

Abstract
Genetic mosaicism is defined as the presence of two or more genetically distinct cell populations in a single individual. Ever more disorders are found to be manifestations of mosaicism and together constitute a significant proportion of the morbidity confronting pediatric specialists. An emerging category is that of overgrowth syndromes with skin manifestations and neurological or developmental abnormalities, such as the well-known Proteus syndrome. In recent years, we have seen dramatic advances in our understanding of these disorders and we now know the genetic basis of many of them. This has profound consequences for diagnosis, counselling, and even treatment, with therapies targeted to specific pathways becoming available for clinical use. Recognizing such overgrowth syndromes, therefore, is more important than ever. Fortunately, their skin manifestations can provide important diagnostic clues when evaluated in the entire phenotypic context. In this review, I provide an overview of the most frequently seen mosaic neurocutaneous phenotypes and discuss their molecular basis.
Keywords: mosaicism, skin, brain, overgrowth, intellectual disability
Introduction
The skin and the brain are both derived from the ectoderm. Germline mutations in genes required for proper ectodermal development will therefore often cause skin manifestations in addition to neurological problems. The cutaneous symptoms can be quite specific and thereby serve as reliable diagnostic criteria and pointers to a specific diagnosis. The café-au-lait spots of Neurofibromatosis type I are a well-known example. It might be less obvious that the shared embryonic origin of skin and brain means that a genetic mosaic can also manifest in those organs. In such cases, the cutaneous abnormalities likewise point to the underlying diagnosis. Several emerging groups of mosaic neurocutaneous disorders are characterized by deregulated growth mainly affecting skin and its appendages, blood vessels, and the brain, although other organs may also be involved. They will be the subjects of this review. Their molecular basis is being rapidly elucidated, offering exciting new insights into human development, while revolutionizing disease classification and providing unprecedented opportunities for treatment. Importantly, they are all characterized by characteristic skin abnormalities which, when recognized, can be of considerable help in establishing a clinical diagnosis.
Genetic Mosaicism
Before embarking upon a discussion of neurocutaneous mosaic overgrowth syndromes, a brief explanation of genetic mosaicism is in order, as understanding this phenomenon is required to appreciate the phenotypes associated with it.
A genetic mosaic is formally defined as the presence of two or more genetically distinct clonal cell populations within one individual.1 As such, it is distinct from chimerism, which is the merger of two distinct individuals into a single organism and can manifest in any organ including the skin.2 Mosaicism may be congenital or acquired; melanocytic nevi and seborrheic keratoses are good examples of the latter.3 4 The genetic events giving rise to mosaicism are diverse and include point mutations, insertions, deletions, chromosome rearrangements, duplications, and so on.5 These changes can, of course, affect autosomes as well as sex chromosomes. For X-linked loci, random X-inactivation can cause germ line mutations to result in mosaic phenotypes; this phenomenon is referred to as “functional mosaicism” to distinguish it from a true mosaic.6 A key concept to grasp is that the extent and nature of mosaic phenotypes are determined by the moment at which the causative genetic event occurs. If very early, affecting a cell that is still pluripotent and that will have many progeny, the phenotype will be extensive, with tissues from different germ layers and large areas affected. If late, and in a cell that is already committed to a certain fate, the abnormalities will be less extensive, to the point of being almost undetectable. Generally, mosaic disorders are not transmissible, unless the mutation is also present in the germ line. However, many mutations that are survivable in the mosaic state are lethal if present in all cells and would thus result in early spontaneous abortion.7
Cutaneous Manifestations of Genetic Mosaicism
In the skin, genetic mosaicism can give rise to abnormalities that follow readily recognizable patterns, which are of considerable diagnostic value. These Blaschko lines, named after the Austrian dermatologist Alfred Blaschko who first reported them in 1901, come in four defined varieties.5 What they represent is not known; the best guess is that they define embryonic cellular migration patterns. The type 1 pattern is linear and likely defines the paths taken by epidermal progenitor cells, as epidermal nevi are always found in type 1 patterns. The other patterns might indicate other (mesenchymal) cell populations, which is consistent with the observation that vascular malformations almost invariably manifest in type 2 Blaschko lines, which form a checkerboard pattern. The cells in a Blaschko line are genetically distinct from those in the surrounding unaffected skin and form a clonal population (i.e., they are derived from a single parent cell). As such, they form a nevus.8 Most of the nevi in the overgrowth syndromes to be discussed here will follow Blaschko type 1 or type 2 patterns.
Activating Mutations in Oncogenes Underlie Mosaic Neurocutaneous Overgrowth Phenotypes
The disorders to be discussed are all caused by activating mutations affecting interconnected pathways that are prominently involved in cancer. While some of those phenotypes do predispose to malignancies, the majority do not. It is not understood why this should be so. Perhaps the risk of developing a malignancy is determined by how committed a progenitor or stem cell is to a particular fate—the more committed, the less the risk. It would be interesting to test this idea, as it is in line with the notion that cancers arise from, and are maintained by, cancer stem cells.9
Distinct nodes in these signaling pathways are implicated in mosaic overgrowth syndromes, starting at receptor proteins on the cell surface and ending—for now—in protein kinase(s) (complexes) that regulate cellular growth, such as mTORC1 and ERKs. In this review, I will follow the signaling pathways from receptor to inside the cell so as to illustrate how the clinical phenotypes follow the molecular biology (Fig. 1).
Fig. 1.
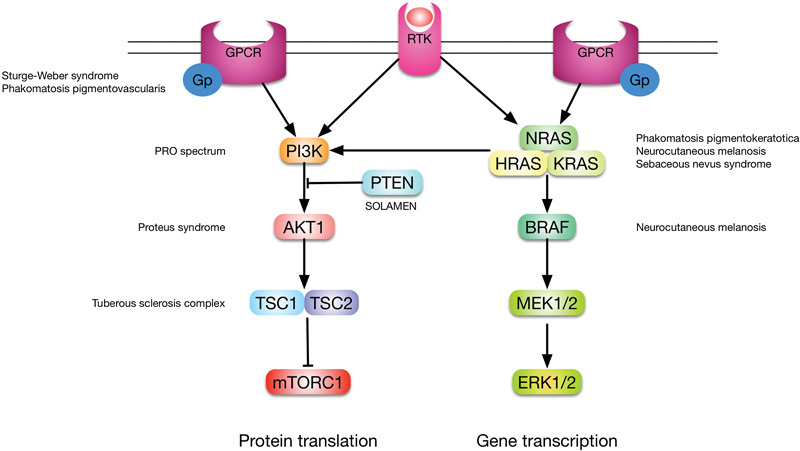
Simplified overview of the pathways discussed in this review and the disorders associated with mosaic mutations in their components. Note that there is considerable cross-talk, which explains why overgrowth syndromes have so many overlapping clinical features.
Mosaic-Activating G-Protein α-Subunit Mutations
Guanine nucleotide-binding (G-) proteins form heterotrimeric complexes composed of α-, β-, and γ-subunits and transduce signals received by G-protein–coupled receptors (GPCR). This is the largest family of transmembrane receptors in humans, with more than 800 genes encoding a GPCR.10 Examples include Rhodopsin and olfactory receptors. G-protein α-subunit encoding genes seem to be particularly prone to disease-causing mutations. McCune-Albright syndrome was the first disorder to be associated with G proteins, being caused by mosaic-activating mutations in GNAS1, a complex imprinted locus that encodes, among other transcripts, the Gsα-subunit.11 The mutations render the G protein constitutively active and initiate a multitude of downstream signaling events. McCune-Albright syndrome is associated with skin hyperpigmentation, indicative of a role for GPCR signaling in pigment production. Indeed, the gene involved in type 1 ocular albinism is a GPCR called GPR143 and is required for melanosome transport in pigment cells.12 Perhaps unsurprisingly, therefore, activating mutations in other Gα-subunit proteins are now emerging as the cause of neurocutaneous overgrowth syndromes, which are associated with abnormal pigmentation and also with vascular malformations.
Sturge-Weber Syndrome
Sturge-Weber syndrome (SWS) is defined as the combination of a facial port-wine stain (PWS, capillary malformation) with capillary/venous malformations in the eye and leptomeninges (Fig. 2). The intracerebral vascular lesions usually manifest as recalcitrant seizures and can be associated with significant developmental delay. The malformations in the eye can lead to glaucoma. Overgrowth of underlying bone and soft tissues can be associated.13 Historically, a facial capillary malformation was considered to have a significant chance of being part of SWS if it affects the region innervated by the first branch of the trigeminal nerve. However, the vascular malformations in SWS seem to follow the distribution of the embryonic facial vasculature rather than the trigeminal nerve branches.14 Forehead and upper eyelid involvement should prompt ophthalmological evaluation. The risk of neurological involvement might also be higher in children with this particular distribution of the PWS, but whether a screening magnetic resonance imaging is then indicated remains to be settled.
Fig. 2.
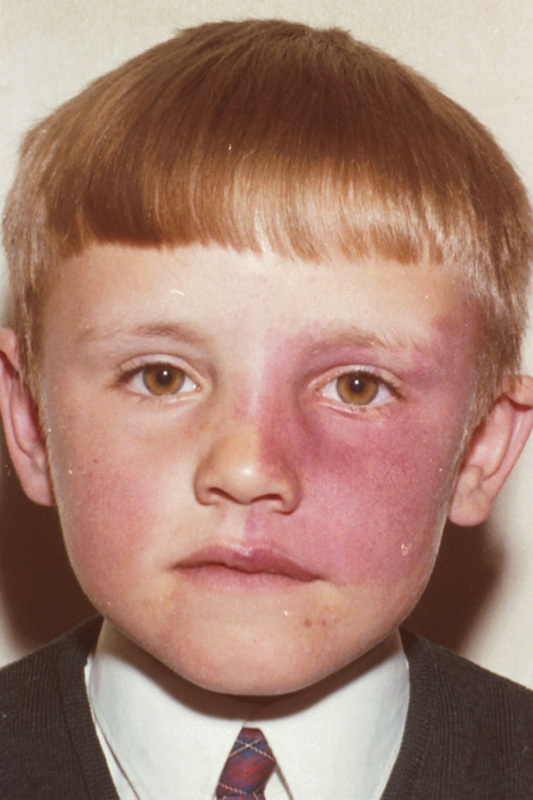
Facial port-wine stain in a distribution that suggests ocular and possibly cerebral involvement. Note that the upper and lower eyelids are affected (courtesy Prof. Peter Steijlen, Maastricht University Medical Center).
Both isolated and SWS-associated facial PWS were recently found to be due to a mosaic-activating mutation (c.548G > A, p.Arg183Gln) in GNAQ, which codes for the Gqα-subunit.15 As expected in a mosaic, the mutations were found in affected skin and brain tissue but not in clinically unaffected areas. Of note, the same mutation had been previously reported in blue nevi and uveal melanoma, its occurrence in the latter suggesting strong oncogenic potential.16
Phakomatosis Pigmentovascularis
Phakomatosis pigmentovascularis (PPV) is defined as the coexistence of widespread vascular (usual capillary) malformations and large melanocytic nevi. Epilepsy and developmental delay may also be present. Different types are distinguished based on the precise nature of the vascular and pigmentary changes. There are five types, defined by the nature of the vascular malformation and the pigmentary lesion.17 The most common one is type 2, which combines a capillary malformation with Mongolian spots, which are deep melanocytic lesions.17 For a long time, PPV was considered as an example of nonallelic twin spotting. In this phenomenon, mitotic chromosomal rearrangements unmask mutations on two different chromosomes, resulting in neighboring body areas being affected by distinct abnormalities. However, recently it was reported that PPV type 2 is due to an activating mutation in GNA11 coding for the Gq α11 subunit (c.626A > T, p.Gln209Leu), in both the vascular and pigmented lesions (Waelchli et al, personal communication). Based on this observation, the most parsimonious explanation for twin spotting seems to be that a single mutation affects a multipotent progenitor cell prior to specification of the neural crest. Should the mutation occur at a later stage, in cells that have committed to a particular lineage, a single type of nevus will result. Indeed, a related twin spotting phenotype called phakomatosis pigmentokeratotica was found to be also due to a single mutation (see later).
As an aside, the author would like to suggest that classic Klippel-Trenaunay syndrome, as first reported by Klippel and Trenaunay, is caused by somatic activating GNAQ or GNA11 mutations. Patients manifesting both SWS and KTS have been reported.18 Other phenotypes might rather be in the PRO spectrum. The time may be ripe to do away with KTS as a nosologic entity as was suggested a few years ago based on the analysis of phenotypes classified previously as KTS.19
Mosaic RASopathies
A large and fascinating group of disorders, collectively referred to as the RASopathies, is caused by activating mutations affecting RAS signaling components (Fig. 1). The RAS pathway is a key growth regulator downstream of several growth factor receptors. As such, its inappropriate activation in the germ line has widespread consequences and can cause pervasive developmental disorders. These share defining features of varying intellectual disability, specific dysmorphic traits, vascular malformations, and characteristic abnormalities of skin and hair. Recently, mosaic RASopathies have been defined, and although their phenotypes do show some overlap with those that are caused by germ line RAS pathway mutations, there are surprising and as yet unexplained differences. However, they can all be associated with mental disability and neurological abnormalities.
Phakomatosis Pigmentokeratotica
Phakomatosis pigmentokeratotica (PP) was recognized by Happle et al as a distinct epidermal nevus syndrome in 1996.20 It is characterized by coexisting and neighboring sebaceous and melanocytic nevi, and a variety of other abnormalities, including neurological ones, can be associated. Like phakomatosis pigmentovascularis, PP was considered as a classic twin spotting phenotype. However, it was recently found that an activating HRAS mutation (c.182A > G, p.Gln61Arg) is responsible, with the seemingly disparate cutaneous manifestations being caused by the mutation affecting a pluripotent progenitor cell that gave rise to both epidermal cells and melanocytes.21 Consistent with this interpretation, RAS pathway gene mutations have also been found in epidermal and melanocytic nevi consisting of a single cell type.
Sebaceous Nevus Syndrome
Sebaceous nevus is a common condition, affecting approximately 1 in 1,000 persons. Clinically, it appears like a hairless, yellowish orange cutaneous plaque with a papillomatous surface and following type 1 Blaschko lines (Fig. 3). It is named after the prominent sebaceous glands that make up the bulk of the lesion and is most often located on the scalp for reasons yet unknown. Of note, various types of neoplasm may develop in a sebaceous nevus, including malignant ones such as basal cell carcinoma. The nevus can be associated with skeletal, ocular, or cerebral effects and in that case Schimmelpenning (-Feuerstein-Mims) syndrome or linear sebaceous nevus syndrome can be diagnosed.22 As is to be expected when multiple tissues are affected, the skin lesions tend to be more extensive in cases of Schimmelpenning syndrome. About 7% of persons with the disorder have neurological abnormalities such as seizures and developmental delay, associated with hemimegalencephaly. In 2012, Groesser et al found that the majority of epithelial cells derived from the skin lesions in patients with both sebaceous nevus and Schimmelpenning syndrome have activating HRAS mutations (mostly c.37G > C, p. Gly13Arg).21 Of note, the mutations are oncogenic and the sebaceous nevi do have a ±8% risk of developing malignant tumors, typically basal cell carcinomas.23 The extracutaneous lesions do not seem to have malignant potential. As indicated earlier, this might be due to stem cells being affected in the skin but not in other tissues.
Fig. 3.
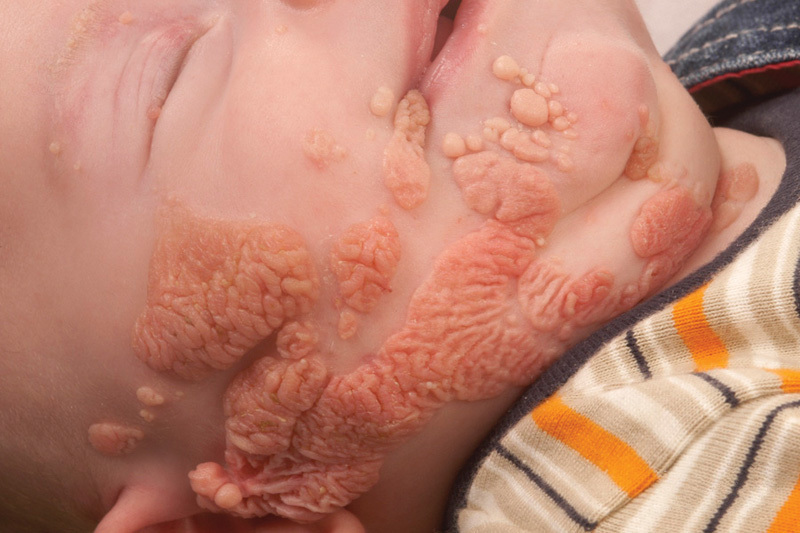
An extensive sebaceous nevus affecting the right half of the face. This patient later developed a malignant tumor of the right parotid gland, presumably because it also carried the causal mutation.
Mosaic RASopathies Are Quite Different from Their Germ Line Counterparts
Interestingly, the KRAS and HRAS mutations found in phakomatosis pigmentovascularis and sebaceous nevus lesions seem to be mostly unique to those disorders, but a few patients have been found carrying the HRAS mutation (c.34G > A, p.Gly12Ser) that when present in the germ line causes Costello syndrome, a notably different phenotype.24 In the latter disorder, characteristic skin changes are present, but sebaceous gland abnormalities have never been reported. Likewise, germ line KRAS mutations cause Noonan and cardio-facio-cutaneous syndromes, which again show very little overlap with the mosaic phenotype.25 26 At present, it is not clear why this difference exists. It has been speculated that the cell types affected by the mutations, the degree of pathway activation resulting from them, as well as the time at which the mutation occurs during embryogenesis all determine the eventual phenotype.27 It would be of considerable interest to address this question (e.g., using inducible conditional knockouts in mice), as it seems to point to key mechanisms of mammalian embryonic development.
Congenital Melanocytic Nevi and Neurocutaneous Melanosis
The melanocytic nevi (“moles”) that everybody knows are manifestations of postzygotic mosaicism, that is, they arise during life as a consequence of acquired somatic mutations. At least 60% of acquired melanocytic nevi (AMN) carry a highly oncogenic mutation in the BRAF gene, which on the protein level results in the substitution of valine 600 for a glutamic acid (colloquially referred to as V600E3). Sometimes, however, melanocytic nevi are congenital, present at birth. They do not follow classic Blaschko lines. This might be reflective of melanocyte precursors migrating along different paths from ectodermal progenitor cells. Having arisen, presumably, from mutations affecting a committed melanocytic progenitor cell, congenital melanocytic nevi (CMN) can have varying sizes and distributions depending on the timing of the mutational event—the earlier it takes place, the more widespread the nevus is going to be. The larger it is, the higher the risk of developing melanoma. The most dramatic manifestations are giant CMN and neurocutaneous melanosis (Fig. 4). In the latter, melanocytic nevi are scattered throughout leptomeninges, and brain abnormalities such as hemimegalencephaly can also occur. If there are neurological symptoms, such as seizures, the prognosis tends to be poor.28
Fig. 4.
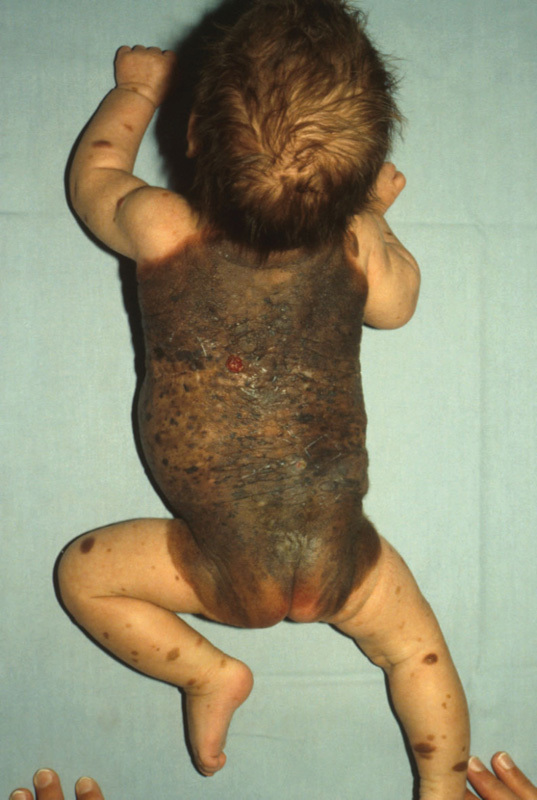
Neurocutaneous melanosis. The red nodule on the upper back is a melanoma.
The majority of cases are due to specific activating NRAS mutations (c.181C > A, p.Gln61Lys, and c.182A > G, p.Gln61Arg),29 although BRAF mutations have been reported.30 It is not clear why NRAS mutations should be more prevalent in CMN, whereas BRAF mutations underlie the majority of AMN. It also is of interest to note that melanocytic nevi with RAS pathway mutations invariably are junctional (superficial), whereas such lesions due to GNAQ/GNA11 mutations are located much deeper, in the dermis. This difference is suggestive that G-protein activation might affect melanocyte migration in addition to proliferation.
PIK3CA-Related Overgrowth Spectrum
PIK3CA-related overgrowth spectrum (PROS) is an emerging spectrum of congenital overgrowth disorders caused by a range of mosaic-activating mutations in PIK3CA, coding for p110α.31 32 This protein is a subunit of the phosphatidylinositol-4,5-bisphosphate 3-kinase complex which transduces growth factor signaling downstream of receptors such as the insulin receptor by phosphorylating AKT proteins (Fig. 1). The mutations that have been functionally characterized to date result in moderate to strong constitutive activation of PIK3CA,33 presumably leading to continuous deregulated growth of the affected tissue(s). The PROS spectrum is wide, ranging from very limited phenotypes such as isolated macrodactyly to extensive and progressive malformations (Table 1), reflecting the consequences of mutations that occur at different times during embryogenesis and in progenitor cells with various degrees of pluripotency. There also seems to be a genotype–phenotype correlation, but this needs to be confirmed. Further complexity is suggested by the observation that overgrowth affects the lower extremities more often than the upper, moving from distal to proximal, and is more often left sided than right.32 Within PROS, some more or less distinct entities can be recognized, despite considerable overlap. Cutaneous abnormalities with developmental delay and/or neurological abnormalities are typically present in macrocephaly–capillary malformation (MCAP) and congenital lipomatous overgrowth vascular malformations, epidermal nevi, and spinal/skeletal anomalies (CLOVES). Other disorders in the spectrum are associated with connective tissue overgrowth and do not usually present with developmental delay or neurological abnormalities, presumably because they arise from mutations affecting progenitor cells committed to a mesenchymal fate.
Table 1. Major clinical manifestations of the PRO spectrum.
| Disorder | Major clinical manifestations |
|---|---|
| MCAP | Macrocephaly, capillary malformations, brain overgrowth, distal limb defects, mental disability |
| CLOVES | Mixed vascular malformations, adipose tissue overgrowth, scoliosis and skeletal overgrowth |
| FAO/HMML | Progressive segmental overgrowth of fibrous and adipose tissue and bone |
| ILM | Large lymphatic malformation limited to a single body site and without associated symptoms |
| Macrodactyly | Localized overgrowth of a single or a few digits |
| Hemimegalencephaly | Overgrowth of one cerebral hemisphere |
| Muscle hemihyperplasia | Asymmetric muscular overgrowth |
| Facial infiltrating lipomatosis | Diffuse infiltration of facial tissues by mature adipose tissue |
| Epidermal nevi | Congenital verrucous skin plaques following Blaschko lines |
| Seborrheic keratosis | Localized verrucous plaque occurring later in life, usually round and dark brown |
| Benign lichenoid keratosis | Considered as a variant of seborrheic keratosis, flat red-brown skin plaque |
Abbreviations: CLOVES, capillary malformations, lipomatous overgrowth of adipose tissue, and scoliosis; FAO/HMML, fibroadipose overgrowth/hemihyperplasia-multiple lipomatosis; ILM, isolated large lymphatic malformation; MCAP, macrocephaly-capillary malformation.
Source: Adapted from Keppler-Noreuil et al.32
Macrocephaly–Capillary Malformation
MCAP combines vascular (mostly capillary) malformation with early overgrowth of bone and connective tissue with distinctive cerebral abnormalities, typically megalencephaly and polymicrogyria. Distal limb anomalies, in particular syndactyly of fingers or toes, are also a key feature. The capillary malformations have a distinct midline distribution, which facilitates recognition of the disorder (Fig. 5). Malformations of larger blood vessels and infantile hemangiomas have also been reported. The majority of patients with MCAP have some form of developmental delay, with a wide range in severity. The abnormal brain development can lead to seizures, and their presence is related to the degree of developmental delay.34 Malignancy is rare, despite the fact that PIK3CA mutations are common in breast cancer.35 It is important to note that MCAP can occasionally be caused by germline PIK3CA mutations.31
Fig. 5.
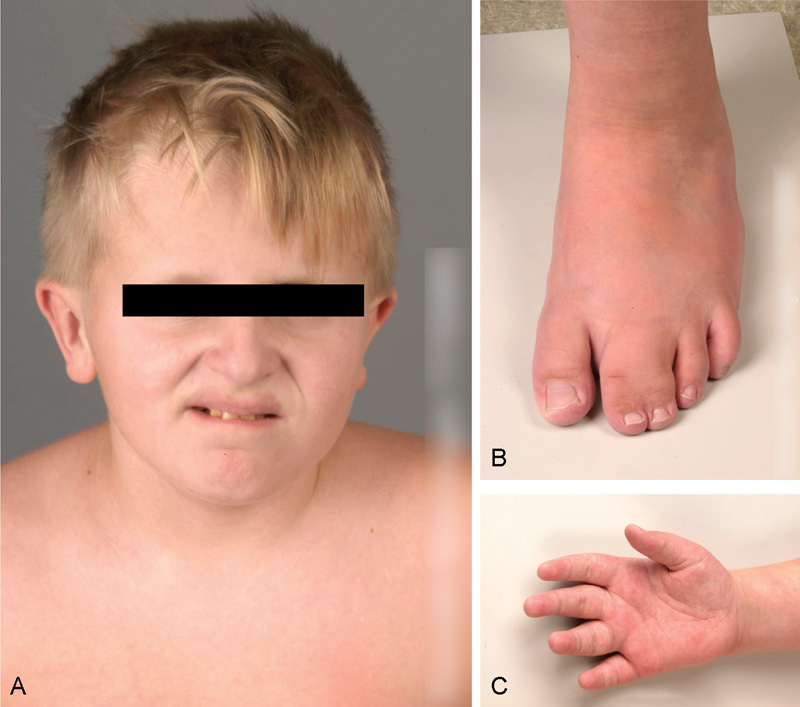
MCAP (A) Macrocephaly and midline capillary malformation affecting the upper lip. (B and C) Syndactyly. This patient had developmental delay due to megalencephaly and polymicrogyria.
Capillary Malformations, Lipomatous Overgrowth of Adipose Tissue, and Scoliosis (CLOVES)
This rare entity somewhat resembles Proteus syndrome (PS) (see subsection “Proteus Syndrome”) but is clearly distinct from it. Patients generally present with mixed (i.e., capillary, venous, arterial, and/or lymphatic, but most often lymphatic-venous) predominantly truncal vascular malformations, lipomatous overgrowth of adipose tissue, varying degrees of scoliosis, and skeletal overgrowth that is progressive but not distorting as in PS36 (Fig. 6). The various malformations can be quite extensive and lead to considerable disability, much more so than in MCAP. Developmental delay and neurological abnormalities have been reported in CLOVES, due to cerebral malformations such as polymicrogyria.37
Fig. 6.
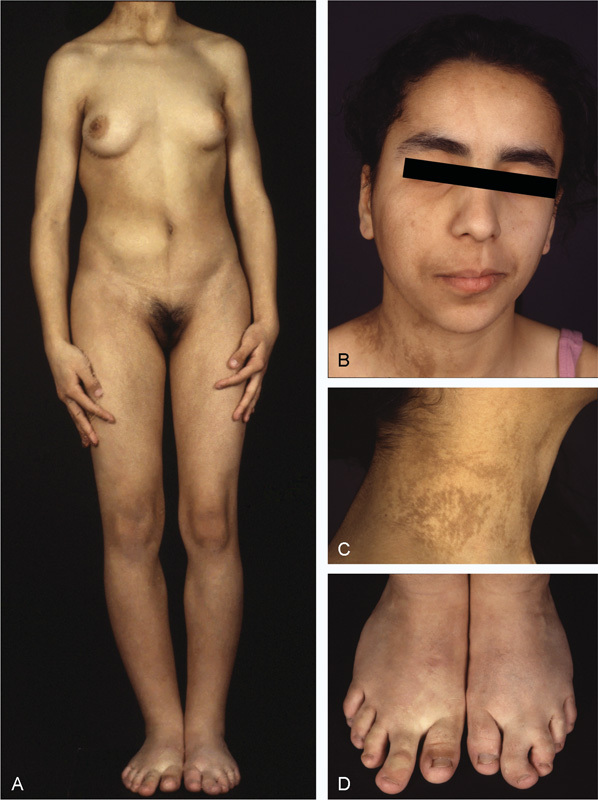
CLOVES due to a confirmed mosaic PIK3CA activating mutation. (A) Asymmetric overgrowth of the limbs with megadactyly. Breast development likewise is asymmetric. A body wall lipoma is visible on the right abdomen as is a scoliosis. (B) Asymmetrical bony overgrowth of the right facial half. (C) A verrucous epidermal nevus following type 1 Blaschko patterns. (D) Megadactyly of both feet, with partial 2–3 syndactyly on the left foot.
PTEN-Related Overgrowth
The PTEN protein is a phosphatase that, among other emerging functions, suppresses PI3K-AKT signaling by counteracting PI3K-mediated AKT phosphorylation.38 Heterozygous inactivating germ line mutations in PTEN are responsible for ∼90% of Cowden syndrome (CS) cases.39 The clinical spectrum of CS is broad, but key manifestations include macrocephaly, benign facial hair follicle tumors called tricholemmoma, verrucous hyperkeratotic skin plaques on the face and limbs, and a strong predisposition to multiple cancer types, most prominently of colon, thyroid, and the female breast.40 PTEN is thus considered as a tumor suppressor. Mild to moderate developmental delay has been reported in association with CS.
PTEN mutations can also cause macrocephaly/autism syndrome, in which most CS manifestations are lacking.41 Of note, a recent review of imaging studies revealed that 54% of patients with CS had vascular abnormalities affecting soft tissues that could manifest with skin discoloration.42 However, capillary malformations were not seen, but this is not the case for a peculiar manifestation of CS that results from postnatal loss of the healthy PTEN allele in a mosaic pattern.
Segmental Overgrowth, Lipomatosis, Arteriovenous Malformation, and Epidermal Nevus
In segmental overgrowth, lipomatosis, arteriovenous malformation, and epidermal nevus (SOLAMEN), classic manifestations of Cowden syndrome are combined with segmental overgrowth, extensive arteriovenous and lymphatic malformations, body wall lipomas, and epidermal nevi. The disorder is sometimes confused with PS. Caux et al first identified SOLAMEN in 2007 and demonstrated that its symptoms are due to mosaic loss of the healthy PTEN allele in people with a germline PTEN mutation.43 This phenomenon is known as type 2 mosaicism and has been extensively documented for other genetic disorders.44 For patients with SOLAMEN, it is not clear whether the healthy allele had already been lost during embryogenesis or postnatally. However, the vascular abnormalities were either congenital or appeared very early in life in the patients reported by Caux et al, suggestive for prenatal loss. Interestingly, the patients did not appear to develop malignancies in the tissues affected by the PTEN loss, raising questions as to PTEN's role as a tumor suppressor.
AKT-Related Overgrowth
The AKT family members are kinases that regulate mTORC1 activity by phosphorylating and thereby inhibiting the TSC1/2 complex.45 In 2011, Lindhurst et al reported that PS is caused by a mosaic-activating AKT1 mutation (c.49G > A, p.Glu17Lys).46 PS probably was the disorder that Joseph Merrick, the Elephant Man, suffered from47 and is associated with prominent skin abnormalities, including vascular malformations. The other AKT family members, AKT2 and AKT3, are implicated in deregulated growth syndromes31 48 but do not seem to be associated with vascular development.
Proteus Syndrome
While often confused with SOLAMEN or PROS, PS is associated with very specific abnormalities that constitute reliable diagnostic criteria.49 Intelligence is usually normal; however, developmental delay has been reported. The disorder can be difficult to diagnose because of its protean manifestations—it is named after the Greek god Proteus, who could change his shape at will. However, PS does have a few defining characteristics that facilitate diagnosis. First, bony overgrowth in PS tends to distort the bones it affects. In contrast, the other overgrowth syndromes leave overall bone shape intact. Second, the feet in PS can be affected by a cerebriform connective tissue nevus, which seems to be a unique manifestation of the disorder.50 Third, PS is associated with specific dysmorphic traits including dolichocephaly with a long face, down slanting palpebral fissures, low nasal bridge with anteverted nostrils, and an open mouth.49 An important symptom that emerged rather recently is an increased risk of deep venous thrombosis and pulmonary thromboembolism, requiring careful anticoagulation around surgical procedures.51
Type 2 Mosaicism in Tuberous Sclerosis Complex
Tuberous sclerosis complex (TSC) is an autosomal dominantly inherited disorder with pleiotropic manifestations in several organs, including the skin, lung, heart, brain, and kidney. Central nervous symptoms include epilepsy and developmental delay, while angiomyolipomas, cysts, and renal cell carcinomas can affect the kidney. Skin symptoms are prominent, the most typical ones being facial angiofibromas, which are highly vascularized benign hair follicle tumors, and connective tissue nevi.52 Its causative genes, TSC1 and TSC2, are directly downstream of AKT and code for a protein complex that suppresses mTORC1 activity.45 In TSC, mTORC1 is deregulated, and consequently, TSC has considerable overlap with the syndromes discussed in this review. It stands to reason that in TSC, as in CS, the remaining healthy allele might be lost in a mosaic pattern resulting in segmental worsening of disease symptoms as in SOLAMEN, manifesting with vascular malformations and abnormal growth. Vascular malformations have occasionally been reported in TSC,53 54 but overgrowth has never been described.
However, the author is aware of a patient with a germline TSC2 mutation who also has overgrowth of the lower extremities, combined with cutaneous capillary malformations and macrocephaly. It is tempting to speculate that this patient has incurred mosaic loss of the healthy TSC2 allele in the affected tissues, and it would be of interest to test this hypothesis in TSC patients with similar manifestations.
Oncogenes versus Tumor Suppressors in Capillary Malformations
Out of several unanswered questions, an interesting one to ask is why capillary malformations are such a prominent manifestation of neurocutaneous syndromes caused by mosaic-activating oncogene mutations and not of disorders due to germ line loss of tumor suppressors. In the latter, CM appears preferentially in the rare case of postzygotic mosaic loss of the healthy allele, as in SOLAMEN.
Summary and Conclusion
Now that the molecular basis for many mosaic overgrowth syndromes is known, we could start to think about treating them. Several older and newer drugs that have been developed to treat cancer target the relevant pathways. For instance, mTORC1 can be inhibited by rapamycin.55 There are specific inhibitors for BRAF, such as vemurafenib,56 and PIK3CA, such as taselisib.57 Although the congenital abnormalities associated with the overgrowth syndromes described earlier may not be amenable to pharmacological correction, the mutations having interfered with embryogenesis, the postnatal progression of symptoms might be amenable to pharmacological correction. There are obvious concerns about the effects of powerful growth-inhibiting drugs during childhood. However, as the signaling systems that are being targeted are abnormally active in the affected tissues only, it is to be expected that, with proper dosage, normal growth could remain unaffected. Whether neurological symptoms or developmental delay would be improved by such treatment remains to be seen. The experience with rapamycin in TSC suggests that this might indeed happen. In TSC, mTORC1 is deregulated resulting in abnormal growth and development of several tissues including the brain, in which typical neuronal migration defects occur, giving rise to epilepsy and developmental delay. The epilepsy can be quite resistant to anticonvulsants; however, it was recently shown that rapamycin, which inhibits mTORC1 activity, could help control convulsions.58 In animal models of the disease, rapamycin ameliorates the cognitive defects associated with TSC.
Thus, proper recognition and subsequent molecular diagnosis of mosaic neurocutaneous overgrowth syndromes is more important than ever. As genetic analysis in mosaic disorders is technically challenging, referral to a specialist center with appropriate expertise and capabilities is essential.
Acknowledgments
The author's work in Singapore is supported by the Skin Research Institute of Singapore. His work in Dundee is supported by the Wellcome Trust and by DEBRA UK. His work is also supported by Tenovus Scotland and the University of Dundee.
References
- 1.Happle R. Cutaneous manifestation of lethal genes. Hum Genet. 1986;72(3):280. doi: 10.1007/BF00291899. [DOI] [PubMed] [Google Scholar]
- 2.Findlay G H, Moores P P. Pigment anomalies of the skin in the human chimaera: their relation to systematized naevi. Br J Dermatol. 1980;103(5):489–498. doi: 10.1111/j.1365-2133.1980.tb01663.x. [DOI] [PubMed] [Google Scholar]
- 3.Pollock P M, Harper U L, Hansen K S. et al. High frequency of BRAF mutations in nevi. Nat Genet. 2003;33(1):19–20. doi: 10.1038/ng1054. [DOI] [PubMed] [Google Scholar]
- 4.Logié A, Dunois-Lardé C, Rosty C. et al. Activating mutations of the tyrosine kinase receptor FGFR3 are associated with benign skin tumors in mice and humans. Hum Mol Genet. 2005;14(9):1153–1160. doi: 10.1093/hmg/ddi127. [DOI] [PubMed] [Google Scholar]
- 5.Vreeburg M, van Steensel M AM. Genodermatoses caused by genetic mosaicism. Eur J Pediatr. 2012;171(12):1725–1735. doi: 10.1007/s00431-012-1855-9. [DOI] [PubMed] [Google Scholar]
- 6.Happle R. Lyonization and the lines of Blaschko. Hum Genet. 1985;70(3):200–206. doi: 10.1007/BF00273442. [DOI] [PubMed] [Google Scholar]
- 7.Happle R. Lethal genes surviving by mosaicism: a possible explanation for sporadic birth defects involving the skin. J Am Acad Dermatol. 1987;16(4):899–906. doi: 10.1016/s0190-9622(87)80249-9. [DOI] [PubMed] [Google Scholar]
- 8.Happle R. What is a nevus? A proposed definition of a common medical term. Dermatology. 1995;191(1):1–5. doi: 10.1159/000246468. [DOI] [PubMed] [Google Scholar]
- 9.Lapidot T, Sirard C, Vormoor J. et al. A cell initiating human acute myeloid leukaemia after transplantation into SCID mice. Nature. 1994;367(6464):645–648. doi: 10.1038/367645a0. [DOI] [PubMed] [Google Scholar]
- 10.Kobilka B K. G protein coupled receptor structure and activation. . Biochim Biophys Acta. 2007;1768(4):794–807. doi: 10.1016/j.bbamem.2006.10.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Weinstein L S, Shenker A, Gejman P V, Merino M J, Friedman E, Spiegel A M. Activating mutations of the stimulatory G protein in the McCune-Albright syndrome. N Engl J Med. 1991;325(24):1688–1695. doi: 10.1056/NEJM199112123252403. [DOI] [PubMed] [Google Scholar]
- 12.Palmisano I, Bagnato P, Palmigiano A. et al. The ocular albinism type 1 protein, an intracellular G protein-coupled receptor, regulates melanosome transport in pigment cells. Hum Mol Genet. 2008;17(22):3487–3501. doi: 10.1093/hmg/ddn241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Comi A M. Update on Sturge-Weber syndrome: diagnosis, treatment, quantitative measures, and controversies. Lymphat Res Biol. 2007;5(4):257–264. doi: 10.1089/lrb.2007.1016. [DOI] [PubMed] [Google Scholar]
- 14.Waelchli R, Aylett S E, Robinson K, Chong W K, Martinez A E, Kinsler V A. New vascular classification of port-wine stains: improving prediction of Sturge-Weber risk. Br J Dermatol. 2014;171(4):861–867. doi: 10.1111/bjd.13203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Shirley M D, Tang H, Gallione C J. et al. Sturge-Weber syndrome and port-wine stains caused by somatic mutation in GNAQ. N Engl J Med. 2013;368(21):1971–1979. doi: 10.1056/NEJMoa1213507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Van Raamsdonk C D, Bezrookove V, Green G. et al. Frequent somatic mutations of GNAQ in uveal melanoma and blue naevi. Nature. 2009;457(7229):599–602. doi: 10.1038/nature07586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fernández-Guarino M, Boixeda P, de Las Heras E, Aboin S, García-Millán C, Olasolo P J. Phakomatosis pigmentovascularis: clinical findings in 15 patients and review of the literature. J Am Acad Dermatol. 2008;58(1):88–93. doi: 10.1016/j.jaad.2007.08.012. [DOI] [PubMed] [Google Scholar]
- 18.Vissers W, Van Steensel M, Steijlen P, Renier W, Van De Kerkhof P, Van Der Vleuten C. Klippel-Trenaunay syndrome and Sturge-Weber syndrome: variations on a theme? Eur J Dermatol. 2003;13(3):238–241. [PubMed] [Google Scholar]
- 19.Oduber C EU, van der Horst C MAM, Sillevis Smitt J H. et al. A proposal for classification of entities combining vascular malformations and deregulated growth. Eur J Med Genet. 2011;54(3):262–271. doi: 10.1016/j.ejmg.2011.02.007. [DOI] [PubMed] [Google Scholar]
- 20.Happle R, Hoffmann R, Restano L, Caputo R, Tadini G. Phacomatosis pigmentokeratotica: a melanocytic-epidermal twin nevus syndrome. Am J Med Genet. 1996;65(4):363–365. doi: 10.1002/(SICI)1096-8628(19961111)65:4<363::AID-AJMG27>3.0.CO;2-R. [DOI] [PubMed] [Google Scholar]
- 21.Groesser L, Herschberger E, Sagrera A. et al. Phacomatosis pigmentokeratotica is caused by a postzygotic HRAS mutation in a multipotent progenitor cell. J Invest Dermatol. 2013;133(8):1998–2003. doi: 10.1038/jid.2013.24. [DOI] [PubMed] [Google Scholar]
- 22.Brandling-Bennett H A, Morel K D. Epidermal nevi. Pediatr Clin North Am. 2010;57(5):1177–1198. doi: 10.1016/j.pcl.2010.07.004. [DOI] [PubMed] [Google Scholar]
- 23.Moody M N, Landau J M, Goldberg L H. Nevus sebaceous revisited. Pediatr Dermatol. 2012;29(1):15–23. doi: 10.1111/j.1525-1470.2011.01562.x. [DOI] [PubMed] [Google Scholar]
- 24.Aoki Y, Niihori T, Kawame H. et al. Germline mutations in HRAS proto-oncogene cause Costello syndrome. Nat Genet. 2005;37(10):1038–1040. doi: 10.1038/ng1641. [DOI] [PubMed] [Google Scholar]
- 25.Søvik O, Schubbert S, Houge G. et al. De novo HRAS and KRAS mutations in two siblings with short stature and neuro-cardio-facio-cutaneous features. BMJ Case Rep. 2009;2009 doi: 10.1136/bcr.07.2008.0550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Niihori T, Aoki Y, Narumi Y. et al. Germline KRAS and BRAF mutations in cardio-facio-cutaneous syndrome. Nat Genet. 2006;38(3):294–296. doi: 10.1038/ng1749. [DOI] [PubMed] [Google Scholar]
- 27.Groesser L, Herschberger E, Ruetten A. et al. Postzygotic HRAS and KRAS mutations cause nevus sebaceous and Schimmelpenning syndrome. Nat Genet. 2012;44(7):783–787. doi: 10.1038/ng.2316. [DOI] [PubMed] [Google Scholar]
- 28.Siddiqui M A, Siddiqui S, Zaman N, Ahmad I, Ullah E. Neurocutaneous melanosis: Review of a rare non-familial neuroectodermal dysplasia with newer association of cerebellopontine angle cistern lipoma. Neuroradiol J. 2015;28(2):222–226. doi: 10.1177/1971400915581746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kinsler V A, Thomas A C, Ishida M. et al. Multiple congenital melanocytic nevi and neurocutaneous melanosis are caused by postzygotic mutations in codon 61 of NRAS. J Invest Dermatol. 2013;133(9):2229–2236. doi: 10.1038/jid.2013.70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Salgado C M, Basu D, Nikiforova M. et al. BRAF mutations are also associated with neurocutaneous melanocytosis and large/giant congenital melanocytic nevi. Pediatr Dev Pathol. 2015;18(1):1–9. doi: 10.2350/14-10-1566-OA.1. [DOI] [PubMed] [Google Scholar]
- 31.Rivière J-B, Mirzaa G M, O'Roak B J. et al. De novo germline and postzygotic mutations in AKT3, PIK3R2 and PIK3CA cause a spectrum of related megalencephaly syndromes. Nat Genet. 2012;44(8):934–940. doi: 10.1038/ng.2331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Keppler-Noreuil K M, Rios J J, Parker V ER. et al. PIK3CA-related overgrowth spectrum (PROS): diagnostic and testing eligibility criteria, differential diagnosis, and evaluation. Am J Med Genet A. 2015;167A(2):287–295. doi: 10.1002/ajmg.a.36836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Loconte D C, Grossi V, Bozzao C. et al. Molecular and functional characterization of three different postzygotic mutations in PIK3CA-related overgrowth spectrum (PROS) patients: effects on PI3K/AKT/mTOR signaling and sensitivity to PIK3 inhibitors. PLoS ONE. 2015;10(4):e0123092. doi: 10.1371/journal.pone.0123092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mirzaa G M, Conway R L, Gripp K W. et al. Megalencephaly-capillary malformation (MCAP) and megalencephaly-polydactyly-polymicrogyria-hydrocephalus (MPPH) syndromes: two closely related disorders of brain overgrowth and abnormal brain and body morphogenesis. Am J Med Genet A. 2012;158A(2):269–291. doi: 10.1002/ajmg.a.34402. [DOI] [PubMed] [Google Scholar]
- 35.Fu X, Osborne C K, Schiff R. Biology and therapeutic potential of PI3K signaling in ER+/HER2-negative breast cancer. Breast. 2013;22 02:S12–S18. doi: 10.1016/j.breast.2013.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Alomari A I. Characterization of a distinct syndrome that associates complex truncal overgrowth, vascular, and acral anomalies: a descriptive study of 18 cases of CLOVES syndrome. Clin Dysmorphol. 2009;18(1):1–7. doi: 10.1097/MCD.0b013e328317a716. [DOI] [PubMed] [Google Scholar]
- 37.Gucev Z S, Tasic V, Jancevska A. et al. CLOVE syndrome (congenital lipomatous overgrowth, vascular malformations, and epidermal nevi): CNS malformations and seizures may be a component of this disorder. Am J Med Genet A. 2008;146A:2688–2690. doi: 10.1002/ajmg.a.32515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Milella M, Falcone I, Conciatori F. et al. PTEN: multiple functions in human malignant tumors. Front Oncol. 2015;5:24. doi: 10.3389/fonc.2015.00024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Pilarski R, Eng C. Will the real Cowden syndrome please stand up (again)? Expanding mutational and clinical spectra of the PTEN hamartoma tumour syndrome. J Med Genet. 2004;41(5):323–326. doi: 10.1136/jmg.2004.018036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bubien V, Bonnet F, Brouste V. et al. High cumulative risks of cancer in patients with PTEN hamartoma tumour syndrome. J Med Genet. 2013;50(4):255–263. doi: 10.1136/jmedgenet-2012-101339. [DOI] [PubMed] [Google Scholar]
- 41.Goffin A, Hoefsloot L H, Bosgoed E, Swillen A, Fryns J P. PTEN mutation in a family with Cowden syndrome and autism. Am J Med Genet. 2001;105(6):521–524. doi: 10.1002/ajmg.1477. [DOI] [PubMed] [Google Scholar]
- 42.Tan W-H, Baris H N, Burrows P E. et al. The spectrum of vascular anomalies in patients with PTEN mutations: implications for diagnosis and management. J Med Genet. 2007;44(9):594–602. doi: 10.1136/jmg.2007.048934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Caux F, Plauchu H, Chibon F. et al. Segmental overgrowth, lipomatosis, arteriovenous malformation and epidermal nevus (SOLAMEN) syndrome is related to mosaic PTEN nullizygosity. Eur J Hum Genet. 2007;15(7):767–773. doi: 10.1038/sj.ejhg.5201823. [DOI] [PubMed] [Google Scholar]
- 44.Happle R. Superimposed segmental manifestation of both rare and common cutaneous disorders: a new paradigm. Actas Dermosifiliogr. 2009;100 01:77–85. doi: 10.1016/s0001-7310(09)73171-0. [DOI] [PubMed] [Google Scholar]
- 45.Huang J, Manning B D. The TSC1-TSC2 complex: a molecular switchboard controlling cell growth. Biochem J. 2008;412(2):179–190. doi: 10.1042/BJ20080281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lindhurst M J, Sapp J C, Teer J K. et al. A mosaic activating mutation in AKT1 associated with the Proteus syndrome. N Engl J Med. 2011;365(7):611–619. doi: 10.1056/NEJMoa1104017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Tibbles J A, Cohen M M Jr. The Proteus syndrome: the Elephant Man diagnosed. Br Med J (Clin Res Ed) 1986;293(6548):683–685. doi: 10.1136/bmj.293.6548.683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hussain K, Challis B, Rocha N. et al. An activating mutation of AKT2 and human hypoglycemia. Science. 2011;334(6055):474. doi: 10.1126/science.1210878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Cohen M M Jr. Proteus syndrome review: molecular, clinical, and pathologic features. Clin Genet. 2014;85(2):111–119. doi: 10.1111/cge.12266. [DOI] [PubMed] [Google Scholar]
- 50.Beachkofsky T M, Sapp J C, Biesecker L G, Darling T N. Progressive overgrowth of the cerebriform connective tissue nevus in patients with Proteus syndrome. J Am Acad Dermatol. 2010;63(5):799–804. doi: 10.1016/j.jaad.2009.12.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Biesecker L. The challenges of Proteus syndrome: diagnosis and management. Eur J Hum Genet. 2006;14(11):1151–1157. doi: 10.1038/sj.ejhg.5201638. [DOI] [PubMed] [Google Scholar]
- 52.Jacks S K, Witman P M. Tuberous sclerosis complex: an update for dermatologists. Pediatr Dermatol. 2015 doi: 10.1111/pde.12567. [DOI] [PubMed] [Google Scholar]
- 53.Navarre P, Poitras B. Lymphoedema in tuberous sclerosis: case report and review of the literature. J Pediatr Orthop. 2014;34(6):e27–e32. doi: 10.1097/BPO.0000000000000240. [DOI] [PubMed] [Google Scholar]
- 54.Wong H, Hadi M, Khoury T, Geary D, Rubin B, Filler G. Management of severe hypertension in a child with tuberous sclerosis-related major vascular abnormalities. J Hypertens. 2006;24(3):597–599. doi: 10.1097/01.hjh.0000209994.33680.11. [DOI] [PubMed] [Google Scholar]
- 55.Sampson J R. Therapeutic targeting of mTOR in tuberous sclerosis. Biochem Soc Trans. 2009;37(Pt 1):259–264. doi: 10.1042/BST0370259. [DOI] [PubMed] [Google Scholar]
- 56.Chapman P B, Hauschild A, Robert C. et al. Improved survival with vemurafenib in melanoma with BRAF V600E mutation. N Engl J Med. 2011;364(26):2507–2516. doi: 10.1056/NEJMoa1103782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Lopez S, Schwab C L, Cocco E. et al. Taselisib, a selective inhibitor of PIK3CA, is highly effective on PIK3CA-mutated and HER2/neu amplified uterine serous carcinoma in vitro and in vivo. Gynecol Oncol. 2014;135(2):312–317. doi: 10.1016/j.ygyno.2014.08.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Sadowski K, Kotulska-Jóźwiak K, Jóźwiak S. Role of mTOR inhibitors in epilepsy treatment. Pharmacol Rep. 2015;67(3):636–646. doi: 10.1016/j.pharep.2014.12.017. [DOI] [PubMed] [Google Scholar]