

Abstract
Nephrotic syndrome (NS) is a common pediatric kidney disease and is defined as massive proteinuria, hypoalbuminemia, and edema. Dysfunction of the glomerular filtration barrier, which is made up of endothelial cells, glomerular basement membrane, and visceral epithelial cells known as podocytes, is evident in children with NS. While most children have steroid-responsive nephrotic syndrome (SSNS), approximately 20% have steroid-resistant nephrotic syndrome (SRNS) and are at risk for progressive kidney dysfunction. While the cause of SSNS is still not well understood, there has been an explosion of research into the genetic causes of SRNS in the past 15 years. More than 30 proteins regulating the function of the glomerular filtration barrier have been associated with SRNS including podocyte slit diaphragm proteins, podocyte actin cytoskeletal proteins, mitochondrial proteins, adhesion and glomerular basement membrane proteins, transcription factors, and others. A genetic cause of SRNS can be found in approximately 70% of infants presenting in the first 3 months of life and 50% of infants presenting between 4 and 12 months, with much lower likelihood for older patients. Identification of the underlying genetic etiology of SRNS is important in children because it allows for counseling of other family members who may be at risk, predicts risk of recurrent disease after kidney transplant, and predicts response to immunosuppressive therapy. Correlations between genetic mutation and clinical phenotype as well as genetic risk factors for SSNS and SRNS are reviewed in this article.
Keywords: nephrotic syndrome, end-stage kidney disease, podocyte, slit diaphragm, proteinuria
Introduction
Nephrotic syndrome (NS) is one of the most common pediatric kidney diseases seen in pediatric nephrology clinics, affecting approximately 16 per 100,000 children.1 It is the most prevalent diagnosis in children receiving maintenance dialysis in the North American Pediatric Renal Trials and Collaborative Studies registry.2 NS is defined as a constellation of clinical signs and symptoms including massive proteinuria (>40 mg/m2/h or urine protein to creatinine ratio of >2 g/g) hypoalbuminemia (<2.5 g/dL), and edema. Life-threatening complications including infection and thrombosis can be observed in affected children owing to specific protein losses in the urine.3 NS can also be categorized by age of presentation with congenital nephrotic syndrome (CNS) presenting in the first 3 months of life, infantile NS presenting between the age of 4 and 12 months, and childhood NS in children older than 1 year.4
Most children older than 1 year with NS (∼80%) have steroid-sensitive nephrotic syndrome (SSNS) and go into remission after treatment with corticosteroids demonstrating complete resolution of their symptoms.5 Unfortunately, approximately 20% of children older than 1 year and a majority of children younger than 1 year presenting with NS will have steroid-resistant nephrotic syndrome (SRNS). While some children with SRNS will go into remission with alternate immunosuppressive therapy, others remain nephrotic and have a high risk of progressive loss of kidney function to the point of needing kidney dialysis or transplantation.6 Landmark genetic studies over the past 15 years have identified more than 30 monogenic causes of SRNS, mostly in genes that regulate the function of the glomerular filtration barrier (GFB) (Table 1). Identification of the underlying genetic etiology of SRNS is important in children because it allows for counseling of other family members who may be affected, predicts risk of recurrent disease after kidney transplant, predicts response to immunosuppressive therapy and may also present unique opportunity for secondary prevention of progression to end-stage kidney disease by putting in place measures that can slow down progression of kidney disease such as control of hypertension and reduction of asymptomatic proteinuria.7
Table 1. Steroid resistant nephrotic syndrome.
| Gene | Protein | Mode of inheritance | Syndrome or extrarenal manifestations |
|---|---|---|---|
| Slit diaphragm associated | |||
| CD2AP | CD2-associated protein | Autosomal recessive/autosomal dominant | |
| NPHS1 | Nephrin | Autosomal recessive | |
| NPHS2 | Podocin | Autosomal recessive | |
| PLCE1 | Phospholipase C, ε1 | Autosomal recessive | |
| TRPC6 | Transient receptor potential cation channel, subfamily C, member 6 | Autosomal dominant | |
| Actin cytoskeleton | |||
| ACTN4 | α-Actinin 4 | Autosomal dominant | |
| ANLN | Anillin | Autosomal dominant | |
| ARHGAP24 | Rho GTPase activating protein 24 | Autosomal dominant | |
| ARHGDIA | RhoGDP dissociation inhibitor α | Autosomal recessive | |
| INF2 | Inverted formin 2 | Autosomal dominant | Charcot–Marie–Tooth |
| MYO1E | Nonmuscle myosin 1e | Autosomal recessive | |
| Mitochondrial proteins | |||
| ADCK4 | aarF domain containing kinase 4 | Autosomal recessive | |
| COQ2 | Coenzyme Q2 4-hydroxybenzoate polyprenyl transferase | Autosomal recessive | Seizures, encephalopathy |
| COQ6 | Coenzyme Q6 monooxygenase | Autosomal recessive | Sensorineural deafness |
| MTTL1 | tRNA-LEU | Unknown | Mental retardation, diabetes mellitus, MELAS syndrome |
| PDSS2 | Prenyl diphosphate synthase subunit 2 | Autosomal recessive | Encephalomyopathy, Leigh syndrome |
| Adhesion and glomerular basement membrane proteins | |||
| COL4A3 | α3 type IV collagen | Autosomal recessive | Sensorineural deafness |
| COL4A4 | α4 type IV collagen | Autosomal recessive | Sensorineural deafness |
| COL4A5 | α5 type IV collagen | X-linked | Sensorineural deafness |
| ITGA3 | Integrin α3 | Autosomal recessive | Interstitial lung disease, epidermolysis bullosa |
| ITGB4 | Integrin β4 | Autosomal recessive | Epidermolysis bullosa |
| LAMB2 | Laminin β2 | Autosomal recessive | Pierson syndrome |
| Nuclear transcription factors | |||
| LMX1B | LIM homeobox transcription factor 1β | Autosomal dominant | Nail–patella syndrome |
| NXF5 | Nuclear RNA export factor 5 | X-linked | Cardiac conduction disorder |
| SMARCL1 | SMARCA-like protein | Autosomal recessive | Schimke immuno-osseous dysplasia |
| WT1 | Wilms tumor 1 | Autosomal dominant | Denys-Drash, Frasier syndrome |
| Others | |||
| CFH | Complement factor H | Autosomal recessive | Atypical hemolytic uremic syndrome |
| CUBN | Cubilin | Autosomal recessive | Megaloblastic anemia |
| DGKE | Diacylglycerol kinase ε | Autosomal recessive | Atypical hemolytic uremic syndrome |
| MEFV | Pyrin | Autosomal recessive | Mediterranean fever |
| NEIL1 | Nei endonuclease VIII-like 1 | Autosomal recessive | |
| PMM2 | Phosphomannomutase 2 | Autosomal recessive | Congenital defects of glycosylation |
| PTPRO | GLEPP1 | Autosomal recessive | |
| SCARB2 | Lysosomal integral membrane protein type 2 | Autosomal recessive | Action myoclonus, renal failure syndrome |
| WDR73 | WD repeat domain 73 | Autosomal recessive | Galloway-Mowat syndrome |
| ZMPSTE24 | Zinc metalloproteinase STE24 | Autosomal recessive | Mandibuloacral dysplasia |
Abbreviation: MELAS, mitochondrial myopathy, encephalopathy, lactic acidosis, and strokelike episodes.
Glomerular Filtration Barrier
The kidney GFB is composed of three layers that functionally prevent passage of plasma proteins into the urine: endothelium, glomerular basement membrane (GBM), and visceral epithelial cells also known as podocytes. Podocytes are highly differentiated cells that have a complex cellular structure made up of a cell body, major processes, and foot processes.8 Adjacent foot processes are connected by a slit diaphragm, which is a vital component of the kidney filtration barrier (Fig. 1). Abnormalities in podocyte foot process structure are observed on renal pathology examination in children with NS. Minimal change disease (MCD) is the most common histopathologic finding in children with SSNS, although this can be observed in children with SRNS as well.5 Kidney biopsies from patients with MCD have a normal appearance by light microscopy.9 By electron microscopy (EM), broadening and retraction of podocyte foot processes “effacement” is evident. Foot process effacement is the hallmark pathologic finding in NS and is present in most, but not all, affected patients.10 11 In children with SRNS, focal segmental glomerulosclerosis (FSGS) is the most common pathological correlate. In addition to podocyte effacement by EM, biopsy samples from children with FSGS demonstrate light microscopic findings of segmental hyalinosis and sclerosis within some glomeruli or segmental glomerular collapse.12 Diffuse mesangial sclerosis (DMS) is another pathologic variant of NS that is characterized by early-onset, rapid progression and distinct histopathology including mesangial matrix expansion, hypertrophied podocytes that surround the glomerular tuft like a crown and thickened basement membranes.13
Fig. 1.
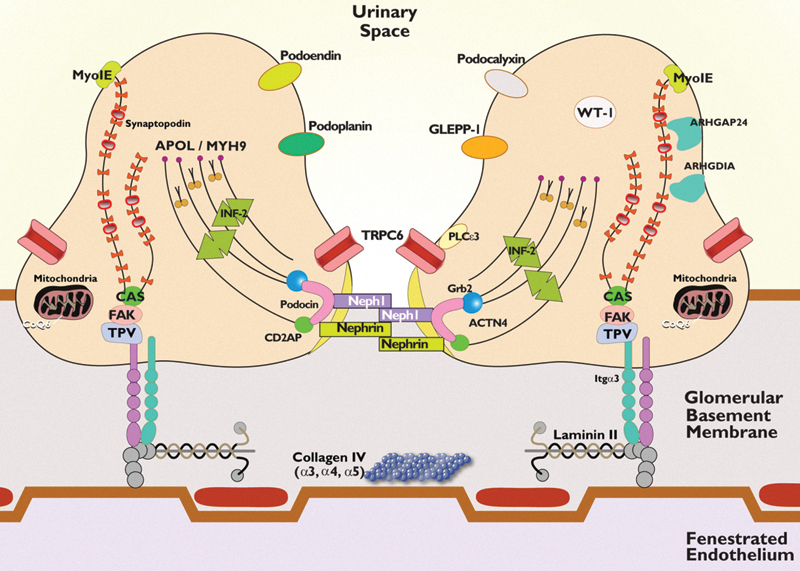
Schematic of the podocyte and the glomerular filtration barrier (GFB). The GFB is composed of the podocytes, the glomerular basement membrane (GBM), and the fenestrated endothelial cells. The podocyte has a unique actin cytoskeleton made up of F-actin and nonmuscle myosin such as nonmuscle myosin heavy chain 9 (MYH9) and myosin 1E (MYO1E). Its other structural components include actin-binding proteins such as synaptopodin and α-actinin 4 (ACTN4) and actin polymerization regulatory protein inverted formin 2 (INF2). Wilms tumor 1 gene is a nuclear transcription factor important in podocyte gene development. The cell–cell junction of the podocyte (the slit diaphragm) is formed by nephrin, podocin, CD2-associated protein, and NEPH1. Podocin is associated with lipid rafts, a signaling domain of the slit diaphragm. It recruits nephrin and NEPH1 to form a signaling complex with other molecules such as transient receptor potential cation channel 6 (TRPC6), growth factor receptor–bound protein 2, and phospholipase C epsilon-1 (PLCE1) at the slit diaphragm. The apical membrane of the podocyte is formed by negatively charged molecules such as podocalyxin, podoplanin, podoendin, and glomerular epithelial protein-1. RhoA-activated Rac1 GTPase-activating protein (ARHGAP24) and its regulator RhoGDIα (ARHGDIA) are critical for maintaining podocyte cell shape and membrane dynamics. COQ6 and other regulators of the coenzyme Q10 biosynthesis pathway are important in podocyte energy regulation. The basal part of the podocyte contains α3β1 integrin and α- and β-dystroglycans that anchor the podocyte to the GBM. Talin, paxillin, and vinculin (TPV) interact with different laminins in the GBM, especially laminin β2. Mutations in many of the aforementioned proteins have been associated with nephrotic syndrome.
In 1998, mutations in nephrin (NPHS1), a protein that makes up an essential component of the podocyte slit diaphragm, were found to be causative for a subset of patients with CNS.14 This discovery led to an explosion of research into the structure and function of the podocyte in NS. Causative mutations have since been found in several vital pathways including those involved in slit diaphragm structure and function, podocyte actin cytoskeletal organization, co-enzyme Q biosynthesis, lysosomal pathways, and adhesion to the GBM.
Slit Diaphragm Proteins
Mutations in nephrin (NPHS1), a structural component of the glomerular slit diaphragm, account for between 40 and 60% of infants with CNS.14 15 16 Absence of the slit diaphragm and its size selective barrier properties leads to massive loss of proteins into the urine that begins in utero. Associated perinatal findings can include prematurity, enlarged placenta, and elevated maternal serum α-fetoprotein.17 Affected infants develop clinical NS in the first 3 months of life with progression to end-stage kidney disease in the first few years of life. There is a high incidence of disease in patients with Finnish ancestry owing to founder mutations in this population (nt121delCT or R1109X) that lead to premature stop codons and absence of detectable protein at the slit diaphragm.18 Less severe NPHS1 mutations have been reported in other populations, including the New Zealand Maori population founder mutation, which are reported to be associated with prolonged renal survival, up to 30 years in some affected individuals.19
Podocin (NPHS2) is a transmembrane protein that is required for recruitment of nephrin to slit diaphragms.20 Patients with homozygous or compound heterozygous mutations commonly present with isolated SRNS before the age of 6 years with FSGS on renal biopsy. Affected children rapidly progress to end-stage renal disease (ESRD). Podocin mutations account for 10.6% of infants with CNS and 10 to 40% of older children with both familial and sporadic SRNS.15 21 22 23 More than 125 NPHS2 mutations have been described, including an R138Q founder mutation that is prevalent in the European population.20 23 Several polymorphisms have also been described including R229Q, which is considered a nonneutral polymorphism that leads to decreased binding of podocin to nephrin in vitro.24 When associated with another NPHS2 mutation, it may lead to a wide spectrum of disease phenotypes with a typically slower progression of renal disease.23 The R229Q polymorphism has also predisposes patients with thin basement membrane disease to proteinuria and progression to ESRD.25 26
Transient receptor potential cation channel type 6 (TRPC6) is a nonselective calcium channel that localizes to podocyte foot processes and interacts with nephrin and podocin.27 Mutations in TRPC6 are associated with autosomal-dominant SRNS with onset typically in the third or fourth decade of life; however, early childhood onset has also been described.28 29 TRPC6 mutations are a rare cause of SRNS and account for 2 to 4% of familial disease and <2% of sporadic cases.15 30 31 Most described TRPC6 mutations lead to enhanced influx of calcium into podocytes; however, it is unclear how this leads to the development of FSGS and NS.28 Mutations in TRPC6 have also been found to promote angiotensin II–dependent increases in calcium signaling, perhaps enhancing the deleterious effects of angiotensin II on the podocyte.32
CD2-associated protein (CD2AP) is an adapter molecule originally identified as a ligand for the T-cell-adhesion protein CD2. In the podocyte, CD2AP acts as a bridge between the slit diaphragm and the actin cytoskeleton.33 Homozygous mutations in CD2AP cause CNS in mice.33 Mutations in CD2AP have only very rarely been described in human SRNS.34 35
Phospholipase C epsilon 1 (PLCε1) is a signaling protein for many G protein–coupled receptors that catalyzes the hydrolysis of polyphosphoinositides to generate second messengers that influence cell growth, differentiation, and gene expression.36 PLCε1 interacts with nephrin at the slit diaphragm through the adaptor protein IQGAP1 and may be required for normal glomerular development.37 PLCε1 mutations are a major cause of isolated DMS, occurring in 28 to 33% of affected families.38 39 Mutations are also observed in patients with sporadic and familial FSGS, although at lesser frequency.15 38 The age of disease onset for children with PLCε1 mutations is earlier for those with splice site mutations compared with C-terminal truncating mutations or missense mutations.15 At least two patients have been described in the literature with PLCε1 mutations who responded to treatment with corticosteroids or cyclosporine.37 Interestingly, several unaffected patients have been described in the literature with homozygous PLCε1 mutations that were associated with disease in other family members.38 40 Modifier genes or a “second-hit” may be required for development of NS in patients with PLCε1 mutations; however, the modifier genes are yet to be identified.
Actin Cytoskeleton
Podocytes require a highly organized actin cytoskeleton to maintain their specialized shape. During the development of NS, there is reorganization of the actin cytoskeleton from a core of parallel actin bundles surrounded by a branched subcortical actin network into a dense branched network with loss of actin bundles.41 Mutations in several proteins that regulate podocyte actin cytoskeletal arrangement have been described in association with SRNS.
Alpha-actinin 4 (ACTN4) is an F-actin-binding and crosslinking protein with widespread distribution, including the podocyte foot process.42 Mutations in ACTN4 are associated with autosomal-dominant, adult-onset SRNS with FSGS on renal biopsy.43 Mutations in ACTN4 lead to altered binding affinity for actin and abnormal adhesion of podocytes to the GBM.44 45
Inverted formin 2 (IFN2) is a member of the formin family of actin-regulating proteins that accelerates actin polymerization. Mutations in INF2 can be found in 9 to 17% of families with autosomal-dominant FSGS and rarely in patients with sporadic FSGS.30 46 47 48 Affected patients develop SRNS in their teens through early adulthood.46 The majority of mutations in INF2 cluster in the diaphanous inhibitory domain.46 48 In addition to its actin regulatory function, IFN2 interacts with nephrin and PLCε1 through the adaptor protein IQGAP1, and disruption of this interaction may play a role in the pathogenesis of FSGS in affected patients.47 Mutations in INF2 are also responsible for disease in patients with Charcot–Marie–Tooth disease with FSGS.49 Affected patients develop FSGS at a median age of 18 years with onset of peripheral-nerve dysfunction at a median age of 13 years.
Nonmuscle myosin 1E (Myo1E) is an actin binding molecular motor expressed in podocyte foot processes.50 Mutations in MYO1E cause autosomal-recessive SRNS with FSGS on renal biopsy.51 Affected patients present with SRNS in the first decade of life. EM in affected patients is unique in that it demonstrates focal thickening and disorganization of the basement membranes with multilamination of the GBM, similar to the appearance of the GBM in Alport syndrome.50 51
The organization of F-actin in podocytes, as well as all other cells, is controlled by RhoGTPases known as RhoA, Rac, and Cdc42.52 RhoA activity induces actin bundle formation, whereas Rac and Cdc42 activity favor formation of a branched actin network and the balance between the two is vital for the maintenance of podocyte structure and the GFB.53 RhoGTPase activity is tightly controlled in the cell by modifier proteins known as guanine nucleotide exchange factors, GTPase activating proteins (GAP), and guanine nucleotide exchange inhibitors (GDI).52 Mutations in at least two of these modifier proteins cause SRNS. Mutations in ARHGAP24 (Rho GTPase activating protein 24) lead to increased Rac activity in podocytes and familial autosomal-dominant FSGS.54 Affected individuals develop ESRD between the age of 12 and 29 years. Mutations in ARHGDIA (RhoGDP dissociation inhibitor α) also lead to increased Rac activity in podocytes.55 56 Mutations in ARHGDIA cause CNS or early-onset SRNS within the first 2 years of life. Affected individuals have DMS on biopsy and may have associated neurological deficits.55 56
More recently, mutations were reported in anillin (ANLN), an F-actin binding cell cycle gene, as a new cause of FSGS.57 ANLN is very important in cytokinesis during development and this may explain the reason why mutations in this gene are rare in the general population. The mechanisms by which mutant ANLN causes FSGS are unknown, but initial data showed that mutant ANLN displays reduced binding ability to the slit diaphragm-associated scaffold protein CD2AP.57
Mitochondrial Proteins
Coenzyme Q10 (ubiquinone) is a lipid-soluble component of cell membranes that is important in electron transport in the respiratory chain of the mitochondrial inner membrane. Primary coenzyme Q10 deficiency is primarily characterized by neurological and muscular symptoms with rare renal involvement. Mutations in several genes associated with biosynthesis of coenzyme Q10 have been associated with SRNS including COQ2, COQ6, prenyl diphosphate synthase subunit 2 (PDSS2), and aarF domain containing kinase 4 (ADCK4). Mutations in any of these proteins leads to decreased coenzyme Q10 levels and reduced mitochondrial respiratory enzyme activity, suggesting that regulation of energy metabolism is important for normal podocyte function. Mutations in COQ2 cause collapsing FSGS, a variant of FSGS characterized by podocyte proliferation and dedifferentiation, in the first decade of life.58 59 EM demonstrates increased number of abnormal mitochondria.59 Some affected patients with seizures and cardiomyopathy have been described.60 Mutations in COQ6 cause onset of SRNS within the first few years of life associated with sensorineural deafness.61 Mutations in ADCK cause SRNS in the first two decades of life.62 Mutations in PDSS2 were found in a patient with onset of SRNS at age 7 months and associated seizures.63 In addition, homozygous single-nucleotide polymorphisms genotypes for variant alleles for the PDSS2 gene are more common in European patients with FSGS than controls, suggesting that PDSS2 may also be an FSGS susceptibility gene.64 Interestingly, coenzyme Q10 levels were decreased in cell lines derived from patients with FSGS, independent of the PDSS2 haplotype.64 In one large study, mutations in the Q10 biosynthesis pathway were found in approximately 1% of SRNS cases overall.15 If a mutation in the coenzyme Q10 biosynthetic pathway is identified, early supplementation with coenzyme Q10 may be beneficial.61 65
Rare patients with FSGS and mitochondrial cytopathies have been described with mutations in tRNA-LEU, a mitochondrial-specific transfer RNA.66 67 Patients are variably affected with MELAS syndrome (mitochondrial encephalomyopathy, lactic acidosis, and stroke-like episodes).
Adhesion and GBM Proteins
The mature GBM consists of type IV collagen (α3, α4, and α5 isoforms), laminin-521 (α5β2γ1), entactin/nidogen, and agrin.68 Podocytes must adhere tightly to the GBM to maintain the GFB.69 The major adhesion receptor in podocytes is integrin α3β1, which connects laminin-521 in the GBM to the podocyte actin cytoskeleton through various adaptor proteins.69 Mutations in several adhesion proteins as well as components of the GBM have been demonstrated in patients with SRNS.
Mutations in LAMB2 encoding laminin β2 cause Pierson syndrome, an autosomal-recessive disorder characterized by CNS with DMS on renal biopsy, microcoria, and neurodevelopmental deficits.70 Isolated CNS is also caused by LAMB2 mutations in 2 to 5.5% of affected patients.15 16 Later-onset disease within the first decade of life has also been described with FSGS on renal biopsy.71 Genotype influences the phenotype in individuals with LAMB2 mutations. Truncating or splice site mutations are more likely to be associated with complete Pierson syndrome, whereas missense mutations or small deletions are more likely to be associated with the absence of neurologic abnormalities and higher age of onset of renal disease.71
Mutations in ITGA3 encoding integrin α3 cause CNS associated with interstitial lung disease and a mild variant of epidermolysis bullosa.72 Mutations in another podocyte integrin, ITGB4, were identified in an infant with CNS, pyloric atresia, and epidermolysis bullosa.73
Hemizygous or heterozygous mutations in COL4A5 encoding the α5 isoform of type IV collagen classically are associated with X-linked Alport syndrome, a disorder characterized by hematuria, progressive renal dysfunction, sensorineural deafness, and ocular abnormalities.74 Homozygous or heterozygous mutations in COL4A3 and COL4A4 cause autosomal-recessive Alport syndrome. Mutations in COL4A3–5 have been found in up to 10% of families presenting with SRNS and FSGS on renal biopsy, even in the absence of sensorineural deafness and the typical basement membrane abnormalities seen by EM in Alport patients.75 76 77
Nuclear Transcription Factors
The Wilms tumor 1 (WT1) gene encodes a zinc finder DNA-binding protein that acts as a transcriptional activator or repressor that is vital for kidney and urogenital development.78 NPHS1 is a transcriptional target of WT1, along with several other genes important in renal development.79 80 81 Mutations in WT1 are associated with SRNS in the context of Denys-Drash syndrome, isolated DMS, Frasier syndrome, and isolated FSGS.
Denys-Drash syndrome is the constellation of SRNS in the first year of life, DMS on kidney biopsy, male pseudohermaphroditism, and Wilms tumor.78 Affected infants often have mutations in exon 8 or 9 that code for zinc-fingers 2 and 3.82 83 WT1 mutations are found in around 8 to 13% of patients with CNS and around 8.5% of patients with isolated DMS on renal biopsy.15 31 39 Mutations in the second splice donor site in intron 9 are observed in patients with Frasier syndrome.84 Patients with Frasier syndrome present with normal female external genitalia, streak gonads, XY karyotype, SRNS with FSGS on renal biopsy, and gonadoblastoma.78 Primary amenorrhea may be the presenting symptom of this disorder.85 WT1 mutations have also been described in sporadic nonsyndromic FSGS.86
SMARCAL1 encodes an ATP-driven annealing helicase that plays an essential role in DNA–nucleosome interactions and chromatin remodeling during gene regulation and is expressed broadly in the kidney, including in podocytes.87 Mutations in SMARCAL1 cause Schimke immuno-osseous dysplasia, a rare autosomal-recessive multisystem disorder with disproportionate growth failure, impaired T-cell function, and SRNS with FSGS on renal biopsy.88
LIM homeobox transcription factor 1β (LMX1B) protein is expressed in podocytes during development and is essential for the maintenance of differentiated podocytes through effects on actin cytoskeletal organization and transcriptional regulation of COL4A3, COL4A4, NPHS2, and CD2AP.89 Mutations in LMX1B cause nail-patella syndrome, a rare autosomal-dominant disorder characterized by dystrophic nails, hypoplasia or absence of the patellae, dysplasia of the elbows, and iliac horns and SRNS with FSGS in less than 50% of affected patients.90 91 Affected patients generally have mild renal disease with a 5 to 10% risk of progression to ESRD. Mutations in LMX1B have also been described in patients with sporadic nonsyndromic autosomal-dominant FSGS.15 92
Nuclear RNA export factor 5 (NXF5) encodes a nuclear pore complex-associated protein that is involved in export of mRNAs from the nucleus. Mutations in NXF5 cause X-linked FSGS with associated heart block.93
Other Monogenic Causes of SRNS
Galloway-Mowat syndrome is a clinically heterogeneous syndrome classically presenting with microcephaly, structural brain anomalies, CNS, and developmental delay.94 Recently, mutations in WD repeat domain 73 (WDR73), a WD40 repeat containing protein, were found in two families with Galloway-Mowat syndrome with later-onset NS between the ages of 5 and 8 years.95 WDR73 is detectable in podocytes and brain tissue and mutations may manifest as defects in microtubule regulation.95 Other causative mutations for children with Galloway-Mowat syndrome are likely to be found.
Mutations in the CUBN gene encoding cubilin cause a hereditary form of megaloblastic anemia secondary to vitamin B12 deficiency. Cubilin is essential for albumin reabsorption by proximal tubule cells.96 Some affected patients have SRNS that may be treatable with vitamin B12.97
Mutations in lysosomal proteins can also cause SRNS. SCARB2 encodes a lysosomal membrane protein and when mutated causes action myoclonus renal failure syndrome, a progressive neurologic diseases with collapsing variant of FSGS.98 99
Rare patients with CNS and mutations in phosphomannomutase 2 (PMM2)100 have been described. Rare cases of childhood-onset SRNS have also been described in patients with mutations in PTPRO,101 DGKE,102 and NEIL1.103 Adult-onset SRNS has been described in patients with mutations in ZMPSTE24 causing mandibuloacral dysplasia, a rare autosomal-recessive disorder characterized by skeletal abnormalities of the mandible and clavicles and acro-osteolysis.104 Additional patients with adult-onset SRNS have been associated with mutations in CFH 105 and MEFV.106
Genetic Risk Factors for SRNS
Variants in APOL1, denoted G1 (missense S342G and I384M) and G2 (six base pair deletion near the C-terminus), are common in patients of African ancestry and are found in approximately 35% of African Americans.107 108 The APOL1 protein confers a form of innate immunity against the organism that causes African sleeping sickness, with increased activity in the G1 and G2 variants that may have led to positive selection pressure in this population. Individuals homozygous or compound heterozygous for these risk alleles have a 17-fold increased risk of developing FSGS and an estimated 4% lifetime risk of developing FSGS.109
Glypicans are members of the cell surface heparan sulfate proteoglycan family expressed in podocytes and endothelial cells. Variants in GPC5, encoding glypican-5, are associated with increased susceptibility for developing NS.110
Monogenic Causes of SSNS
Familial SSNS has been rarely described in the literature.111 Mutations in EMP2 were recently described in three unrelated families with autosomal-recessive SSNS.112 Age of onset was within the first 3 years of life and one biopsied patient demonstrated MCD. However, one family was reported to have SRNS due to EMP2 mutation as well. The exact mechanism by which mutations in EMP2 cause NS are unknown; however, EMP2 localizes to glomeruli and regulates CAVEOLIN-1, a protein involved in podocyte endocytosis.
Genetic Risk Factors for SSNS
HLA-DQA1 missense coding variants were identified in a large multiethnic cohort as a candidate risk locus for SSNS.113 Variants at this locus account for only approximately 4.6% of the risk for SSNS, suggesting the presence of other risk loci. PLCG2 is a transmembrane signaling enzyme that is important in the regulation of the immune system. Variants of PLCG2 are also associated with increased risk of SSNS.113 Both of these risk loci highlight the importance of the immune system in the pathogenesis of SSNS.
Recommendation for Genetic Testing in Children with NS
There are several benefits of having a genetic diagnosis for children with NS. With genetic information, counseling can be provided to family members who may also be at risk of having or passing on disease-associated genes. It also allows for appropriate choice of donor for kidney transplantation. Finally, a genetic diagnosis may provide prognostic information about the expected course of disease, predict the risk of recurrence posttransplant, and avoid unnecessary exposure to immunosuppressive agents. Genetic testing is recommended for all children with NS presenting in the first year of life.7 A monogenic cause can be found in 69% of children with CNS and in 50% of children presenting between 4 and 12 months.15 Genetic testing should also be performed for children with SRNS and other associated malformations or syndromes. Finally, genetic testing is recommended for children with a family history of NS or progressive kidney disease. Genetic testing for children with SSNS is not recommended at this time.
Acknowledgments
RAG is supported by NIH/NIDDK DK098135-01A1, DK094987. RAG is the recipient of a Doris Duke Clinical Scientist Development Award and acknowledge that part of this work was supported by the Doris Duke Charitable Foundation Grant # 2009033.
References
- 1.McKinney P A, Feltbower R G, Brocklebank J T, Fitzpatrick M M. Time trends and ethnic patterns of childhood nephrotic syndrome in Yorkshire, UK. Pediatr Nephrol. 2001;16(12):1040–1044. doi: 10.1007/s004670100021. [DOI] [PubMed] [Google Scholar]
- 2.North American Pediatric Renal Trials and Collaborative Studies Annual Dialysis Report 2011 Available at: https://web.emmes.com/study/ped/annlrept/annualrept2011.pdf. Accessed December 4, 2014
- 3.Rheault M N, Wei C C, Hains D S, Wang W, Kerlin B A, Smoyer W E. Increasing frequency of acute kidney injury amongst children hospitalized with nephrotic syndrome. Pediatr Nephrol. 2014;29(1):139–147. doi: 10.1007/s00467-013-2607-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kari J A. Changing trends of histopathology in childhood nephrotic syndrome in western Saudi Arabia. Saudi Med J. 2002;23(3):317–321. [PubMed] [Google Scholar]
- 5.The primary nephrotic syndrome in children . Identification of patients with minimal change nephrotic syndrome from initial response to prednisone. A report of the International Study of Kidney Disease in Children. J Pediatr. 1981;98(4):561–564. doi: 10.1016/s0022-3476(81)80760-3. [DOI] [PubMed] [Google Scholar]
- 6.Gipson D S, Chin H, Presler T P. et al. Differential risk of remission and ESRD in childhood FSGS. Pediatr Nephrol. 2006;21(3):344–349. doi: 10.1007/s00467-005-2097-0. [DOI] [PubMed] [Google Scholar]
- 7.Gbadegesin R A, Winn M P, Smoyer W E. Genetic testing in nephrotic syndrome—challenges and opportunities. Nat Rev Nephrol. 2013;9(3):179–184. doi: 10.1038/nrneph.2012.286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Grahammer F, Schell C, Huber T B. The podocyte slit diaphragm—from a thin grey line to a complex signalling hub. Nat Rev Nephrol. 2013;9(10):587–598. doi: 10.1038/nrneph.2013.169. [DOI] [PubMed] [Google Scholar]
- 9.Vernier R L, Farquhar M G, Brunson J G, Good R A. Chronic renal disease in children; correlation of clinical findings with morphologic characteristics seen by light and electron microscopy. AMA J Dis Child. 1958;96(3):306–343. [PubMed] [Google Scholar]
- 10.Kalluri R. Proteinuria with and without renal glomerular podocyte effacement. J Am Soc Nephrol. 2006;17(9):2383–2389. doi: 10.1681/ASN.2006060628. [DOI] [PubMed] [Google Scholar]
- 11.Good K S, O'Brien K, Schulman G, Kerjaschki D, Fogo A B. Unexplained nephrotic-range proteinuria in a 38-year-old man: a case of “no change disease”. Am J Kidney Dis. 2004;43(5):933–938. doi: 10.1053/j.ajkd.2003.06.006. [DOI] [PubMed] [Google Scholar]
- 12.D'Agati V D, Fogo A B, Bruijn J A, Jennette J C. Pathologic classification of focal segmental glomerulosclerosis: a working proposal. Am J Kidney Dis. 2004;43(2):368–382. doi: 10.1053/j.ajkd.2003.10.024. [DOI] [PubMed] [Google Scholar]
- 13.Habib R, Gubler M C, Antignac C, Gagnadoux M F. Diffuse mesangial sclerosis: a congenital glomerulopathy with nephrotic syndrome. Adv Nephrol Necker Hosp. 1993;22:43–57. [PubMed] [Google Scholar]
- 14.Kestilä M, Lenkkeri U, Männikkö M. et al. Positionally cloned gene for a novel glomerular protein—nephrin—is mutated in congenital nephrotic syndrome. Mol Cell. 1998;1(4):575–582. doi: 10.1016/s1097-2765(00)80057-x. [DOI] [PubMed] [Google Scholar]
- 15.Sadowski C E, Lovric S, Ashraf S. et al. A single-gene cause in 29.5% of cases of steroid-resistant nephrotic syndrome. J Am Soc Nephrol. 2015;26(6):1279–1289. doi: 10.1681/ASN.2014050489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Machuca E, Benoit G, Nevo F. et al. Genotype-phenotype correlations in non-Finnish congenital nephrotic syndrome. J Am Soc Nephrol. 2010;21(7):1209–1217. doi: 10.1681/ASN.2009121309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Patrakka J, Martin P, Salonen R. et al. Proteinuria and prenatal diagnosis of congenital nephrosis in fetal carriers of nephrin gene mutations. Lancet. 2002;359(9317):1575–1577. doi: 10.1016/S0140-6736(02)08504-5. [DOI] [PubMed] [Google Scholar]
- 18.Patrakka J, Kestilä M, Wartiovaara J. et al. Congenital nephrotic syndrome (NPHS1): features resulting from different mutations in Finnish patients. Kidney Int. 2000;58(3):972–980. doi: 10.1046/j.1523-1755.2000.00254.x. [DOI] [PubMed] [Google Scholar]
- 19.Wong W, Morris M C, Kara T. Congenital nephrotic syndrome with prolonged renal survival without renal replacement therapy. Pediatr Nephrol. 2013;28(12):2313–2321. doi: 10.1007/s00467-013-2584-7. [DOI] [PubMed] [Google Scholar]
- 20.Boute N, Gribouval O, Roselli S. et al. NPHS2, encoding the glomerular protein podocin, is mutated in autosomal recessive steroid-resistant nephrotic syndrome. Nat Genet. 2000;24(4):349–354. doi: 10.1038/74166. [DOI] [PubMed] [Google Scholar]
- 21.Hinkes B, Vlangos C, Heeringa S. et al. Specific podocin mutations correlate with age of onset in steroid-resistant nephrotic syndrome. J Am Soc Nephrol. 2008;19(2):365–371. doi: 10.1681/ASN.2007040452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hinkes B G, Mucha B, Vlangos C N. et al. Nephrotic syndrome in the first year of life: two thirds of cases are caused by mutations in 4 genes (NPHS1, NPHS2, WT1, and LAMB2) Pediatrics. 2007;119(4):e907–e919. doi: 10.1542/peds.2006-2164. [DOI] [PubMed] [Google Scholar]
- 23.Bouchireb K, Boyer O, Gribouval O. et al. NPHS2 mutations in steroid-resistant nephrotic syndrome: a mutation update and the associated phenotypic spectrum. Hum Mutat. 2014;35(2):178–186. doi: 10.1002/humu.22485. [DOI] [PubMed] [Google Scholar]
- 24.Tsukaguchi H, Sudhakar A, Le T C. et al. NPHS2 mutations in late-onset focal segmental glomerulosclerosis: R229Q is a common disease-associated allele. J Clin Invest. 2002;110(11):1659–1666. doi: 10.1172/JCI16242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tonna S, Wang Y Y, Wilson D. et al. The R229Q mutation in NPHS2 may predispose to proteinuria in thin-basement-membrane nephropathy. Pediatr Nephrol. 2008;23(12):2201–2207. doi: 10.1007/s00467-008-0934-7. [DOI] [PubMed] [Google Scholar]
- 26.Voskarides K, Arsali M, Athanasiou Y, Elia A, Pierides A, Deltas C. Evidence that NPHS2-R229Q predisposes to proteinuria and renal failure in familial hematuria. Pediatr Nephrol. 2012;27(4):675–679. doi: 10.1007/s00467-011-2084-6. [DOI] [PubMed] [Google Scholar]
- 27.Reiser J, Polu K R, Möller C C. et al. TRPC6 is a glomerular slit diaphragm-associated channel required for normal renal function. Nat Genet. 2005;37(7):739–744. doi: 10.1038/ng1592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Winn M P, Conlon P J, Lynn K L. et al. A mutation in the TRPC6 cation channel causes familial focal segmental glomerulosclerosis. Science. 2005;308(5729):1801–1804. doi: 10.1126/science.1106215. [DOI] [PubMed] [Google Scholar]
- 29.Gigante M, Caridi G, Montemurno E. et al. TRPC6 mutations in children with steroid-resistant nephrotic syndrome and atypical phenotype. Clin J Am Soc Nephrol. 2011;6(7):1626–1634. doi: 10.2215/CJN.07830910. [DOI] [PubMed] [Google Scholar]
- 30.Barua M, Brown E J, Charoonratana V T, Genovese G, Sun H, Pollak M R. Mutations in the INF2 gene account for a significant proportion of familial but not sporadic focal and segmental glomerulosclerosis. Kidney Int. 2013;83(2):316–322. doi: 10.1038/ki.2012.349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Santín S, Bullich G, Tazón-Vega B. et al. Clinical utility of genetic testing in children and adults with steroid-resistant nephrotic syndrome. Clin J Am Soc Nephrol. 2011;6(5):1139–1148. doi: 10.2215/CJN.05260610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Eckel J, Lavin P J, Finch E A. et al. TRPC6 enhances angiotensin II-induced albuminuria. J Am Soc Nephrol. 2011;22(3):526–535. doi: 10.1681/ASN.2010050522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kim J M, Wu H, Green G. et al. CD2-associated protein haploinsufficiency is linked to glomerular disease susceptibility. Science. 2003;300(5623):1298–1300. doi: 10.1126/science.1081068. [DOI] [PubMed] [Google Scholar]
- 34.Gigante M, Pontrelli P, Montemurno E. et al. CD2AP mutations are associated with sporadic nephrotic syndrome and focal segmental glomerulosclerosis (FSGS) Nephrol Dial Transplant. 2009;24(6):1858–1864. doi: 10.1093/ndt/gfn712. [DOI] [PubMed] [Google Scholar]
- 35.Benoit G, Machuca E, Nevo F, Gribouval O, Lepage D, Antignac C. Analysis of recessive CD2AP and ACTN4 mutations in steroid-resistant nephrotic syndrome. Pediatr Nephrol. 2010;25(3):445–451. doi: 10.1007/s00467-009-1372-x. [DOI] [PubMed] [Google Scholar]
- 36.Smrcka A V, Brown J H, Holz G G. Role of phospholipase Cε in physiological phosphoinositide signaling networks. Cell Signal. 2012;24(6):1333–1343. doi: 10.1016/j.cellsig.2012.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hinkes B, Wiggins R C, Gbadegesin R. et al. Positional cloning uncovers mutations in PLCE1 responsible for a nephrotic syndrome variant that may be reversible. Nat Genet. 2006;38(12):1397–1405. doi: 10.1038/ng1918. [DOI] [PubMed] [Google Scholar]
- 38.Boyer O, Benoit G, Gribouval O. et al. Mutational analysis of the PLCE1 gene in steroid resistant nephrotic syndrome. J Med Genet. 2010;47(7):445–452. doi: 10.1136/jmg.2009.076166. [DOI] [PubMed] [Google Scholar]
- 39.Gbadegesin R, Hinkes B G, Hoskins B E. et al. Mutations in PLCE1 are a major cause of isolated diffuse mesangial sclerosis (IDMS) Nephrol Dial Transplant. 2008;23(4):1291–1297. doi: 10.1093/ndt/gfm759. [DOI] [PubMed] [Google Scholar]
- 40.Gilbert R D, Turner C L, Gibson J. et al. Mutations in phospholipase C epsilon 1 are not sufficient to cause diffuse mesangial sclerosis. Kidney Int. 2009;75(4):415–419. doi: 10.1038/ki.2008.573. [DOI] [PubMed] [Google Scholar]
- 41.Shirato I, Sakai T, Kimura K, Tomino Y, Kriz W. Cytoskeletal changes in podocytes associated with foot process effacement in Masugi nephritis. Am J Pathol. 1996;148(4):1283–1296. [PMC free article] [PubMed] [Google Scholar]
- 42.Drenckhahn D, Franke R P. Ultrastructural organization of contractile and cytoskeletal proteins in glomerular podocytes of chicken, rat, and man. Lab Invest. 1988;59(5):673–682. [PubMed] [Google Scholar]
- 43.Kaplan J M, Kim S H, North K N. et al. Mutations in ACTN4, encoding alpha-actinin-4, cause familial focal segmental glomerulosclerosis. Nat Genet. 2000;24(3):251–256. doi: 10.1038/73456. [DOI] [PubMed] [Google Scholar]
- 44.Dandapani S V, Sugimoto H, Matthews B D. et al. Alpha-actinin-4 is required for normal podocyte adhesion. J Biol Chem. 2007;282(1):467–477. doi: 10.1074/jbc.M605024200. [DOI] [PubMed] [Google Scholar]
- 45.Weins A, Schlondorff J S, Nakamura F. et al. Disease-associated mutant alpha-actinin-4 reveals a mechanism for regulating its F-actin-binding affinity. Proc Natl Acad Sci U S A. 2007;104(41):16080–16085. doi: 10.1073/pnas.0702451104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Brown E J, Schlöndorff J S, Becker D J. et al. Mutations in the formin gene INF2 cause focal segmental glomerulosclerosis. Nat Genet. 2010;42(1):72–76. doi: 10.1038/ng.505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Boyer O, Benoit G, Gribouval O. et al. Mutations in INF2 are a major cause of autosomal dominant focal segmental glomerulosclerosis. J Am Soc Nephrol. 2011;22(2):239–245. doi: 10.1681/ASN.2010050518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Gbadegesin R A, Lavin P J, Hall G. et al. Inverted formin 2 mutations with variable expression in patients with sporadic and hereditary focal and segmental glomerulosclerosis. Kidney Int. 2012;81(1):94–99. doi: 10.1038/ki.2011.297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Boyer O, Nevo F, Plaisier E. et al. INF2 mutations in Charcot-Marie-Tooth disease with glomerulopathy. N Engl J Med. 2011;365(25):2377–2388. doi: 10.1056/NEJMoa1109122. [DOI] [PubMed] [Google Scholar]
- 50.Krendel M, Kim S V, Willinger T. et al. Disruption of Myosin 1e promotes podocyte injury. J Am Soc Nephrol. 2009;20(1):86–94. doi: 10.1681/ASN.2007111172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Mele C, Iatropoulos P, Donadelli R. et al. MYO1E mutations and childhood familial focal segmental glomerulosclerosis. N Engl J Med. 2011;365(4):295–306. doi: 10.1056/NEJMoa1101273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Etienne-Manneville S, Hall A. Rho GTPases in cell biology. Nature. 2002;420(6916):629–635. doi: 10.1038/nature01148. [DOI] [PubMed] [Google Scholar]
- 53.Kistler A D, Altintas M M, Reiser J. Podocyte GTPases regulate kidney filter dynamics. Kidney Int. 2012;81(11):1053–1055. doi: 10.1038/ki.2012.12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Akilesh S, Suleiman H, Yu H. et al. Arhgap24 inactivates Rac1 in mouse podocytes, and a mutant form is associated with familial focal segmental glomerulosclerosis. J Clin Invest. 2011;121(10):4127–4137. doi: 10.1172/JCI46458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Gupta I R, Baldwin C, Auguste D. et al. ARHGDIA: a novel gene implicated in nephrotic syndrome. J Med Genet. 2013;50(5):330–338. doi: 10.1136/jmedgenet-2012-101442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Gee H Y, Saisawat P, Ashraf S. et al. ARHGDIA mutations cause nephrotic syndrome via defective RHO GTPase signaling. J Clin Invest. 2013;123(8):3243–3253. doi: 10.1172/JCI69134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Gbadegesin R A, Hall G, Adeyemo A. et al. Mutations in the gene that encodes the F-actin binding protein anillin cause FSGS. J Am Soc Nephrol. 2014;25(9):1991–2002. doi: 10.1681/ASN.2013090976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Quinzii C, Naini A, Salviati L. et al. A mutation in para-hydroxybenzoate-polyprenyl transferase (COQ2) causes primary coenzyme Q10 deficiency. Am J Hum Genet. 2006;78(2):345–349. doi: 10.1086/500092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Diomedi-Camassei F, Di Giandomenico S, Santorelli F M. et al. COQ2 nephropathy: a newly described inherited mitochondriopathy with primary renal involvement. J Am Soc Nephrol. 2007;18(10):2773–2780. doi: 10.1681/ASN.2006080833. [DOI] [PubMed] [Google Scholar]
- 60.Scalais E, Chafai R, Van Coster R. et al. Early myoclonic epilepsy, hypertrophic cardiomyopathy and subsequently a nephrotic syndrome in a patient with CoQ10 deficiency caused by mutations in para-hydroxybenzoate-polyprenyl transferase (COQ2) Eur J Paediatr Neurol. 2013;17(6):625–630. doi: 10.1016/j.ejpn.2013.05.013. [DOI] [PubMed] [Google Scholar]
- 61.Heeringa S F, Chernin G, Chaki M. et al. COQ6 mutations in human patients produce nephrotic syndrome with sensorineural deafness. J Clin Invest. 2011;121(5):2013–2024. doi: 10.1172/JCI45693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Ashraf S, Gee H Y, Woerner S. et al. ADCK4 mutations promote steroid-resistant nephrotic syndrome through CoQ10 biosynthesis disruption. J Clin Invest. 2013;123(12):5179–5189. doi: 10.1172/JCI69000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.López L C, Schuelke M, Quinzii C M. et al. Leigh syndrome with nephropathy and CoQ10 deficiency due to decaprenyl diphosphate synthase subunit 2 (PDSS2) mutations. Am J Hum Genet. 2006;79(6):1125–1129. doi: 10.1086/510023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Gasser D L, Winkler C A, Peng M. et al. Focal segmental glomerulosclerosis is associated with a PDSS2 haplotype and, independently, with a decreased content of coenzyme Q10. Am J Physiol Renal Physiol. 2013;305(8):F1228–F1238. doi: 10.1152/ajprenal.00143.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Montini G, Malaventura C, Salviati L. Early coenzyme Q10 supplementation in primary coenzyme Q10 deficiency. N Engl J Med. 2008;358(26):2849–2850. doi: 10.1056/NEJMc0800582. [DOI] [PubMed] [Google Scholar]
- 66.Kurogouchi F, Oguchi T, Mawatari E. et al. A case of mitochondrial cytopathy with a typical point mutation for MELAS, presenting with severe focal-segmental glomerulosclerosis as main clinical manifestation. Am J Nephrol. 1998;18(6):551–556. doi: 10.1159/000013406. [DOI] [PubMed] [Google Scholar]
- 67.Yorifuji T, Kawai M, Momoi T. et al. Nephropathy and growth hormone deficiency in a patient with mitochondrial tRNA(Leu(UUR)) mutation. J Med Genet. 1996;33(7):621–622. doi: 10.1136/jmg.33.7.621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Miner J H. The glomerular basement membrane. Exp Cell Res. 2012;318(9):973–978. doi: 10.1016/j.yexcr.2012.02.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Sachs N, Sonnenberg A. Cell-matrix adhesion of podocytes in physiology and disease. Nat Rev Nephrol. 2013;9(4):200–210. doi: 10.1038/nrneph.2012.291. [DOI] [PubMed] [Google Scholar]
- 70.Zenker M, Aigner T, Wendler O. et al. Human laminin beta2 deficiency causes congenital nephrosis with mesangial sclerosis and distinct eye abnormalities. Hum Mol Genet. 2004;13(21):2625–2632. doi: 10.1093/hmg/ddh284. [DOI] [PubMed] [Google Scholar]
- 71.Matejas V, Hinkes B, Alkandari F. et al. Mutations in the human laminin beta2 (LAMB2) gene and the associated phenotypic spectrum. Hum Mutat. 2010;31(9):992–1002. doi: 10.1002/humu.21304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Has C, Spartà G, Kiritsi D. et al. Integrin α3 mutations with kidney, lung, and skin disease. N Engl J Med. 2012;366(16):1508–1514. doi: 10.1056/NEJMoa1110813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Kambham N, Tanji N, Seigle R L. et al. Congenital focal segmental glomerulosclerosis associated with beta4 integrin mutation and epidermolysis bullosa. Am J Kidney Dis. 2000;36(1):190–196. doi: 10.1053/ajkd.2000.8293. [DOI] [PubMed] [Google Scholar]
- 74.Gross O, Perin L, Deltas C. Alport syndrome from bench to bedside: the potential of current treatment beyond RAAS blockade and the horizon of future therapies. Nephrol Dial Transplant. 2014;29 04:iv124–iv130. doi: 10.1093/ndt/gfu028. [DOI] [PubMed] [Google Scholar]
- 75.Malone A F, Phelan P J, Hall G. et al. Rare hereditary COL4A3/COL4A4 variants may be mistaken for familial focal segmental glomerulosclerosis. Kidney Int. 2014;86(6):1253–1259. doi: 10.1038/ki.2014.305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Miner J H. Pathology vs. molecular genetics: (re)defining the spectrum of Alport syndrome. Kidney Int. 2014;86(6):1081–1083. doi: 10.1038/ki.2014.326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Pierides A, Voskarides K, Athanasiou Y. et al. Clinico-pathological correlations in 127 patients in 11 large pedigrees, segregating one of three heterozygous mutations in the COL4A3/ COL4A4 genes associated with familial haematuria and significant late progression to proteinuria and chronic kidney disease from focal segmental glomerulosclerosis. Nephrol Dial Transplant. 2009;24(9):2721–2729. doi: 10.1093/ndt/gfp158. [DOI] [PubMed] [Google Scholar]
- 78.Niaudet P, Gubler M C. WT1 and glomerular diseases. Pediatr Nephrol. 2006;21(11):1653–1660. doi: 10.1007/s00467-006-0208-1. [DOI] [PubMed] [Google Scholar]
- 79.Wagner N, Wagner K D, Xing Y, Scholz H, Schedl A. The major podocyte protein nephrin is transcriptionally activated by the Wilms' tumor suppressor WT1. J Am Soc Nephrol. 2004;15(12):3044–3051. doi: 10.1097/01.ASN.0000146687.99058.25. [DOI] [PubMed] [Google Scholar]
- 80.Guo G, Morrison D J, Licht J D, Quaggin S E. WT1 activates a glomerular-specific enhancer identified from the human nephrin gene. J Am Soc Nephrol. 2004;15(11):2851–2856. doi: 10.1097/01.ASN.0000143474.91362.C4. [DOI] [PubMed] [Google Scholar]
- 81.Ratelade J, Arrondel C, Hamard G. et al. A murine model of Denys-Drash syndrome reveals novel transcriptional targets of WT1 in podocytes. Hum Mol Genet. 2010;19(1):1–15. doi: 10.1093/hmg/ddp462. [DOI] [PubMed] [Google Scholar]
- 82.Pelletier J, Bruening W, Kashtan C E. et al. Germline mutations in the Wilms' tumor suppressor gene are associated with abnormal urogenital development in Denys-Drash syndrome. Cell. 1991;67(2):437–447. doi: 10.1016/0092-8674(91)90194-4. [DOI] [PubMed] [Google Scholar]
- 83.Lipska B S, Ranchin B, Iatropoulos P. et al. Genotype-phenotype associations in WT1 glomerulopathy. Kidney Int. 2014;85(5):1169–1178. doi: 10.1038/ki.2013.519. [DOI] [PubMed] [Google Scholar]
- 84.Barbaux S, Niaudet P, Gubler M C. et al. Donor splice-site mutations in WT1 are responsible for Frasier syndrome. Nat Genet. 1997;17(4):467–470. doi: 10.1038/ng1297-467. [DOI] [PubMed] [Google Scholar]
- 85.Gwin K, Cajaiba M M, Caminoa-Lizarralde A, Picazo M L, Nistal M, Reyes-Múgica M. Expanding the clinical spectrum of Frasier syndrome. Pediatr Dev Pathol. 2008;11(2):122–127. doi: 10.2350/07-01-0209.1. [DOI] [PubMed] [Google Scholar]
- 86.Hall G, Gbadegesin R A, Lavin P. et al. A novel missense mutation of Wilms' Tumor 1 causes autosomal dominant FSGS. J Am Soc Nephrol. 2015;26(4):831–843. doi: 10.1681/ASN.2013101053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Sarin S, Javidan A, Boivin F. et al. Insights into the renal pathogenesis in Schimke immuno-osseous dysplasia: a renal histological characterization and expression analysis. J Histochem Cytochem. 2015;63(1):32–44. doi: 10.1369/0022155414558335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Boerkoel C F, Takashima H, John J. et al. Mutant chromatin remodeling protein SMARCAL1 causes Schimke immuno-osseous dysplasia. Nat Genet. 2002;30(2):215–220. doi: 10.1038/ng821. [DOI] [PubMed] [Google Scholar]
- 89.Burghardt T, Kastner J, Suleiman H. et al. LMX1B is essential for the maintenance of differentiated podocytes in adult kidneys. J Am Soc Nephrol. 2013;24(11):1830–1848. doi: 10.1681/ASN.2012080788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Lemley K V. Kidney disease in nail-patella syndrome. Pediatr Nephrol. 2009;24(12):2345–2354. doi: 10.1007/s00467-008-0836-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Sweeney E, Fryer A, Mountford R, Green A, McIntosh I. Nail patella syndrome: a review of the phenotype aided by developmental biology. J Med Genet. 2003;40(3):153–162. doi: 10.1136/jmg.40.3.153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Boyer O, Woerner S, Yang F. et al. LMX1B mutations cause hereditary FSGS without extrarenal involvement. J Am Soc Nephrol. 2013;24(8):1216–1222. doi: 10.1681/ASN.2013020171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Esposito T, Lea R A, Maher B H. et al. Unique X-linked familial FSGS with co-segregating heart block disorder is associated with a mutation in the NXF5 gene. Hum Mol Genet. 2013;22(18):3654–3666. doi: 10.1093/hmg/ddt215. [DOI] [PubMed] [Google Scholar]
- 94.Galloway W H, Mowat A P. Congenital microcephaly with hiatus hernia and nephrotic syndrome in two sibs. J Med Genet. 1968;5(4):319–321. doi: 10.1136/jmg.5.4.319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Colin E, Huynh Cong E, Mollet G. et al. Loss-of-function mutations in WDR73 are responsible for microcephaly and steroid-resistant nephrotic syndrome: Galloway-Mowat syndrome. Am J Hum Genet. 2014;95(6):637–648. doi: 10.1016/j.ajhg.2014.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Amsellem S, Gburek J, Hamard G. et al. Cubilin is essential for albumin reabsorption in the renal proximal tubule. J Am Soc Nephrol. 2010;21(11):1859–1867. doi: 10.1681/ASN.2010050492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Hauck F H, Tanner S M, Henker J, Laass M W. Imerslund-Gräsbeck syndrome in a 15-year-old German girl caused by compound heterozygous mutations in CUBN. Eur J Pediatr. 2008;167(6):671–675. doi: 10.1007/s00431-007-0571-3. [DOI] [PubMed] [Google Scholar]
- 98.Balreira A, Gaspar P, Caiola D. et al. A nonsense mutation in the LIMP-2 gene associated with progressive myoclonic epilepsy and nephrotic syndrome. Hum Mol Genet. 2008;17(14):2238–2243. doi: 10.1093/hmg/ddn124. [DOI] [PubMed] [Google Scholar]
- 99.Berkovic S F, Dibbens L M, Oshlack A. et al. Array-based gene discovery with three unrelated subjects shows SCARB2/LIMP-2 deficiency causes myoclonus epilepsy and glomerulosclerosis. Am J Hum Genet. 2008;82(3):673–684. doi: 10.1016/j.ajhg.2007.12.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Vande Walle J, Donckerwolcke R, Boer P, van Isselt H W, Koomans H A, Joles J A. Blood volume, colloid osmotic pressure and F-cell ratio in children with the nephrotic syndrome. Kidney Int. 1996;49(5):1471–1477. doi: 10.1038/ki.1996.207. [DOI] [PubMed] [Google Scholar]
- 101.Ozaltin F, Ibsirlioglu T, Taskiran E Z. et al. Disruption of PTPRO causes childhood-onset nephrotic syndrome. Am J Hum Genet. 2011;89(1):139–147. doi: 10.1016/j.ajhg.2011.05.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Ozaltin F, Li B, Rauhauser A. et al. DGKE variants cause a glomerular microangiopathy that mimics membranoproliferative GN. J Am Soc Nephrol. 2013;24(3):377–384. doi: 10.1681/ASN.2012090903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Sanna-Cherchi S, Burgess K E, Nees S N. et al. Exome sequencing identified MYO1E and NEIL1 as candidate genes for human autosomal recessive steroid-resistant nephrotic syndrome. Kidney Int. 2011;80(4):389–396. doi: 10.1038/ki.2011.148. [DOI] [PubMed] [Google Scholar]
- 104.Agarwal A K, Zhou X J, Hall R K. et al. Focal segmental glomerulosclerosis in patients with mandibuloacral dysplasia owing to ZMPSTE24 deficiency. J Investig Med. 2006;54(4):208–213. doi: 10.2310/6650.2006.05068. [DOI] [PubMed] [Google Scholar]
- 105.Sethi S, Fervenza F C, Zhang Y, Smith R J. Secondary focal and segmental glomerulosclerosis associated with single-nucleotide polymorphisms in the genes encoding complement factor H and C3. Am J Kidney Dis. 2012;60(2):316–321. doi: 10.1053/j.ajkd.2012.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Fisher P W, Ho L T, Goldschmidt R, Semerdjian R J, Rutecki G W. Familial Mediterranean fever, inflammation and nephrotic syndrome: fibrillary glomerulopathy and the M680I missense mutation. BMC Nephrol. 2003;4:6. doi: 10.1186/1471-2369-4-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Friedman D J, Kozlitina J, Genovese G, Jog P, Pollak M R. Population-based risk assessment of APOL1 on renal disease. J Am Soc Nephrol. 2011;22(11):2098–2105. doi: 10.1681/ASN.2011050519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Limou S, Nelson G W, Kopp J B, Winkler C A. APOL1 kidney risk alleles: population genetics and disease associations. Adv Chronic Kidney Dis. 2014;21(5):426–433. doi: 10.1053/j.ackd.2014.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Kopp J B, Nelson G W, Sampath K. et al. APOL1 genetic variants in focal segmental glomerulosclerosis and HIV-associated nephropathy. J Am Soc Nephrol. 2011;22(11):2129–2137. doi: 10.1681/ASN.2011040388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Okamoto K, Tokunaga K, Doi K. et al. Common variation in GPC5 is associated with acquired nephrotic syndrome. Nat Genet. 2011;43(5):459–463. doi: 10.1038/ng.792. [DOI] [PubMed] [Google Scholar]
- 111.Chehade H, Cachat F, Girardin E. et al. Two new families with hereditary minimal change disease. BMC Nephrol. 2013;14:65. doi: 10.1186/1471-2369-14-65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Gee H Y, Ashraf S, Wan X. et al. Mutations in EMP2 cause childhood-onset nephrotic syndrome. Am J Hum Genet. 2014;94(6):884–890. doi: 10.1016/j.ajhg.2014.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Gbadegesin R A Adeyemo A Webb N J et al. HLA-DQA1 and PLCG2 are candidate risk loci for childhood-onset steroid-sensitive nephrotic syndrome J Am Soc Nephrol 2014; In press. Doi: 10.1681/ASN.2014030247 [DOI] [PMC free article] [PubMed] [Google Scholar]