

Abstract
Rubinstein–Taybi syndrome (RSTS) is a rare, congenital, plurimalformative, and neurodevelopmental disorder. Clinical diagnosis can be complicated by the heterogeneous clinical presentation and the lack of a consensus list of diagnostic criteria, and it is confirmed by molecular tests in approximately 55 to 78% of cases. The etiology is partially known with mutations in two functionally related genes: CREBBP and EP300. Notwithstanding the knowledge on clinical, genetic, and allelic heterogeneity, no clear genotype–phenotype correlation has yet been established. Standardized guidelines for the management of pediatric patients are available and therapy for RSTS patients is currently only symptomatic. In this article, several clinic and genetic aspects of RSTS are critically reviewed.
Keywords: Rubinstein–Taybi syndrome, clinical and molecular diagnosis, genotype–phenotype correlation, management, therapy
Introduction
Originally observed approximately 60 years ago by three orthopedic surgeons, the Rubinstein–Taybi Syndrome (RSTS; OMIM #180849, #613684) derives the name from the pediatrician and the radiologist, respectively, who described it for the first time in 1963.1
One of the over 8,000 known rare diseases, RSTS affects males and females equally with a birth prevalence of 1:100,000 to 1:125,000. Notwithstanding its rarity, more than 1,000 clinical cases have been up to now reported in literature and details of clinical features have been collected, revealing a wide range of multiple congenital anomalies. Despite the presence of pathognomonic signs and symptoms of variable entity concerning cognitive impairment (the estimated incidence of RSTS in patient with intellectual disability [ID] older than 5 years is two- to four fold higher than in general population),2 postnatal growth deficiency, craniofacial dysmorphisms and skeletal abnormalities (such as broad thumbs and large toes), several additional signs and symptoms have been reported, which complicate and make the clinical picture heterogeneous, sometimes making the clinical diagnosis more difficult.
The genetic bases of RSTS were elucidated in 1995, when the cAMP response element-binding protein (CREB)-binding protein (CREBBP), located on chromosome 16p13.3, was identified by positional cloning.3 The heterogeneous etiology of RSTS was confirmed in 2005, as the leading functional candidate E1A-associated protein p300 (EP300), located on chromosome 22q13.2, was found mutated in RSTS-affected patients.4
To date, approximately 230 causative mutations in CREBBP (www.lovd.nl/CREBBP) 5 6 7 8 and 28 genetic defects in EP300 (http://chromium.liacs.nl/LOVD2/home.php?select_db=EP300) 9 10 11 12, almost all private, have been described, accounting for approximately 50 to 70% and 5 to 8% of cases, respectively.
RSTS is an autosomal-dominant disease; almost all cases are sporadic and result from de novo mutations. To the best of our knowledge, only seven familial RSTS cases have been up to now described: five inherited as autosomal-dominant trait (two of them showing incomplete penetrance)1 13 14 15 16 and two somatic mosaicism in very mildly affected fathers of RSTS children.13 17 In addition, two cases of germline mosaicisms have been speculated.13 18 Owing to the finding of such mosaicisms, the estimated risk for a couple with a previous child affected by RSTS is roughly estimated in the range of 0.5 to 1%.
The purpose of the present article is to summarize current knowledge about the clinical presentation, the clinical and differential diagnosis as well as the genetic defects underlying RSTS, the genotype–phenotype correlation, and the notions on current and future therapeutic approaches.
Clinical Diagnosis and Management
Nowadays, the diagnosis of RSTS is essentially clinic and established by clinical geneticists after psycho/physical and dysmorphological evaluation.
The clinical diagnosis is often made at birth or in early childhood and it is based on characteristic facial appearance (in particular down slanting of palpebral fissures and a broad nasal bridge/beaked nose), broad short thumbs and/or big toes, a reduced growth, and early medical problems consisting of a failure to thrive caused by gastroesophageal reflux (GER), recurrent infections, and hypotonia (Table 1).
Table 1. Signs and symptoms in RSTS patients.
| Craniofacial dysmorphisms | Skeletal malformations |
|---|---|
| Microcephaly | Broad short halluces/big toes with radial deviation |
| Low anterior hairline | 5th finger clinodactyly, preaxial polydactyly |
| Nevus flammeus at forehead | Delayed bone age |
| Down slanting palpebral fissures | Increased fractures |
| High-arched eyebrows, long eyelashes | Orthopedic problems (scoliosis, kyphosis, lordosis) |
| Ptosis, epicanthus, strabismus | Joint anomalies (patella/hip dislocation, femur head inflammation, femoral epiphysis) |
| Broad nasal bridge, beaked nose | Cervical vertebral anomalies (C1–C2 instability, fusion, hypoplasia of the dens, stenosis) |
| Prominent columella | Neuroradiological issues (corpus callosum dysgenesis, Chiari type I malformation, Dandy–Walker malformation, hydrocephalus, tethered cord) |
| High arched palate | |
| Micrognathia | Additional signs/symptoms |
| Dental malposition, talon cups, enamel hypoplasia | Ocular anomalies (strabismus, refractive errors, obstruction of tear ducts, glaucoma, coloboma, cataract) |
| Short upper lip | Hear anomalies (hypoacusia, middle ear infections) |
| Grimacing smile | Renal malformations (pyelectasia, double district, horseshoe kidney) |
| Low-set ears | Heart defects (PDA, VSD, ASD, aortic coarctation and aortic/pulmonic stenosis, BAV, pseudotruncus, dextrocardia, vascular rings, hypoplastic left heart, conduction disorders) |
| Growth delay | Vascular anomalies (spontaneous dissection of the supra-aortic arteries, aneurysm of the anterior cerebral artery) |
| Growth delay in infancy | Genital anomaly (cryptorchidism) |
| Lack of puberal spurt | Gastrointestinal problems (GER, constipation, megacolon/Hirschsprung disease) |
| Excessive weight gain at puberty | Endocrine anomalies (hypothyroidism, hypoplasia, growth hormone deficiency, pituitary hypoplasia) |
| Extranumerary nipples | |
| Psychomotor development delay | Skin anomalies (hirsutism, keloids, pilomatrixomas, hirsutism, ingrown toenails, paronychia) |
| Hypotonia | Increased risk of benign and malignant tumors |
| Intellectual disability | Nocturnal obstructive apnea |
| Speech delay | Recurrent infections |
| Behavioral/neurological problems (mood swings, obsessive-compulsive disorders, attention deficit, motor stereotypies, seizures, poor coordination) |
Abbreviations: ASD, atrial septal defect; BAV, bicuspid aortic valve; GER, gastroesophageal reflux; PDA, patent ductus arteriosus; RSTS, Rubinstein–Taybi syndrome; VSD, ventricular septal defect.
Note: Frequent clinical characteristics are in bold, and those useful for an early clinical diagnosis are in bold italics.
Still now some patients with RSTS are diagnosed in adulthood as a consequence of a misleading diagnostic classification that hampers a promptly and personalized patient care and increases costs for diagnostic workup. As a matter of fact, an early clinical diagnosis of RSTS is sometimes difficult due to the lack, as for other rare diseases, of a consensus definition of the diagnostic criteria (i.e., quantity and type of clinical symptoms and features). Moreover, the clinical diagnosis is made more complex by the wide spectrum of anomalies, their variable degree, and the different combination of signs and symptoms, as indicated in the following section.
In addition, some features of RSTS become more evident or worsen with age. This is the case of facial traits (detailed later), growth delay, and behavioral and psychiatric problems.19 Prenatal development is usually believed normal, although in a recent work performed in a cohort of 46 RSTS patients, intrauterine growth retardation was reported in approximately 38% of analyzed patients.7 Growth delay in infancy was reported, mainly due to feeding problems, followed by the lack of pubertal spurt which results in short stature and a final average height of 162.6 cm in males and 151.0 cm in females.20 Patients with RSTS can be characterized by an excessive weight gain at puberty (earlier in males than in females) with sometimes obesity. Recently, specific growth charts have been revised for appropriate growth evaluation of RSTS patients.20
In this respect, an updated proposal for medical guidelines in RSTS patients has been recently published.21 Based on literature data and personal experience of the authors, medical evaluations were recommended from 6 to 30 months at 6-month intervals and later after 3 years of age and at the adolescent age (Table 2). The management, adjusted by age, consists in: audiological and orthopedic evaluations at each age, dental evaluation from 1 year of age, ophthalmologic evaluation from 6 months and then at 1-year interval, and endocrinological evaluation at 30 months. In addition to all these medical examinations, neuropsychiatric, cardiologic, and dermatologic evaluations, as well as brain and medullary NMR, renal ultrasonography and genetic counselling were proposed for the diagnosis and follow-up at adolescence age when pressure measurement was also advised (Table 2).
Table 2. Recent proposal for medical guidelines in RSTS patients.
| Diagnosis | 6 M | 12 M | 18 M | 24 M | 30 M | >36 M (yearly) | Adolescent age | |
|---|---|---|---|---|---|---|---|---|
| Brain and medullary NMRa | X | X | ||||||
| Neuropsychiatric evaluation | X | X | ||||||
| Dermatologic evaluationa | X | X | ||||||
| Genetic counseling | X | X | ||||||
| Cardiologic evaluationa | X | X | ||||||
| Renal US scana | X | X | ||||||
| Pressure measurement | X | X | ||||||
| Endocrinological evaluationa | X | X | X | |||||
| Ophthalmologic evaluation | X | X | X | X | X | X | ||
| Odontoiatric evaluation | X | X | X | X | X | X | ||
| Orthopedic evaluation | X | X | X | X | X | X | X | X |
| Audiologic evaluation | X | X | X | X | X | X | X | X |
Abbreviations: M, months; NMR, nuclear magnetic resonance; RSTS, Rubinstein–Taybi syndrome; US, ultrasonography.
Note: Medical evaluations and examinations are in order of number and earliness in the first 3 years of age.
Follow-up if necessary.
No standardized guidelines for the management of adult RSTS patients are yet available. Despite more than 90% of individuals with RSTS survive to adulthood,22 few reports of adult patients have been reported, revealing excessive weight gain, relevant medical problems, worsening of neuropsychiatric and behavioral problems (anxiety, mood instability, and aggressiveness in 37% of cases), and decreased abilities over time (32% of individuals).23 24 25 26 27 28 29 30 Life expectancy is generally normal but may be reduced in RSTS individuals particularly susceptible to infections or with severe congenital heart defects.
Clinical Presentation
As a multisystem disorder, RSTS is mainly characterized by manifestations that should be grouped into three categories: craniofacial dysmorphisms, skeletal malformations, and delay of growth and psychomotor development (Table 1). In addition, several major malformations affecting ocular system, heart, kidney, and genitalia and medical complications primarily involving eyes, teeth, hearing, skeletal system, and heart have been reported (Table 1).
Facial anomalies are always present. A low anterior hairline is typical, occasionally with a nevus flammeus at forehead. Microcephaly seems also to be frequent (35–94%).
Down slanting of the palpebral fissures is a frequent sign (82% of cases) and can be associated with high-arched and thick eyebrows, long eyelashes, ptosis, and epicanthus.
A protruding beaked nose (92%) with a prominent columella is characteristic. Ears are usually low-set and dysplastic. High arched palate, malocclusion, supernumerary teeth, frequent talon cusps (73%), enamel hypoplasia were reported,31 32 and thin upper lip with a characteristic “grimacing” smile was observed.
Micrognathia, together with obesity and larynx weakness, can promote nocturnal obstructive apnea and complications due to anesthetic and intubation, as well as recurrent respiratory infections (75%).33 34
Hypoacusia (24%) and recurrent middle ear infections are reported.35 36
Among ocular anomalies, strabismus (60–71%), refractive errors (41–56%), lacrimal duct obstructions (38–47%), glaucoma, unilateral/bilateral iris/retinal/optic nerve coloboma (9–11%), and cataract (peripheral avascularity) in order of prevalence have been described.37 38
Skeletal malformations mainly concern thumbs and big toes, which are frequently broad and short (96%) and laterally deviated in one-third of cases. Broadening of distal phalanges of all fingers, not always taken into account and hence likely underestimated, was recently found in many patients (36%) and hence should be considered a typical and frequent sign of RSTS.7 Other features of hands and feet include abducted thumb, clinodactyly of fifth finger, and, rarely, preaxial polydactyly.
Other skeletal anomalies include delayed bone age (74%) and increased fractures, orthopedic problems (scoliosis, kyphosis, lordosis), joint anomalies (patella/hip dislocation, inflammation of the femur head, and rarely slipped capital femoral epiphysis), and cervical vertebral abnormalities (instability of C1–C2, fusion of the cervical vertebrae, hypoplasia of the dens).39 40 41
Possible cervical myelopathy due to stenosis at the craniovertebral junction, complex neuroradiological issues such as corpus callosum dysgenesis (17%),42 43 Chiari type I malformation with or without syringomyelia,43 44 45 46 Dandy–Walker malformation and hydrocephalus,47 48 and tethered cord45 49 have been reported in few cases.
Renal malformations (52%), including pyelectasia, double district, and horseshoe kidney,7 50 and several congenital heart defects (24–38%), including atrial/ventricular septal defect, patent ductus arteriosus, coarctation of the aorta, pulmonic stenosis, bicuspid aortic valve, pseudotruncus, aortic stenosis, dextrocardia, vascular rings, hypoplastic left heart, and conduction disorders (24–38%),51 52 have been described.
Vascular anomalies have also been occasionally noticed (spontaneous dissection of the supraaortic arteries53 and aneurysm of the anterior cerebral artery54).
Cryptorchidism is frequently described in males as genital anomaly (78–100%).
Gastrointestinal problems include early GER (68%), constipation (40–74%), and megacolon/Hirschsprung disease.50 55
In addition, hypothyroidism,56 57 thyroid hypoplasia, growth hormone deficiency,58 and pituitary hypoplasia46 have been reported as endocrine anomalies.
Skin is also affected, and keloids (24%), single and multiple pilomatrixoma, hirsutism, ingrown toenails, and paronychia are described.59 60 61
Finally, increased risk (5%) of benign (meningioma, odontoma, choristoma, dermoid cyst, and pilomatrixoma) and malignant (oligodendroglioma, medulloblastoma, neuroblastoma, pheochromocytoma, rhabdomyosarcoma, leiomyosarcoma, seminoma, embryonal carcinoma, leukemia, and lymphoma) tumors, mostly affecting the central nervous system and arising from the neural crest, have been reported in patients with RSTS.24 28 62 63 64 65 66 67 68 Recently, synchronous ovarian and endometrial carcinoma were also reported.69
As previously described, growth delay involves approximately 21% of patients, whereas ID, the most disabling aspect of the disease, is always present (100%). Almost all patients suffered from a variable degree of cognitive impairment (from mild to profound). Psychometric test evidences an intelligence quotient (IQ) score ranging from 25 to 79 with an average value of approximately 51.19 70 In our knowledge, a low-normal psychomotor development was until now found in only two RSTS patients who are carrier of the same RSTS-causing mutation.7
Speech delay (90%) and hypotonia (70%) are also frequent.
Despite a preservation of good communication skills and friendly and sociable features, RSTS patients show behavioral and neurological problems. In particular, mood swings, obsessive-compulsive disorders, attention deficit, motor stereotypies, autistic features (i.e., seizures [25%]), and poor coordination have been described. Electroencephalography frequently shows nonspecific abnormalities (57–66%).71 72
Differential Diagnosis
The peculiar facial features and abnormalities of the hands and feet generally allow the clinical diagnosis of RSTS. However, owing to the high variability in the clinical presentation (ranging from mild to severe phenotype) and the sharing of some RSTS features (microcephaly, facial dysmorphisms, growth delay, short stature, skeletal dysplasia, ID, and expressive language delay) with other diseases, RSTS enters the differential diagnosis with the genitopatellar syndrome (GTPTS; OMIM #606170), the Floating–Harbor syndrome (FLHS; OMIM #136140), and the Cornelia de Lange syndrome (CdLS; OMIM #122470)9 (Fig. 1).
Fig. 1.
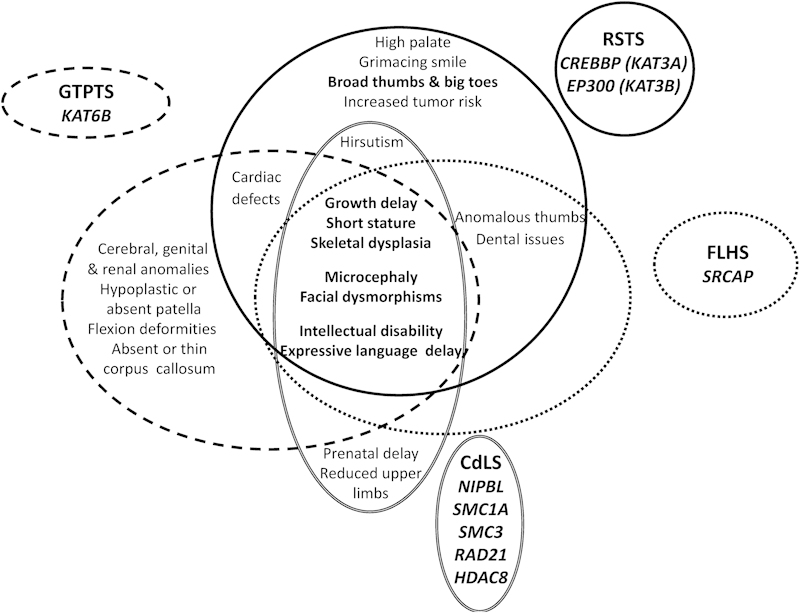
Schematic representation of clinical features shared by Rubinstein–Taybi, genitopatellar, floating harbor, and Cornelia de Lange syndromes. The main clinical characteristics of RSTS, GTPTS, FLHS, and CdLS are grouped by continuous, dashed, dotted, and double lines, respectively. CdLS, Cornelia de Lange syndrome; FLHS, Floating–Harbor syndrome ; GTPTS, genitopatellar syndrome; RSTS, Rubinstein–Taybi syndrome;
GTPTS is characterized by microcephaly, broad nose and small chin, severe psychomotor retardation, genitourinary anomalies, and absent or hypoplastic kneecap, which is a distinctive feature of GTPTS.
The clinical features of the FLHS are much more similar to RSTS. Nevertheless, besides the presence of short stature and delayed bone and language development, facial dysmorphisms, differently from RSTS, become less severe with advancing age.
GTPTS and FLHS are caused by mutations in KAT6B and SRCAP genes, respectively, as recently highlighted through massive sequencing techniques.73 74 75 Both genes code for proteins functionally related to CBP/p300; KAT6B is a histone acetyltransferase (HAT) involved in the early step of embryonic development and SRCAP is the catalytic component of a chromatin remodeling complex of which CBP is coactivator.
CdLS is a multisystem malformation disease characterized by pre- and postnatal growth delay, facial dysmorphisms (low anterior hairline, arched eyebrows, synophrys, anteverted nares, maxillary prognathism, long philtrum, thin lips, and “carp” mouth), ID, hirsutism, and typical skeletal reduction defects at the upper limbs. CdLS-causative mutations have been found in five genes: NIPBL, SMC1A, SMC3, and RAD21, encoding components of the cohesin complex involved in chromosome segregation during mitosis, and the histone deacetylase HDAC8. A similar localization of NIPBL, SMC1A, SMC3, and p300 proteins on enhancer regions has been described as well as a similar involvement of HDAC8 and p300 on p53 transcriptional activity.76 77 78
Genetic Defects
Before the identification of CREBBP and later of EP300 as causative genes, clinical diagnosis of RSTS was supported only by radiological examination (i.e., X-ray of the hands and feet).
As the molecular bases were elucidated and the molecular screening in these two genes has been performed, the mutational spectra were understood. A high degree of allelic heterogeneity is upheld by the numerous (∼230 and 28) genetic alterations found, respectively, in the CREBBP and EP300 genes (Fig. 2).
Fig. 2.
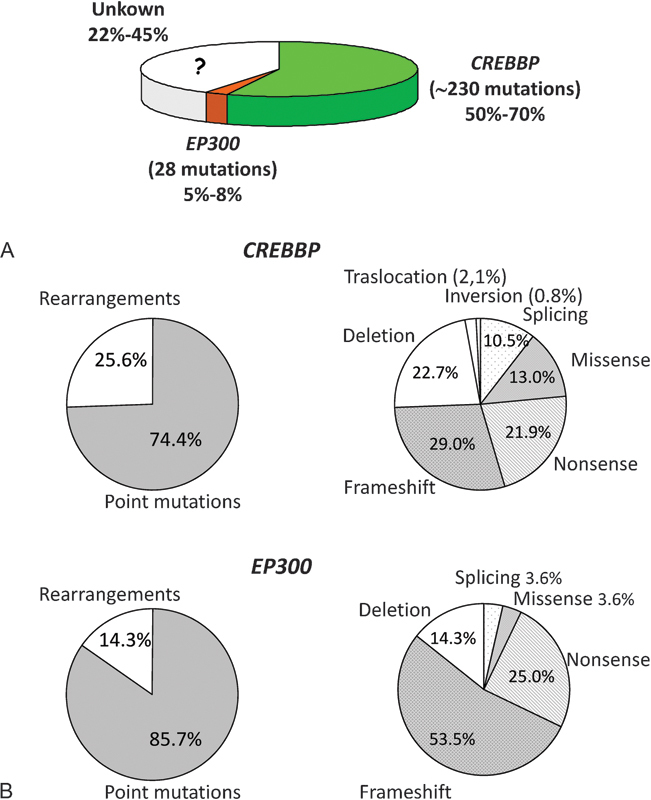
Mutational spectra of RSTS. (A) Genetic and allelic heterogeneity of RSTS. The relative frequency of CREBBP and EP300 mutations found in RSTS patients is reported in the pie chart. (B) Frequency of mutations found in CREBBP (upper panel) and EP300 (lower panel). Proportion of point mutations versus rearrangements is on the left. Details of specific type of point mutations versus rearrangements are on the right.
Owing to the different mutational “burden,” CREBBP, involved in approximately 50 to 70% of RSTS patients,72 79 80 81 and EP300, mutated in approximately 5 to 8%4 10 82 of all analyzed cases, are currently defined as the “major” and “minor” gene, respectively.
Thus, the clinical diagnosis of RSTS is confirmed by genetic screening in 55 to 78%, while the molecular basis remains unknown in a consistent fraction of RSTS patients (Fig. 2).
The difference in the mutation detection relies on the laboratory techniques used. The ideal molecular diagnostic flowchart should provide a multiapproach study ranging from molecular-cytogenetic analyses to molecular-biology screening focusing first on CREBBP gene scanning and on EP300 gene screening in CREBBP-negative patients. In particular, the reference centers for the molecular diagnosis of RSTS (whose list is available at the Orphanet website: http://www.orpha.net/consor/cgi-bin/index.php) should be taken into account the frequency of different type of genetic defects and the cost of specific diagnostic tests.
Since in both CREBBP and EP300 genes point mutations (including frameshift, nonsense, missense, and splicing in order of prevalence) are the majority of genetic defects (74.4–85.7% of cases) (Fig. 2), denaturing high-performance liquid chromatography and direct sequencing analyses, for longtime used in the molecular screening, and the innovative next generation sequencing approach, that recently enables the detection of a mutation in EP300,9 should be chosen as screening techniques.
Nevertheless, since these approaches are time consuming, the identification of chromosome rearrangements (i.e., deletions, translocations, and inversions, representing 14.3–25.6% of all reported abnormalities) (Fig. 2) should be preferred as first step of genetic test. Intragenic differently sized deletions are easily detected by fluorescence in situ hybridization, multiplex ligation-dependent probe amplification, long-range PCR, and by the more expensive quantitative (multiplex) PCR and array-comparative genomic hybridization. Translocations, the first type of rearrangement associated with RSTS identified,83 84 intergenic deletions, and inversions are rare in RSTS (Fig. 2), so the karyotype analysis, which enables the detection of these chromosomal aberrations, is not the suitable molecular diagnostic tool.
CREBBP and EP300
CREBBP, located in a 156-kb region on chromosome 16p13.3, and its paralog EP300, located in a region of approximately 88 kb on chromosome 22q13.2, consist of an equal number of exons (31) that are transcribed into mRNAs of comparable length coding for the CBP (2442 aa) and its structural and functional homologue p300 (2414 aa), respectively.
Both are nuclear proteins ubiquitously expressed that possess a HAT and non-HAT activity and multiple protein-binding domains: three cysteine/histidine-rich regions (CH1–CH2–CH3), the CREB interacting domain (KIX), and the acetyl-lysine binding domain (BROMO). As acetyltransferases belong to the KAT3 family, CREBBP and EP300 have been renamed as KAT3A and KAT3B, respectively.85
Acting as epigenetic regulators by promoting chromatin remodeling and as network “hubs” by interacting with more than 400 protein partners, they regulate gene expression and coordinate many cellular pathways during embryonic development and postnatal life such as cell growth and differentiation. By promoting the acetylation of more than 70 non-histone proteins, CBP/p300 is involved into the DNA's replication and repair. They also possess a polyubiquitin-ligase activity that is responsible for the rapid turnover of p53 and they have been postulated to act as transcriptional repressors of the H3K9me3-specific methyltransferase which leads to condensation of pericentromeric chromatin structure.86
In vivo studies on nullizygous, hemizygous, conditional knockouts, and knock-in mice, aimed to understand the pathogenetic role of CBP and p300 in RSTS, showed that they have both partially overlapping and few distinct functions. In particular, the role of p300 in cognition seems to be less relevant or more easily compensated as compared with CBP.
Currently, two pathogenetic mechanisms have been proposed. The loss of function (i.e., haploinsufficiency) is sustained by the identification of heterozygous CREBBP/EP300 mutations in RSTS patients and by a mimicked RSTS phenotype in hemizygous (cbp+/−) mice.87 In addition, a dominant negative inhibition relies to the finding that injection of KIX domain into fibroblast nuclei blocks transcriptional activation of CRE-lacZ reporter gene (i.e., the abnormal product derived from the mutant allele inhibits the wild-type product as a result of competitive binding for CREB) and that cbp (+/truncated) mice retaining the KIX domain exhibit a more severe phenotype than cbp (+/−) mice.88
A deeper assessment of molecular mechanisms underlying RSTS could help in deciphering the extreme RSTS clinical heterogeneity. In this context, since genetic defects in known genes account for about two-thirds of all reported cases leaving remaining patients without molecular diagnosis, the discovery of novel RSTS-causative genes should provide insights into the molecular pathophysiology of the syndrome, disclosing the role of currently unknown proteins, their possible interaction with CBP and/or p300, and ultimately leading to a better understanding of the pathway/s that are perturbed in RSTS and a better comprehension of genotype–phenotype correlation.
Genotype–Phenotype Association
Despite the great number of clinically diagnosed patients and numerous genetic defects reported, no clear genotype–phenotype correlation has been up to now established.
The lack of a consensus list of the diagnostic criteria and score for the multiple signs and symptoms found in RSTS patients does not allow a consistent clinical classification of analyzed patients. Moreover, many RSTS individuals enrolled for the molecular screening lack a detailed clinical description. In addition, many reported genotype–phenotype relationship relies on the analysis of small case studies. Taken together, these issues make it difficult to draw a clear, systematic, and statistically significant analysis and correlation.
Nevertheless, as the discovery of the EP300 as novel gene involved in RSTS and hence of the genetic heterogeneity underlying RSTS, insights into the correlation between a specific genotype (i.e., affected gene) and a specific phenotype (i.e., type and/or severity of clinical sign(s) or the global severity of clinical presentation) have been addressed.
Recent studies on 28 clinical and molecular characterized EP300-mutated patients evidenced a milder phenotype than that observed in CREBBP-mutated probands, although in one patient the genetic defect was incompatible with life.9 In particular, faint skeletal anomalies at thumbs and toes are reported as well as mild/absent IDs. Among behavioral problems, anxiety is reported as frequent and pilomatrixomas and nevi are often described as cutaneous anomalies. In addition, preeclampsia, as common complication during pregnancy, has frequently reported in mother of child carrying mutation of EP300 gene.10 23 82 89
Concerning genetic defects in CREBBP, single studies, involving few RSTS patients, showed mild RSTS phenotype in patients with mosaic microdeletions90 and the absence of microcephaly and mental retardation/growth delay in patients carrying a same recurrent missense mutation localized in the BROMO domain. By contrast, a severe neuropsychiatric involvement (i.e., hyperactivity, attention deficiency, self-injurious behavior, aggressiveness toward people, and autistic-like behavior) was reported in a patient with the unique missense mutation in the KIX domain. A major involvement of the HAT domain in RSTS dysmorphic/malformative phenotype was also hypothesized.7
Although not supported by other studies, a severe phenotype has been described in RSTS patients with deletion of CREBBP and contiguous genes.91 Moreover, association between lower IQ and autistic features with large deletions are also reported72 and recently, a slightly increase in cardiac malformations and growth retardation was described in patients carrying deletions of CREBBP and neighboring genes.8 Anyhow, no specific, clear, and convincing correlation between size of CREBBP deletion/involvement of genes other than CREBBP and patient's clinical presentation has been to date established.8
The possibility to cluster RSTS patients according to a specific clinical presentation (in terms of type/severity of clinical sign/s and/or symptom/s) and to correlate a “sub”-phenotype to a specific genetic defect could lead to an optimization of the molecular diagnostic practice resulting in a rapid and early molecular diagnosis (in the perinatal period) of the newborns affected by RSTS, with global improvement of the clinical management and benefits for patients.
Therapeutic Approach
Currently, therapy for RSTS patients is only symptomatic and includes logotherapy, physiotherapy, ergotherapy, tube feeding, antibiotic prophylaxis for airway infections, and surgery in case of heart or kidney defects or life-threatening malformations.61
Nevertheless, several efforts have been made to develop therapeutic approaches aimed to improve symptoms of RSTS.
In this respect, evidences of a mitigated skeletal defects in cbp+/− new-born mice come from a replacement therapy based on the in utero administration of the bone morphogenetic protein.92
Evidences of a therapeutic strategy enabling to rescue the epigenetic alterations due to the CBP/p300 acetyltransferase dysfunction/deficiency were also obtained. Treatments with histone deacetylase inhibitors (HDACi) such as suberoylanilide hydroxamic acid and trichostatin A were shown to improve deficits in synaptic plasticity and cognition in several CBP mutant mice93 and HAT activity of CBP in RSTS lymphoblastoid human cell lines.94
A phase II clinical trial using sodium valproate, acting as a class I HDACi, is in progress (https://clinicaltrials.gov/ct2/show/NCT01619644). Owing to the lack of specificity of HDACi which could change the general epigenetic pattern of histone acetylation, alternative methods are worth considering, such as the use of HDACi selective for CBP/p300 and the activation of CBP/p300 by overexpression or by pharmacologic methods.86
Progression in the comprehension of functional effects of mutations will provide the basis for a possible future molecular treatment of RSTS patients.
References
- 1.Rubinstein J H, Taybi H. Broad thumbs and toes and facial abnormalities. A possible mental retardation syndrome. Am J Dis Child. 1963;105:588–608. doi: 10.1001/archpedi.1963.02080040590010. [DOI] [PubMed] [Google Scholar]
- 2.Padfield C J, Partington M W, Simpson N E. The Rubinstein-Taybi syndrome. Arch Dis Child. 1968;43(227):94–101. doi: 10.1136/adc.43.227.94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Petrij F, Giles R H, Dauwerse H G. et al. Rubinstein-Taybi syndrome caused by mutations in the transcriptional co-activator CBP. Nature. 1995;376(6538):348–351. doi: 10.1038/376348a0. [DOI] [PubMed] [Google Scholar]
- 4.Roelfsema J H, White S J, Ariyürek Y. et al. Genetic heterogeneity in Rubinstein-Taybi syndrome: mutations in both the CBP and EP300 genes cause disease. Am J Hum Genet. 2005;76(4):572–580. doi: 10.1086/429130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Coupry I, Monnet L, Attia A A, Taine L, Lacombe D, Arveiler B. Analysis of CBP (CREBBP) gene deletions in Rubinstein-Taybi syndrome patients using real-time quantitative PCR. Hum Mutat. 2004;23(3):278–284. doi: 10.1002/humu.20001. [DOI] [PubMed] [Google Scholar]
- 6.Stef M, Simon D, Mardirossian B. et al. Spectrum of CREBBP gene dosage anomalies in Rubinstein-Taybi syndrome patients. Eur J Hum Genet. 2007;15(8):843–847. doi: 10.1038/sj.ejhg.5201847. [DOI] [PubMed] [Google Scholar]
- 7.Spena S, Milani D, Rusconi D. et al. Insights into genotype-phenotype correlations from CREBBP point mutation screening in a cohort of 46 Rubinstein-Taybi syndrome patients. Clin Genet. 2014 doi: 10.1111/cge.12537. [DOI] [PubMed] [Google Scholar]
- 8.Rusconi D, Negri G, Colapietro P. et al. Characterization of 14 novel deletions underlying Rubinstein-Taybi syndrome: an update of the CREBBP deletion repertoire. Hum Genet. 2015;134(6):613–626. doi: 10.1007/s00439-015-1542-9. [DOI] [PubMed] [Google Scholar]
- 9.Woods S A, Robinson H B, Kohler L J, Agamanolis D, Sterbenz G, Khalifa M. Exome sequencing identifies a novel EP300 frame shift mutation in a patient with features that overlap Cornelia de Lange syndrome. Am J Med Genet A. 2014;164A(1):251–258. doi: 10.1002/ajmg.a.36237. [DOI] [PubMed] [Google Scholar]
- 10.Negri G, Milani D, Colapietro P. et al. Clinical and molecular characterization of Rubinstein-Taybi syndrome patients carrying distinct novel mutations of the EP300 gene. Clin Genet. 2015;87(2):148–154. doi: 10.1111/cge.12348. [DOI] [PubMed] [Google Scholar]
- 11.Bounakis N, Karampalis C, Sharp H, Tsirikos A I. Surgical treatment of scoliosis in Rubinstein-Taybi syndrome type 2: a case report. J Med Case Reports. 2015;9:10. doi: 10.1186/1752-1947-9-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Solomon B D, Bodian D L, Khromykh A. et al. Expanding the phenotypic spectrum in EP300-related Rubinstein-Taybi syndrome. Am J Med Genet A. 2015;167A(5):1111–1116. doi: 10.1002/ajmg.a.36883. [DOI] [PubMed] [Google Scholar]
- 13.Chiang P W, Lee N C, Chien N, Hwu W L, Spector E, Tsai A C. Somatic and germ-line mosaicism in Rubinstein-Taybi syndrome. Am J Med Genet A. 2009;149A(7):1463–1467. doi: 10.1002/ajmg.a.32948. [DOI] [PubMed] [Google Scholar]
- 14.Padfield C J, Partington M W, Simpson N E. The Rubinstein-Taybi syndrome. Arch Dis Child. 1968;43(227):94–101. doi: 10.1136/adc.43.227.94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Verma I C. Rubinstein Taybi syndrome. Case report. Indian Pediatr. 1970;7(12):672–674. [PubMed] [Google Scholar]
- 16.Cotsirilos P, Taylor J C, Matalon R. Dominant inheritance of a syndrome similar to Rubinstein-Taybi. Am J Med Genet. 1987;26(1):85–93. doi: 10.1002/ajmg.1320260115. [DOI] [PubMed] [Google Scholar]
- 17.Bartsch O, Kress W, Kempf O, Lechno S, Haaf T, Zechner U. Inheritance and variable expression in Rubinstein-Taybi syndrome. Am J Med Genet A. 2010;152A(9):2254–2261. doi: 10.1002/ajmg.a.33598. [DOI] [PubMed] [Google Scholar]
- 18.Tajir M, Fergelot P, Lancelot G. et al. Germline mosaicism in Rubinstein-Taybi syndrome. Gene. 2013;518(2):476–478. doi: 10.1016/j.gene.2012.12.105. [DOI] [PubMed] [Google Scholar]
- 19.Yagihashi T, Kosaki K, Okamoto N. et al. Age-dependent change in behavioral feature in Rubinstein-Taybi syndrome. Congenit Anom (Kyoto) 2012;52(2):82–86. doi: 10.1111/j.1741-4520.2012.00356.x. [DOI] [PubMed] [Google Scholar]
- 20.Beets L, Rodríguez-Fonseca C, Hennekam R C. Growth charts for individuals with Rubinstein-Taybi syndrome. Am J Med Genet A. 2014;164A(9):2300–2309. doi: 10.1002/ajmg.a.36654. [DOI] [PubMed] [Google Scholar]
- 21.Milani D, Manzoni F M, Pezzani L. et al. Rubinstein-Taybi syndrome: clinical features, genetic basis, diagnosis, and management. Ital J Pediatr. 2015;41(1):4. doi: 10.1186/s13052-015-0110-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Blum R W. Transition to adult health care: setting the stage. J Adolesc Health. 1995;17(1):3–5. doi: 10.1016/1054-139X(95)00073-2. [DOI] [PubMed] [Google Scholar]
- 23.Bartsch O, Labonté J, Albrecht B. et al. Two patients with EP300 mutations and facial dysmorphism different from the classic Rubinstein-Taybi syndrome. Am J Med Genet A. 2010;152A(1):181–184. doi: 10.1002/ajmg.a.33153. [DOI] [PubMed] [Google Scholar]
- 24.Roelfsema J H, Peters D J. Rubinstein-Taybi syndrome: clinical and molecular overview. Expert Rev Mol Med. 2007;9(23):1–16. doi: 10.1017/S1462399407000415. [DOI] [PubMed] [Google Scholar]
- 25.Wieczorek D, Bartsch O, Lechno S. et al. Two adults with Rubinstein-Taybi syndrome with mild mental retardation, glaucoma, normal growth and skull circumference, and camptodactyly of third fingers. Am J Med Genet A. 2009;149A(12):2849–2854. doi: 10.1002/ajmg.a.33129. [DOI] [PubMed] [Google Scholar]
- 26.Nakai K, Yoneda K, Moriue T, Kubota Y. Striate palmoplantar keratoderma in a patient with Rubinstein-Taybi syndrome. J Eur Acad Dermatol Venereol. 2009;23(3):333–335. doi: 10.1111/j.1468-3083.2008.02852.x. [DOI] [PubMed] [Google Scholar]
- 27.van Genderen M M, Kinds G F, Riemslag F CC, Hennekam R CM. Ocular features in Rubinstein-Taybi syndrome: investigation of 24 patients and review of the literature. Br J Ophthalmol. 2000;84(10):1177–1184. doi: 10.1136/bjo.84.10.1177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Miller R W, Rubinstein J H. Tumors in Rubinstein-Taybi syndrome. Am J Med Genet. 1995;56(1):112–115. doi: 10.1002/ajmg.1320560125. [DOI] [PubMed] [Google Scholar]
- 29.Stevens C A, Pouncey J, Knowles D. Adults with Rubinstein-Taybi syndrome. Am J Med Genet A. 2011;155A(7):1680–1684. doi: 10.1002/ajmg.a.34058. [DOI] [PubMed] [Google Scholar]
- 30.Levitas A S, Reid C S. Rubinstein-Taybi syndrome and psychiatric disorders. J Intellect Disabil Res. 1998;42(Pt 4):284–292. doi: 10.1046/j.1365-2788.1998.00136.x. [DOI] [PubMed] [Google Scholar]
- 31.Hennekam R C, Stevens C A, Van de Kamp J J. Etiology and recurrence risk in Rubinstein-Taybi syndrome. Am J Med Genet Suppl. 1990;6:56–64. doi: 10.1002/ajmg.1320370610. [DOI] [PubMed] [Google Scholar]
- 32.Bloch-Zupan A, Stachtou J, Emmanouil D, Arveiler B, Griffiths D, Lacombe D. Oro-dental features as useful diagnostic tool in Rubinstein-Taybi syndrome. Am J Med Genet A. 2007;143A(6):570–573. doi: 10.1002/ajmg.a.31622. [DOI] [PubMed] [Google Scholar]
- 33.Zucconi M, Ferini-Strambi L, Erminio C, Pestalozza G, Smirne S. Obstructive sleep apnea in the Rubinstein-Taybi syndrome. Respiration. 1993;60(2):127–132. doi: 10.1159/000196186. [DOI] [PubMed] [Google Scholar]
- 34.Choi H S, Yu J J, Kim Y H, Ko J K, Park I S. Pulmonary hypertension due to obstructive sleep apnea in a child with Rubinstein-Taybi syndrome. Korean J Pediatr. 2012;55(6):212–214. doi: 10.3345/kjp.2012.55.6.212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Peñaranda A, Cerón M. Rubinstein-Taybi syndrome and mixed bilateral hypoacousia case report. Otol Neurotol. 2007;28(4):501–503. doi: 10.1097/MAO.0b013e31803115d4. [DOI] [PubMed] [Google Scholar]
- 36.Naimi D R, Munoz J, Rubinstein J, Hostoffer R W Jr. Rubinstein-Taybi syndrome: an immune deficiency as a cause for recurrent infections. Allergy Asthma Proc. 2006;27(3):281–284. doi: 10.2500/aap.2006.27.2864. [DOI] [PubMed] [Google Scholar]
- 37.Marabotti A, Giannecchini G, Cariello A. et al. Stenosis of the lachrymal system in Rubinstein-Taybi syndrome. Ophthalmologica t. 2002;216(4):272–276. doi: 10.1159/000063837. [DOI] [PubMed] [Google Scholar]
- 38.van Genderen M M, Kinds G F, Riemslag F C, Hennekam R C. Ocular features in Rubinstein-Taybi syndrome: investigation of 24 patients and review of the literature. Br J Ophthalmol. 2000;84(10):1177–1184. doi: 10.1136/bjo.84.10.1177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Robson M J, Brown L M, Sharrard W J. Cervical spondylolisthesis and other skeletal abnormalities in Rubinstein-Taybi syndrome. J Bone Joint Surg Br. 1980;62(3):297–299. doi: 10.1302/0301-620X.62B3.7410460. [DOI] [PubMed] [Google Scholar]
- 40.Yamamoto T, Kurosawa K, Masuno M. et al. Congenital anomaly of cervical vertebrae is a major complication of Rubinstein-Taybi syndrome. Am J Med Genet A. 2005;135(2):130–133. doi: 10.1002/ajmg.a.30708. [DOI] [PubMed] [Google Scholar]
- 41.Marzuillo P, Grandone A, Luongo C. et al. Brain magnetic resonance in the routine management of Rubinstein-Taybi syndrome (RTS) can prevent life-threatening events and neurological deficits. Am J Med Genet A. 2014;164A(8):2129–2132. doi: 10.1002/ajmg.a.36585. [DOI] [PubMed] [Google Scholar]
- 42.Rubinstein J H. Broad thumb-hallux (Rubinstein-Taybi) syndrome 1957-1988. Am J Med Genet Suppl. 1990;6:3–16. doi: 10.1002/ajmg.1320370603. [DOI] [PubMed] [Google Scholar]
- 43.Wójcik C, Volz K, Ranola M. et al. Rubinstein-Taybi syndrome associated with Chiari type I malformation caused by a large 16p13.3 microdeletion: a contiguous gene syndrome? Am J Med Genet A. 2010;152A(2):479–483. doi: 10.1002/ajmg.a.33303. [DOI] [PubMed] [Google Scholar]
- 44.Parsley L, Bellus G, Handler M, Tsai A C. Identical twin sisters with Rubinstein-Taybi syndrome associated with Chiari malformations and syrinx. Am J Med Genet A. 2011;155A(11):2766–2770. doi: 10.1002/ajmg.a.34227. [DOI] [PubMed] [Google Scholar]
- 45.Giussani C, Selicorni A, Fossati C. et al. The association of neural axis and craniovertebral junction anomalies with scoliosis in Rubinstein-Taybi syndrome. Childs Nerv Syst. 2012;28(12):2163–2168. doi: 10.1007/s00381-012-1893-7. [DOI] [PubMed] [Google Scholar]
- 46.Marzuillo P, Grandone A, Coppola R. et al. Novel cAMP binding protein-BP (CREBBP) mutation in a girl with Rubinstein-Taybi syndrome, GH deficiency, Arnold Chiari malformation and pituitary hypoplasia. BMC Med Genet. 2013;14:28. doi: 10.1186/1471-2350-14-28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Barson A J. Proceedings: Rubinstein-Taybi syndrome. Arch Dis Child. 1974;49(6):495. doi: 10.1136/adc.49.6.495-f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Agarwal R, Aggarwal R, Kabra M, Deorari A K. Dandy-Walker malformation in Rubinstein-Taybi syndrome: a rare association. Clin Dysmorphol. 2002;11(3):223–224. doi: 10.1097/00019605-200207000-00017. [DOI] [PubMed] [Google Scholar]
- 49.Tanaka T Ling B C Rubinstein J H Crone K R Rubinstein-Taybi syndrome in children with tethered spinal cord J Neurosurg 2006105(4, Suppl):261–264. [DOI] [PubMed] [Google Scholar]
- 50.Hennekam R CM. Hoboken, NJ: Wiley-Blackwell; 2010. Rubinstein-Taybi syndrome; pp. 705–715. [Google Scholar]
- 51.Stevens C A, Bhakta M G. Cardiac abnormalities in the Rubinstein-Taybi syndrome. Am J Med Genet. 1995;59(3):346–348. doi: 10.1002/ajmg.1320590313. [DOI] [PubMed] [Google Scholar]
- 52.Hanauer D, Argilla M, Wallerstein R. Rubinstein-Taybi syndrome and hypoplastic left heart. Am J Med Genet. 2002;112(1):109–111. doi: 10.1002/ajmg.10617. [DOI] [PubMed] [Google Scholar]
- 53.Fischer S, Bäzner H, Henkes H. Cervical artery dissection in a young patient with Rubinstein-Taybi syndrome. Clin Neuroradiol. 2013;23(1):41–44. doi: 10.1007/s00062-011-0100-7. [DOI] [PubMed] [Google Scholar]
- 54.Ishizaka S, Sou G, Morofuji Y. et al. Dissecting aneurysm of the anterior cerebral artery with Rubinstein-Taybi syndrome—a case report [in Japanese] Brain Nerve. 2010;62(10):1083–1088. [PubMed] [Google Scholar]
- 55.Isidor B, Podevin G, Camby C. et al. Rubinstein-Taybi syndrome and Hirschsprung disease in a patient harboring an intragenic deletion of the CREBBP gene. Am J Med Genet A. 2010;152A(7):1847–1848. doi: 10.1002/ajmg.a.33480. [DOI] [PubMed] [Google Scholar]
- 56.Olson D P, Koenig R J. Thyroid function in Rubinstein-Taybi syndrome. J Clin Endocrinol Metab. 1997;82(10):3264–3266. doi: 10.1210/jcem.82.10.4273. [DOI] [PubMed] [Google Scholar]
- 57.Kurtoglu S, Akcakus M, Gunes T, Cetin N, Topaloglu N. Congenital hypothyroidism associated with Rubinstein-Taybi syndrome. J Pediatr Endocrinol Metab. 2003;16(3):457–459. doi: 10.1515/jpem.2003.16.3.457. [DOI] [PubMed] [Google Scholar]
- 58.Tornese G, Marzuillo P, Pellegrin M C. et al. A case of Rubinstein-Taybi syndrome associated with growth hormone deficiency in childhood. Clin Endocrinol (Oxf) 2015 doi: 10.1111/cen.12748. [DOI] [PubMed] [Google Scholar]
- 59.Bayle P, Bazex J, Lamant L, Lauque D, Durieu C, Albes B. Multiple perforating and non perforating pilomatricomas in a patient with Churg-Strauss syndrome and Rubinstein-Taybi syndrome. J Eur Acad Dermatol Venereol. 2004;18(5):607–610. doi: 10.1111/j.1468-3083.2004.00991.x. [DOI] [PubMed] [Google Scholar]
- 60.van de Kar A L, Houge G, Shaw A C. et al. Keloids in Rubinstein-Taybi syndrome: a clinical study. Br J Dermatol. 2014;171(3):615–621. doi: 10.1111/bjd.13124. [DOI] [PubMed] [Google Scholar]
- 61.Papathemeli D, Schulzendorff N, Kohlhase J, Göppner D, Franke I, Gollnick H. Pilomatricomas in Rubinstein-Taybi syndrome. J Dtsch Dermatol Ges. 2015;13(3):240–242. doi: 10.1111/ddg.12504. [DOI] [PubMed] [Google Scholar]
- 62.Siraganian P A, Rubinstein J H, Miller R W. Keloids and neoplasms in the Rubinstein-Taybi syndrome. Med Pediatr Oncol. 1989;17(6):485–491. doi: 10.1002/mpo.2950170526. [DOI] [PubMed] [Google Scholar]
- 63.de Kort E, Conneman N, Diderich K. A case of Rubinstein-Taybi syndrome and congenital neuroblastoma. Am J Med Genet A. 2014;164A(5):1332–1333. doi: 10.1002/ajmg.a.36399. [DOI] [PubMed] [Google Scholar]
- 64.Ihara K, Kuromaru R, Takemoto M, Hara T. Rubinstein-Taybi syndrome: a girl with a history of neuroblastoma and premature thelarche. Am J Med Genet. 1999;83(5):365–366. [PubMed] [Google Scholar]
- 65.Bourdeaut F, Miquel C, Richer W. et al. Rubinstein-Taybi syndrome predisposing to non-WNT, non-SHH, group 3 medulloblastoma. Pediatr Blood Cancer. 2014;61(2):383–386. doi: 10.1002/pbc.24765. [DOI] [PubMed] [Google Scholar]
- 66.Evans G, Burnell L, Campbell R, Gattamaneni H R, Birch J. Congenital anomalies and genetic syndromes in 173 cases of medulloblastoma. Med Pediatr Oncol. 1993;21(6):433–434. doi: 10.1002/mpo.2950210608. [DOI] [PubMed] [Google Scholar]
- 67.Skousen G J, Wardinsky T, Chenaille P. Medulloblastoma in patient with Rubinstein-Taybi syndrome. Am J Med Genet. 1996;66(3):367. doi: 10.1002/(SICI)1096-8628(19961218)66:3<367::AID-AJMG27>3.0.CO;2-J. [DOI] [PubMed] [Google Scholar]
- 68.Taylor M D, Mainprize T G, Rutka J T, Becker L, Bayani J, Drake J M. Medulloblastoma in a child with Rubenstein-Taybi syndrome: case report and review of the literature. Pediatr Neurosurg. 2001;35(5):235–238. doi: 10.1159/000050428. [DOI] [PubMed] [Google Scholar]
- 69.Johannesen E J, Williams T, Miller D C, Tuller E. Synchronous ovarian and endometrial carcinomas in a patient with Rubinstein-Taybi syndrome: a case report and literature review. Int J Gynecol Pathol. 2015;34(2):132–135. doi: 10.1097/PGP.0000000000000125. [DOI] [PubMed] [Google Scholar]
- 70.Hennekam R C, Baselier A C, Beyaert E. et al. Psychological and speech studies in Rubinstein-Taybi syndrome. Am J Ment Retard. 1992;96(6):645–660. [PubMed] [Google Scholar]
- 71.Wiley S, Swayne S, Rubinstein J H, Lanphear N E, Stevens C A. Rubinstein-Taybi syndrome medical guidelines. Am J Med Genet A. 2003;119A(2):101–110. doi: 10.1002/ajmg.a.10009. [DOI] [PubMed] [Google Scholar]
- 72.Schorry E K, Keddache M, Lanphear N. et al. Genotype-phenotype correlations in Rubinstein-Taybi syndrome. Am J Med Genet A. 2008;146A(19):2512–2519. doi: 10.1002/ajmg.a.32424. [DOI] [PubMed] [Google Scholar]
- 73.Hood R L, Lines M A, Nikkel S M. et al. Mutations in SRCAP, encoding SNF2-related CREBBP activator protein, cause Floating-Harbor syndrome. Am J Hum Genet. 2012;90(2):308–313. doi: 10.1016/j.ajhg.2011.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Campeau P M, Kim J C, Lu J T. et al. Mutations in KAT6B, encoding a histone acetyltransferase, cause Genitopatellar syndrome. Am J Hum Genet. 2012;90(2):282–289. doi: 10.1016/j.ajhg.2011.11.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Simpson M A, Deshpande C, Dafou D. et al. De novo mutations of the gene encoding the histone acetyltransferase KAT6B cause Genitopatellar syndrome. Am J Hum Genet. 2012;90(2):290–294. doi: 10.1016/j.ajhg.2011.11.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Chen X, Xu H, Yuan P. et al. Integration of external signaling pathways with the core transcriptional network in embryonic stem cells. Cell. 2008;133(6):1106–1117. doi: 10.1016/j.cell.2008.04.043. [DOI] [PubMed] [Google Scholar]
- 77.Scolnick D M, Chehab N H, Stavridi E S. et al. CREB-binding protein and p300/CBP-associated factor are transcriptional coactivators of the p53 tumor suppressor protein. Cancer Res. 1997;57(17):3693–3696. [PubMed] [Google Scholar]
- 78.Yan W, Liu S, Xu E. et al. Histone deacetylase inhibitors suppress mutant p53 transcription via histone deacetylase 8. Oncogene. 2013;32(5):599–609. doi: 10.1038/onc.2012.81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Coupry I, Roudaut C, Stef M. et al. Molecular analysis of the CBP gene in 60 patients with Rubinstein-Taybi syndrome. J Med Genet. 2002;39(6):415–421. doi: 10.1136/jmg.39.6.415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Bentivegna A, Milani D, Gervasini C. et al. Rubinstein-Taybi syndrome: spectrum of CREBBP mutations in Italian patients. BMC Med Genet. 2006;7:77. doi: 10.1186/1471-2350-7-77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Bartsch O, Schmidt S, Richter M. et al. DNA sequencing of CREBBP demonstrates mutations in 56% of patients with Rubinstein-Taybi syndrome (RSTS) and in another patient with incomplete RSTS. Hum Genet. 2005;117(5):485–493. doi: 10.1007/s00439-005-1331-y. [DOI] [PubMed] [Google Scholar]
- 82.Bartholdi D, Roelfsema J H, Papadia F. et al. Genetic heterogeneity in Rubinstein-Taybi syndrome: delineation of the phenotype of the first patients carrying mutations in EP300. J Med Genet. 2007;44(5):327–333. doi: 10.1136/jmg.2006.046698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Imaizumi K, Kuroki Y. Rubinstein-Taybi syndrome with de novo reciprocal translocation t(2;16)(p13.3;p13.3) Am J Med Genet. 1991;38(4):636–639. doi: 10.1002/ajmg.1320380430. [DOI] [PubMed] [Google Scholar]
- 84.Tommerup N, van der Hagen C B, Heiberg A. Tentative assignment of a locus for Rubinstein-Taybi syndrome to 16p13.3 by a de novo reciprocal translocation, t(7;16)(q34;p13.3) Am J Med Genet. 1992;44(2):237–241. doi: 10.1002/ajmg.1320440223. [DOI] [PubMed] [Google Scholar]
- 85.Allis C D, Berger S L, Cote J. et al. New nomenclature for chromatin-modifying enzymes. Cell. 2007;131(4):633–636. doi: 10.1016/j.cell.2007.10.039. [DOI] [PubMed] [Google Scholar]
- 86.Park E, Kim Y, Ryu H, Kowall N W, Lee J, Ryu H. Epigenetic mechanisms of Rubinstein-Taybi syndrome. Neuromolecular Med. 2014;16(1):16–24. doi: 10.1007/s12017-013-8285-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Wang J, Weaver I C, Gauthier-Fisher A. et al. CBP histone acetyltransferase activity regulates embryonic neural differentiation in the normal and Rubinstein-Taybi syndrome brain. Dev Cell. 2010;18(1):114–125. doi: 10.1016/j.devcel.2009.10.023. [DOI] [PubMed] [Google Scholar]
- 88.Oike Y, Hata A, Mamiya T. et al. Truncated CBP protein leads to classical Rubinstein-Taybi syndrome phenotypes in mice: implications for a dominant-negative mechanism. Hum Mol Genet. 1999;8(3):387–396. doi: 10.1093/hmg/8.3.387. [DOI] [PubMed] [Google Scholar]
- 89.Foley P, Bunyan D, Stratton J, Dillon M, Lynch S A. Further case of Rubinstein-Taybi syndrome due to a deletion in EP300. Am J Med Genet A. 2009;149A(5):997–1000. doi: 10.1002/ajmg.a.32771. [DOI] [PubMed] [Google Scholar]
- 90.Gervasini C, Castronovo P, Bentivegna A. et al. High frequency of mosaic CREBBP deletions in Rubinstein-Taybi syndrome patients and mapping of somatic and germ-line breakpoints. Genomics. 2007;90(5):567–573. doi: 10.1016/j.ygeno.2007.07.012. [DOI] [PubMed] [Google Scholar]
- 91.Bartsch O, Rasi S, Delicado A. et al. Evidence for a new contiguous gene syndrome, the chromosome 16p13.3 deletion syndrome alias severe Rubinstein-Taybi syndrome. Hum Genet. 2006;120(2):179–186. doi: 10.1007/s00439-006-0215-0. [DOI] [PubMed] [Google Scholar]
- 92.Shim J H, Greenblatt M B, Singh A. et al. Administration of BMP2/7 in utero partially reverses Rubinstein-Taybi syndrome-like skeletal defects induced by Pdk1 or Cbp mutations in mice. J Clin Invest. 2012;122(1):91–106. doi: 10.1172/JCI59466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Vecsey C G, Hawk J D, Lattal K M. et al. Histone deacetylase inhibitors enhance memory and synaptic plasticity via CREB:CBP-dependent transcriptional activation. J Neurosci. 2007;27(23):6128–6140. doi: 10.1523/JNEUROSCI.0296-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Lopez-Atalaya J P, Gervasini C, Mottadelli F. et al. Histone acetylation deficits in lymphoblastoid cell lines from patients with Rubinstein-Taybi syndrome. J Med Genet. 2012;49(1):66–74. doi: 10.1136/jmedgenet-2011-100354. [DOI] [PubMed] [Google Scholar]