

Abstract
Familial adenomatous polyposis (FAP), caused by a germline mutation in the adenomatous polyposis coli (APC) gene on chromosome 5q21, is an autosomal dominant disorder characterized by hundreds to thousands of adenomas throughout the gastrointestinal tract. A variety of extraintestinal manifestations, including thyroid, soft tissue, and brain tumors, may also be present. These patients inevitably develop colorectal carcinoma by the fourth decade of life. In this review, the pathology, epidemiology, and genetic features of FAP are discussed.
Keywords: familial adenomatous polyposis, Turcot syndrome, Gardner syndrome
Introduction
Familial adenomatous polyposis (FAP) is an autosomal dominant disorder, caused by an inherited mutation in the adenomatous polyposis coli (APC) gene on 5q21, with a frequency that ranges from 1 in 6,850 to 29,000 people. Approximately 10 to 30% of patients will develop FAP spontaneously without a family history.1 There is often genotypic variation, as mutations can occur at several different sites in the gene, leading to divergent phenotypes. Environmental and dietary factors are also thought to contribute to the variation in clinical expression.2
FAP patients usually have greater than 100 gastrointestinal polyps, often in the range of thousands (Fig. 1). Although adenomas are usually absent until puberty, the number and size of adenomas increase drastically until the gastrointestinal tract becomes completely filled by these dysplastic polyps. By the age of 30 years, almost three-quarters of afflicted patients develop colorectal carcinoma, and the average age of death is 42 years.3 The first and second most common causes of FAP-related mortality are metastatic colorectal carcinoma and desmoid tumors, respectively.4 Common symptoms include abdominal pain, diarrhea, hematochezia, and melena. Patients may also present with severe dehydration due to electrolyte imbalances and depletion from diarrhea and mucous discharge. Adenomas at the ampulla of Vater can obstruct bile flow and pancreaticobiliary enzymes, leading to acute pancreatitis.5
Fig. 1.
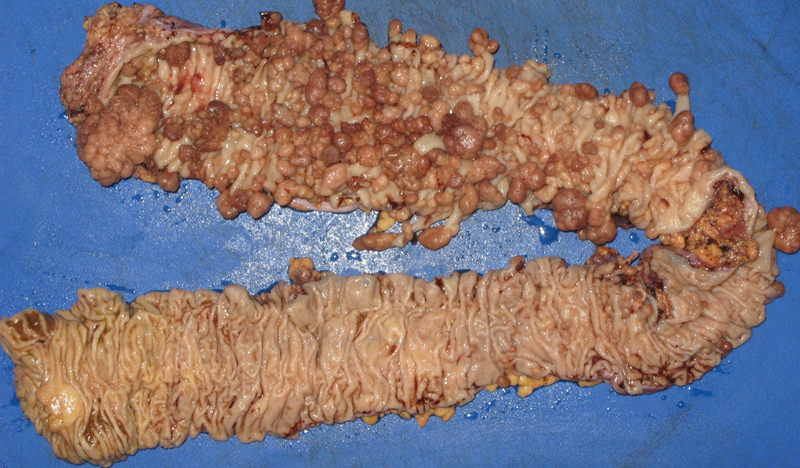
Colectomy specimen from a patient with FAP, showing innumerable tubular adenomas. (Photograph courtesy of Robert Weir, PA.)
The diagnosis of FAP is not particularly challenging, since symptomatic patients will undergo colonoscopic evaluation and the endoscopic impression is fairly unique. After molecular confirmation, family members should be placed under the appropriate screening protocol. Children of FAP patients should be screened by genetic testing and begin flexible sigmoidoscopy by age 10 years, with annual subsequent exams.6 One of the most cost-effective methods for FAP screening is a fundoscopic examination looking for congenital hypertrophy of the retinal pigment epithelium (CHRPE). This is seen in up to 90% of FAP patients, even including infants.7 In addition to gastrointestinal polyps and malignancy, other associated pathologic findings include hepatoblastomas, desmoid tumors, and brain cancer, which can be fatal. Therefore, even a prophylactic colectomy is not completely effective in reducing these patients' mortality risk.8
Pathologic Features
FAP is characterized by hundreds of adenomas throughout the luminal gastrointestinal tract that develop during early adolescence. Starting from the caudal end of the tract and working downward, fundic gland polyps (FGPs) are benign lesions commonly seen within the stomach; however, up to 25% of FGPs in FAP patients harbor foveolar dysplasia.9 Sporadic and FAP-associated FGPs also demonstrate differences at the molecular level. Sporadic FGPs often show somatic mutations in the β-catenin gene at exon 3. This mutation is not seen within FGPs of FAP patients. Instead, the common genetic mutation in this latter group is a second somatic APC hit on top of a germline APC mutation. While there are similarities in the mutational pathways between sporadic and FAP-associated FGPs, there are clear differences in the initial mutation.10
Adenomas can be seen in the small intestine, with approximately two-thirds located in the periampullary region.4 11 Progression of adenomas in the small intestine to adenocarcinoma is the third leading cause of mortality in FAP patients.
The pathognomonic feature of FAP is the presence of hundreds of adenomas within the colon and rectum. The rectum is almost always involved, and the length of the large intestine is carpeted by adenomas. There is variation in size and shape, ranging from less than 1 mm to greater than 1 cm in size. The number of polyps can be less than 100 to more than 5,000. The adenomas are histologically similar to spontaneous tubular adenomas, with elongated, hyperchromatic nuclei lining the crypts. There is no surface maturation, and some adenomas may focally demonstrate fingerlike villous extensions. Rather than the typical polypoid configuration, some adenomas are flat and can grow in a horizontal fashion. A proctocolectomy is necessary, since the adenomas invariably progress to colorectal carcinoma by the age of 35 to 40 years. These cancers are more likely to be seen on the left side of the colon.12 13 14
Nongastrointestinal Diseases
There is extensive phenotypic diversity in FAP patients. Lesions may involve several organs outside the gastrointestinal tract. Along with the initial mutation, there are likely additional factors that contribute to these variations. Bile from FAP patients has been theorized to cause more mutations than bile from non-FAP patients, potentially affecting the APC gene or other genes in the progression of dysplasia to carcinoma. Others have postulated that the hormonal or environmental milieu may influence FAP manifestations due to a strong predilection of thyroid cancer in female FAP patients and desmoid tumors (abdominal fibromatosis) discovered after trauma.15 16 17
A common extraintestinal lesion in FAP patients is CHRPE. This benign finding occurs in approximately 70 to 80% of patients and is often present at birth. It is asymptomatic and does not demonstrate progression into a malignancy.18 19 20 CHRPE can be visualized on fundoscopic examination of the eye. Bilateral, multiple CHRPEs are a specific feature for FAP, with 95 to 100% specificity.21 It is an excellent diagnostic modality for testing family members before moving to genetic testing. The histologic features of CHRPE demonstrate hypertrophy of the retinal pigmented epithelium (RPE) with cells that are up to twice as large as normal RPE cells. These RPE cells are filled with melanin granules and lose their nuclear basal polarity. Electron microscopy shows that the pigmented cells of a CHRPE contain melanosomes rather than lipofuscin.22
Thyroid cancer is also associated with FAP. The most common thyroid cancer among FAP patients is papillary thyroid carcinoma. They are often multifocal and confined to one thyroid lobe. From a prognosis standpoint, FAP-associated thyroid cancers behave nonaggressively, with a low incidence of metastatic disease and mortality rates within a decade; sometimes, they appear as a rare variant, known as cribriform-morular papillary thyroid carcinoma, that is almost always seen only in patients with FAP.23 24 25 26 27 Papillary thyroid carcinomas demonstrate a characteristic papillary architecture with pathognominic cytologic features such as nuclear clearing, pseudostratification, nuclear grooves, and pseudoinclusions.28
Desmoid tumors, or fibromatosis, are a rare tumor caused by mutations in the β-catenin gene.29 They are typically present in the abdomen and can involve the mesentery or abdominal wall; less commonly, they are seen within the limbs.30 Approximately 15% of FAP patients have desmoid tumors.31 These tumors are locally invasive and are the second leading cause of death among FAP patients.4 This is due to the numerous complications that can occur from the locally aggressive nature of these tumors or the surgical interventions necessary to remove these lesions, which do not always succeed in removing the entire tumor. Complications can include bowel obstruction, perforation, bleeding, and fistula formation.30 Desmoid tumors are poorly circumscribed tumors with either a well-circumscribed or infiltrative growth pattern at its interface with the normal surrounding tissue. There is also considerable variability in cellularity between a bland, hypocellular fibroblastic proliferation composed of spindle cells with bland nuclei, and a more cellular proliferation. Rare mitoses and perivascular chronic inflammation may be present, and the stroma has an eosinophilic, dense, collagenous appearance. Blood vessels within the tumor may be dilated.32
Hepatoblastoma, a rare liver tumor affecting children, can be fatal if left untreated. FAP patients have a 1:235 incidence of hepatoblastoma, compared with the general population incidence of 1:100,000 to 1:1,000,000.33 There are two main variants of hepatoblastoma: epithelial and mesenchymal. The epithelial type shows tumor cells arranged in a variety of patterns, forming trabeculae, tubules, acini, or papillae. This type shows recapitulation of the liver architecture. Depending on the predominant pattern, epithelial hepatoblastomas can be further subclassified into different patterns: fetal, embryonal, anaplastic, and macrotrabecular. The mixed epithelial and mesenchymal type is characterized by an epithelial component and foci of mesenchymal differentiation, forming cartilage, bone, or skeletal muscle. Hepatoblastomas are characterized by constitutive activation of the Wnt/β-catenin signaling pathway, leading to increased cellular proliferation. Other characteristic chromosomal abnormalities include overexpression, deletions, translocations, and trisomies.34
Variants of FAP
Turcot Syndrome
In 1959, Turcot described two cases of malignant central nervous system (CNS) tumors arising in patients with diffuse colonic polyposis.35 Since then Turcot syndrome has become synonymous with gastrointestinal polyposis (FAP or Lynch syndrome) patients who develop tumors of the CNS, most commonly medulloblastoma. Medulloblastomas arise from embryonal cells and are aggressive brain tumors found primarily in children. Of all primary CNS tumors in childhood, they comprise about 20%. Patients usually present with seemingly benign symptoms of headache, malaise, and vomiting due to increased intracranial pressure. It was determined that patients with an APC gene mutation between codons 697 and 1224 are at a 13-fold increased risk of developing medulloblastoma. The incidence of medulloblastomas in FAP patients is greatest before the age of 20 years.36
Histological classification of medulloblastomas includes classic medulloblastoma as well as four other variants: medulloblastoma with extensive nodularity, desmoplastic nodular, anaplastic, and large cell. Classic medulloblastomas are the most common form and are characterized by sheets of densely packed, small, mitotically active, basophilic cells with high nuclear-to-cytoplasmic ratio. Medulloblastoma with extensive nodularity has an expanded lobular architecture because of reticulin-free zones and a streaming pattern similar in pattern to a central neurocytoma. This is in contrast to the desmoplastic nodular variant, which has far fewer reticulin-rich components.37 Desmoplastic nodular and medulloblastoma with extensive nodularity are thought to have more favorable outcomes compared with classic medulloblastoma.37 38 Anaplastic medulloblastoma has marked nuclear pleomorphism, nuclear molding, cell wrapping, high mitotic activity, and prominent atypia. Because of sizable overlap between large cell medulloblastomas and anaplastic medulloblastomas, it is not uncommon for these two variants to be in a combined category. Large cell variant is classified by spherical cells with round nuclei, open chromatin, and prominent central nucleoli.37
While histology has guided medulloblastoma research in the past, within the last decade, four recognized molecular subgroups of medulloblastoma have emerged: Wnt, SHH, Group 3, and Group 4. Of these, SHH is more common in infants (<3 years old) and adults (>16 years old) compared with Group 3 and 4, which are most common in children (>3 years old).39 40 The Wnt subgroup has a loss in chromosome 6 and has no amplification of MYC or MYCN, which have been found to be markers of poorer prognosis.39 This tumor type often displays classic histology, a higher female incidence, and excellent long-term prognosis.39 41 42 The SHH subgroup is found to have high expression of MYCN41; however, it appears that several genes are involved in the development of this tumor. Histologically, this subgroup is an amalgam of all variants; however, the desmoplastic nodular variant has been classified as SHH.42 Tumors in Group 3 have high expression of MYC,39 41 are more likely to metastasize and reoccur,41 and generally have a poor prognosis. The genetic underpinning of Group 4 is thought to be isochromosome 17q, which is found in 80% of cases.42 Group 3 and Group 4 are less understood in comparison to the other subgroups. Subgroup affiliation and metastatic status have been found to be the most powerful predictive prognostic biomarkers, an important future tool in predicting patient outcomes and survival.43 These will likely be the masthead under which future treatments are defined.
Therapeutic approach to medulloblastomas is a combination of chemotherapy, surgical resection, and radiotherapy. Five-year overall survival with standard treatment is between 70 and 85%. Of the subgroups, the classic histology has a 5-year event-free survival of 84%, desmoplastic tumors 77%, and large-cell anaplastic tumors 57%.44 MYC and MYCN are poor prognostic factors and, as such, correlate to the poorer prognosis of Group 3 (MYC) and SHH (MYCN).39 As research into the molecular pathways of these tumors continues to evolve, targeted treatment modalities may keep pace, especially in cases of highly aggressive forms of medulloblastoma.
Gardner Syndrome
Gardner syndrome is characterized by the gastrointestinal findings of FAP (small bowel and colorectal adenomas) along with fibromatosis and thyroid tumors. Osteomas, lipomas, and dental abnormalities may also be characteristic. These extraintestinal manifestations are pathognomonic for a diagnosis of Gardner syndrome. Osteomas occur within the mandible, skull, and long bones. Dental abnormalities include impacted teeth and additional numbers of teeth. Epidermoid cysts are a common dermatologic manifestation, involving the scalp, face, and limbs.45 46 47
Attenuated Adenomatous Polyposis
Attenuated adenomatous polyposis coli (AAPC) is a milder form of FAP with a decreased number of adenomas throughout the gastrointestinal tract. These patients have adenomas predominantly on the right side of the colon, which appear to be flat rather than polypoid on colonoscopic examination. The number of adenomas is commonly in the range of 1 to 50, and rarely more than 100. In comparison, FAP patients can have thousands of polyps. Also, the incidence and risk for colorectal carcinoma in AAPC is decreased and appears later in life compared with FAP patients; AAPC patients are afflicted by colon cancer approximately 15 years later than FAP patients but 10 years earlier than patients with sporadic colon cancer. Therefore, AAPC can be seen as an intermediate in biological aggressiveness, with FAP patients on one end of the spectrum and normal patients on the other.48
Genetic Features
The molecular pathogenesis that culminates in colorectal adenocarcinoma is versatile, with different pathways leading to this outcome. The two most common pathways are the APC/β-catenin pathway portrayed by the adenoma-to-carcinoma sequence and the microsatellite instability (MSI) pathway. The MSI pathway is caused by mutations in the proteins that are responsible for DNA repair. Each pathway is caused by several mutations in different genes. There are also epigenetic changes, such as methylation-induced gene silencing, which play a significant role in the pathobiology.49
The classic adenoma-to-carcinoma sequence postulates that there is either a germline or somatic loss of a single normal copy of the APC gene, which encodes a tumor suppressor protein responsible for degrading β-catenin. When β-catenin is not bound to APC, it migrates to the nucleus and activates gene transcription, leading to proliferation of MYC and cyclin D1. The second hit or inactivation of the remaining normal APC allele leads to the dysplastic lesion of adenomas. Additional accumulation of genetic mutations at KRAS, inactivation of TP53, and loss of herozygosity at 18q21 lead to carcinoma. That stepwise sequence is supported by the assertion that KRAS mutations are seen in fewer than 10% of subcentimeter adenomas but greater than 50% of adenomas larger than 1 cm, and in half of invasive adenocarcinomas.49
In the MSI pathway, there is a defect in the mismatch repair genes (which include MLH1, MSH2, MSH6, and PMS2). As a result, microsatellite repeats accumulate without being repaired. Microsatellites are typically noncoding regions; however, some microsatellite repeats are in the coding or promoter region of genes responsible for cell growth regulation and proliferation. Also, BRAF mutations and CpG island hypermethylation, which silences genes, are also common genetic and epigenetic events in the MSI pathway.49 The dysplastic lesion arising from this pathway (analogous to the adenoma of the APC/β-catenin pathway) is the sessile serrated adenoma. These lesions are commonly located in the right side of the colon and demonstrate serrations that extend to the base of the crypt, with dilation and outpouchings parallel to the basement membrane. The carcinomas that develop in this pathway have unique histologic features, including extensive mutinous differentiation, a Crohn-like inflammatory response with a peritumoral lymphoid cuff, or signet ring cell differentiation.50 These changes underlie carcinogenesis in patients with Lynch syndrome (hereditary nonpolyposis colorectal cancer) and are not considered a feature of FAP.
There is a third pathway in the pathobiology of colon cancer determined by CpG island methylation without MSI. Interestingly, many tumors from this pathway have KRAS mutations but most do not harbor TP53 or BRAF mutations.49
APC Gene
During the 1970s, a patient with Gardner syndrome was studied by researchers. Cytogenetic studies and restriction fragment length polymorphism analysis on this patient discovered a deletion on chromosome 5 in the q21–q22 region. A decade later, additional research discovered that this region coded for the APC gene, a tumor suppressor gene responsible for regulating β-catenin. If APC function is compromised, there is constitutive activation of β-catenin, leading to loss of cell cycle checkpoints and unregulated cellular proliferation. In the early 1990s, four additional mutations were seen in APC that lead to stop codons and premature termination.2
The APC gene is located in the q21 region of chromosome 5. It consists of 21 exons, which are retained in mRNA transcript 17. This codes for more than 2,000 amino acids, which may explain why there are a large number of possible mutations.51 52 53 APC is a ubiquitous gene, seen in all tissues, and most of the gene is taken up by exon 15, which comprises more than 70% of the coding sequence and harbors 45% of germline mutations.48 When there is alternative splicing of different exons, an attenuated version of FAP can be seen. Therefore, posttranscriptional modification is paramount in determining the severity of disease expression.2
Due to a germline mutation in FAP patients, there is only a single active APC allele. An additional hit or inactivation of the remaining allele initiates the early steps in the progression toward carcinoma. Even small lesions, such as focal adenomatous dysplasia, have been shown to demonstrate inactivation of the second APC allele, confirming it as an early marker in dysplasia. Besides single base pair deletions, which often become stop codons, small deletions and insertions of approximately one to four base pairs lead to significant frame shifts.48 51 52
The initial discovery toward understanding the function of the APC protein was due to immunoprecipitation, as anti-APC antibodies caused the precipitation of β-catenin. This leads to a better understanding of their roles in the Wnt signaling pathway, which is responsible for several cellular processes related to proliferation, apoptosis, and differentiation. Besides its role in the Wnt signaling pathway, APC is also involved in the cell cycle by acting as a checkpoint from the G0/G1 to the S phase.48
Normally, β-catenin is bound to E-cadherin, an intercellular adhesion molecule located on the surface of the cell membrane. When β-catenin is not bound to E-cadherin, it is quickly degraded by a complex of proteins. These proteins include APC, protein phosphatase 2A, protein kinases, and axin. This protein complex phosphorylates β-catenin, tagging it for degradation and removal by a ubiquitin-mediated process. When the Wnt receptor has been stimulated by the appropriate ligand, the protein complex is inactivated. This lets β-catenin travel in the cytoplasm without being degraded. It enters the nucleus and interacts with Tcf, a group of transcription factors. The appropriate transcript and proteins are produced, and increased cellular proliferation takes place. For patients with FAP, the APC protein is dysfunctional; therefore, the degradation protein complex is unable to bind to β-catenin. Once again, β-catenin is free to enter into the nucleus and bind with Tcf. Several proteins are transcribed, including Wnt, cyclin D1, and c-MYC.48
Depending on where a mutation occurs along APC, several varying clinical phenotypes may result. The final gene product length determines the severity of gastrointestinal tract disease (number and size of polyps) and the presence of CHRPE or desmoid tumors. Mutations at the 5′ end of the gene have less severe manifestations than mutations at the 3′ end. This attenuated form of FAP is known as attenuated adenomatous polyposis. The manifestations of these molecular biologic changes at the protein level are numerous. For example, abnormal DNA synthesis and activated cellular replication lead to increased ornithine decarboxylase, which is an enzyme involved in mucosal proliferation.48
Molecular Diagnosis
Definitive diagnosis of FAP requires molecular analysis of APC. There are several screening methods to establish this. High-resolution melting is a modality for screening whereby the melting process converts double-stranded DNA into single-stranded DNA. This denaturation process is carefully observed, and the melting profiles are compared. Individual fragments have unique melting profiles that indicate mutations in the sequence. Since this is a screening test, all positive results by high-resolution melting necessitate confirmation by sequencing.2
In looking for a specific mutation, either multiplex ligation-dependent probe amplification (MLPA) or next-generation sequencing can be used. MLPA is based on using probes to hybridize to specific fragments of DNA. These products are amplified by polymerase chain reaction and subsequently quantified. This process can analyze up to 40 DNA fragments. Next-generation sequencing can, with a complete detailed sequence, identify both new and small mutations.2
Screening
Once patients are confirmed to have FAP, patients and family members should be regularly screened. This approach led to a 55% decrease in the occurrence of colorectal carcinoma and a marked improvement in survival in this population.54 55 Annual sigmoidoscopy or colonoscopy is recommended for FAP patients until the appropriate time for a colectomy is determined. Even after a total colectomy, an endoscopic/colonoscopic procedure is recommended every 6 to 12 months after surgery to assess the anastomosis site, pouch, and residual rectum.56
References
- 1.Petersen G M, Slack J, Nakamura Y. Screening guidelines and premorbid diagnosis of familial adenomatous polyposis using linkage. Gastroenterology. 1991;100(6):1658–1664. doi: 10.1016/0016-5085(91)90666-9. [DOI] [PubMed] [Google Scholar]
- 2.Plawski A, Banasiewicz T, Borun P. et al. Familial adenomatous polyposis of the colon. Hered Cancer Clin Pract. 2013;11(1):15. doi: 10.1186/1897-4287-11-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bussey H JR. Baltimore, MD: Johns Hopkins University Press; 1975. Familial Polyposis Coli: Family Studies, Histopathology, Differential Diagnosis, and Results of Treatment; pp. 73–90. [Google Scholar]
- 4.Arvanitis M L, Jagelman D G, Fazio V W, Lavery I C, McGannon E. Mortality in patients with familial adenomatous polyposis. Dis Colon Rectum. 1990;33(8):639–642. doi: 10.1007/BF02150736. [DOI] [PubMed] [Google Scholar]
- 5.Sanner R F Jr. Diffuse polyposis of the colon with severe electrolyte depletion. Arch Surg. 1973;107(6):903–905. doi: 10.1001/archsurg.1973.01350240067019. [DOI] [PubMed] [Google Scholar]
- 6.Lynch H T, Smyrk T, Watson P. et al. Hereditary colorectal cancer. Semin Oncol. 1991;18(4):337–366. [PubMed] [Google Scholar]
- 7.Iwama T, Mishima Y, Okamoto N, Inoue J. Association of congenital hypertrophy of the retinal pigment epithelium with familial adenomatous polyposis. Br J Surg. 1990;77(3):273–276. doi: 10.1002/bjs.1800770312. [DOI] [PubMed] [Google Scholar]
- 8.Iwama T, Mishima Y. Mortality in young first-degree relatives of patients with familial adenomatous polyposis. Cancer. 1994;73(8):2065–2068. doi: 10.1002/1097-0142(19940415)73:8<2065::aid-cncr2820730809>3.0.co;2-o. [DOI] [PubMed] [Google Scholar]
- 9.Wu T T, Kornacki S, Rashid A, Yardley J H, Hamilton S R. Dysplasia and dysregulation of proliferation in foveolar and surface epithelia of fundic gland polyps from patients with familial adenomatous polyposis. Am J Surg Pathol. 1998;22(3):293–298. doi: 10.1097/00000478-199803000-00003. [DOI] [PubMed] [Google Scholar]
- 10.Abraham S C, Nobukawa B, Giardiello F M, Hamilton S R, Wu T T. Sporadic fundic gland polyps: common gastric polyps arising through activating mutations in the beta-catenin gene. Am J Pathol. 2001;158(3):1005–1010. doi: 10.1016/s0002-9440(10)64047-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bertoni G, Sassatelli R, Nigrisoli E. et al. High prevalence of adenomas and microadenomas of the duodenal papilla and periampullary region in patients with familial adenomatous polyposis. Eur J Gastroenterol Hepatol. 1996;8(12):1201–1206. doi: 10.1097/00042737-199612000-00013. [DOI] [PubMed] [Google Scholar]
- 12.Björk J, Akerbrant H, Iselius L, Alm T, Hultcrantz R. Epidemiology of familial adenomatous polyposis in Sweden: changes over time and differences in phenotype between males and females. Scand J Gastroenterol. 1999;34(12):1230–1235. doi: 10.1080/003655299750024751. [DOI] [PubMed] [Google Scholar]
- 13.Hornick J L, Odze R. Philadelphia, PA: Saunders Elsevier; 2009. Polyps of the large intestine; pp. 481–533. [Google Scholar]
- 14.Matsumoto T, Iida M, Tada S, Mibu R, Yao T, Fujishima M. Early detection of nonpolypoid cancers in the rectal remnant in patients with familial adenomatous polyposis/Gardner's syndrome. Cancer. 1994;74(1):12–15. doi: 10.1002/1097-0142(19940701)74:1<12::aid-cncr2820740104>3.0.co;2-9. [DOI] [PubMed] [Google Scholar]
- 15.Scates D K, Spigelman A D, Phillips R KS, Venitt S. DNA adducts detected by 32P-postlabelling, in the intestine of rats given bile from patients with familial adenomatous polyposis and from unaffected controls. Carcinogenesis. 1992;13(4):731–735. doi: 10.1093/carcin/13.4.731. [DOI] [PubMed] [Google Scholar]
- 16.Giardiello F M, Hamilton S R, Krush A J. et al. Treatment of colonic and rectal adenomas with sulindac in familial adenomatous polyposis. N Engl J Med. 1993;328(18):1313–1316. doi: 10.1056/NEJM199305063281805. [DOI] [PubMed] [Google Scholar]
- 17.Bell B, Mazzaferri E L. Familial adenomatous polyposis (Gardner's syndrome) and thyroid carcinoma. A case report and review of the literature. Dig Dis Sci. 1993;38(1):185–190. doi: 10.1007/BF01296795. [DOI] [PubMed] [Google Scholar]
- 18.Ruhswurm I, Zehetmayer M, Dejaco C, Wolf B, Karner-Hanusch J. Ophthalmic and genetic screening in pedigrees with familial adenomatous polyposis. Am J Ophthalmol. 1998;125(5):680–686. doi: 10.1016/s0002-9394(98)00005-1. [DOI] [PubMed] [Google Scholar]
- 19.Romania A, Zakov Z N, McGannon E, Schroeder T, Heyen F, Jagelman D G. Congenital hypertrophy of the retinal pigment epithelium in familial adenomatous polyposis. Ophthalmology. 1989;96(6):879–884. doi: 10.1016/s0161-6420(89)32822-3. [DOI] [PubMed] [Google Scholar]
- 20.Traboulsi E I, Krush A J, Gardner E J. et al. Prevalence and importance of pigmented ocular fundus lesions in Gardner's syndrome. N Engl J Med. 1987;316(11):661–667. doi: 10.1056/NEJM198703123161104. [DOI] [PubMed] [Google Scholar]
- 21.Morton D G, Gibson J, Macdonald F. et al. Role of congenital hypertrophy of the retinal pigment epithelium in the predictive diagnosis of familial adenomatous polyposis. Br J Surg. 1992;79(7):689–693. doi: 10.1002/bjs.1800790733. [DOI] [PubMed] [Google Scholar]
- 22.Parsons M A, Rennie I G, Rundle P A, Dhingra S, Mudhar H, Singh A D. Congenital hypertrophy of retinal pigment epithelium: a clinico-pathological case report. Br J Ophthalmol. 2005;89(7):920–921. doi: 10.1136/bjo.2004.061887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bülow C Bülow S; Leeds Castle Polyposis Group. Is screening for thyroid carcinoma indicated in familial adenomatous polyposis? The Leeds Castle Polyposis Group Int J Colorectal Dis 1997124240–242. [DOI] [PubMed] [Google Scholar]
- 24.Truta B, Allen B A, Conrad P G. et al. Genotype and phenotype of patients with both familial adenomatous polyposis and thyroid carcinoma. Fam Cancer. 2003;2(2):95–99. doi: 10.1023/a:1025762706854. [DOI] [PubMed] [Google Scholar]
- 25.Perrier N D van Heerden J A Goellner J R et al. Thyroid cancer in patients with familial adenomatous polyposis World J Surg 1998227738–742., discussion 743 [DOI] [PubMed] [Google Scholar]
- 26.Harach H R, Williams G T, Williams E D. Familial adenomatous polyposis associated thyroid carcinoma: a distinct type of follicular cell neoplasm. Histopathology. 1994;25(6):549–561. doi: 10.1111/j.1365-2559.1994.tb01374.x. [DOI] [PubMed] [Google Scholar]
- 27.Cameselle-Teijeiro J, Chan J K. Cribriform-morular variant of papillary carcinoma: a distinctive variant representing the sporadic counterpart of familial adenomatous polyposis-associated thyroid carcinoma? Mod Pathol. 1999;12(4):400–411. [PubMed] [Google Scholar]
- 28.Baloch Z, Livolsi V. Philadelphia, PA: Lippincott Williams & Wilkins; 2010. Pathology of thyroid and parathyroid disease. In: Mills SE, ed. Sternberg's Diagnostic Surgical Pathology. 5th ed; pp. 460–492. [Google Scholar]
- 29.Miyoshi Y, Iwao K, Nawa G, Yoshikawa H, Ochi T, Nakamura Y. Frequent mutations in the beta-catenin gene in desmoid tumors from patients without familial adenomatous polyposis. Oncol Res. 1998;10(11–12):591–594. [PubMed] [Google Scholar]
- 30.Clark S K, Neale K F, Landgrebe J C, Phillips R K. Desmoid tumours complicating familial adenomatous polyposis. Br J Surg. 1999;86(9):1185–1189. doi: 10.1046/j.1365-2168.1999.01222.x. [DOI] [PubMed] [Google Scholar]
- 31.Sturt N J, Gallagher M C, Bassett P. et al. Evidence for genetic predisposition to desmoid tumours in familial adenomatous polyposis independent of the germline APC mutation. Gut. 2004;53(12):1832–1836. doi: 10.1136/gut.2004.042705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Brooks J SJ. Philadelphia, PA: Lippincott Williams & Wilkins; 2010. Disorders of soft tissue; pp. 124–197. [Google Scholar]
- 33.Hughes L J, Michels V V. Risk of hepatoblastoma in familial adenomatous polyposis. Am J Med Genet. 1992;43(6):1023–1025. doi: 10.1002/ajmg.1320430621. [DOI] [PubMed] [Google Scholar]
- 34.Washington K, Harris E. Philadelphia, PA: Lippincott Williams & Wilkins; 2010. Masses of the liver; pp. 1535–1599. [Google Scholar]
- 35.Turcot J, Despres J P, St Pierre F. Malignant tumors of the central nervous system associated with familial polyposis of the colon: report of two cases. Dis Colon Rectum. 1959;2(5):465–468. doi: 10.1007/BF02616938. [DOI] [PubMed] [Google Scholar]
- 36.Attard T M, Giglio P, Koppula S, Snyder C, Lynch H T. Brain tumors in individuals with familial adenomatous polyposis: a cancer registry experience and pooled case report analysis. Cancer. 2007;109(4):761–766. doi: 10.1002/cncr.22475. [DOI] [PubMed] [Google Scholar]
- 37.Louis D N, Ohgaki H, Wiestler O D. et al. The 2007 WHO classification of tumours of the central nervous system. Acta Neuropathol. 2007;114(2):97–109. doi: 10.1007/s00401-007-0243-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Leary S ES, Zhou T, Holmes E, Geyer J R, Miller D C. Histology predicts a favorable outcome in young children with desmoplastic medulloblastoma: a report from the children's oncology group. Cancer. 2011;117(14):3262–3267. doi: 10.1002/cncr.25856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kool M, Korshunov A, Remke M. et al. Molecular subgroups of medulloblastoma: an international meta-analysis of transcriptome, genetic aberrations, and clinical data of WNT, SHH, Group 3, and Group 4 medulloblastomas. Acta Neuropathol. 2012;123(4):473–484. doi: 10.1007/s00401-012-0958-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gajjar A J, Robinson G W. Medulloblastoma-translating discoveries from the bench to the bedside. Nat Rev Clin Oncol. 2014;11(12):714–722. doi: 10.1038/nrclinonc.2014.181. [DOI] [PubMed] [Google Scholar]
- 41.Northcott P A, Korshunov A, Witt H. et al. Medulloblastoma comprises four distinct molecular variants. J Clin Oncol. 2011;29(11):1408–1414. doi: 10.1200/JCO.2009.27.4324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Northcott P A, Korshunov A, Pfister S M, Taylor M D. The clinical implications of medulloblastoma subgroups. Nat Rev Neurol. 2012;8(6):340–351. doi: 10.1038/nrneurol.2012.78. [DOI] [PubMed] [Google Scholar]
- 43.Shih D J, Northcott P A, Remke M. et al. Cytogenetic prognostication within medulloblastoma subgroups. J Clin Oncol. 2014;32(9):886–896. doi: 10.1200/JCO.2013.50.9539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Gajjar A, Chintagumpala M, Ashley D. et al. Risk-adapted craniospinal radiotherapy followed by high-dose chemotherapy and stem-cell rescue in children with newly diagnosed medulloblastoma (St Jude Medulloblastoma-96): long-term results from a prospective, multicentre trial. Lancet Oncol. 2006;7(10):813–820. doi: 10.1016/S1470-2045(06)70867-1. [DOI] [PubMed] [Google Scholar]
- 45.Gardner E J, Richards R C. Multiple cutaneous and subcutaneous lesions occurring simultaneously with hereditary polyposis and osteomatosis. Am J Hum Genet. 1953;5(2):139–147. [PMC free article] [PubMed] [Google Scholar]
- 46.Bilkay U, Erdem O, Ozek C. et al. Benign osteoma with Gardner syndrome: review of the literature and report of a case. J Craniofac Surg. 2004;15(3):506–509. doi: 10.1097/00001665-200405000-00032. [DOI] [PubMed] [Google Scholar]
- 47.Gardner E J. Follow-up study of a family group exhibiting dominant inheritance for a syndrome including intestinal polyps, osteomas, fibromas and epidermal cysts. Am J Hum Genet. 1962;14(4):376–390. [PMC free article] [PubMed] [Google Scholar]
- 48.Fenoglio-Preiser C M, Noffsinger A E, Stemmerman G N, Lantz P E, Isaacson P G. Philadelphia, PA: Lippincott Williams & Wilkins; 2008. Gastrointestinal Pathology: An Atlas and Text. 3rd ed. [Google Scholar]
- 49.Turner J R. Philadelphia, PA: Saunders Elsevier; 2010. The gastrointestinal tract; pp. 763–831. [Google Scholar]
- 50.Cooper H S. Philadelphia, PA: Lippincott Williams & Wilkins; 2010. Intestinal neoplasms; pp. 1368–1431. [Google Scholar]
- 51.Groden J, Thliveris A, Samowitz W. et al. Identification and characterization of the familial adenomatous polyposis coli gene. Cell. 1991;66(3):589–600. doi: 10.1016/0092-8674(81)90021-0. [DOI] [PubMed] [Google Scholar]
- 52.Nishisho I, Nakamura Y, Miyoshi Y. et al. Mutations of chromosome 5q21 genes in FAP and colorectal cancer patients. Science. 1991;253(5020):665–669. doi: 10.1126/science.1651563. [DOI] [PubMed] [Google Scholar]
- 53.Kinzler K W, Nilbert M C, Su L K. et al. Identification of FAP locus genes from chromosome 5q21. Science. 1991;253(5020):661–665. doi: 10.1126/science.1651562. [DOI] [PubMed] [Google Scholar]
- 54.Bülow S. Results of national registration of familial adenomatous polyposis. Gut. 2003;52(5):742–746. doi: 10.1136/gut.52.5.742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Heiskanen I, Luostarinen T, Järvinen H J. Impact of screening examinations on survival in familial adenomatous polyposis. Scand J Gastroenterol. 2000;35(12):1284–1287. doi: 10.1080/003655200453638. [DOI] [PubMed] [Google Scholar]
- 56.Rex D K Johnson D A Anderson J C Schoenfeld P S Burke C A Inadomi J M; American College of Gastroenterology. American College of Gastroenterology guidelines for colorectal cancer screening 2009 [corrected] Am J Gastroenterol 20091043739–750. [DOI] [PubMed] [Google Scholar]