

Abstract
Pitt–Hopkins syndrome is an emerging neurodevelopmental disorder caused by haploinsufficiency of the TCF4 gene on chromosome 18q21. It is characterized by severe intellectual disability, seizures, microcephaly, constipation and a distinctive facial gestalt. Although the overlapping phenotype of microcephaly, epilepsy, absent speech and constipation represents a challenge for the differential diagnosis with Angelman syndrome, Rett syndrome and Mowat–Wilson syndrome, distinctive of Pitt–Hopkins syndrome are breathing abnormalities, that can occur as either hyperventilation episodes or apnea crises, and a typical facial dysmorphism, including bitemporal narrowing, squared forehead, deep-set eyes, peculiar nose conformation, with broad nasal bridge, down-turned nasal tip and flaring nostrils, typical shape of the mouth, with a tented and M shaped upper lip, and widely spaced teeth. The occurrence of these signs in variable association of uncoordinated movements, microcephaly of postnatal onset, eye abnormalities, constipation, epilepsy and subtle brain abnormalities is highly predictive of a TCF4 mutation, making it possible to plan a genetic test of choice among severe encephalopathies. Angelman syndrome represents the nosological condition closest to Pitt–Hopkins syndrome.
Keywords: Pitt-Hopkins syndrome, TCF4, differential diagnosis
Introduction
Pitt–Hopkins syndrome (PTHS; MIM # 610954) is an emerging neurodevelopmental disorder falling within syndromic forms of severe intellectual disability (ID). First described clinically in 1978 in two unrelated patients with recurrent episodes of hyperventilation and distinctive facial appearance,1 only a few cases were reported2 3 4 5 until 2007, when haploinsufficiency of the gene encoding the basic helix-loop-helix (bHLH) transcription factor 4 (TCF4, OMIM *602272) was identified as the underlying cause of this condition.6 7
Following the identification of the causative gene, approximately 200 patients have been reported so far1 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18(Pitt–Hopkins Syndrome Support Group: http://groups.google.com/group/pitt-hopkins/about?pli=1).
Clinically, PTHS is a severe, but not progressive, encephalopathy, characterized by ID with nearly absent speech and a distinctive facial gestalt. Highly predictive of a TCF4 mutation are breathing abnormalities, which can occur as either hyperventilation episodes or apnea crises. Common clinical signs also include uncoordinated movements, microcephaly of postnatal onset, eye abnormalities, constipation, epilepsy, and subtle brain abnormalities.
The overlapping phenotype of severe ID with absent speech, epilepsy, and constipation makes it difficult to differentiate the PTHS phenotype from that of Angelman syndrome (AS; MIM # 105830), MECP2-associated Rett syndrome (MIM # 300005), and Mowat–Wilson syndrome (MWS; MIM # 235730), and most reported patients underwent molecular tests for all these conditions first. However, there is evidence that a distinctive PTHS phenotype exists. Comprehensive clinical analysis of literature patients and of our personal patients led us to define a checklist of the most consistent phenotypic manifestations of this condition. On this basis, we have developed a clinical scoring system, in an attempt not only to drive a first choice molecular testing for PTHS but also to allow a clinically based diagnosis in patients without a proven mutation in TCF4.16 Relying on key morphological, neurological, and behavioral diagnostic features, another clinical diagnostic score for PTHS was recently designed by Whalen et al,17 to distinguish PTHS among patients with the Rett-like clinical phenotype.
Of importance, both clinical variability and genetic heterogeneity exist within PTHS, most likely, as a consequence of the variable impairment of the protein function caused by different TCF4 mutations, and the involvement of different causative genes, respectively.
In this review, we made an attempt to highlight the following issues: revision of the biological properties of TCF4; the distinctive features of the PTHS phenotype; the standardized clinical criteria for enrolling patients into the targeted genetic test; the mutational spectrum affecting TCF4, with the aim to suggest the most sensitive diagnostic procedure; and the relevant clinical elements for the differential diagnosis with conditions characterized by overlapping clinical manifestations.
The TCF4 Gene
The TCF4 gene (MIM# 610954) is located on chromosome 18q21.2, and it has 20 exons (the first and the last are noncoding) that span 360 kb. It encodes a bHLH transcription factor TCF4, a protein with transcription-regulatory activities that is highly expressed during early human development throughout the central nervous system, the sclerotome, peribronchial and kidney mesenchyme, and the genital bud,13 playing an important role in cellular proliferation, lineage commitment, and cellular differentiation.
Several alternatively spliced TCF4 variants have been described, allowing for the translation of at least 18 protein isoforms, with different N-terminal sequences. TCF4-A and TCF4-B (the longest one) are the most extensively studied isoforms.19 20
TCF4 protein has two activation domains (AD1 and AD2), likely involved in modulating transcriptional activity, a nuclear localization signal (NLS), and the bHLH domain. The N-terminal region of this domain is responsible for the interaction with DNA and specifically with the so-called E-boxes (Ephrussi boxes), characterized by the consensus sequence CANNTG. The remaining part of the bHLH motif, along with an adjacent C-terminal domain, favors homo- and heterodimerization of TCF4 with other bHLH proteins.20
TCF4 has been claimed to be involved in a series of developmental processes, including lymphoid development, epithelial–mesenchymal transition, and the development of several regions of the nervous system. Many proneural factors have been identified to form heterodimers with TCF4 to regulate gene expression during neurodevelopment, such as ATOH1, NEUROG2, and ASCL1. ASCL1 is likely involved in the noradrenergic neuronal development in the brain stem and has been hypothesized that the breathing anomalies that characterize PTHS may result from impaired noradrenergic neuronal development after defective TCF4 interaction with the ASCL1–PHOX–RET pathway.13 Some of the genes that appear to be regulated by TCF4 are known to be responsible for syndromic conditions included in the differential diagnosis of PTHS: UBE3A (AS); ZEB2 (MWS); MEF2C (Rett-like syndrome); and CNTNAP2 and NRXN1, both associated with a variant autosomal recessive form of the PTHS phenotype.
Even though PTHS is caused by the haploinsufficiency of TCF4, it has been demonstrated that mutations involving different regions of the gene have variable impact on the protein function: several deletions and truncating mutations do not result in complete loss of function of the protein, while missense mutations are more likely deleterious if they involve the bHLH domain.21
Notably, the TCF4 gene appears to be involved in other medical conditions. Genome-wide association studies (GWAS) found significant associations between a group of polymorphic TCF4 variants and schizophrenia,22 primary sclerotizing cholangitis,23 and Fuchs' endothelial dystrophy.24
The PTHS Phenotype
Relying on personal observation of a total of 32 patients, of whom 18 were previously reported,16 and on literature reports, referring to additional 96 individuals,17 the most relevant component manifestations of the PTHS phenotype are described as following.
The Facial Dysmorphism
The majority of patients with a proven mutation in TCF4 present with a distinctive craniofacial dysmorphism, characterized by brachycephaly; bitemporal narrowing; squared forehead; deep-set eyes; a peculiar nose conformation, with broad nasal bridge, down-turned nasal tip, and flaring nostrils; and by a highly distinctive mouth conformation, with a tented and M-shaped upper lip, with protruding and thick lower lip vermillion and widely spaced teeth. Typical craniofacial features also include full cheeks associated with prominent lower face and cup-shaped and fleshy ears (Fig. 1).
Fig. 1.
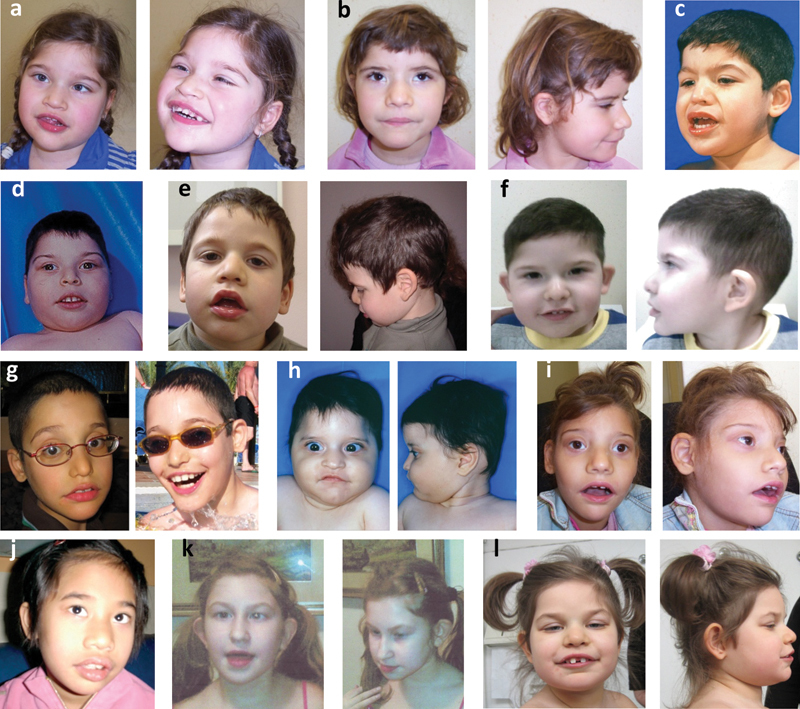
Comprehensive facial appearance in a group of PTHS patients with a proven mutation in TCF4. Please note that one patient (no. k), lacks the typical facial dysmorphism, and yet she presents with strabismus (already presented in “Marangi et al16”).
The Pattern of Growth
Measures at birth, including head circumference, are within normal parameters in the majority of children with few exceptions. However, true microcephaly, or progressive decrease of head circumference, occurs in approximately 60% of patients postnatally, associated with normal body weight and height. Moderate short stature is observed in a small proportion of patients in the postnatal period, mainly in association with a relatively large chromosome deletion on 18q21.2.
ID/Speech Delay and Behavior
ID is severe and language is almost absent in most patients. Of importance, a relatively mild cognitive impairment was described in few cases, associated with either a balanced translocation allowing residual function of a large part of the TCF4 protein25 or with hypomorphic missense variants outside of the bHLH domain which is not productive of major functional deficiencies.21
Smiling appearance and lovable behavior characterize the majority of patients. However, anxiety, agitation, and true pervasive developmental disorder represent other relevant component manifestations of the behavioral phenotype. Stereotypic movements, including hand clapping and flapping and repeated hand–mouth movements, are frequent features as well.
Major Malformations
While major visceral malformations are unusual, subtle central nervous system abnormalities are detected in 60 to 70% of patients by brain magnetic resonance imaging (MRI), consisting mainly of corpus callosum hypoplasia/aplasia, ventricular dilatation, and thin hindbrain.
Ophthalmological Signs
Ocular abnormalities in a variable association of strabismus, myopia, and astigmatism were found to affect nearly all of our patients (32), and a large proportion of patients in the literature. Rare ophthalmological features include maculopathy, electroretinography anomalies, optical atrophy, and nystagmus.
Other Neurological Signs
Additional and frequent neurological signs include hypotonia, which is diagnosed in more than 90% of patients; motor delay; ataxic gait/motor incoordination; and epilepsy. In particular, the seizure disorder affects on average 50% of patients and it occurs with variable clinical presentation, ranging from spasms to episodes of absence and tonic–clonic seizures. Seizures are usually easily controlled by anticonvulsant drugs. None of the reported patients had Hirschsprung disease; however, the majority of them suffer from constipation, which can be severe in some cases.
Breathing Abnormalities
Episodes of hyperventilation, which may be followed by apnea, are highly suggestive of PTHS when they occur in combination with the characteristic facial dysmorphisms and with additional features, as discussed later. Breathing anomalies are recorded in over half of patients, with onset at different ages, ranging from 7 months to 7 years.
A Clinical Score System for the Molecular Diagnosis of PTHS
Comprehensive clinical analysis of either literature patients or our personal patients with a proven mutation in TCF4 led us to define a checklist of the most consistent phenotypic manifestations of this condition, with the aim to facilitate the clinical diagnosis of PTHS, and to drive a first choice molecular test for TCF4.16 They are the facial dysmorphism; ID of severe degree; microcephaly of postnatal onset; seizures; ataxic gait or motor incoordination; breathing abnormalities; constipation; ophthalmologic anomalies; and subtle brain abnormalities. In more detail, a score was assigned to each of them according to the following criteria: (1) distinctive facial appearance—four points to a full facial phenotype, two points to facial features partially consistent with PTHS, no points to absent facial gestalt; (2) ID—two points to moderate or severe ID, no point to mild ID; (3) language impairment—two points to absent speech, one to speech limited to few words or short sentences; and (4) one point to each of the following signs—normal measurements at birth; microcephaly or progressive deceleration of head circumference growth in the postnatal period; electroencephalographic (EEG) pattern abnormalities with or without epileptic fits; ataxic gait or general motor incoordination; breathing abnormalities including either hyperventilation or apnea episodes; constipation; ophthalmologic abnormalities, including strabismus, myopia, and astigmatism; and subtle brain MRI abnormalities, consisting mainly of corpus callosum hypoplasia, enlargement of ventricles, and thin hindbrain.
Mutational Spectrum Affecting the TCF4 Gene and the Most Effective Diagnostic Procedure
Different genomic mutations affecting the TCF4 gene have been described in association with PTHS. They include the following:
Whole gene deletions, which are identified in approximately 30% of cases. Larger rearrangements can be several megabases in size, and the involvement of contiguous genes can further worsen the phenotype.
Partial gene deletion, usually detectable by MLPA analysis, in less than 10% of cases and involving one or more of the exons from 7 to 20.
Balanced translocations, disrupting the coding sequence of the gene and identifiable only by standard karyotype or FISH analysis. In our series of patients, 2 out 32 genomic mutations were t(14;18)(q13.1;q21.2) and t(2;18)(q37;q21.2) de novo balanced translocations, respectively, with breakpoints falling within the second half of the gene, resulting in classical PTHS phenotype. Of note, a milder PTHS phenotype was described in the patient reported by Kalscheuer et al25 in association with a balanced t(18;20)translocation. It was explained by the evidence that the translocation left preserved exons 4 to 20 of TCF4, in which the main functions of the gene reside.
Missense mutations (20–35% of cases), which mainly involve the bHLH domain of TCF4.
Nonsense and frameshift mutations, observed in approximately 40% of cases, which are spread throughout the gene between exons 7 and 18.
Splice site mutations (∼10%), mainly affecting the donor and acceptor consensus splice sites and usually resulting in the shift of the reading frame. In fact, no splice mutations affecting exon 15, the only in-frame exon, have been reported in PTHS patients to date.
Suggested by the relative prevalence of the different genomic mutations, the most effective diagnostic procedure allowing for confirmation of a typical PTHS clinical phenotype should include in order (1) direct sequencing/MLPA of TCF4; (2) array-CGH, whenever a complete loss of TCF4 is detected by MLPA, with the purpose of defining the whole gene content of the deletion; array-CGH is recommended as second diagnostic step in case with normal genetic test for TCF4 as well, to search for different genomic copy number variations mimicking PTHS; (3) conventional cytogenetics, to search for balanced translocations disrupting the coding region of TCF4.
It must be specified that patients negative for all the aforementioned genomic mutations can be currently enrolled in Next Generation Analysis (NGS) techniques, by either resequencing of target genes or whole exome sequencing, to search for genetic heterogeneity of the PTHS phenotype. However, the diagnostic relevance of NGS results can be limited to specific subtypes of mutations.
Differential Diagnosis
Clinical differential diagnosis of PTHS includes distinguishing PTHS versus different epileptic encephalopathies with severe ID, mainly versus MWS, Rett syndrome, and AS. Relevant for the differential diagnosis are the following considerations.
Mowat–Wilson Syndrome
MWS is associated with a very distinctive pattern of facial anomalies, characterized by sparse eyebrows in early infancy, the typical and unusual uplifted earlobe configuration, hypertelorism, and linear mandible arch. A variety of major malformations, including heart defects, genitourinary anomalies, and true Hirschsprung disease, are more frequently detected in MWS rather than in PTHS.
Angelman Syndrome
AS represents the neurodevelopmental disorder most closely resembling PTHS, due to the overlapping features of severe ID with absent speech, epilepsy, microcephaly, ataxic gait, and happy disposition. However, more consistent with AS are episodes of unmotivated laughing, severe seizure disorder, and severe microcephaly as well, other than highly distinctive EEG features. More importantly, AS subjects lack the typical facial features observed in PTHS, mainly with respect to the ocular anomalies, the typical nose conformation with broad nasal bridge and flared nostrils, and the prominent M-shaped upper lip. Of note, Takano et al14 recently reported that 2% of patients initially suspected as having AS were found to have PTHS with a mutation in TCF4. However, several facial characteristics highly consistent with PTHS can be appreciated in these patients on the published clinical pictures, confirming the pivotal role of the facial dysmorphism in moving the diagnosis of PTHS.
Rett Syndrome
Owing to episodes of hyperventilation/apnea and stereotypic movements, PTHS was initially considered to be part of the clinical spectrum of Rett syndrome. However, the typical MECP2-associated Rett syndrome represents the neurodevelopmental disorder least close to PTHS clinically. First, Rett syndrome usually presents as a progressive encephalopathy, in which an apparently normal early development is followed by arrest and regression of language and motor skills. In addition, brain abnormalities are unusual in Rett syndrome and, importantly, patients lack the facial characteristics associated with PTHS.
NRXN1- and CNTNAP2-Associated Autosomal Recessive Intellectual Disability Disorders
The autosomal recessive disorder caused by mutations in NRXN1 was reported in three individuals in association with severe global developmental delay, lack of speech, stereotypies, and episodic breathing differences.26 27 A similar clinical phenotype was described in association with a homozygous mutation in CNTNAP2 as well.26 Although both conditions are widely referred to as AR forms of PTHS, affected individuals lack the characteristic facial features seen in PTHS and have abnormal sleep–wake cycles, which are not commonly reported with PTHS.
Discussion
ID affects approximately 1 to 3% of the general population. Although most severe forms of ID are genetic in origin, with mutations ranging from large cytogenetic abnormalities to point mutations, the genetic etiology of ID remains currently unexplained in approximately 50% of cases. The underlying genomic defect in these cases is a variant in a single pathogenic gene, most likely, that can go undetected by targeted gene sequencing. Extensive clinical characterization of rare genetic conditions can allow not only for successful targeted gene testing but also for clinical validation of certain gene variants that can be diagnosed by NGS techniques, including whole exome sequencing.
PTHS is a rare nonprogressive encephalopathy, described so far in approximately 200 reported subjects, whose causative gene, encoding the bHLH transcription factor 4 (TCF4, MIM *602272), was identified only in 2007.6 7 However, it currently represents an emerging diagnosis within syndromic forms of ID that share severe cognitive impairment, absent speech, seizures, and a constellation of additional clinical manifestations. One can speculate that several PTHS cases are currently going undetected, most likely, due to the very recent definition of PTHS at a nosological level, and to the clinical overlap with different conditions, including mainly AS, but also MWS and Rett syndrome.
As highlighted in the present review, the PTHS phenotype represents a well-defined clinical and genetic entity. Consistent clinical signs include a highly distinctive facial dysmorphism, with bitemporal narrowing, full cheeks associated with a prominent lower face, deep-set eyes, large nasal bridge, large mouth with M-shaped upper lip and a protruding and thick lower lip vermillion, and cup-shaped ears. Strongly predictive of a mutation in TCF4 are breathing abnormalities, which can occur as either hyperventilation episodes or apnea crisis. Of relevance for the clinically based hypothesis of PTHS, breathing anomalies are recorded in over half of patients, with onset at different ages, ranging from 7 months to 7 years. For that reason, and also in considering that they can go undetected when limited to apnea episodes, the absence of a breathing abnormality should not eliminate consideration of the diagnosis of PTHS.
A constellation of additional features, including sweet personality, subtle brain abnormalities, seizures, postnatal microcephaly, ataxic gait/motor incoordination, and eye abnormalities, such as strabismus, myopia, and astigmatism, are detected in most patients. Although quite unspecific, these latter features turn out to be very significant for the clinical diagnosis when they occur in association. As a matter of fact, many clinical signs in PTHS can be found in different genetic conditions, including mainly the aforementioned AS, MWS, and Rett syndrome, and the majority of PTHS patients reported so far underwent the genetic test for all these conditions first. However, extensive genotype–phenotype correlation analyses led us to define a clinical score system for targeted genetic testing for PTHS,16 with a score ≥13 highly predictive of a variant in TCF4 (Table 1). It must be specified that the most sensitive feature for the clinical suspicion of PTHS is the facial dysmorphism. However, in considering that some patients in literature, including one of our patients,15 lack the typical facial characteristics, a clinically based score system can help. To look for sensitivity of our score system, we also performed a comparison with the resulting scores obtained in patients suffering from different conditions, including MWS patients with ZEB2 mutation, who had a score ≤9; AS patients with positive 15q11 methylation-based MLPA analysis (score on average ≤9, but ranging from 10 to 11 in some); Rett syndrome patients with MECP2 mutation (score ≤9) (Fig. 1).
Table 1. Checklist for the clinical score.
| Clinical signs | Points |
|---|---|
| Moderate to severe mental retardation | 2 |
| Absent speech | 2 |
| Severe speech impairment with more than 10 words vocabulary and/or capacity to form two- to three-word sentences | 1 |
| Normal growth parameters at birth | 1 |
| Postnatal microcephaly or progressive slowing down of head circumference | 1 |
| Epilepsy/EEG abnormalities | 1 |
| Ataxic gait/motor incoordination | 1 |
| Breathing anomalies: hyperventilation fits or apnea episodes | 1 |
| Mild to severe constipation | 1 |
| Brain MRI abnormalities (corpus callosum hypoplasia, enlargement of ventricles, and thin hindbrain) | 1 |
| Ophthalmologic abnormalities (strabismus, myopia, and astigmatism) | 1 |
| Typical PTHS facial features | 4 |
| Facial features only partially consistent with PTHS | 2 |
| Maximum score | 16 |
Abbreviations: EEG, electroencephalographic; MRI, magnetic resonance imaging; PTHS, Pitt–Hopkins syndrome.
A summary of the most consistent clinical signs is shown in Table 2, along with their individual frequency.
Table 2. Overview of clinical features of PTHS patients, based on personal observation (32) and the studies by Whalen et al17 and Sweatt18 .
| Clinical signs | Total (%) |
|---|---|
| Typical facial appearance: | >95 |
| • Deep-set eyes | |
| • Broad nasal bridge | |
| • Beaked nose | |
| • Down-turned nasal tip | |
| • Pointed nasal tip | |
| • Flaring nostrils | |
| • Wide mouth | |
| • Widely spaced teeth | |
| • Cupid's bow upper lip | |
| • Protruding lower face | |
| • Cup-shaped ears | |
| • Fleshy ears | |
| Severe mental retardation with severe speech impairment | 100 |
| Hypotonia | 70–90 |
| Normal growth parameters at birth | 80–90 |
| Postnatal growth retardation | 10–30 |
| Postnatal microcephaly (or progressive slowing down of head circumference growth) | 25–75 |
| Breathing anomalies | 50–80 |
| • Hyperventilation episodes | 40–60 |
| • Apnea crises | 40–60 |
| Motor incoordination | 50–80 |
| • Severe ataxia | |
| • Broad-based gait | |
| • Incoordination of hand–mouth movements | |
| Ocular anomalies | 50–80 |
| • Strabismus | |
| • Myopia | |
| • Astigmatism | |
| Constipation | 60–80 |
| Seizures (or EEG anomalies)a | 20–90 |
| Happy disposition | 80–95 |
| Stereotypic movements | 70–95 |
| • Bruxism | |
| • Lateral movements | |
| • Hand clapping | |
| • Hand flapping | |
| • Hand–mouth movements | |
| Hand anomalies | 50–70 |
| • Tapering fingers | |
| • Clinodactyly | |
| • Fetal finger pads | |
| • Single palmar crease | |
| • Absent thumb flexion crease | |
| Brain abnormalities | 50–85 |
| • Broadening of ventricles | 20–35 |
| • Corpus callosum hypoplasia | 20–45 |
Abbreviations: EEG, electroencephalographic; MRI, magnetic resonance imaging; PTHS, Pitt–Hopkins syndrome.
Differences due to including or not including isolated EEG anomalies.
A diagnostic strategy in PTHS based on a clinical score system was also suggested by Whalen et al,17 with some differences.
Although it was suggested that PTHS mainly evolves from a differential diagnosis with Rett syndrome,17 we found that AS represents the nosological condition closest to PTHS, as also confirmed by our clinical score system (Fig. 2). Shared clinical manifestations include severe ID with absent speech, epilepsy, microcephaly, ataxic gait, and happy disposition. However, more specific for AS are episodes of unmotivated laughing, a more severe seizure disorder which is usually associated, importantly, with highly distinctive EEG features. In addition, AS subjects lack the typical facial features observed in PTHS, mainly with respect to the typical nose and mouth conformation. Of note, Takano et al14 have reported that 2% of patients initially suspected as having AS were found to have PTHS with a mutation in TCF4. Confirming the pivotal role of the facial dysmorphism, these patients had several PTHS-specific facial characteristics when critically analyzed later.
Fig. 2.
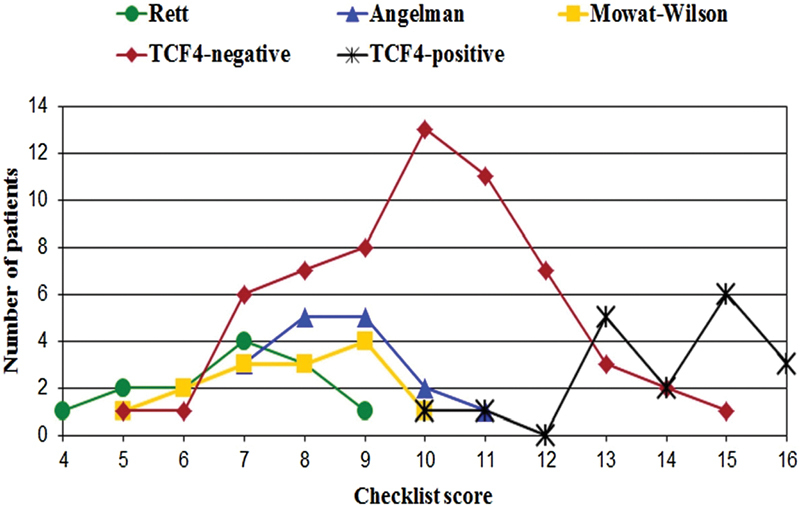
Distribution of scores for Rett syndrome patients (no. 13), Angelman syndrome patients (no. 16), Mowat–Wilson syndrome patients (no. 14), patients with a negative TCF4 molecular test (no. 60), and PTHS patients with a proven TCF4 alteration (no. 18), using the proposed clinical scoring system (already presented in “Marangi et al16).
With respect to phenotype–genotype correlations, several literature reports point at significant variation of the clinical phenotype depending on the PTHS-associated mutations in TCF4, which can lead to variable impairment of the protein function ranging from hypomorphic to dominant-negative effects. In particular, missense mutations outside of the bHLH domain were demonstrated to cause no major functional deficiency,21 while a balanced t(18;20) translocation was inferred to cause a milder phenotype because exons 4 to 20 of TCF4, in which the main functions of the gene reside, were fully preserved on 18q.25 On the contrary, large 18p deletions, encompassing MBD1 and MBD2 genes responsible for a Rett-like condition, can even worsen the neurological phenotype.28 All these considerations should prompt the clinicians proceeding with the diagnosis of PTHS by simultaneous evaluation of the clinical phenotype and of the underlying genomic defect. As here reported, more than 70% of the PTHS-associated mutations are intragenic variants detectable by gene sequencing.
In conclusion, we suggest that the most sensitive diagnostic procedure allowing for both the clinical and the genetic diagnosis of PTHS should include the following steps: clinical evaluation, including evaluation of the facial phenotype; direct sequencing/MLPA of TCF4; array-CGH; and conventional chromosome analysis.
A final consideration is in order. It is well known that a certain degree of genetic heterogeneity exists for well-defined clinical conditions, including PTHS, most likely, as a consequence of either a mosaic status for a pathogenic mutation in TCF4 that can escape detection by conventional Sanger sequencing or depending on mutations in different genes involved in the same molecular pathway. As NGS techniques can hopefully fill this diagnostic gap, the present clinical review of PTHS is also provided as useful tool for validating clinically the wide range of genomic variations that can be detected by NGS techniques.
Acknowledgments
We would like to acknowledge Prof. Giovanni Neri for critically reading the manuscript, and for his helpful suggestions. The authors gratefully acknowledge the collaboration of the study participants and their families. We are grateful to Dr Donatella Milani for her trustful support.
Footnotes
Competing Interests None. Funding This work was supported by telethon GEP 14089 (M. Z.) and by the MIUR-University Grant 2013 (M. Z.).
References
- 1.Pitt D, Hopkins I. A syndrome of mental retardation, wide mouth and intermittent overbreathing. Aust Paediatr J. 1978;14(3):182–184. doi: 10.1111/jpc.1978.14.3.182. [DOI] [PubMed] [Google Scholar]
- 2.Singh H A. Mental retardation, macrostomia and hyperpnoea syndrome. J Paediatr Child Health. 1993;29(2):156–157. doi: 10.1111/j.1440-1754.1993.tb00472.x. [DOI] [PubMed] [Google Scholar]
- 3.Van Balkom I D, Quartel S, Hennekam R C. Mental retardation, “coarse” face, and hyperbreathing: confirmation of the Pitt-Hopkins syndrome. Am J Med Genet. 1998;75(3):273–276. doi: 10.1002/(sici)1096-8628(19980123)75:3<273::aid-ajmg9>3.0.co;2-r. [DOI] [PubMed] [Google Scholar]
- 4.Orrico A, Galli L, Zappella M. et al. Possible case of Pitt-Hopkins syndrome in sibs. Am J Med Genet. 2001;103(2):157–159. doi: 10.1002/ajmg.1523. [DOI] [PubMed] [Google Scholar]
- 5.Peippo M M, Simola K O, Valanne L K. et al. Pitt-Hopkins syndrome in two patients and further definition of the phenotype. Clin Dysmorphol. 2006;15(2):47–54. doi: 10.1097/01.mcd.0000184973.14775.32. [DOI] [PubMed] [Google Scholar]
- 6.Amiel J, Rio M, de Pontual L. et al. Mutations in TCF4, encoding a class I basic helix-loop-helix transcription factor, are responsible for Pitt-Hopkins syndrome, a severe epileptic encephalopathy associated with autonomic dysfunction. Am J Hum Genet. 2007;80(5):988–993. doi: 10.1086/515582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zweier C, Peippo M M, Hoyer J. et al. Haploinsufficiency of TCF4 causes syndromal mental retardation with intermittent hyperventilation (Pitt-Hopkins syndrome) Am J Hum Genet. 2007;80(5):994–1001. doi: 10.1086/515583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Brockschmidt A, Todt U, Ryu S. et al. Severe mental retardation with breathing abnormalities (Pitt-Hopkins syndrome) is caused by haploinsufficiency of the neuronal bHLH transcription factor TCF4. Hum Mol Genet. 2007;16(12):1488–1494. doi: 10.1093/hmg/ddm099. [DOI] [PubMed] [Google Scholar]
- 9.Zweier C, Sticht H, Bijlsma E K. et al. Further delineation of Pitt-Hopkins syndrome: phenotypic and genotypic description of 16 novel patients. J Med Genet. 2008;45(11):738–744. doi: 10.1136/jmg.2008.060129. [DOI] [PubMed] [Google Scholar]
- 10.Andrieux J, Lepretre F, Cuisset J M. et al. Deletion 18q21.2q21.32 involving TCF4 in a boy diagnosed by CGH-array. Eur J Med Genet. 2008;51(2):172–177. doi: 10.1016/j.ejmg.2007.12.002. [DOI] [PubMed] [Google Scholar]
- 11.Giurgea I, Missirian C, Cacciagli P. et al. TCF4 deletions in Pitt-Hopkins syndrome. Hum Mutat. 2008;29(11):E242–E251. doi: 10.1002/humu.20859. [DOI] [PubMed] [Google Scholar]
- 12.Rosenfeld J A, Leppig K, Ballif B C. et al. Genotype-phenotype analysis of TCF4 mutations causing Pitt-Hopkins syndrome shows increased seizure activity with missense mutations. Genet Med. 2009;11(11):797–805. doi: 10.1097/GIM.0b013e3181bd38a9. [DOI] [PubMed] [Google Scholar]
- 13.de Pontual L, Mathieu Y, Golzio C. et al. Mutational, functional, and expression studies of the TCF4 gene in Pitt-Hopkins syndrome. Hum Mutat. 2009;30(4):669–676. doi: 10.1002/humu.20935. [DOI] [PubMed] [Google Scholar]
- 14.Takano K, Lyons M, Moyes C, Jones J, Schwartz C E. Two percent of patients suspected of having Angelman syndrome have TCF4 mutations. Clin Genet. 2010;78(3):282–288. doi: 10.1111/j.1399-0004.2010.01380.x. [DOI] [PubMed] [Google Scholar]
- 15.Marangi G, Ricciardi S, Orteschi D. et al. The Pitt-Hopkins syndrome: report of 16 new patients and clinical diagnostic criteria. Am J Med Genet A. 2011;155A(7):1536–1545. doi: 10.1002/ajmg.a.34070. [DOI] [PubMed] [Google Scholar]
- 16.Marangi G, Ricciardi S, Orteschi D. et al. Proposal of a clinical score for the molecular test for Pitt-Hopkins syndrome. Am J Med Genet A. 2012;158A(7):1604–1611. doi: 10.1002/ajmg.a.35419. [DOI] [PubMed] [Google Scholar]
- 17.Whalen S, Héron D, Gaillon T. et al. Novel comprehensive diagnostic strategy in Pitt-Hopkins syndrome: clinical score and further delineation of the TCF4 mutational spectrum. Hum Mutat. 2012;33(1):64–72. doi: 10.1002/humu.21639. [DOI] [PubMed] [Google Scholar]
- 18.Sweatt J D. Pitt-Hopkins syndrome: intellectual disability due to loss of TCF4-regulated gene transcription. Exp Mol Med. 2013;45:e21. doi: 10.1038/emm.2013.32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sepp M, Kannike K, Eesmaa A, Urb M, Timmusk T. Functional diversity of human basic helix-loop-helix transcription factor TCF4 isoforms generated by alternative 5′ exon usage and splicing. PLoS ONE. 2011;6(7):e22138. doi: 10.1371/journal.pone.0022138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Forrest M P, Hill M J, Quantock A J, Martin-Rendon E, Blake D J. The emerging roles of TCF4 in disease and development. Trends Mol Med. 2014;20(6):322–331. doi: 10.1016/j.molmed.2014.01.010. [DOI] [PubMed] [Google Scholar]
- 21.Sepp M, Pruunsild P, Timmusk T. Pitt-Hopkins syndrome-associated mutations in TCF4 lead to variable impairment of the transcription factor function ranging from hypomorphic to dominant-negative effects. Hum Mol Genet. 2012;21(13):2873–2888. doi: 10.1093/hmg/dds112. [DOI] [PubMed] [Google Scholar]
- 22.Stefansson H, Ophoff R A, Steinberg S. et al. Common variants conferring risk of schizophrenia. Nature. 2009;460(7256):744–747. doi: 10.1038/nature08186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ellinghaus D, Folseraas T, Holm K. et al. Genome-wide association analysis in primary sclerosing cholangitis and ulcerative colitis identifies risk loci at GPR35 and TCF4. Hepatology. 2013;58(3):1074–1083. doi: 10.1002/hep.25977. [DOI] [PubMed] [Google Scholar]
- 24.Baratz K H, Tosakulwong N, Ryu E. et al. E2-2 protein and Fuchs's corneal dystrophy. N Engl J Med. 2010;363(11):1016–1024. doi: 10.1056/NEJMoa1007064. [DOI] [PubMed] [Google Scholar]
- 25.Kalscheuer V M, Feenstra I, Van Ravenswaaij-Arts C M. et al. Disruption of the TCF4 gene in a girl with mental retardation but without the classical Pitt-Hopkins syndrome. Am J Med Genet A. 2008;146A(16):2053–2059. doi: 10.1002/ajmg.a.32419. [DOI] [PubMed] [Google Scholar]
- 26.Zweier C, de Jong E K, Zweier M. et al. CNTNAP2 and NRXN1 are mutated in autosomal-recessive Pitt-Hopkins-like mental retardation and determine the level of a common synaptic protein in Drosophila. Am J Hum Genet. 2009;85(5):655–666. doi: 10.1016/j.ajhg.2009.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Harrison V, Connell L, Hayesmoore J, McParland J, Pike M G, Blair E. Compound heterozygous deletion of NRXN1 causing severe developmental delay with early onset epilepsy in two sisters. Am J Med Genet A. 2011;155A(11):2826–2831. doi: 10.1002/ajmg.a.34255. [DOI] [PubMed] [Google Scholar]
- 28.Kato Z, Morimoto W, Kimura T, Matsushima A, Kondo N. Interstitial deletion of 18q: comparative genomic hybridization array analysis of 46, XX,del(18)(q21.2.q21.33) Birth Defects Res A Clin Mol Teratol. 2010;88(2):132–135. doi: 10.1002/bdra.20633. [DOI] [PubMed] [Google Scholar]