, especially if a known mutation exists; however, de novo mutations could potentially present for diagnosis. In these cases in which no familial association is known, it is particularly important to be aware of clues to a syndrome so that genetic screening can ensue.
Multiple Endocrine Neoplasia Type 1 (MEN1)
MEN1 (also known as Wermer syndrome) is inherited in an autosomal dominant fashion. Tumors in patients with the syndrome harbor a somatic mutation in addition to the germline mutation.1 The syndrome is most frequently associated with parathyroid adenomas, pancreatoenteric neuroendocrine tumors, anterior pituitary tumors, and adrenal cortical tumors; nonendocrine manifestations include lipomas, angiofibromas, collagenomas, and ependymomas.2 The diagnosis of MEN1 should be considered in a patient with a family history of a gene mutation, two of the three main neuroendocrine tumors associated with the syndrome (parathyroid adenoma, pancreatoenteric neuroendocrine tumor, and/or anterior pituitary tumor), a first-degree relative with MEN1 and presentation of one of the main tumor types, early onset of neuroendocrine tumors, or multiple neuroendocrine tumors within one organ.2 10 12 A cohort of patients present with familial isolated hyperparathyroidism, and studies have shown varying frequency of MEN mutations in these patients, ranging from 0 to 20%.3 4 Other syndromes causing the familial occurrence of hyperparathyroidism include MEN2, hyperparathyroidism jaw tumor syndrome, and familial benign hypocalciuric hypercalcemia.5 When patients present with isolated hyperparathyroidism and a family history of the same, appropriate imaging and clinical tests should be employed to rule out other symptomology, and if no other cause is identified, MEN1 testing should be the first genetic test performed.5
Identification of the MEN1 gene was achieved through positional cloning of chromosome 11q13 in 1997, and the protein was named menin by the group who identified the gene.1 The original 12 mutations discovered consisted of frameshift and nonsense mutations, in-frame deletions, and missense alterations; they were felt to most likely result in loss of function of the protein product which is consistent with a tumor suppressor mechanism.1 Later studies in pancreatic cells and human fibroblasts supported this theory.6 When the MEN1 gene was inserted into a pancreatic tumor cell line with decreased menin protein, cell growth stopped with normalization of menin levels.7 Conversely, in fibroblasts with menin downregulation, cells proliferated in an uncontrolled manner through telomerase reactivation.8 Additionally, menin has been shown to play a role in apoptosis. Mouse fibroblast studies have demonstrated that when menin is nonfunctional, apoptosis is impaired, suggesting that in tumors of MEN1 patients the same is likely true.9
In patients with MEN1, parathyroid adenoma is the most frequent tumor and is present in 90% of patients.10 Pancreatoenteric tumors occur in 30 to 70% of patients, anterior pituitary tumors in 30 to 40%, and adrenal cortical tumors in 40%.10 With aging, increased tumors are expressed in a given patient.10 Goudet et al11 queried the MEN1-GTE database of 924 patients, and 160 patients were found to present with MEN1 prior to age 21 years. In patients in this age group, primary hyperparathyroidism was the most common manifestation of MEN1, seen in 75% of patients, and the mean age of diagnosis of hyperparathyroidism was 16 years, with earliest presentation at age 4 years. Interestingly, an adrenal neuroendocrine tumor was the earliest presenting lesion in a 3-year-old. Of the patients in the group, four had malignant neuroendocrine tumors, one of which, a thymic tumor, caused death. Because of the potential for early presentation of tumors in MEN1, genetic testing of patients with first-degree relatives with MEN1 should be performed very early in life.10 12
Once a patient has been identified as having a MEN1 gene mutation, aggressive screening should ensue, along with intensive follow-up. In patients with known mutations, screening should include biomarkers appropriate for parathyroid adenomas, pancreatoenteric tumors, and pituitary tumors on a yearly basis.10 Additionally, because some pancreatoenteric, pituitary, thymic, and bronchial neuroendocrine tumors are nonfunctioning, imaging is recommended as a complimentary study on a 1- to 3-year basis depending on the tumor of interest.10 12
Hyperparathyroidism is frequently the first manifestation of MEN1. Due to the increased parathyroid hormone, hypercalcemia can be seen, as well as osteoporosis and renal stones. In patients with MEN1, multiple adenomas develop in the parathyroid glands, as opposed to single adenomas. Asymmetry of parathyroid glands is frequently seen, and most glands, given enough time, will likely develop adenomas.13 Although both subtotal and complete parathyroidectomy with partial reimplantation of a gland have been advocated, a recent prospective study showed no significant difference in outcomes in patients who underwent the procedures.14 While the four parathyroid glands normally reside along the posterior aspect of the thyroid, ectopic parathyroid glands are not uncommon. In one study of 231 patients, 16% were found to have ectopic parathyroid glands that were located both above and below the normal locations to include glands within the mediastinum, thyroid, submandibular gland, and thymus, and around the carotid artery and esophagus.15
Parathyroid adenomas with concurrent hyperparathyroidism are almost universally present in patients with MEN1, but they are rarely the cause of death.16 17 Histologically, parathyroid adenomas comprise a circumscribed proliferation of chief cells (Fig. 1). The chief cells are responsible for parathyroid hormone production and stain with antibodies to it. In many cases, the tumor has a capsule that separates the neoplasm from the adjacent normal parathyroid tissue. Follicle formation can be seen and can occasionally mimic thyroid.18
Fig. 1.
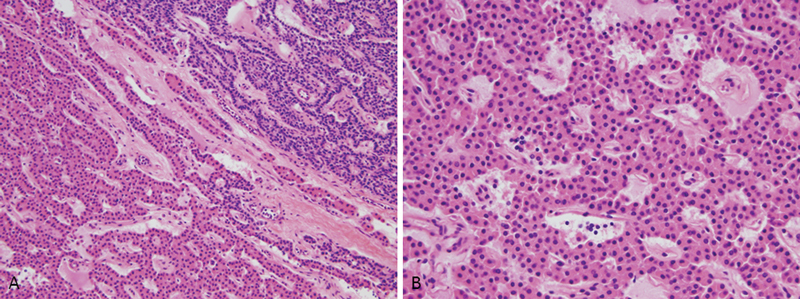
(A) Normal parathyroid gland, upper right, with adenoma in lower left. (B) High power of parathyroid adenoma with monotonous cell population.
The most frequent cause of MEN1-related death is complications secondary to pancreatoenteric neuroendocrine tumors.16 17 A large cohort study by Goudet et al found that ulceration secondary to increased gastrin was a significant cause of death prior to 1990, but since then, death related to malignant features of the neuroendocrine tumors with local growth or metastatic disease was more common.16 This finding was substantiated in a later study by Ito et al.17 This shift is secondary to the advent of proton-pump inhibitors, which prevent peptic ulcer disease and its subsequent complications.17 Gastrinomas in patients with MEN1 tend to arise in the duodenum, can be quite small, associate with gastrin cell hyperplasias, and are frequently multiple.19 20 21 These tumors produce gastrin and can lead to Zollinger–Ellison syndrome. Histologically, they comprise proliferations of monotonous neuroendocrine cells. Although these tumors are histologically bland, microtumors in the duodenum of MEN1 patients can present with lymph node metastasis.20 Genetic studies of these small duodenal tumors have shown loss of heterozygosity, supporting the concept that these are true neoplastic proliferations rather than hyperplasias.22 Thevenon et al studied the interacting domains of menin and discovered a JunD mutation in 23 of 38 patients with pancreatoenteric tumors who died.23 A later study found that mutations of the CHES1 domain in pancreatic neuroendocrine tumors were associated with functional tumors that were clinically aggressive.24 This suggests that certain mutations may warrant closer follow-up.23 24 Clinically nonfunctional tumors are particularly dangerous because of their lack of a biochemical hallmark that can allow early detection of the tumor. These tumors have been documented in pediatric patients.25
Neuroendocrine tumors of the pancreas frequently have an organoid growth pattern accompanied by the previously described cellular features (Fig. 2). Like other neuroendocrine-type tumors, chromogranin and synaptophysin immunohistochemical stains are positive, and antibodies to particular tumor byproducts can also stain the neoplasms. Unfortunately, histologically bland tumors can metastasize, making predictions of tumor behavior based solely on histologic appearance difficult.26 27
Fig. 2.

(A) Normal pancreas with acinar and islet cells. (B) Normal pancreas with synaptophysin stain highlighting islet cells. (C) Pancreatic neuroendocrine tumor. (D) Pancreatic neuroendocrine tumor with diffuse synaptophysin staining.
Pituitary adenomas in MEN1 patients have some associated features but are histologically indistinguishable from those that occur in nonfamilial cases.28 The normal anterior pituitary, where these tumors arise, has clusters of pituitary cells known as basophils, acidophils, and chromophobes. An adenoma forms from one of these cells types, causing it to dominate the other types. A special stain for reticulin can aid in identifying adenomas because it is decreased in the adenoma compared with the surrounding normal tissue (Fig. 3).29 Patients can present with symptoms related to hormone production by the lesion, or the tumors can manifest as mass-forming nonfunctional lesions. In cases where tumors become large, they can cause mass effect in the region of the sella, leading to vision disturbance, nausea and vomiting, and headache. In a study of 77 patients with MEN1-related pituitary adenomas in the Group d'Etat Registry, tumors producing more than one hormone product were more frequent in the MEN1 group than in the control group. Additionally, the MEN1 group had a higher frequency of multiple tumors and presented with tumors of increased size at an earlier age.28
Fig. 3.

(A) Pituitary adenoma with diffuse sheetlike growth. (B) Loss of reticulin staining in pituitary adenoma. (C) Pituitary adenoma with synaptophysin staining. (D) Pituitary adenoma with prolactin staining.
Multiple Endocrine Neoplasia Type 2 (MEN2)
Multiple endocrine neoplasia type 2 (MEN2) can be separated into two subtypes: MEN2A and MEN2B. The American Thyroid Association (ATA) has incorporated the older designation of familial medullary thyroid carcinoma into the MEN2A category in the most recent guidelines.30 Although the subtypes are unique based on the type of tumor(s) presenting in the patient, all are caused by mutations in the RET gene and are inherited in an autosomal dominant fashion. MEN2A is typified by parathyroid adenomas, medullary thyroid carcinomas, and pheochromocytomas; MEN2B is typified by medullary thyroid carcinomas, pheochromocytomas, mucosal ganglioneuromas, and a marfanoid habitus.2 30 Additionally, MEN2A can associate with cutaneous lichen amyloidosis or Hirschsprung disease, and these are considered variants within MEN2A.2 30 Through linkage studies, chromosome 10 was elucidated as the location of RET, the causal gene of MEN2A.31 32 It was identified in 1985 and subsequently was found to be upregulated in patients with tumors associated with MEN2.33 34 Further work to find the MEN2 and RET connection ensued. A genotype–phenotype correlation with age of tumor presentation and presenting tumors does exist and is related to the exon location of the RET mutation.2 30 35
Medullary thyroid carcinoma is the common link between clinical manifestations of the MEN2 subtypes; the tumor has a 90% penetrance.2 In the general population, medullary thyroid carcinoma is a rare disease. In a patient presenting with no family history of medullary thyroid carcinoma or MEN2, the frequency of RET germline mutations has been reported to be between 1.5 and 7.3% in studies done after 1990.36 37 Because of this, the ATA has recommended that patients who present with “presumed sporadic medullary thyroid carcinoma” undergo germline testing for RET mutations.30 In patients with known familial MEN2, the ATA 2015 Guidelines recommend risk stratification based on the patient's RET mutation. The risk stratification then correlates to screening plans and recommendations for age of thyroidectomy as well as age to initiate adrenal screening.30 Children with MEN2B with the most common M918T mutation should undergo prophylactic thyroidectomy before 1 year of age, although this is challenging because 75% are the result of de novo mutations.30 Brauckhoff et al38 noted that fewer than 20% of MEN2B patients presented with the typical features of the syndrome (marfanoid hypermobility and scoliosis, medullary thyroid carcinoma, and apparent oral neuromas) before 1 year of age, but 86% of patients did have a lack of tears. Although the study was small and retrospective, the unusual finding of decreased tears in an infant could potentially alert clinicians caring for these patients that a syndrome may be present. MEN2A screening and prophylactic therapy is also determined by mutation location, but thyroidectomy is recommended at a slightly later age, by age 5 years, for high-risk mutations, and screening with calcitonin and consideration of thyroidectomy begins at the age of 5 years in the moderate-risk groups.30 MEN2A is inherited in about 90% of cases.39 The low rate of de novo mutations makes genetic screening of infants in these families a more effective tool in identifying the vast majority of patients with the mutation.
Medullary thyroid carcinoma is a tumor that develops from the C-cells of the thyroid, which are located predominantly in the upper halves of the right and left lobes of the gland.40 These cells secrete calcitonin, which makes it a good biomarker in the screening of patients after thyroidectomy for residual disease or prior to thyroidectomy to determine when the procedure is necessary. The tumor is characterized by nests of malignant, proliferating C-cells in fibrous stroma with and without amyloid. The cells are positive for calcitonin by immunohistochemistry (Fig. 4).18 Like other forms of thyroid carcinoma, the first site of metastatic disease is the cervical lymph nodes.
Fig. 4.

(A) Normal thyroid on right with adjacent medullary thyroid carcinoma. (B) Calcitonin staining, negative in normal thyroid (except for normal C-cells) and positive in medullary thyroid carcinoma. (C) High-power view of medullary thyroid carcinoma.
In MEN2 the medullary carcinoma develops out of neoplastic C-cell hyperplasia, which is found in the background thyroid accompanying medullary thyroid carcinoma; however, C-cell hyperplasia is present in sporadic medullary thyroid cancers as well and cannot be used as a diagnostic indicator of MEN2.41 42 In two studies of prophylactic thyroidectomies of patients with germline RET mutations, 90 to 100% of patients had C-cell hyperplasia and 62 to 79% had medullary thyroid carcinoma, thus substantiating the practice in patients with MEN2.43 44 Perry et al distinguished between “physiologic” and neoplastic C-cell hyperplasia based on the cytologic atypia that is appreciated in the latter and found that neoplastic C-cell hyperplasia is typically bilateral, whereas reactive C-cell hyperplasia is not.45 Conversely, in nonneoplastic C-cell hyperplasia, the cells are bland and usually require immunohistochemical stains to distinguish them from follicular cells.45 Studies have shown that neoplastic C-cell hyperplasia can exist in both MEN2-associated and sporadic cases, but it is more frequent in MEN2 patients41 42 Once the basement membrane of the follicle surrounding the proliferation of C-cells has been penetrated, the lesion becomes a microcarcinoma, and when it exceeds 1 cm, it becomes a carcinoma.40 46
Pheochromocytomas, and paragangliomas to a lesser extent, are rare tumors that are associated with MEN2A and have a penetrance of 50% in these patients.47 Pheochromocytomas develop in the adrenal medulla and paragangliomas in the paraganglia. The tumors produce catecholamines, and patients frequently present with hypertension, headache, sweating, and palpitations.48 Histologically, the tumors are characterized by the “Zellballen” pattern, consisting of nests of cytologically bland cells that are immunoreactive for chromogranin and synaptophysin.49 50 Interspersed within the nests are sustentacular cells, which are spindled, have scant cytoplasm, and stain with S100 by immunohistochemistry.49 50 MEN2-related pheochromocytomas, when compared with sporadic cases, are much more commonly bilateral and can have associated medullary hyperplasia.48 Recent molecular studies support the idea that hyperplasias are actually neoplastic processes in MEN2 patients; furthermore, the previously used criteria of size less than 1 cm as a defining factor of hyperplasia versus pheochromocytoma should be dropped, as all of these proliferations in MEN2 patients are likely neoplastic.51
Hirschsprung disease and cutaneous lichen amyloidosis are two of the nontumoral associations of MEN2A. Hirschsprung disease is defined as distal intestinal aganglionosis and is diagnosed by suction rectal biopsy showing a lack of ganglion cells within the submucosa. Patients with Hirschsprung disease usually present within the first days of life with intestinal obstruction. RET codon 10 mutations have been found to be responsible for all reported cases of Hirschsprung disease and MEN2A coexpression, but it should be noted that expression of only MEN2A is more frequent than the coexpression in patients with these mutations, and alternately, familial Hirschsprung disease can exist without other manifestations of MEN2A.52 53 Testing for the RET mutation should be considered in patients with Hirschsprung and a family history of Hischsprung disease and/or known familial MEN2A.52 Cutaneous lichen amyloidosis is a dermatologic manifestation that presents with itching in the upper midback, usually between the scapulae. The area subsequently develops papules with hyperkeratosis and hyperpigmentation. Microscopy reveals dermal amyloid deposition.54 55 56 While cutaneous lichen amyloidosis can be difficult to diagnose, it is important to recognize because symptomatology can develop early in life and prior to the diagnosis of medullary thyroid carcinoma.55 56 Both of these nontumoral associations with MEN2 are therefore significant because they can be the presenting manifestation of the syndrome.
Ganglioneuromatoses are proliferations of nerves with admixed ganglion cells that occur in the mucosa of patients with MEN2B.57 While they do not create hormonal disturbances and are not malignant, they can cause significant functional impairment. Gastrointestinal symptomatologies, such as constipation, failure to thrive, and diarrhea, are frequently reported in these patients and in many cases precede the diagnosis of MEN2B.38 53 In infants who undergo suction rectal biopsy for Hirschsprung disease, the finding of hypertrophic submucosal nerves accompanied by increased ganglion cells could be an initial sign of MEN2B, and genetic screening should be considered.58
Multiple Endocrine Neoplasia Type 4 (MEN4)
In the last decade, multiple endocrine neoplasia type 4 (MEN4) has emerged as a genetically distinct form of MEN. The cases that are now placed in this category were previously considered part of the MEN1 syndrome. In 2002, Fritz et al described a unique cancer syndrome that arose de novo in a Sprague Dawley rat colony.59 The rats presented initially with early onset of cataracts and then went on to develop neuroendocrine tumors, including pituitary adenomas, parathyroid adenomas, pheochromocytomas, medullary thyroid hyperplasia, and paragangliomas. The trait was found to be autosomal recessive, and the mice were found to be negative for MEN1 and RET mutations. Between 70 and 90% of familial MEN1 cases are associated with mutations in the MEN1 gene, leaving 10 to 30% of cases negative for mutations.60 61 Pellegata et al combined knowledge of the above-mentioned MENX rat line with knowledge of the human subgroup of patients that had tumors suggestive of MEN1, but negative for MEN1 genetic testing, to discover that a germline mutation in CDKN1B caused a MEN-type syndrome in one patient, her sister, and possibly other family members.62 63 Mutations were found to result in a decrease in the expression of p27 tumor suppressor, causing syndromic manifestations.62 Later studies have shown that while CDKN1B mutations are responsible for some familial cases of MEN1 mutation-negative MEN1-like presentations, isolated familial pituitary adenomas that are MEN1 negative do not harbor CDKN1B mutations, and the mutation does not explain all MEN1-negative families with pituitary and parathyroid involvement.64 65
Since the original mutation was identified in 2006, several investigators have found mutations in the CDKN1B gene in familial cases of neuroendocrine tumors as well as in seemingly sporadic parathyroid adenomas.60 66 67 68 A large study of 196 index cases with MEN1 features but lacking MEN1 mutations was performed by Agarwal et al.60 The cyclin-dependent kinase inhibitor genes were amplified and sequenced, and mutations were discovered that were thought to be pathologic not just in p27, but also in p15, p18, and p21. The group found seven variants that were thought to cause disease in index cases. Three of the seven index patients had family members who were affected by similar tumors, and all index cases had primary hyperparathyroidism.
Patients with MEN4 present in much the same way as those with MEN1. Hyperparathyroidism has been well documented in the majority of cases with CDKN1B mutations, but the age of diagnosis varies from 30 to 74 years.68 Because of the rarity of these patients and the heterogeneity of tumors seen in them, it is difficult to define a phenotype beyond its similarity to MEN1.63 Of note, the youngest patient with a confirmed CDKN1B p27 mutation and a syndrome-related neoplasm was 30 years old.62 68 Other associated tumors in patients with cyclin-dependent kinase inhibitor mutations (including p27, p15, p18, and p21) and relatives with genetically confirmed mutations include neuroendocrine tumors (pituitary adenomas, small-cell neuroendocrine cervical carcinoma, adrenal masses, bronchial carcinoid, papillary thyroid carcinoma, gastric carcinoid, and pancreatoenteric masses) as well as nonneuroendocrine tumors (angiomyolipoma, schwannoma, meningioma, liver hemangioma, prostate cancer, breast cancer, lipoma, and uterine fibroids).60 62 63 64 66 68 69 70
Although the MEN4 genotype is now described as a separate syndrome from MEN1, the presentation of disease appears to be similar. Testing could be considered in all patients with a family history of a known mutation and in patients who initially undergo MEN1 testing based on tumor development but are negative. However, without more extensive information on the syndrome and its penetrance, it is difficult to make a firm recommendation for testing.63 As the literature grows on this topic and a defined phenotype emerges, screening parameters for the genetics and tumors of this subtype of MEN will likely evolve, much as they have for MEN1 and MEN2.
Conclusion
MEN syndrome is actually an umbrella term used to describe a family of tumor syndromes characterized by histologically similar tumors arising in patients and families with mutations in one of three genes: MEN1, RET, or CDKN1B. The histologic monotony of tumors characterizing these syndromes disguises their genetic heterogeneity. The neuroendocrine tumors are the most commonly associated lesions in patients with the MEN syndromes; however, it is important to be aware of the nontumor associations—cutaneous lichen amyloidosis and Hirschsprung disease with MEN2A and ganglioneuromas and lack of tears in MEN2B—because they may be the earliest presenting sign in some patients.38 52 53 55 56 Tumors in MEN patients have been typified by neuroendocrine cell hyperplasias in the background organ. As research continues, it seems likely to be discovered that many of these so-called hyperplasias are actually neoplastic processes, as has been shows in the adrenal medulla and duodenum.22 51
The importance of recognizing MEN is particularly great in patients who present with de novo mutations so that appropriate monitoring and therapy can ensue. Much like the identification of CDKN1B mutations led to a new MEN variant, in the future it is possible that familial cases of neuroendocrine tumors presenting without currently defined genetic mutations will have new gene mutations discovered, possibly leading to additional MEN subtypes. These patients represent a rich research frontier. As with all cancer predisposition syndromes, knowing the risk in an individual patient, the risk to their family members, and the potential risk to offspring is important for planning a treatment and screening strategy and for family planning purposes.
References
- 1.Chandrasekharappa S C, Guru S C, Manickam P. et al. Positional cloning of the gene for multiple endocrine neoplasia-type 1. Science. 1997;276(5311):404–407. doi: 10.1126/science.276.5311.404. [DOI] [PubMed] [Google Scholar]
- 2.Brandi M L, Gagel R F, Angeli A. et al. Guidelines for diagnosis and therapy of MEN type 1 and type 2. J Clin Endocrinol Metab. 2001;86(12):5658–5671. doi: 10.1210/jcem.86.12.8070. [DOI] [PubMed] [Google Scholar]
- 3.Simonds W F, James-Newton L A, Agarwal S K. et al. Familial isolated hyperparathyroidism: clinical and genetic characteristics of 36 kindreds. Medicine (Baltimore) 2002;81(1):1–26. doi: 10.1097/00005792-200201000-00001. [DOI] [PubMed] [Google Scholar]
- 4.Bergman L, Teh B, Cardinal J. et al. Identification of MEN1 gene mutations in families with MEN 1 and related disorders. Br J Cancer. 2000;83(8):1009–1014. doi: 10.1054/bjoc.2000.1380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hannan F M, Nesbit M A, Christie P T. et al. Familial isolated primary hyperparathyroidism caused by mutations of the MEN1 gene. Nat Clin Pract Endocrinol Metab. 2008;4(1):53–58. doi: 10.1038/ncpendmet0718. [DOI] [PubMed] [Google Scholar]
- 6.Lemos M C, Thakker R V. Multiple endocrine neoplasia type 1 (MEN1): analysis of 1336 mutations reported in the first decade following identification of the gene. Hum Mutat. 2008;29(1):22–32. doi: 10.1002/humu.20605. [DOI] [PubMed] [Google Scholar]
- 7.Stålberg P, Grimfjärd P, Santesson M. et al. Transfection of the multiple endocrine neoplasia type 1 gene to a human endocrine pancreatic tumor cell line inhibits cell growth and affects expression of JunD, delta-like protein 1/preadipocyte factor-1, proliferating cell nuclear antigen, and QM/Jif-1. J Clin Endocrinol Metab. 2004;89(5):2326–2337. doi: 10.1210/jc.2003-031228. [DOI] [PubMed] [Google Scholar]
- 8.Lin S Y, Elledge S J. Multiple tumor suppressor pathways negatively regulate telomerase. Cell. 2003;113(7):881–889. doi: 10.1016/s0092-8674(03)00430-6. [DOI] [PubMed] [Google Scholar]
- 9.Schnepp R W, Mao H, Sykes S M. et al. Menin induces apoptosis in murine embryonic fibroblasts. J Biol Chem. 2004;279(11):10685–10691. doi: 10.1074/jbc.M308073200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Thakker R V, Newey P J, Walls G V. et al. Clinical practice guidelines for multiple endocrine neoplasia type 1 (MEN1) J Clin Endocrinol Metab. 2012;97(9):2990–3011. doi: 10.1210/jc.2012-1230. [DOI] [PubMed] [Google Scholar]
- 11.Goudet P, Dalac A, Le Bras M. et al. MEN1 disease occurring before 21 years old: a 160-patient cohort study from the Groupe d'étude des Tumeurs Endocrines. J Clin Endocrinol Metab. 2015;100(4):1568–1577. doi: 10.1210/jc.2014-3659. [DOI] [PubMed] [Google Scholar]
- 12.Newey P J, Thakker R V. Role of multiple endocrine neoplasia type 1 mutational analysis in clinical practice. Endocr Pract. 2011;17 03:8–17. doi: 10.4158/EP10379.RA. [DOI] [PubMed] [Google Scholar]
- 13.Doherty G M, Lairmore T C, DeBenedetti M K. Multiple endocrine neoplasia type 1 parathyroid adenoma development over time. World J Surg. 2004;28(11):1139–1142. doi: 10.1007/s00268-004-7560-8. [DOI] [PubMed] [Google Scholar]
- 14.Lairmore T C Govednik C M Quinn C E Sigmond B R Lee C Y Jupiter D C A randomized, prospective trial of operative treatments for hyperparathyroidism in patients with multiple endocrine neoplasia type 1 Surgery 201415661326–1334., discussion 1334–1335 [DOI] [PubMed] [Google Scholar]
- 15.Phitayakorn R, McHenry C R. Incidence and location of ectopic abnormal parathyroid glands. Am J Surg. 2006;191(3):418–423. doi: 10.1016/j.amjsurg.2005.10.049. [DOI] [PubMed] [Google Scholar]
- 16.Goudet P, Murat A, Binquet C. et al. Risk factors and causes of death in MEN1 disease. A GTE (Groupe d'Etude des Tumeurs Endocrines) cohort study among 758 patients. World J Surg. 2010;34(2):249–255. doi: 10.1007/s00268-009-0290-1. [DOI] [PubMed] [Google Scholar]
- 17.Ito T, Igarashi H, Uehara H, Berna M J, Jensen R T. Causes of death and prognostic factors in multiple endocrine neoplasia type 1: a prospective study: comparison of 106 MEN1/Zollinger-Ellison syndrome patients with 1613 literature MEN1 patients with or without pancreatic endocrine tumors. Medicine (Baltimore) 2013;92(3):135–181. doi: 10.1097/MD.0b013e3182954af1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Baloch Z, Livolsi V. Philadelphia, PA: Lippincott Williams & Wilkins; 2010. Pathology of thyroid and parathyroid disease; pp. 493–544. [Google Scholar]
- 19.Pritchard D M. Pathogenesis of gastrinomas associated with multiple endocrine neoplasia type 1. Gut. 2007;56(5):606–607. doi: 10.1136/gut.2006.113985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Anlauf M, Enosawa T, Henopp T. et al. Primary lymph node gastrinoma or occult duodenal microgastrinoma with lymph node metastases in a MEN1 patient: the need for a systematic search for the primary tumor. Am J Surg Pathol. 2008;32(7):1101–1105. doi: 10.1097/PAS.0b013e3181655811. [DOI] [PubMed] [Google Scholar]
- 21.Pipeleers-Marichal M, Somers G, Willems G. et al. Gastrinomas in the duodenums of patients with multiple endocrine neoplasia type 1 and the Zollinger-Ellison syndrome. N Engl J Med. 1990;322(11):723–727. doi: 10.1056/NEJM199003153221103. [DOI] [PubMed] [Google Scholar]
- 22.Anlauf M, Perren A, Henopp T. et al. Allelic deletion of the MEN1 gene in duodenal gastrin and somatostatin cell neoplasms and their precursor lesions. Gut. 2007;56(5):637–644. doi: 10.1136/gut.2006.108910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Thevenon J, Bourredjem A, Faivre L. et al. Higher risk of death among MEN1 patients with mutations in the JunD interacting domain: a Groupe d'etude des Tumeurs Endocrines (GTE) cohort study. Hum Mol Genet. 2013;22(10):1940–1948. doi: 10.1093/hmg/ddt039. [DOI] [PubMed] [Google Scholar]
- 24.Bartsch D K, Slater E P, Albers M. et al. Higher risk of aggressive pancreatic neuroendocrine tumors in MEN1 patients with MEN1 mutations affecting the CHES1 interacting MENIN domain. J Clin Endocrinol Metab. 2014;99(11):E2387–E2391. doi: 10.1210/jc.2013-4432. [DOI] [PubMed] [Google Scholar]
- 25.Newey P J, Jeyabalan J, Walls G V. et al. Asymptomatic children with multiple endocrine neoplasia type 1 mutations may harbor nonfunctioning pancreatic neuroendocrine tumors. J Clin Endocrinol Metab. 2009;94(10):3640–3646. doi: 10.1210/jc.2009-0564. [DOI] [PubMed] [Google Scholar]
- 26.Reid M D, Balci S, Saka B, Adsay N V. Neuroendocrine tumors of the pancreas: current concepts and controversies. Endocr Pathol. 2014;25(1):65–79. doi: 10.1007/s12022-013-9295-2. [DOI] [PubMed] [Google Scholar]
- 27.Klimstra D S, Arnold R, Capella C, Lyon, France: IARC Press; 2010. Neuroendocrine neoplasms of the pancreas; pp. 322–326. [Google Scholar]
- 28.Trouillas J, Labat-Moleur F, Sturm N. et al. Pituitary tumors and hyperplasia in multiple endocrine neoplasia type 1 syndrome (MEN1): a case-control study in a series of 77 patients versus 2509 non-MEN1 patients. Am J Surg Pathol. 2008;32(4):534–543. doi: 10.1097/PAS.0b013e31815ade45. [DOI] [PubMed] [Google Scholar]
- 29.Scheithauer B. Philadelphia, PA: Lippincott Williams & Wilkins; 2010. The pituitary and sellar region; pp. 460–492. [Google Scholar]
- 30.Wells S A Jr, Asa S L, Dralle H. et al. Revised American Thyroid Association guidelines for the management of medullary thyroid carcinoma. Thyroid. 2015;25(6):567–610. doi: 10.1089/thy.2014.0335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mathew C G, Chin K S, Easton D F. et al. A linked genetic marker for multiple endocrine neoplasia type 2A on chromosome 10. Nature. 1987;328(6130):527–528. doi: 10.1038/328527a0. [DOI] [PubMed] [Google Scholar]
- 32.Simpson N E, Kidd K K, Goodfellow P J. et al. Assignment of multiple endocrine neoplasia type 2A to chromosome 10 by linkage. Nature. 1987;328(6130):528–530. doi: 10.1038/328528a0. [DOI] [PubMed] [Google Scholar]
- 33.Takahashi M, Ritz J, Cooper G M. Activation of a novel human transforming gene, ret, by DNA rearrangement. Cell. 1985;42(2):581–588. doi: 10.1016/0092-8674(85)90115-1. [DOI] [PubMed] [Google Scholar]
- 34.Santoro M, Rosati R, Grieco M. et al. The ret proto-oncogene is consistently expressed in human pheochromocytomas and thyroid medullary carcinomas. Oncogene. 1990;5(10):1595–1598. [PubMed] [Google Scholar]
- 35.Krampitz G W, Norton J A. RET gene mutations (genotype and phenotype) of multiple endocrine neoplasia type 2 and familial medullary thyroid carcinoma. Cancer. 2014;120(13):1920–1931. doi: 10.1002/cncr.28661. [DOI] [PubMed] [Google Scholar]
- 36.Eng C, Mulligan L M, Smith D P. et al. Low frequency of germline mutations in the RET proto-oncogene in patients with apparently sporadic medullary thyroid carcinoma. Clin Endocrinol (Oxf) 1995;43(1):123–127. doi: 10.1111/j.1365-2265.1995.tb01903.x. [DOI] [PubMed] [Google Scholar]
- 37.Elisei R, Romei C, Cosci B. et al. RET genetic screening in patients with medullary thyroid cancer and their relatives: experience with 807 individuals at one center. J Clin Endocrinol Metab. 2007;92(12):4725–4729. doi: 10.1210/jc.2007-1005. [DOI] [PubMed] [Google Scholar]
- 38.Brauckhoff M Machens A Hess S et al. Premonitory symptoms preceding metastatic medullary thyroid cancer in MEN 2B: an exploratory analysis Surgery 200814461044–1050., discussion 1050–1053 [DOI] [PubMed] [Google Scholar]
- 39.Giri D, McKay V, Weber A, Blair J C. Multiple endocrine neoplasia syndromes 1 and 2: manifestations and management in childhood and adolescence. Arch Dis Child. 2015;100(10):994–999. doi: 10.1136/archdischild-2014-307028. [DOI] [PubMed] [Google Scholar]
- 40.Albores-Saavedra J A, Krueger J E. C-cell hyperplasia and medullary thyroid microcarcinoma. Endocr Pathol. 2001;12(4):365–377. doi: 10.1385/ep:12:4:365. [DOI] [PubMed] [Google Scholar]
- 41.Guyétant S, Josselin N, Savagner F, Rohmer V, Michalak S, Saint-André J P. C-cell hyperplasia and medullary thyroid carcinoma: clinicopathological and genetic correlations in 66 consecutive patients. Mod Pathol. 2003;16(8):756–763. doi: 10.1097/01.MP.0000081727.75778.0C. [DOI] [PubMed] [Google Scholar]
- 42.Kaserer K, Scheuba C, Neuhold N. et al. Sporadic versus familial medullary thyroid microcarcinoma: a histopathologic study of 50 consecutive patients. Am J Surg Pathol. 2001;25(10):1245–1251. doi: 10.1097/00000478-200110000-00004. [DOI] [PubMed] [Google Scholar]
- 43.Etit D, Faquin W C, Gaz R, Randolph G, DeLellis R A, Pilch B Z. Histopathologic and clinical features of medullary microcarcinoma and C-cell hyperplasia in prophylactic thyroidectomies for medullary carcinoma: a study of 42 cases. Arch Pathol Lab Med. 2008;132(11):1767–1773. doi: 10.5858/132.11.1767. [DOI] [PubMed] [Google Scholar]
- 44.Heizmann O, Haecker F M, Zumsteg U, Müller B, Oberholzer M, Oertli D. Presymptomatic thyroidectomy in multiple endocrine neoplasia 2a. Eur J Surg Oncol. 2006;32(1):98–102. doi: 10.1016/j.ejso.2005.10.007. [DOI] [PubMed] [Google Scholar]
- 45.Perry A, Molberg K, Albores-Saavedra J. Physiologic versus neoplastic C-cell hyperplasia of the thyroid: separation of distinct histologic and biologic entities. Cancer. 1996;77(4):750–756. doi: 10.1002/(sici)1097-0142(19960215)77:4<750::aid-cncr22>3.0.co;2-z. [DOI] [PubMed] [Google Scholar]
- 46.Matias-Guiu X, DeLellis R A, Moley J F, Lyons: IARC; 2004. Medullary thyroid carcinoma; pp. 86–91. [Google Scholar]
- 47.Lefebvre M, Foulkes W D. Pheochromocytoma and paraganglioma syndromes: genetics and management update. Curr Oncol. 2014;21(1):e8–e17. doi: 10.3747/co.21.1579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Pomares F J, Cañas R, Rodriguez J M, Hernandez A M, Parrilla P, Tebar F J. Differences between sporadic and multiple endocrine neoplasia type 2A phaeochromocytoma. Clin Endocrinol (Oxf) 1998;48(2):195–200. doi: 10.1046/j.1365-2265.1998.3751208.x. [DOI] [PubMed] [Google Scholar]
- 49.DeLellis R, Mangray S. Philadelphia, PA: Lippincott Williams & Wilkins; 2010. The adrenal glands; pp. 545–586. [Google Scholar]
- 50.Lloyd R V. Adrenal cortical tumors, pheochromocytomas and paragangliomas. Mod Pathol. 2011;24 02:S58–S65. doi: 10.1038/modpathol.2010.126. [DOI] [PubMed] [Google Scholar]
- 51.Korpershoek E, Petri B J, Post E. et al. Adrenal medullary hyperplasia is a precursor lesion for pheochromocytoma in MEN2 syndrome. Neoplasia. 2014;16(10):868–873. doi: 10.1016/j.neo.2014.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Coyle D, Friedmacher F, Puri P. The association between Hirschsprung's disease and multiple endocrine neoplasia type 2a: a systematic review. Pediatr Surg Int. 2014;30(8):751–756. doi: 10.1007/s00383-014-3538-2. [DOI] [PubMed] [Google Scholar]
- 53.Cohen M S Phay J E Albinson C et al. Gastrointestinal manifestations of multiple endocrine neoplasia type 2 Ann Surg 20022355648–654., discussion 654–655 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Tanaka A, Arita K, Lai-Cheong J E, Palisson F, Hide M, McGrath J A. New insight into mechanisms of pruritus from molecular studies on familial primary localized cutaneous amyloidosis. Br J Dermatol. 2009;161(6):1217–1224. doi: 10.1111/j.1365-2133.2009.09311.x. [DOI] [PubMed] [Google Scholar]
- 55.Rothberg A E, Raymond V M, Gruber S B, Sisson J. Familial medullary thyroid carcinoma associated with cutaneous lichen amyloidosis. Thyroid. 2009;19(6):651–655. doi: 10.1089/thy.2009.0021. [DOI] [PubMed] [Google Scholar]
- 56.Verga U, Fugazzola L, Cambiaghi S. et al. Frequent association between MEN 2A and cutaneous lichen amyloidosis. Clin Endocrinol (Oxf) 2003;59(2):156–161. doi: 10.1046/j.1365-2265.2003.01782.x. [DOI] [PubMed] [Google Scholar]
- 57.Fenoglio-Preiser C, Nofsinger A, Stemmerman G, Lantz P, Isaacson P. Philadelphia, PA: Lippincott Williams & Wilkins; 2008. Gastrointestinal Pathology: An Atlas and Text. 3rd ed. [Google Scholar]
- 58.Yin M, King S K, Hutson J M, Chow C W. Multiple endocrine neoplasia type 2B diagnosed on suction rectal biopsy in infancy: a report of 2 cases. Pediatr Dev Pathol. 2006;9(1):56–60. doi: 10.2350/06-05-0072.1. [DOI] [PubMed] [Google Scholar]
- 59.Fritz A, Walch A, Piotrowska K. et al. Recessive transmission of a multiple endocrine neoplasia syndrome in the rat. Cancer Res. 2002;62(11):3048–3051. [PubMed] [Google Scholar]
- 60.Agarwal S K, Mateo C M, Marx S J. Rare germline mutations in cyclin-dependent kinase inhibitor genes in multiple endocrine neoplasia type 1 and related states. J Clin Endocrinol Metab. 2009;94(5):1826–1834. doi: 10.1210/jc.2008-2083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Tsukada T, Nagamura Y, Ohkura N. MEN1 gene and its mutations: basic and clinical implications. Cancer Sci. 2009;100(2):209–215. doi: 10.1111/j.1349-7006.2008.01034.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Pellegata N S, Quintanilla-Martinez L, Siggelkow H. et al. Germ-line mutations in p27Kip1 cause a multiple endocrine neoplasia syndrome in rats and humans. Proc Natl Acad Sci U S A. 2006;103(42):15558–15563. doi: 10.1073/pnas.0603877103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Georgitsi M. MEN-4 and other multiple endocrine neoplasias due to cyclin-dependent kinase inhibitors (p27(Kip1) and p18(INK4C)) mutations. Best Pract Res Clin Endocrinol Metab. 2010;24(3):425–437. doi: 10.1016/j.beem.2010.01.001. [DOI] [PubMed] [Google Scholar]
- 64.Georgitsi M, Raitila A, Karhu A. et al. Germline CDKN1B/p27Kip1 mutation in multiple endocrine neoplasia. J Clin Endocrinol Metab. 2007;92(8):3321–3325. doi: 10.1210/jc.2006-2843. [DOI] [PubMed] [Google Scholar]
- 65.Ozawa A, Agarwal S K, Mateo C M. et al. The parathyroid/pituitary variant of multiple endocrine neoplasia type 1 usually has causes other than p27Kip1 mutations. J Clin Endocrinol Metab. 2007;92(5):1948–1951. doi: 10.1210/jc.2006-2563. [DOI] [PubMed] [Google Scholar]
- 66.Molatore S, Marinoni I, Lee M. et al. A novel germline CDKN1B mutation causing multiple endocrine tumors: clinical, genetic and functional characterization. Hum Mutat. 2010;31(11):E1825–E1835. doi: 10.1002/humu.21354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Costa-Guda J, Marinoni I, Molatore S, Pellegata N S, Arnold A. Somatic mutation and germline sequence abnormalities in CDKN1B, encoding p27Kip1, in sporadic parathyroid adenomas. J Clin Endocrinol Metab. 2011;96(4):E701–E706. doi: 10.1210/jc.2010-1338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Tonelli F, Giudici F, Giusti F. et al. A heterozygous frameshift mutation in exon 1 of CDKN1B gene in a patient affected by MEN4 syndrome. Eur J Endocrinol. 2014;171(2):K7–K17. doi: 10.1530/EJE-14-0080. [DOI] [PubMed] [Google Scholar]
- 69.Malanga D, De Gisi S, Riccardi M. et al. Functional characterization of a rare germline mutation in the gene encoding the cyclin-dependent kinase inhibitor p27Kip1 (CDKN1B) in a Spanish patient with multiple endocrine neoplasia-like phenotype. Eur J Endocrinol. 2012;166(3):551–560. doi: 10.1530/EJE-11-0929. [DOI] [PubMed] [Google Scholar]
- 70.Occhi G, Regazzo D, Trivellin G. et al. A novel mutation in the upstream open reading frame of the CDKN1B gene causes a MEN4 phenotype. PLoS Genet. 2013;9(3):e1003350. doi: 10.1371/journal.pgen.1003350. [DOI] [PMC free article] [PubMed] [Google Scholar]