

Abstract
Chronic kidney disease (CKD) in children is an irreversible process that, in some cases, may lead to end-stage renal disease. The majority of children with CKD have a congenital disorder of the kidney or urological tract arising from birth. There is strong evidence for both a genetic and epigenetic component to progression of CKD. Utilization of gene-mapping strategies, ranging from genome-wide association studies to single-nucleotide polymorphism analysis, serves to identify potential genetic variants that may lend to disease variation. Genome-wide association studies evaluating population-based data have identified different loci associated with CKD progression. Analysis of single-nucleotide polymorphisms on an individual level suggests that secondary systemic sequelae of CKD are closely related to dysfunction of the cardiovascular-inflammatory axis and may lead to advanced cardiovascular disease through abnormal vascular calcification and activation of the renin–angiotensin system. Similarly, genetic variants affecting cytokine control, fibrosis, and parenchymal development may modulate CKD through development and acceleration of renal interstitial fibrosis. Epigenetic studies evaluate modification of the genome through DNA methylation, histone modification, or RNA interference, which may be directly influenced by external or environmental factors directing genomic expression. Lastly, improved understanding of the genetic and epigenetic contribution to CKD progression may allow providers to identify a population at accelerated risk for disease progression and apply novel therapies targeted at the genetic mechanism of disease.
Keywords: pediatric chronic kidney disease, epigenetic modifications, single-nucleotide polymorphisms
Introduction
Chronic kidney disease (CKD) is defined as a progressive loss of renal function, measured by a decline in glomerular filtration rate (GFR). Most clinical and genetic epidemiological studies define CKD as estimated GFR less than 60 mL/min/1.73 m2.1 The interplay of heritable genetic traits on progression of CKD toward end-stage renal disease (ESRD) remains an evolving field in adult and pediatric nephrology. CKD in adults occurs primarily in the developed world as a consequence of longstanding disease processes, such as diabetes or hypertension, but may result also from primary glomerulopathies, infections, physical obstruction, interstitial nephritides, and genetic cystic kidney diseases.2 On the contrary, in pediatric patients the etiology of CKD is dominated by congenital structural disorders occurring in the first year of life, primarily due to dysplasia or obstructive causes and monogenetic renal diseases that comprise ∼20% of progressive CKD/ESRD diagnoses.3 For example, specific single-gene genetic mutations causing hereditary proteinuria/nephrotic syndrome (nephrin [NPHS1], podocin [NPHS2], α-actinin-4, CD-2 molecule [CD-2], Wilms tumor 1 [WT1], transient receptor potential cation channel, subfamily C, member 6 [TRPC6], phospholipase C, epsilon 1 [PLCE1], laminin β-2 [LAMB2], LIM homeobox transcription factor 1 β [LMX1B], myosin, heavy chain 9 [MYH9], fibronectin), Alport syndrome (type IV collagen), and hereditary tubulointerstitial diseases (polycystic kidney disease [PKD1, PKD2], juvenile nephronophthisis [NPHP1], UMOD, HNF1 homeobox B [HNF1B])4 are associated with progressive renal disease, often leading to ESRD and need for renal transplantation. However, monogenic causes of CKD are beyond the scope of this review.
Regardless of the cause, chronically injured kidneys are characterized histologically by tubulointerstitial fibrosis and glomerulosclerosis and the extent of tubulointerstitial fibrosis is the best predictor of kidney survival irrespectively of the underlying disease.5 Interestingly, even in the presence of identical underlying disease states with similar histopathology and known genetic loci, CKD progression can be very variable in nature.6 7 This may further be explained by the complexity of CKD progression involving both an inherited predisposition (DNA) and susceptibility to environmental factors. To illustrate, in a 26,000 adult dialysis patient registry, nearly 23% of patients without Mendelian single-gene mutations were found to have another first- or second-degree relative on dialysis,8 and risk for CKD progressing to ESRD is particularly marked with a strong family history (e.g., two or more first-degree relatives) of renal disease.8 9
Emerging evidence suggests that polymorphic genetic variations (i.e., single-nucleotide polymorphisms [SNPs]) may be key factors in CKD progression, in addition to more recently identified epigenetic modifications of the genome. The vast majority of genetic polymorphic research in the field of CKD progression has been performed with adult data and may confer inherent weakness in extrapolation of data directly to pediatric patients. Contrary to adult CKD, pediatric CKD characteristically occurs without comorbidities such as diabetes and cardiovascular disease. Pediatric patients may, conversely, develop early-onset cardiovascular disease, nutritional abnormalities, and growth disruption as sequelae of renal disease. In spite of the differences, it is likely that the information gleaned from adult genetic and epigenetic analyses can reasonably be applied to our understanding of pediatric CKD progression. The following text reviews our current understanding of genetic and epigenetic factors that could be applicable to pediatric CKD progression.
Gene Mapping Strategies in Chronic Kidney Disease
Complex traits, such as CKD, do not have recognizable Mendelian inheritance patterns or concordance between genetic variation and phenotype. Genetic analyses provide an integral role in identifying how progression of CKD differs between individuals, but they require thoughtful collaboration between laboratory, clinical, and epidemiological resources in their utilization to provide meaningful results (Fig. 1).10 Genetic mapping provides a link between the human genotype and phenotype of CKD progression. In understanding the genetics of CKD, an accurate disease phenotype must be established to reliably test an assumed genotype–phenotype relationship, which makes the comparison among studies challenging. Despite limitations, improvement in laboratory techniques and bioinformatics allows for increasingly cost-effective genotyping approaches, ranging from high-density microarrays that efficiently examine several million SNPs in a targeted manner to next-generation sequencing approaches that interrogate an individual's entire genome. Gene mapping strategies such as candidate gene approaches, linkage studies, and genome-wide association studies (GWASs) have been utilized in further delineating the genetic and epigenetic basis of CKD.
Fig. 1.
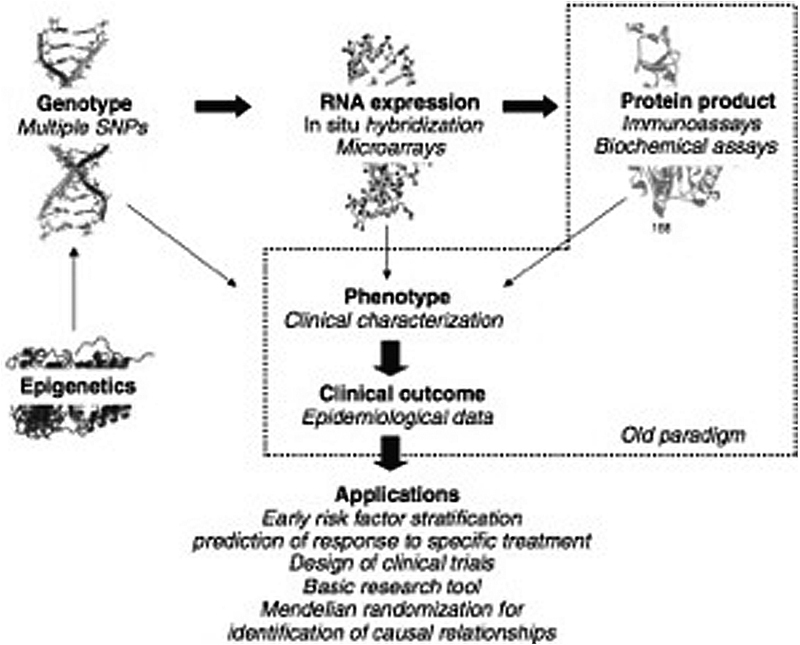
Genetic analyses play a key role in understanding chronic kidney disease progression, but require close interaction with laboratory, clinical, and epidemiological resources to provide clinical application to both the individual patient and population as a whole. (Figure obtained with permission from Axelsson et al.10)
Candidate gene studies utilize analysis of previously identified polymorphisms or regulatory genes with known regulatory functions to search for novel genetic variants associated with common disease progression. Candidate gene studies currently provide much of the pediatric data available for understanding the genetic mechanisms of pediatric CKD progression in relation to cardiovascular disease and progression of tubulointerstitial fibrosis, as described below. This approach to genetic analysis is limited by variability in genotyping methods utilized, phenotypic differences in the population studied, and small sample sizes (particularly in pediatric studies) with subsequent poor power to detect significance.11 Furthermore, study design in candidate gene studies may be prone to influence from investigator biases regarding underlying genetic mechanisms for CKD progression, in that the investigator chooses candidate genes a priori such that a study is hypothesis-driven rather than hypothesis-generating in nature.12
Linkage studies employ family-based collections of DNA to track transmission of genetic variants linked with a common disease and relate inheritance of sparsely distributed polymorphic genetic markers with disease phenotypes among families. Linkage studies have been primarily successful identifying rare genetic markers with disease phenotypes within families such as autosomal dominant polycystic kidney disease.13 This methodology has also been used in understanding genetic factors associated with CKD progression. More than 20 larger scale linkage studies have been conducted to identify loci associated with CKD,14 including genome-wide association linkage studies and genome scan meta-analysis, the latest being an exploratory technique used to quantitatively synthesize linkage results from individual studies to evaluate concordance.15 A challenge in conducting linkage studies lies particularly in recruitment of family members to provide a sufficient population within a sample for tracking genetic risk across generations.14 In an attempt to overcome this limitation, Rao et al16 conducted a meta-analysis of genome-wide linkage scans for renal function traits with data from 14 linkage studies but failed to identify any significant genomic region for renal function traits of GFR, urinary albumin-to-creatinine ratio, serum creatinine, or creatinine clearance.
GWAS is a more powerful approach in diseases like CKD where the genetic risk factors have small or moderate effect sizes; it offers improved resolution for genotyping and makes easier the collection of unrelated renal phenotype cases and controls. GWAS compares the frequency of naturally occurring genetic variants in populations with a disease to those without disease and drives the genetic study of CKD based on the “common disease, common variant” hypothesis.7 This concept is based on the assumption that common diseases are caused by genetic variation occurring within the genome at a frequency greater than 1% within the general population17 and that functional changes associated with specific mutations are mild. Therefore, many common variants are required to segregate in individuals with common diseases in order for phenotypic disease to become clinically apparent. In GWASs, a stringent phenotype definition allows for improved statistical power to more reliably associate phenotype with an otherwise discovered genetic etiology of CKD.18
More than 1,500 GWASs have been published since 2005 with the goal of identifying genetic polymorphisms predisposing to CKD progression,19 and since that time, GWAS data in the field of CKD remain extremely adult-focused with no currently published primary pediatric CKD GWASs. The first GWAS of CKD was performed in 19,877 individuals of European ancestry from the Cohorts for Heart and Aging Research in Genomic Epidemiology Consortium20 to identify genetic risk loci for CKD. A significant risk locus was identified within the UMOD gene. A secondary GWAS further validated the initial association of CKD progression with UMOD, whereby polymorphisms conferring elevated UMOD concentrations increased the 10-year risk of CKD by over 70%.21 Use of GWAS provided evidence that a common UMOD gene variant, which was previously known to cause autosomal dominant medullary cystic kidney disease type 2 and familial juvenile hyperuricemic nephropathy,22 23 was present in up to 18% of a large study population and could contribute to progression of CKD in the general population.20 More recently, data from over 67,000 participants within the CKDGen Consortium were evaluated in meta-analytic discovery genome-wide analysis to identify susceptibility loci for reduced renal function, with additional samples from over 20,000 individuals subsequently reviewed in replication follow-up.24 The discovery analysis from GWAS identified 13 new loci associated with reduced renal function and CKD (longevity assurance homologue 2 of yeast [LASS2], glucokinase regulatory protein [GCKR], Alstrom syndrome protein 1 [ALMS1], transcription factor Dp-2 [TFDP2], disabled 2 mitogen-responsive phosphoprotein [DAB2], solute carrier family 24 member 1 [SLC34A1], vascular endothelial growth factor A [VEGFA], protein kinase AMP-activated, gamma 2 noncatalytic subunit [PRKAG2], phosphatidylinositol-4-phosphate-5-kinase, type 1 β [PIP5K1B], ataxin 2 [ATXN2], dachshund family transcription factor 1 [DACH1], ubiquitin-conjugating enzyme E2Q family member 2 [UBE2Q2], and solute carrier family 7 [SLC7A9]) and 7 loci suspected to affect creatinine production and secretion (carbamoyl-phosphate synthetase 1 [CPS1], solute carrier family 22 [SLC22A2], transmembrane protein 60 [TMEM60], WD repeat domain 37 [WDR37], solute carrier family 6 (neurotransmitter transporter), member 13 [SLC6A13], WD repeat domain 72 [WDR72], and breast carcinoma amplified sequence 2 [BCAS2]), in addition to the previously discovered susceptibility variants for renal function and CKD at UMOD, shroom family member 2 (SHROOM2) and stanniocalcin 1 (STC1) loci (Fig. 2). Of those, it is important to highlight some of the known to be affected in renal disease such as ALMS1 (ciliopathies), SLC7A9 (nephrolithiasis, cystinuria), SLC34A1 (hypophosphatemic nephrolithiasis/osteoporosis), DAB2 (nondiabetic kidney disease in African Americans), VEGFA (stimulation of ureteric bud branching during embryogenesis and affects final nephron number, it is secreted by podocytes), GCKR (involved in formation of primary cilia), PRKAG2 (renal hypertrophy), and DACH1 (branchio-oto-renal syndrome).24 Another gene that was found to be associated with CKD from GWASs is the MYH9-APOL1 (myosin heavy chain 9, apolipoprotein L, 1) gene on chromosome 22,25 26 which is known to be associated with focal segmental glomerulosclerosis, diabetic kidney disease, and ESRD.27 28 Interestingly, APOL1 SNPs associated with kidney disease are common in African lineage but extremely rare in European populations.26
Fig. 2.
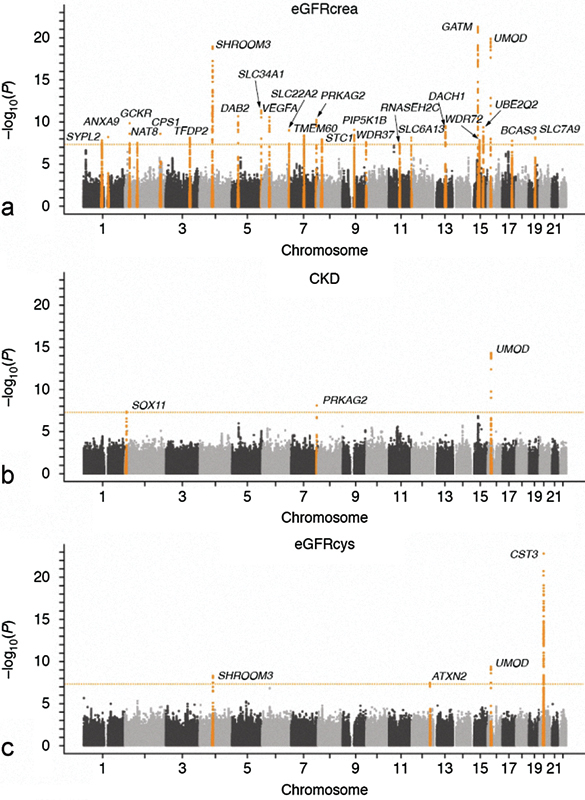
Manhattan plot from Köttgen et al24 illustrating the discovery analysis from a GWAS that identified 13 new genetic loci associated with reduced renal function and chronic kidney disease with an additional 7 loci suspected to affect creatinine production and secretion. Previously discovered susceptibility variants for renal function and chronic kidney disease at UMOD, SHROOM2, and STC1 are also shown.24 The dotted line indicates the genome-wide significance threshold at p = 5 × 10 − 8.
One of the most significant weaknesses in utilization of GWASs is the inability to generalize results for individuals who are not of the primary ethnicity represented in the sample analyzed. In an effort to understand the impact of ethnicity, cross-ethnic analyses have been performed with data from over 8,000 samples within the Candidate-gene Association Resource (CARe) Consortium, demonstrating that genomic risk regions are largely conserved and shared across ethnic groups.29
Genetic Polymorphism in Pediatric Chronic Kidney Disease
In pediatric CKD, it is reasonable to consider that polymorphic mutations linked with common diseases may further alter the amount or activity of progression of CKD. Numerous genetic polymorphisms have been identified that are associated with pediatric CKD progression. These polymorphic variations may account for some of the differences in both focal and systemic disease associated with renal impairment (e.g., cardiovascular disease and development of tubulointerstitial fibrosis) between individual patients with pediatric CKD. We review a selection of key functional polymorphisms in the following sections.
Cardiovascular Polymorphisms in Chronic Kidney Disease Progression
Cardiovascular disease is a well-recognized feature of CKD progression. Hypertension and subsequent prevention of end-organ damage (including left ventricular hypertrophy [LVH]) are primary therapeutic targets in disease progression. Ultimately, cardiovascular-related death remains the leading cause of mortality in children on maintenance dialysis and posttransplantation across the world.30 31 32
Vascular calcification and atherosclerotic cardiovascular disease are well-known complications in the transition from CKD to ESRD and are important risk factors for cardiovascular disease. Fetuin-A (also known as α 2-Heremans-Schmid glycoprotein) is a circulating negative acute-phase protein released in response to an inflammatory stimulus on endothelial surfaces that inhibits calcium phosphate product formation and precipitation on the vascular endothelial surface.33 34 35 CKD patients appear to have significantly lower Fetuin-A levels, which is associated with increased risk for vascular disease and poorer hemodialysis survival in CKD.33 36 The Fetuin-A Thr256Ser polymorphism is linked to increased atherosclerosis and heightened cardiovascular mortality in the ESRD population and may be a marker of accelerated vascular calcification risk.36 Although the aforementioned studies provide insight into genetic cardiovascular risk in CKD, none included data from a pediatric population. Data available from one pediatric sample do demonstrate that lower Fetuin-A levels predict intima-media thickness changes of the carotid arteries in children with CKD.37
The Renin–Angiotensin System
The renin–angiotensin system (RAS) facilitates blood pressure regulation, with activation and upregulation in CKD leading to end-organ damage, including LVH.38 Polymorphic change in the angiotensinogen (ANG) gene is associated with progressive cardiovascular and renal disease. The ANG gene has a Met235Thr polymorphism with Thr/Thr haplotype associated with increased risk for hypertension and cardiac remodeling with LVH in both healthy and ESRD populations secondary to high ANG levels.39 40 41 Insertion/deletion (I/D) polymorphisms in the angiotensin-converting enzyme (ACE) gene have been shown to affect the activity of the RAS.42 The ACE has a known I/D polymorphism in intron 16, and deletion/deletion (D/D) individuals have higher levels of ACE. The ACE D/D polymorphism is linked with LVH,43 hypertension,44 and progression of renal disease in children with known CKD.45 46 47 There is additional evidence that the ACE D/D genotype may confer increased risk for renal parenchymal damage in children with congenital abnormalities of the urologic tract and renal scarring following urinary tract infections.45 48 49 50 The effect of these genes on renal scarring has been postulated due to gene roles in both local vasomotor and inflammatory control within renal parenchyma following injury. Patients with the ACE D/D genotype may have a more favorable response to ACE inhibition with decrease in proteinuria and reduction in rate of GFR decline in contrast to patients with I/D or insertion/insertion genotypes.51 52
ACE activity determines levels of the vasoactive peptide angiotensin-II (ANG II). ANG II has both systemic and intrarenal vasoconstrictor effects. The intrarenal effects of ANG II are driven by the angiotensin type 1 receptor (AT1R) and angiotensin type 2 receptor (AT2R). Receptor polymorphism analyses have yet to provide significant associations between known polymorphisms for the AT1R or AT2R and progression of CKD. The AT1R polymorphism at A1166C has minimal evidence for CKD progression.44 53 Similarly, the AT2R A1332G polymorphism has been investigated with no significant effect on the pathogenesis or outcomes of CKD progression in patients with vesicoureteral reflux in individual studies54 55 56 or meta-analytic review of existing data.53
Inflammatory Polymorphisms in Chronic Kidney Disease Progression
Progressive CKD can be considered a proinflammatory state. In CKD, perturbations in the inflammatory-axis occur secondary to multiple factors (e.g., oxidative stress, decreased clearance of inflammatory cytokines with falling GFR, poor antioxidant intake) and can lead to accelerated atherosclerosis, endothelial damage, and malnutrition via induced anorexia and protein-energy wasting.
Homocysteine is an inflammatory molecule implicated in risk for vascular disease. CKD patients are at risk for hyperhomocysteinemia secondary to nutritional and intradialytic losses of vitamin B12 and folate. The enzyme methylenetetrahydrofolate reductase (MTFHR) is key in homocysteine regulation via reduction of homocysteine to methionine.57 Genetic variation in the MTHFR gene may lead to homocysteine accumulation in patients carrying a genetic polymorphism of the MTHFR gene, Ala677VaL (C/T), conferring reduction of enzyme function.58 Data suggest that pediatric dialysis patients having a MTHFR (C/T) polymorphism are at increased risk for hyperhomocysteinemia, although it is unclear exactly what the implications are for long-term cardiovascular disease progression.59
Interleukin (IL)-10 is an anti-inflammatory cytokine that mediates the inflammatory response through regulation of the adaptive immune system interaction with antigen-presenting cells with subsequent ability to downregulate release of proinflammatory molecules, such as IL-1 β, tumor necrosis factor-α, and IL-6.60 Several IL-10 polymorphisms have been identified within the promoter region of the IL-10 gene; however, data for the G/G-1082 polymorphism are the most robust. The G/G-1082 gene polymorphism results in quantitatively higher levels of IL-10 in comparison to A/A- or A/G-1082 variants.61 62 Patients who are high or intermediate IL-10 producers (G/G and G/A alleles) are more likely to rate higher Karnofsky functional status scores than low producers (A/A alleles).63 Conversely, animal models suggest that constitutively high levels of IL-10 could induce anorexia and weight loss.64 Cumulatively, data demonstrate that under some conditions, IL-10 has a protective effect in counterbalancing the inflammatory response with decreased comorbidity65 and cardiovascular events,66 but unchecked, high levels of IL-10 may be associated with adverse patient outcomes.
Genetic Variants in Fibrosis and Parenchymal Development: Transforming Growth Factor-β
Fibrosis is one of the histological landmarks of progressive CKD. Transforming growth factor-β (TGF-β) is a cytokine that regulates cell growth and extracellular matrix production.67 Prolonged expression of TGF-β has been associated with disruption of renal architecture at the cellular level, leading to progression of CKD, specifically tubulointerstitial fibrosis and extracellular matrix deposition.68 TGF-β mediates progression of renal and cardiac fibrosis in association with activation of ANG II within renal parenchyma and systemic RAS activation, with subsequent development of hypertension69 and secondary LVH.70 ANG II blockade (e.g., with ACE inhibitors or angiotensin receptor blocking agents) has been shown to suppress TGF-β overexpression in kidney and heart.71 TGF-β production may vary between patients, given the presence of several polymorphic variants identified that modulate levels of cytokine production.72 Furthermore, risk for scarring following urinary tract infection is significantly increased in children with the TGF-β1 509T polymorphism, irrespective of presence or absence of vesicoureteral reflux.73
Role of Epigenetics in Chronic Kidney Disease
Epigenetic modifications of DNA occur through DNA methylation, histone modifications, and RNA interference. They serve as key regulators in gene expression and repression to allow for normal cellular functioning. The ability to identify and target epigenetic modifications, in contrast to polymorphic variation, represents an opportunity to pinpoint CKD-related processes that are reversible, developmentally and temporally variable, and susceptible to environmental cues.74 There is some evidence suggesting that some extreme environmental cues, for example, intrauterine growth restriction or starvation, induce long-term epigenetic genomic modifications.75 Epigenetic modifications in the fetal environment are thought to play a key role in the link between low-birth-weight infants and congenital deficits in nephron number,76 which ultimately lead to an increased lifetime risk of hypertension, glomerulosclerosis, and CKD/ESRD.77 Moreover, CKD progression may be influenced by external factors acting on the patient epigenome, opening new options for novel therapeutic approaches in the future. For example, chronic inflammation (as evidenced by hyperhomocysteinemia) and oxidative stress have been hypothesized to act as negative epigenetic risk factors in CKD progression74 and correction of hyperhomocysteinemia with exogenous folate therapy has been shown to correct significant DNA hypomethylation in an adult hemodialysis population.78 Bechtel et al79 noted that cytosine hypermethylation of the RAS protein activator-like 1 (RASAL1) gene was associated with activation of fibroblasts and fibrogenesis in mouse models and that fibrogenesis could be rescued in vitro using a demethylating agent, 5′-azacitidine. Similarly, Ko et al80 describe cytosine methylation analysis of renal epithelial samples in both diseased and control patients with more than 4,000 differentially methylated enhancer regions found in CKD samples compared with controls.
Deregulation of histone modification has been widely implicated in renal disease leading to CKD, for example, congenital renal anomalies, fibrosis, and diabetic renal sequelae.81 82 Angiotensin blockade with Losartan reverses histone H3 modification within the glomeruli83 of diabetic mice, suggesting additional therapeutic benefit of adequacy in drug utilization. Similarly in CKD, TGF-β production increases histone H3 methylation with subsequent upregulation of profibrotic collagen-1α1 within the extracellular matrix and plasminogen activator inhibitor-1 in mesangial cells.84 Inhibition of TGF-β and renal fibroblast activation is potentially possible through blockade of class I histone deacetylates with a selective class I histone deacetylase inhibitor MS-275.85
MicroRNAs (miRs) are small, noncoding RNAs that act as intrinsic regulators of gene expression and affect the expression of genes at the posttranslational level. MiRNAs have been implicated in a variety of biological processes affecting CKD.86 Data specific to miR-21 demonstrate association within CKD through sustained expression contributing to pathways promoting renal fibrosis through the peroxisome proliferator-activated receptor-α.87 miR-192 is associated with development of progressive renal fibrosis and known to be upregulated in ureteral obstruction mouse models as well as rat models with activation of TGF-β.88 In humans, there was significant upregulation of miR-200a, miR-200b, miR-141, and miR-429 intrarenal expression in kidney biopsies of patients with hypertensive nephrosclerosis.89
Conclusion
Pediatric research in genetic and epigenetic factors that contribute to understanding of pediatric CKD progression is greatly lacking. Although the majority of pediatric CKD cases are due to congenital causes, there is still significant variability in rate of CKD progression that may reflect the complexity of CKD pathophysiology involving both inherited predisposition, such as genetic polymorphisms, and environmental factors causing epigenetic modifications. In contrast to adult samples where the onset of CKD may be ambiguous, the pediatric population largely has an identifiable onset of renal insult leading to CKD and genetic samples could readily be obtained at identifiable time points in a child's disease course. Certainly, pediatric CKD research must also take advantage of multicenter cooperative trials to garner adequate sample sizes given the small sample size otherwise found at individual centers.
Characterization of polymorphic variants is important and epigenetic modifications in pediatric CKD progression may prompt the pediatric nephrologist to be increasingly directed at improving modifiable risk factors for disease progression, including those related to cardiovascular status, even for those of mild/moderate CKD status. Increased understanding of the genetic and epigenetic impact on CKD progression may assist in development of genetic screening tools to identify those children comparatively at risk, and this may someday allow nephrologists to provide targeted therapies to prevent progression of CKD, improve health outcomes into adulthood, and enhance quality of life.
Funding
None.
References
- 1.Levey A S, Eckardt K U, Tsukamoto Y. et al. Definition and classification of chronic kidney disease: a position statement from Kidney Disease: Improving Global Outcomes (KDIGO) Kidney Int. 2005;67(6):2089–2100. doi: 10.1111/j.1523-1755.2005.00365.x. [DOI] [PubMed] [Google Scholar]
- 2.System U. Bethesda, MD: National Institutes of Health, National Institute of Diabetes and Digestive and Kidney Diseases; 2011. USRDS 2011 Annual Report: Atlas of Chronic Kidney Disease and End-stage Renal Disease in the United States. [Google Scholar]
- 3.Dressler G R, Woolf A S. Pax2 in development and renal disease. Int J Dev Biol. 1999;43(5):463–468. [PubMed] [Google Scholar]
- 4.Vehaskari V M. Genetics and CKD. Adv Chronic Kidney Dis. 2011;18(5):317–323. doi: 10.1053/j.ackd.2011.07.001. [DOI] [PubMed] [Google Scholar]
- 5.Zeisberg M, Neilson E G. Mechanisms of tubulointerstitial fibrosis. J Am Soc Nephrol. 2010;21(11):1819–1834. doi: 10.1681/ASN.2010080793. [DOI] [PubMed] [Google Scholar]
- 6.Satko S G, Sedor J R, Iyengar S K, Freedman B I. Familial clustering of chronic kidney disease. Semin Dial. 2007;20(3):229–236. doi: 10.1111/j.1525-139X.2007.00282.x. [DOI] [PubMed] [Google Scholar]
- 7.Cargill M, Altshuler D, Ireland J. et al. Characterization of single-nucleotide polymorphisms in coding regions of human genes. Nat Genet. 1999;22(3):231–238. doi: 10.1038/10290. [DOI] [PubMed] [Google Scholar]
- 8.Freedman B I, Volkova N V, Satko S G. et al. Population-based screening for family history of end-stage renal disease among incident dialysis patients. Am J Nephrol. 2005;25(6):529–535. doi: 10.1159/000088491. [DOI] [PubMed] [Google Scholar]
- 9.Lei H H, Perneger T V, Klag M J, Whelton P K, Coresh J. Familial aggregation of renal disease in a population-based case-control study. J Am Soc Nephrol. 1998;9(7):1270–1276. doi: 10.1681/ASN.V971270. [DOI] [PubMed] [Google Scholar]
- 10.Axelsson J, Devuyst O, Nordfors L, Heimbürger O, Stenvinkel P, Lindholm B. Place of genotyping and phenotyping in understanding and potentially modifying outcomes in peritoneal dialysis patients. Kidney Int Suppl. 2006;103(103):S138–S145. doi: 10.1038/sj.ki.5001931. [DOI] [PubMed] [Google Scholar]
- 11.Luttropp K, Stenvinkel P, Carrero J J, Pecoits-Filho R, Lindholm B, Nordfors L. Understanding the role of genetic polymorphisms in chronic kidney disease. Pediatr Nephrol. 2008;23(11):1941–1949. doi: 10.1007/s00467-008-0788-z. [DOI] [PubMed] [Google Scholar]
- 12.Drawz P E, Sedor J R. The genetics of common kidney disease: a pathway toward clinical relevance. Nat Rev Nephrol. 2011;7(8):458–468. doi: 10.1038/nrneph.2011.85. [DOI] [PubMed] [Google Scholar]
- 13.Reeders S T, Breuning M H, Davies K E. et al. A highly polymorphic DNA marker linked to adult polycystic kidney disease on chromosome 16. Nature. 1985;317(6037):542–544. doi: 10.1038/317542a0. [DOI] [PubMed] [Google Scholar]
- 14.Smyth L J, Duffy S, Maxwell A P, McKnight A J. Genetic and epigenetic factors influencing chronic kidney disease. Am J Physiol Renal Physiol. 2014;307(7):F757–F776. doi: 10.1152/ajprenal.00306.2014. [DOI] [PubMed] [Google Scholar]
- 15.Wise L H Lanchbury J S Lewis C M Meta-analysis of genome searches Ann Hum Genet 199963(Pt 3):263–272. [DOI] [PubMed] [Google Scholar]
- 16.Rao M, Mottl A K, Cole S A. et al. Meta-analysis of genome-wide linkage scans for renal function traits. Nephrol Dial Transplant. 2012;27(2):647–656. doi: 10.1093/ndt/gfr255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Altshuler D, Daly M J, Lander E S. Genetic mapping in human disease. Science. 2008;322(5903):881–888. doi: 10.1126/science.1156409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.O'Seaghdha C M, Fox C S. Genetics of chronic kidney disease. Nephron Clin Pract. 2011;118(1):c55–c63. doi: 10.1159/000320905. [DOI] [PubMed] [Google Scholar]
- 19.Tampe B, Zeisberg M. Contribution of genetics and epigenetics to progression of kidney fibrosis. Nephrol Dial Transplant. 2014;29 04:iv72–iv79. doi: 10.1093/ndt/gft025. [DOI] [PubMed] [Google Scholar]
- 20.Köttgen A, Glazer N L, Dehghan A. et al. Multiple loci associated with indices of renal function and chronic kidney disease. Nat Genet. 2009;41(6):712–717. doi: 10.1038/ng.377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Köttgen A, Hwang S J, Larson M G. et al. Uromodulin levels associate with a common UMOD variant and risk for incident CKD. J Am Soc Nephrol. 2010;21(2):337–344. doi: 10.1681/ASN.2009070725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hart T C, Gorry M C, Hart P S. et al. Mutations of the UMOD gene are responsible for medullary cystic kidney disease 2 and familial juvenile hyperuricaemic nephropathy. J Med Genet. 2002;39(12):882–892. doi: 10.1136/jmg.39.12.882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Rampoldi L, Caridi G, Santon D. et al. Allelism of MCKD, FJHN and GCKD caused by impairment of uromodulin export dynamics. Hum Mol Genet. 2003;12(24):3369–3384. doi: 10.1093/hmg/ddg353. [DOI] [PubMed] [Google Scholar]
- 24.Köttgen A, Pattaro C, Böger C A. et al. New loci associated with kidney function and chronic kidney disease. Nat Genet. 2010;42(5):376–384. doi: 10.1038/ng.568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kao W H, Klag M J, Meoni L A. et al. MYH9 is associated with nondiabetic end-stage renal disease in African Americans. Nat Genet. 2008;40(10):1185–1192. doi: 10.1038/ng.232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Genovese G, Friedman D J, Ross M D. et al. Association of trypanolytic ApoL1 variants with kidney disease in African Americans. Science. 2010;329(5993):841–845. doi: 10.1126/science.1193032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Freedman B I, Kopp J B, Langefeld C D. et al. The apolipoprotein L1 (APOL1) gene and nondiabetic nephropathy in African Americans. J Am Soc Nephrol. 2010;21(9):1422–1426. doi: 10.1681/ASN.2010070730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bostrom M A, Freedman B I. The spectrum of MYH9-associated nephropathy. Clin J Am Soc Nephrol. 2010;5(6):1107–1113. doi: 10.2215/CJN.08721209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Liu C T, Garnaas M K, Tin A. et al. Genetic association for renal traits among participants of African ancestry reveals new loci for renal function. PLoS Genet. 2011;7(9):e1002264. doi: 10.1371/journal.pgen.1002264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Groothoff J W, Gruppen M P, Offringa M. et al. Mortality and causes of death of end-stage renal disease in children: a Dutch cohort study. Kidney Int. 2002;61(2):621–629. doi: 10.1046/j.1523-1755.2002.00156.x. [DOI] [PubMed] [Google Scholar]
- 31.McDonald S P Craig J C; Australian and New Zealand Paediatric Nephrology Association. Long-term survival of children with end-stage renal disease N Engl J Med 2004350262654–2662. [DOI] [PubMed] [Google Scholar]
- 32.Samuel S M, Tonelli M A, Foster B J. et al. Survival in pediatric dialysis and transplant patients. Clin J Am Soc Nephrol. 2011;6(5):1094–1099. doi: 10.2215/CJN.04920610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ketteler M, Bongartz P, Westenfeld R. et al. Association of low fetuin-A (AHSG) concentrations in serum with cardiovascular mortality in patients on dialysis: a cross-sectional study. Lancet. 2003;361(9360):827–833. doi: 10.1016/S0140-6736(03)12710-9. [DOI] [PubMed] [Google Scholar]
- 34.Lebreton J P, Joisel F, Raoult J P, Lannuzel B, Rogez J P, Humbert G. Serum concentration of human alpha 2 HS glycoprotein during the inflammatory process: evidence that alpha 2 HS glycoprotein is a negative acute-phase reactant. J Clin Invest. 1979;64(4):1118–1129. doi: 10.1172/JCI109551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Schinke T, Amendt C, Trindl A, Pöschke O, Müller-Esterl W, Jahnen-Dechent W. The serum protein alpha2-HS glycoprotein/fetuin inhibits apatite formation in vitro and in mineralizing calvaria cells. A possible role in mineralization and calcium homeostasis. J Biol Chem. 1996;271(34):20789–20796. doi: 10.1074/jbc.271.34.20789. [DOI] [PubMed] [Google Scholar]
- 36.Stenvinkel P, Wang K, Qureshi A R. et al. Low fetuin-A levels are associated with cardiovascular death: Impact of variations in the gene encoding fetuin. Kidney Int. 2005;67(6):2383–2392. doi: 10.1111/j.1523-1755.2005.00345.x. [DOI] [PubMed] [Google Scholar]
- 37.Ziolkowska H, Brzewski M, Roszkowska-Blaim M. Determinants of the intima-media thickness in children and adolescents with chronic kidney disease. Pediatr Nephrol. 2008;23(5):805–811. doi: 10.1007/s00467-007-0733-6. [DOI] [PubMed] [Google Scholar]
- 38.Weiner D E, Tabatabai S, Tighiouart H. et al. Cardiovascular outcomes and all-cause mortality: exploring the interaction between CKD and cardiovascular disease. Am J Kidney Dis. 2006;48(3):392–401. doi: 10.1053/j.ajkd.2006.05.021. [DOI] [PubMed] [Google Scholar]
- 39.Baudin B. Polymorphism in angiotensin II receptor genes and hypertension. Exp Physiol. 2005;90(3):277–282. doi: 10.1113/expphysiol.2004.028456. [DOI] [PubMed] [Google Scholar]
- 40.Caulfield M, Lavender P, Farrall M. et al. Linkage of the angiotensinogen gene to essential hypertension. N Engl J Med. 1994;330(23):1629–1633. doi: 10.1056/NEJM199406093302301. [DOI] [PubMed] [Google Scholar]
- 41.Wang A Y, Chan J C, Wang M. et al. Cardiac hypertrophy and remodeling in relation to ACE and angiotensinogen genes genotypes in Chinese dialysis patients. Kidney Int. 2003;63(5):1899–1907. doi: 10.1046/j.1523-1755.2003.00933.x. [DOI] [PubMed] [Google Scholar]
- 42.Bantis C, Ivens K, Kreusser W. et al. Influence of genetic polymorphisms of the renin-angiotensin system on IgA nephropathy. Am J Nephrol. 2004;24(2):258–267. doi: 10.1159/000077398. [DOI] [PubMed] [Google Scholar]
- 43.Osono E, Kurihara S, Hayama N. et al. Insertion/deletion polymorphism in intron 16 of the ACE gene and left ventricular hypertrophy in patients with end-stage renal disease. Am J Kidney Dis. 1998;32(5):725–730. doi: 10.1016/s0272-6386(98)70126-x. [DOI] [PubMed] [Google Scholar]
- 44.Elshamaa M F, Sabry S M, Bazaraa H M. et al. Genetic polymorphism of ACE and the angiotensin II type1 receptor genes in children with chronic kidney disease. J Inflamm (Lond) 2011;8(1):20. doi: 10.1186/1476-9255-8-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hohenfellner K, Wingen A M, Nauroth O, Wühl E, Mehls O, Schaefer F. Impact of ACE I/D gene polymorphism on congenital renal malformations. Pediatr Nephrol. 2001;16(4):356–361. doi: 10.1007/s004670100567. [DOI] [PubMed] [Google Scholar]
- 46.Papp F, Friedman A L, Bereczki C. et al. Renin-angiotensin gene polymorphism in children with uremia and essential hypertension. Pediatr Nephrol. 2003;18(2):150–154. doi: 10.1007/s00467-002-1032-x. [DOI] [PubMed] [Google Scholar]
- 47.Haszon I, Friedman A L, Papp F. et al. ACE gene polymorphism and renal scarring in primary vesicoureteric reflux. Pediatr Nephrol. 2002;17(12):1027–1031. doi: 10.1007/s00467-002-0968-1. [DOI] [PubMed] [Google Scholar]
- 48.Brock J W III Adams M Hunley T Wada A Trusler L Kon V Potential risk factors associated with progressive renal damage in childhood urological diseases: the role of angiotensin-converting enzyme gene polymorphism J Urol 1997158(3 Pt 2):1308–1311. [DOI] [PubMed] [Google Scholar]
- 49.al-Eisa A, Haider M Z, Srivastva B S. Angiotensin-converting enzyme gene insertion/deletion polymorphism and renal damage in childhood uropathies. Pediatr Int. 2000;42(4):348–353. doi: 10.1046/j.1442-200x.2000.01242.x. [DOI] [PubMed] [Google Scholar]
- 50.Zaffanello M, Tardivo S, Cataldi L, Fanos V, Biban P, Malerba G. Genetic susceptibility to renal scar formation after urinary tract infection: a systematic review and meta-analysis of candidate gene polymorphisms. Pediatr Nephrol. 2011;26(7):1017–1029. doi: 10.1007/s00467-010-1695-7. [DOI] [PubMed] [Google Scholar]
- 51.Perna A, Ruggenenti P, Testa A. et al. ACE genotype and ACE inhibitors induced renoprotection in chronic proteinuric nephropathies1. Kidney Int. 2000;57(1):274–281. doi: 10.1046/j.1523-1755.2000.00818.x. [DOI] [PubMed] [Google Scholar]
- 52.Ruggenenti P, Perna A, Zoccali C. et al. Chronic proteinuric nephropathies. II. Outcomes and response to treatment in a prospective cohort of 352 patients: differences between women and men in relation to the ACE gene polymorphism. Gruppo Italiano di Studi Epidemologici in Nefrologia (Gisen) J Am Soc Nephrol. 2000;11(1):88–96. doi: 10.1681/ASN.V11188. [DOI] [PubMed] [Google Scholar]
- 53.Braliou G G, Grigoriadou A M, Kontou P I, Bagos P G. The role of genetic polymorphisms of the Renin-Angiotensin System in renal diseases: A meta-analysis. Comput Struct Biotechnol J. 2014;10(16):1–7. doi: 10.1016/j.csbj.2014.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Yoneda A, Cascio S, Green A, Barton D, Puri P. Angiotensin II type 2 receptor gene is not responsible for familial vesicoureteral reflux. J Urol. 2002;168(3):1138–1141. doi: 10.1016/S0022-5347(05)64611-3. [DOI] [PubMed] [Google Scholar]
- 55.Hohenfellner K, Hunley T E, Schloemer C. et al. Angiotensin type 2 receptor is important in the normal development of the ureter. Pediatr Nephrol. 1999;13(3):187–191. doi: 10.1007/s004670050589. [DOI] [PubMed] [Google Scholar]
- 56.Hohenfellner K, Hunley T E, Yerkes E, Habermehl P, Hohenfellner R, Kon V. Angiotensin II, type 2 receptor in the development of vesico-ureteric reflux. BJU Int. 1999;83(3):318–322. doi: 10.1046/j.1464-410x.1999.00923.x. [DOI] [PubMed] [Google Scholar]
- 57.Rao M Jaber B L Balakrishnan V S; DialGene ConsortiumGene polymorphism association studies in dialysis: cardiovascular disease Semin Dial 2005183217–225. [DOI] [PubMed] [Google Scholar]
- 58.Zychma M J Gumprecht J Grzeszczak W Zukowska-Szczechowska E; End-Stage Renal Disease Study GroupMethylenetetrahydrofolate reductase gene C677T polymorphism, plasma homocysteine and folate in end-stage renal disease dialysis and non-dialysis patients Nephron 2002921235–239. [DOI] [PubMed] [Google Scholar]
- 59.Merouani A, Lambert M, Delvin E E, Genest J Jr, Robitaille P, Rozen R. Plasma homocysteine concentration in children with chronic renal failure. Pediatr Nephrol. 2001;16(10):805–811. doi: 10.1007/s004670100648. [DOI] [PubMed] [Google Scholar]
- 60.McCarthy A M, Lindgren S, Mengeling M A, Tsalikian E, Engvall J C. Effects of diabetes on learning in children. Pediatrics. 2002;109(1):E9. doi: 10.1542/peds.109.1.e9. [DOI] [PubMed] [Google Scholar]
- 61.Girndt M Heine G H Köhler H; DialGene ConsortiumGene polymorphism association studies in dialysis: anemia and host immunity Semin Dial 2006193227–231. [DOI] [PubMed] [Google Scholar]
- 62.Fuller R, Nopoulos P, Arndt S, O'Leary D, Ho B C, Andreasen N C. Longitudinal assessment of premorbid cognitive functioning in patients with schizophrenia through examination of standardized scholastic test performance. Am J Psychiatry. 2002;159(7):1183–1189. doi: 10.1176/appi.ajp.159.7.1183. [DOI] [PubMed] [Google Scholar]
- 63.Blackman J A, Lindgren S D, Bretthauer J. The validity of continuing developmental follow-up of high-risk infants to age 5 years. Am J Dis Child. 1992;146(1):70–75. doi: 10.1001/archpedi.1992.02160130072024. [DOI] [PubMed] [Google Scholar]
- 64.Moser J J, Veale P M, McAllister D L, Archer D P. A systematic review and quantitative analysis of neurocognitive outcomes in children with four chronic illnesses. Paediatr Anaesth. 2013;23(11):1084–1096. doi: 10.1111/pan.12255. [DOI] [PubMed] [Google Scholar]
- 65.Balakrishnan V S, Guo D, Rao M. et al. Cytokine gene polymorphisms in hemodialysis patients: association with comorbidity, functionality, and serum albumin. Kidney Int. 2004;65(4):1449–1460. doi: 10.1111/j.1523-1755.2004.00531.x. [DOI] [PubMed] [Google Scholar]
- 66.Girndt M, Kaul H, Sester U. et al. Anti-inflammatory interleukin-10 genotype protects dialysis patients from cardiovascular events. Kidney Int. 2002;62(3):949–955. doi: 10.1046/j.1523-1755.2002.00504.x. [DOI] [PubMed] [Google Scholar]
- 67.August P, Suthanthiran M. Transforming growth factor beta signaling, vascular remodeling, and hypertension. N Engl J Med. 2006;354(25):2721–2723. doi: 10.1056/NEJMcibr062143. [DOI] [PubMed] [Google Scholar]
- 68.Yamamoto T, Noble N A, Miller D E, Border W A. Sustained expression of TGF-beta 1 underlies development of progressive kidney fibrosis. Kidney Int. 1994;45(3):916–927. doi: 10.1038/ki.1994.122. [DOI] [PubMed] [Google Scholar]
- 69.Li B Khanna A Sharma V Singh T Suthanthiran M August P TGF-beta1 DNA polymorphisms, protein levels, and blood pressure Hypertension 199933(1, Pt 2):271–275. [DOI] [PubMed] [Google Scholar]
- 70.Schultz JelJ, Witt S A, Glascock B J. et al. TGF-beta1 mediates the hypertrophic cardiomyocyte growth induced by angiotensin II. J Clin Invest. 2002;109(6):787–796. doi: 10.1172/JCI14190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Zoja C, Corna D, Camozzi D. et al. How to fully protect the kidney in a severe model of progressive nephropathy: a multidrug approach. J Am Soc Nephrol. 2002;13(12):2898–2908. doi: 10.1097/01.asn.0000034912.55186.ec. [DOI] [PubMed] [Google Scholar]
- 72.Grainger D J, Heathcote K, Chiano M. et al. Genetic control of the circulating concentration of transforming growth factor type beta1. Hum Mol Genet. 1999;8(1):93–97. doi: 10.1093/hmg/8.1.93. [DOI] [PubMed] [Google Scholar]
- 73.Hussein A, Askar E, Elsaeid M, Schaefer F. Functional polymorphisms in transforming growth factor-beta-1 (TGFbeta-1) and vascular endothelial growth factor (VEGF) genes modify risk of renal parenchymal scarring following childhood urinary tract infection. Nephrol Dial Transplant. 2010;25(3):779–785. doi: 10.1093/ndt/gfp532. [DOI] [PubMed] [Google Scholar]
- 74.Dwivedi R S, Herman J G, McCaffrey T A, Raj D S. Beyond genetics: epigenetic code in chronic kidney disease. Kidney Int. 2011;79(1):23–32. doi: 10.1038/ki.2010.335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Jang H, Serra C. Nutrition, epigenetics, and diseases. Clin Nurs Res. 2014;3(1):1–8. doi: 10.7762/cnr.2014.3.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Woroniecki R, Gaikwad A B, Susztak K. Fetal environment, epigenetics, and pediatric renal disease. Pediatr Nephrol. 2011;26(5):705–711. doi: 10.1007/s00467-010-1714-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Hostetter T H, Rennke H G, Brenner B M. Compensatory renal hemodynamic injury: a final common pathway of residual nephron destruction. Am J Kidney Dis. 1982;1(5):310–314. doi: 10.1016/s0272-6386(82)80032-2. [DOI] [PubMed] [Google Scholar]
- 78.Ingrosso D, Cimmino A, Perna A F. et al. Folate treatment and unbalanced methylation and changes of allelic expression induced by hyperhomocysteinaemia in patients with uraemia. Lancet. 2003;361(9370):1693–1699. doi: 10.1016/S0140-6736(03)13372-7. [DOI] [PubMed] [Google Scholar]
- 79.Bechtel W, McGoohan S, Zeisberg E M. et al. Methylation determines fibroblast activation and fibrogenesis in the kidney. Nat Med. 2010;16(5):544–550. doi: 10.1038/nm.2135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Ko Y A, Mohtat D, Suzuki M. et al. Cytosine methylation changes in enhancer regions of core pro-fibrotic genes characterize kidney fibrosis development. Genome Biol. 2013;14(10):R108. doi: 10.1186/gb-2013-14-10-r108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Witasp A, Ekström T J, Schalling M, Lindholm B, Stenvinkel P, Nordfors L. How can genetics and epigenetics help the nephrologist improve the diagnosis and treatment of chronic kidney disease patients? Nephrol Dial Transplant. 2014;29(5):972–980. doi: 10.1093/ndt/gfu021. [DOI] [PubMed] [Google Scholar]
- 82.Reddy M A, Natarajan R. Epigenetics in diabetic kidney disease. J Am Soc Nephrol. 2011;22(12):2182–2185. doi: 10.1681/ASN.2011060629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Reddy M A, Sumanth P, Lanting L. et al. Losartan reverses permissive epigenetic changes in renal glomeruli of diabetic db/db mice. Kidney Int. 2014;85(2):362–373. doi: 10.1038/ki.2013.387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Sun G, Reddy M A, Yuan H, Lanting L, Kato M, Natarajan R. Epigenetic histone methylation modulates fibrotic gene expression. J Am Soc Nephrol. 2010;21(12):2069–2080. doi: 10.1681/ASN.2010060633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Liu N, He S, Ma L. et al. Blocking the class I histone deacetylase ameliorates renal fibrosis and inhibits renal fibroblast activation via modulating TGF-beta and EGFR signaling. PLoS ONE. 2013;8(1):e54001. doi: 10.1371/journal.pone.0054001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Wing M R, Ramezani A, Gill H S, Devaney J M, Raj D S. Epigenetics of progression of chronic kidney disease: fact or fantasy? Semin Nephrol. 2013;33(4):363–374. doi: 10.1016/j.semnephrol.2013.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Chau B N, Xin C, Hartner J. et al. MicroRNA-21 promotes fibrosis of the kidney by silencing metabolic pathways. Sci Transl Med. 2012;4(121):121ra18. doi: 10.1126/scitranslmed.3003205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Chung A C, Huang X R, Meng X, Lan H Y. miR-192 mediates TGF-beta/Smad3-driven renal fibrosis. J Am Soc Nephrol. 2010;21(8):1317–1325. doi: 10.1681/ASN.2010020134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Wang G, Kwan B C, Lai F M. et al. Intrarenal expression of miRNAs in patients with hypertensive nephrosclerosis. Am J Hypertens. 2010;23(1):78–84. doi: 10.1038/ajh.2009.208. [DOI] [PubMed] [Google Scholar]