

Abstract
A programmable pre-assembly method is described and shown to produce near-infrared fluorescent molecular probes with tunable multivalent binding properties. The modular assembly process threads one or two copies of a tetralactam macrocycle onto a fluorescent PEGylated squaraine scaffold containing a complementary number of docking stations. Appended to the macrocycle periphery are multiple copies of a ligand that is known to target a biomarker. The structure and high purity of each threaded complex was determined by independent spectrometric methods and also by gel electrophoresis. Especially helpful were diagnostic red-shift and energy transfer features in the absorption and fluorescence spectra. The threaded complexes were found to be effective multivalent molecular probes for fluorescence microscopy and in vivo fluorescence imaging of living subjects. Two multivalent probes were prepared and tested for targeting of bone in mice. A pre-assembled probe with twelve bone-targeting iminodiacetate ligands produced more bone accumulation than an analogous pre-assembled probe with six iminodiacetate ligands. Notably, there was no loss in probe fluorescence at the bone target site after 24 hours in the living animal, indicating that the pre-assembled fluorescent probe maintained very high mechanical and chemical stability on the skeletal surface. The study shows how this versatile pre-assembly method can be used in a parallel combinatorial manner to produce libraries of near-infrared fluorescent multivalent molecular probes for different types of imaging and diagnostic applications, with incremental structural changes in the number of targeting groups, linker lengths, linker flexibility, and degree of PEGylation.
Graphical abstract
Introduction
Molecular probes with selective affinity for biomarkers that are expressed on the exterior surface of biological targets are commonly employed for diagnostics, imaging, and targeted therapy.1,2 The most common strategy in the literature uses a high affinity monovalent probe with a single targeting ligand, and under saturating conditions the probe selectivity is defined by the difference in biomarker expression levels.3 A second approach uses a multivalent probe that is equipped with multiple copies of the targeting ligand.4 If the linker between the appended targeting ligands is long enough, there is potential for crosslinking of separate biomarkers and an increase in probe avidity to the target site due to positive binding cooperativity.5 Even if the linker between the appended targeting ligands is too short for biomarker crosslinking, there is still a chance for increased probe avidity to a single biomarker due to the high local ligand concentration and long residence time enforced by the multivalent probe structure.6–8 The probe structural factors that promote strong and selective multivalent association are still being debated in the literature, but two of the most obvious are the number of appended targeting ligands and the structure of the linkers that connect them.9,10 The average distance between two biomarkers depends on the expression level and, in principle, it should be possible to match this average distance with a complementary multivalent probe. In practice, however, it is extremely hard to rationally design a multivalent probe for high performance targeting in complex biological media.11–13 Even if the biomarker expression level is known, there is the in vivo requirement that useful multivalent probes must have appropriate pharmacokinetic properties that enable them to access desired anatomical locations before probe clearance from the blood stream.
An alternative approach to probe development by rational design is a screening paradigm that prepares a relatively large number of different multivalent molecular probes and subsequently identifies, through a screening assay, the library members with the best targeting properties. The first step in this process is the library synthesis, and there are essentially three ways to prepare a library of multivalent probes. (1) Covalent synthesis of discrete molecules with dendritic scaffolds that present multiple copies of the targeting ligands.14,15 This method produces single molecules with precise composition, but the reaction yields are often low at high ligand loading levels and require excess reagents which may make probe purification a wasteful and time consuming process. (2) Statistical attachment of targeting ligands to a polymeric scaffold or nanoparticle surface.16,17 While this approach may be amenable to high throughput production of probe variants, each probe will have a polydisperse composition. This will likely make it difficult to achieve a reproducible synthesis that satisfies government regulatory agencies.18 There are also pharmacokinetic limitations since the in vivo targeting properties of relatively large polymers or nanoparticles may be dominated by non-specific accumulation effects such as the enhanced permeation and retention effect.19,20 Other problems with relatively large polymers or nanoparticles are slow rates of diffusion to size-restricted sites, and limited excretion through the kidneys (typically, polymer molecular weight must be less than 40 kDa for kidney excretion).21 (3) Programmable self-assembly of multivalent probes. This concept is relatively new and has not yet been demonstrated in vivo. One published method uses sequence-selective nucleobase pairing to precisely position targeting ligands on a DNA or PNA scaffold.22–24 Other approaches use supramolecular chemistry to assemble macrocyclic hosts, such as cyclodextrins with attached ligands, onto appropriate scaffolds.25,26 While these literature self-assembly methods have attractive features, they also have limitations, especially if the goal is to produce targeted probes for molecular imaging. The most obvious shortcoming for imaging applications is the transparent nature of the self-assembly building blocks, which means that a reporter group still has to be connected to the probe which lengthens and complicates the fabrication process.
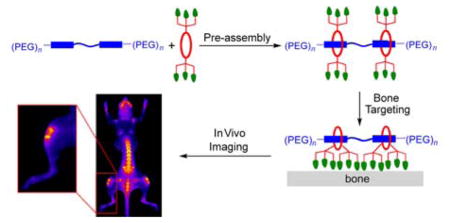
Here, we describe a new programmable pre-assembly method that can rapidly produce libraries of fluorescent multivalent molecular probes with tunable binding properties. Moreover, the fluorescence emission wavelength is in the near-infrared window, which is optimal for in vivo imaging.27–29 The modular assembly process threads one or two copies of a macrocycle onto a PEGylated squaraine scaffold containing a complementary number of fluorescent docking stations (Figure 1). Appended to the macrocycle periphery are multiple copies of a ligand that is known to target a biomarker. A pre-assembled targeted fluorescent probe is created in essentially quantitative yield by simply mixing a sample of squaraine scaffold and macrocycle in appropriate molar ratio. In principle, a sub-library of m macrocycles and sub-library of n squaraine scaffolds can be mixed in parallel combinatorial fashion to create a library of m x n fluorescent probes, each with incremental structural changes such as ligand copy number, linker distance, linker flexibility, and degree of PEGylation to control biodistribution.30, 31 The macrocycle threading process that is the basis of the pre-assembly method is shown in Figure 2 and we call it Synthavidin (Synthetic Avidin) technology.32 The macrocycle is a water-soluble tetralactam with anthracene sidewalls, and previous studies have shown that it can be threaded by a PEGylated squaraine dye to give a highly stable complex with nanomolar dissociation constant.33–35 Detailed thermodynamic and kinetic measurements have identified the structural factors that favor the very strong association and control the rates of macrocycle threading. In short, the two oxygen atoms on the encapsulated squaraine dye form hydrogen bonds to the four macrocycle NH residues and there is coplanar stacking of the squaraine aromatic surfaces with the anthracene sidewalls of the macrocycle. The threading kinetics are insensitive to the length of the two PEG chains that extend from each end of the squaraine structure. Indeed, there is no decrease in threading rates when the PEG chains are extended by several hundred atoms, but the threading kinetics are greatly affected by the steric size of the second N-substituent attached to the squaraine termini, and an N-propyl group produces a set of kinetic and thermodynamic properties that are well-suited for programmable pre-assembly of threaded probes with robust structure.34 To be more specific, mixing the two components in Figure 2 at high micromolar concentration in water at room temperature leads to quantitative formation of the threaded complex. An aqueous stock solution of the pre-assembled complex can be stored without change for long periods and dilution of an aliquot to nanomolar concentrations in fetal bovine serum produces no measurable dissociation after 24 h. The threaded squaraine complexes have near-infrared fluorescence properties that are well-suited for biological imaging. With log ε > 5 and fluorescence quantum yields of ~0.1 in water, they are relatively bright when compared to other near-infrared dyes. Since the encapsulated squaraine dye is protected from the solvent the fluorescence brightness hardly changes with pH or polarity of the surrounding medium, a major advantage for quantitative imaging. The squaraine absorption/emission bands are narrow with absorption maximum near 675 nm and emission maximum around 715 nm. This corresponds to the common Cy5.5 filter set on microscopes, microarrays, and imaging stations, and the narrow bands enable effective multicolor imaging.36, 37 The unusually large Stokes shift of ~40 nm means there is relatively little loss in fluorescence efficiency due to the inner field effect (absorption of the light emitted by another molecule). Furthermore, a “mega-Stokes shift” of 325 nm can be achieved by exploiting energy transfer within the threaded complex. Thus, excitation of the anthracene sidewalls in the surrounding macrocycle with blue light produces efficient internal energy transfer to the encapsulated squaraine. Observation of this energy transfer is a convenient way to confirm that the complex has retained its threaded structure.
Figure 1.
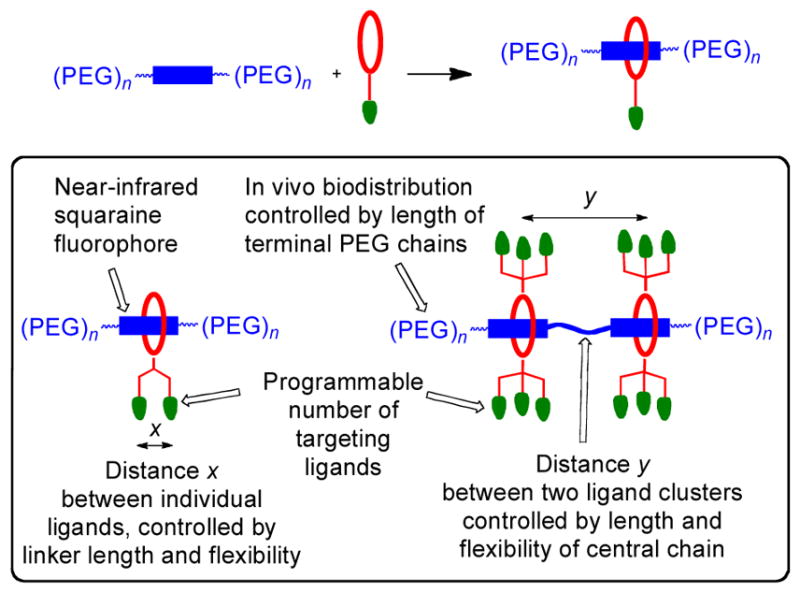
[top] Programmable pre-assembly method that threads a macrocycle (red) with an appended targeting ligand (green) onto a PEGylated-squaraine scaffold (blue) to create a fluorescent probe for selective biomarker recognition. [bottom] Summary of probe structural features that can be varied by the pre-assembly method.
Figure 2.
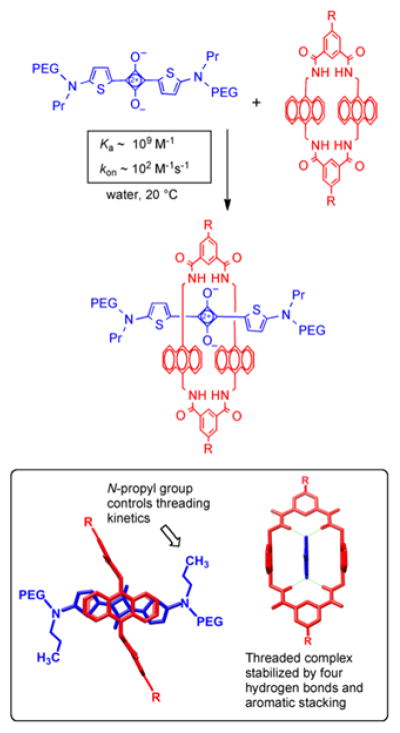
Supramolecular basis of Synthavidin technology. [top] Threading of tetralactam macrocycle (red) with a squaraine dye that is flanked by long PEG chains (blue) gives the pre-assembled complex. [bottom] Two views of the threaded complex highlighting the structural features controlling the threading kinetics and thermodynamics.
The primary aim of this study was to demonstrate for the first time that our Synthavidin method for macrocycle threading could be used for programmed pre-assembly of multivalent fluorescent probes that enable effective targeting of specific anatomical locations in a living subject. To achieve this crucial aim we needed to develop robust synthetic methods to produce the appropriate building blocks (squaraine scaffolds with one or two docking stations, and macrocycles decorated with multiple copies of a targeting ligand) and convenient structural elucidation methods to prove that quantitative probe pre-assembly had occurred. We also had to confirm that the pre-assembled multivalent probes were mechanically and chemically stable during different types of biological imaging experiments. Finally, we wanted to demonstrate the practical utility of this new class of pre-assembled fluorescent probes by testing the hypothesis that multivalent molecular targeting of bone in living subjects increases when the molecular probes have an increased number of appended bone targeting ligands.
Results and Discussion
Probe Design
Shown in Figure 3 are the two squaraine scaffolds and two macrocycles that were used to pre-assemble four near-infrared fluorescent probes as threaded complexes. The scaffold S has one squaraine docking station, whereas, the scaffold S3S has two squaraine docking stations separated by a triethylene glycol chain. The scaffolds were threaded with either the macrocycle 6C which has six appended carboxylate groups (designated as C), or the macrocycle 6B which has six appended iminodiacetate groups (designated as B and also colored green in the simplified schematic structures). Previously, we reported the structure and properties of threaded, single-station complex 6C ⊃S.33 As a fluorescent probe in cell culture, it exhibited no specific cell affinity and was observed to slowly enter living cells over several hours. Thus, in this present mouse imaging study, the threaded complex 6C ⊃S and new two-station version 2(6C) ⊃S3S were expected to act as non-targeted control probes that cleared from the blood stream through the kidneys with little retention in other organs.38 The other pre-assembled probe system is the related pair of 6B ⊃S and 2(6B) ⊃S3S which have six and twelve appended iminodiacetate groups, respectively. Three years ago, we discovered that fluorescent probes with appended iminodiacetate groups have affinity for regions of living bone that are undergoing high turnover.39 The iminodiacetate groups associate with the hydroxyapatite mineral within the bone.40 Moreover, a comparative in vivo imaging study showed that a fluorescent probe with four iminodiacetate groups exhibited more bone accumulation in living mice than a probe with two iminodiacetate groups. Thus, in this present mouse imaging study, the threaded complexes 6B ⊃S and two-station version 2(6B) ⊃S3S were designed to act as multivalent bone-targeting probes. Our starting hypothesis was that the dodecavalent probe 2(6B) ⊃S3S would produce more bone accumulation than hexavalent 6B ⊃S.
Figure 3.

Parallel pre-assembly of four near-infrared fluorescent probes by mixing separate samples of squaraine scaffold (S or S3S) and macrocycle (6C or 6B) in water at 1:1 or 1:2 molar ratio.
Synthesis of Building Blocks and Probe Pre-assembly
The single-station squaraine scaffold S and double-station S3S were synthesized in straightforward fashion by conducting copper catalyzed alkyne/azide cycloaddition reactions using a common squaraine bisalkyne precursor (see Supporting Information). The two macrocycles 6B and 6C were also prepared in a few steps using established chemistry, and the high purity of each of the four building blocks was confirmed by standard spectroscopic methods.
The macrocycle threading process is conveniently monitored by absorption or fluorescence spectroscopy, and threading is indicated by several diagnostic changes in spectral properties, such as a 20–30 nm red-shift in squaraine absorption/emission maxima and also efficient internal energy transfer from the macrocycle (ex: 390 nm) to the encapsulated squaraine dye (em: 715 nm). Previously, we showed that mixing S and 6C in water at 20 °C formed the single-station untargeted complex 6C ⊃S with an association constant (Ka) of 1 × 109 M−1 and threading rate kon of 130 M−1s−1.33 Essentially the same values of Ka and kon were observed when S and 6B were mixed in water to form the single-station targeted complex 6B ⊃S. This was expected since the structures of the two macrocycles differ only by a relatively minor alteration of the peripheral ligands located away from the internal macrocycle cavity that contacts the squaraine docking station. With the two-station complexes, 2(6C) ⊃S3S and 2(6B) ⊃S3S, the rate of the double macrocycle threading process was observed to be 20–50 times slower but it still produced the threaded complexes in nearly quantitative yield. The most likely major reason for the slower threading rates is that the unthreaded two-station scaffold S3S is folded in aqueous solution, as reflected by its highly blue-shifted absorption band (605 nm) and very low fluorescence quantum yield. Sequential threading of two macrocycles onto S3S produced incremental red-shifting of the squaraine absorption maxima and an increase in fluorescence emission intensity (Figure S11–14). In addition, there was an incremental increase in internal energy transfer efficiency from the macrocycle to the encapsulated squaraine (Figure S15–16). These photophysical differences are reflected by the spectral data in Table 1, which shows that the fluorescence quantum yields for the doubly threaded, two-station complexes 2(6C) ⊃S3S and 2(6B) ⊃S3S are higher than the singly threaded complexes of the two-station scaffold (i.e., 6C ⊃S3S and 6B ⊃S3S). The kinetics for sequential threading of two macrocycles onto S3S were investigated in more detail by conducting step-wise threading experiments. The experiments started by adding one molar equivalent of macrocycle (6C or 6B) to a solution of the two-station scaffold S3S, and once threading was complete a second molar equivalent of the macrocycle was added to the solution. In Figure 4 are plots showing the squaraine fluorescence enhancement caused by this step-wise macrocycle threading. Each stage of the two-stage plots was treated as a separate bimolecular association event, and fitting to a second order kinetic model gave kon for the two individual threading steps (Figure S20 and Table S2). With both macrcocycles, the values of kon for each step were similar (1 – 4 M−1s−1 at 20 °C) and there was essentially quantitative production of 2(6C) ⊃S3S or 2(6B) ⊃S3S within a few hours. Based on these threading rates, a standard set of pre-assembly conditions was employed to prepare a stock solution of each of the four pre-assembled complexes in Figure 3. In each case, the squaraine scaffold (250 μM) was mixed with one or two molar equivalents of the appropriate macrocycle and the aqueous solution incubated for eight hours at room temperature. This waiting period ensured completion of the pre-assembly process, which was confirmed by monitoring the absorption and fluorescence signatures described above.
Table 1.
Spectral data for absorption and emission maxima bands in H2O (3.0 μM).
| Compound | λabs (nm) | λem (nm) | log ε | Φfa |
|---|---|---|---|---|
| S3S | 605 | -- | 5.38 | <0.003 |
| 6C ⊃S | 677 | 710 | 5.02 | 0.18 |
| 6B ⊃S | 675 | 710 | 5.14 | 0.10 |
| 6C ⊃S3S | 662 | 711 | -- | 0.01 |
| 6B ⊃S3S | 669 | 712 | -- | 0.03 |
| 2(6C) ⊃S3S | 673 | 716 | 5.36 | 0.02 |
| 2(6B) ⊃S3S | 675 | 716 | 5.40 | 0.05 |
Relative to a standard solution of methylene blue in H2O (Φf = 0.02).
Figure 4.

Fluorescence enhancement due to step-wise threading of two macrocycles onto the two-station scaffold S3S in water. The experiments started by adding one molar equivalent of macrocycle (6C or 6B) to a solution of S3S (250 μM), and once threading was complete (t = 200 minutes) a second molar equivalent of the macrocycle was added to the solution. Each data point is an average of three experiments with error bars corresponding the standard deviation from the mean. Ex 670 nm, Em = 715 nm, T = 25 °C.
Three other structural elucidation techniques (1H NMR spectroscopy, MALDI-TOF mass spectrometry and gel electrophoresis) were investigated as independent methods to confirm that probe pre-assembly had occurred. The 1H NMR spectrum of single-station complex 6C ⊃S contains a set of sharp signals with chemical shift changes that are highly consistent with the threaded structure 6C ⊃S (Figure S3). However, 1H NMR spectra for the other three threaded complexes were less informative (Figure S4–S6). The spectrum of 2(6C) ⊃S3S was sharp but the patterns were complicated because of the unsymmetric structure of the dimeric squaraine scaffold and also the existence of multiple squaraine conformational isomers (due to relative orientation of the thiophene rings). 1H NMR spectra for the two targeted probes 6B ⊃S and 2(6B) ⊃S3S in water were very broad and this was attributed to proton exchange within the peripheral iminodiacetate groups. Overall, 1H NMR was not a generally effective method for structural characterization of the threaded complexes in water. More compelling qualitative evidence for the existence of the threaded structures was gained by MALDI-TOF mass spectrometry. Although the relatively harsh ionization conditions of MALDI mass spectrometry make it a challenging method for detection of noncovalent assemblies, the technique is attractive because it can be exploited for mass spectrometric imaging.41 After some experimentation, good levels of complex ionization were obtained by incorporating the complexes within a dried spot of MALDI matrix containing a large concentration of 2,4-dihydroxybenozoic acid, and with each separate sample of complex there was clear mass spectral evidence for the presence of the threaded structure with correct stoichiometry. There were also peaks corresponding to the unthreaded components, most likely induced by the highly acidic matrix and the laser induced desorption ionization process.41 Therefore, we searched for a structure characterization method that analyzed the threaded complexes under conditions that closely matched the pre-assembled stock solutions, and found success with agarose gel electrophoresis followed by near-infrared fluorescence imaging. Shown in Figure 5 are images of two representative gels that were run with loading wells containing pre-determined mixtures of singly or doubly threaded two-station complexes (i.e., the mixtures were made by adding aliquots from independently prepared stock solutions). The gel images show that the singly threaded and doubly threaded two-station complexes can be separated according to their different mass-to-charge ratios and that the relative amounts in a mixture can be quantified by fluorescence imaging of the gel. The gel images also show that the fluorescence bands for the doubly threaded, two-station complexes 2(6C) ⊃S3S and 2(6B) ⊃S3S are brighter than the singly threaded homologues 6C ⊃S3S and 6B ⊃S3S, which matches the order of fluorescence quantum yields in Table 1. Focusing on gel lanes b and f in Figure 5, which contained only aliquots from the stock solutions of pre-assembled 2(6C) ⊃S3S or 2(6B) ⊃S3S, respectively, the bands show clearly that the purity of each solution was > 90% doubly threaded, two-station complex. Additional gel electrophoresis experiments proved that the pre-assembled stock solutions of threaded one-station complexes, 6C ⊃S and 6B ⊃S, were also very high purity (Figure S19). The results indicate that agarose gel electrophoresis followed by fluorescence imaging is a convenient and general method for confirming that a pre-assembled threaded complex has formed in high yield.
Figure 5.

Gel electrophoresis characterization of samples containing pre-determined mixtures of singly and/or doubly threaded two-station complexes in the loading wells. After applying a voltage of 110 V for 1.5 h across a running buffer of tris (8.9 mM)/borate (8.9 mM)/EDTA (200 μM), pH = 8.3, the agarose gel was imaged using a fluorescence Cy5.5 excitation/emission filter set.
Preliminary Biocompatibility Studies
Before embarking on extensive in vitro and in vivo imaging studies we conducted a limited set of assays to prove probe biocompatibility. Cell vitality assays using cultured Chinese Hamster Ovary (CHO) cells found that 24 h incubations with the free building blocks or the threaded complexes produced negligible cell toxicity at concentrations up to 50 μM (Figure S21). Furthermore, there was negligible loss of fluorescence for free S or pre-assembled 6B ⊃S after a 30 minute incubation with rat liver microsomes, indicating high metabolic stability (Figure S22). Finally, a fluorescence binding assay showed that pre-assembled 6C ⊃S has only moderate affinity for albumin proteins (Kd ~ 20 μM with bovine serum albumin, see Figure S23). Together these results suggested that low micromolar doses of the pre-assembled probes were not likely to cause cell or animal toxicity, and that liver metabolism or sequestration by albumin proteins in the blood stream was not likely to be a factor preventing the pre-assembled probes from reaching their anatomical targets in a living subject. This general conclusion agrees with the high biocompatibilities observed previously with structurally related squaraine rotaxane probes.42–43
In Vitro Studies of Bone Targeting by Pre-Assembled Multivalent Probes
An initial set of test tube studies measured the relative abilities of each pre-assembled probe to adsorb to powdered mouse bone. Separate aqueous solutions (4 μM) of non-targeted, control probe 6C ⊃S, and the two targeted probes, 6B ⊃S and 2(6B) ⊃S3S, were treated with increasing amounts of bone powder and shaken for 1 h. The bone powder was separated from each sample by centrifugation, and the amount of probe remaining in the supernatant was determined by absorption spectroscopy. As shown in Figure 6 the non-targeted 6C ⊃S had very little affinity for the powdered bone. In contrast, large fractions of the targeted probes adsorbed to the powdered bone. For example, the presence of 200 mg bone powder removed >75% of hexavalent 6B ⊃S and >90% of dodecavalent 2(6B) ⊃S3S from the respective solutions, indicating higher bone affinity with the dodecavalent probe, 2(6B) ⊃S3S. The same affinity trend was observed in fluorescence microscopy experiments that assessed the ability of the three probes to stain histology sections of mouse bone. Thin slices of mouse tibia tissue were mounted on slides and then treated with one of the probe solutions (10 μM). As shown in Figure 7, there was negligible staining of the sections by non-targeted 6C ⊃S, weak staining by the hexavalent probe 6B ⊃S, and relatively intense staining by the dodecavalent probe 2(6B) ⊃S3S. Previously, we have shown that bone section staining with a fluorescent probe containing multiple iminodiacetate groups is lost if the bone section is decalcified by pre-treatment with EDTA.39
Figure 6.

Loss of probe absorption intensity due to probe binding with bone powder that was subsequently removed from solution. Varying amounts of powder were incubated with 4 μM of either S ⊃ 6C, S ⊃6B, or 2(6B) ⊃S3S in water for 1 h at room temperature and absorbance spectra of the supernatant for each sample was acquired after removal of the powder.
Figure 7.
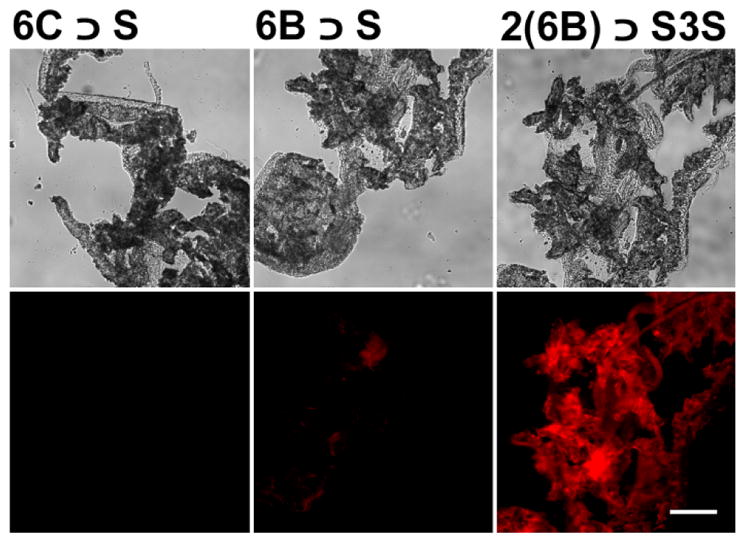
Representative histology slices of tibia extracted from a healthy SKH1 mouse. The bone sections were incubated with untargeted control 6C ⊃S, 6B ⊃S, or 2(6B) ⊃S3S in water (10 μM) for 1 h, and then micrographs were acquired using brightfield [top] or Cy5.5 fluorescence [bottom] filter sets. Scale bar = 0.16 mm for all panels.
In Vivo Studies of Bone Targeting by Pre-Assembled Multivalent Probes
A series of in vivo imaging experiments were designed to evaluate the relative abilities of each pre-assembled probe to target the skeletons of living mice. The first set of experiments used separate cohorts of mice that were given an intravenous injection of one of the probes (10 nmol) in water. Three hours later, the animals were euthanized, the skin and organs removed and the skeletons were subjected to planar, whole-body fluorescence imaging with a Cy5.5 fluorescence filter set. The ex vivo mouse imaging showed that dodecavalent probe 2(6B) ⊃S3S produced much higher bone signal than hexavalent probe 6B ⊃S, with strong localization at skeletal regions that have high bone turnover, such as tibial and femoral heads, lumbar vertebrae, and scapulae (Figure 8). Furthermore, the strong probe staining enabled magnified fluorescence imaging of these skeletal regions (see Figure S26 for magnified images). The non-targeted control probe 6C ⊃S exhibited no bone affinity, and organ biodistribution measurements showed that all three probes were cleared from the body through the kidneys, with minimal retention in other organs (Figure S24). The higher bone staining with 2(6B) ⊃S3S compared to 6B ⊃S suggests that the dodecavalent probe associates more strongly with the bone surface than the hexavalent probe, a trend that is consistent with the bone/probe binding data in Figure 6. In addition, it appears that association of the dodecavalent probe to the bone surface switches on the probe’s fluorescence. The top left panel of Figure 8 shows representative Cy5.5 fluorescence images of the syringes containing the probe solutions just before they were injected into the mice. The aqueous solution of two-station 2(6B) ⊃S3S is much less fluorescent than the solutions containing the one-station probes, 6B ⊃S or 6C ⊃S, which matches the order of fluorescence quantum yields in Table 1. The low fluorescence of 2(6B) ⊃S3S in water is most likely due to partial folding of the two-station probe which causes squaraine self-quenching, and it seems that this quenching effect is alleviated by a factor of 2 to 3 when the two-station probe unfolds on the bone surface in order to maximize multivalent binding (Figure 9). One of the goals of future studies is to determine if this “switch on” fluorescence effect can be achieved with other classes of targeted two-station probes, as it is a useful way to enhance signal to background ratio and improve image contrast.
Figure 8.

[top left] Fluorescence intensity images of syringes containing a single dose of probe (50 μL, 200 μM). [top right and bottom] Representative fluorescence images (dorsal view) of skinless mice that had been dosed with 10 nmol of 6C ⊃S, 6B ⊃S or 2(6B) ⊃S3S and euthanized 3 h later. The Cy5.5 fluorescence pixel intensity scale bar applies to all animal images (N = 3).
Figure 9.
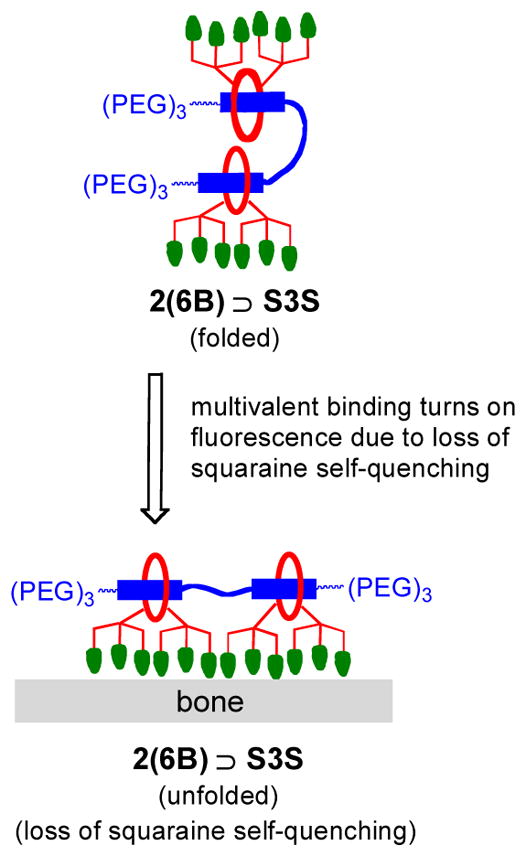
Fluorescence enhancement caused by unfolding of self-quenched two-station probe on the bone surface.
A second set of mouse imaging experiments injected a cohort of mice with a single dose of dodecavalent 2(6B) ⊃S3S probe (10 nmol) and euthanized them only five minutes later. Ex vivo images of the mice showed that a large fraction of the fluorescent probe had either associated with the skeleton or had passed into the kidneys (Figure S25). Thus, association of the targeted 2(6B) ⊃S3S probe to the bone occurs very quickly in the living animal.
A final mouse imaging study determined the longitudinal stability of the dodecavalent 2(6B) ⊃S3S probe at the target bone sites. Two cohorts of mice were given single intravenous doses of dodecavalent 2(6B) ⊃S3S probe (10 nmol). One cohort was sacrificed after 3 h and the other cohort was sacrified after 24 h, and all mice were imaged using the same ex vivo conditions. As shown by the imaging data in Figure 10, there was no change in the NIR fluorescence intensity suggesting that once bound, the 2(6B) ⊃S3S probe does not dissociate from the bone or undergo any measurable decomposition or mechanical unthreading. It is important to realize that unthreading of the pre-assembled 2(6B) ⊃S3S inside the mouse would sequentially produce the singly threaded complex, 6B ⊃S3S, and then the fully unthreaded two-station scaffold S3S. These two structures are less fluorescent than 2(6B) ⊃S3S (Table 1) and in the case of S3S there is no bone targeting capability. Thus, the imaging data in Figure 10 is compelling evidence that the pre-assembled probes maintain very high mechanical, chemical, and metabolic stability over a 24 h time frame on the skeletal surface in living mice.
Figure 10.

[top] Representative whole-body fluorescence images skinless mice that were sacrificed 3 h and 24 h after intravenous injection of 2(6B) ⊃S3S (10 nmol). The fluorescence mean pixel intensity scale applies to both images. [bottom] Graph showing mean Cy5.5 fluorescence pixel intensity for a region of interest on the spine of the mice given a single dose of 2(6B) ⊃S3S. Error bars represent the standard error mean (N = 3).
These imaging results confirm the hypothesis that multivalent molecular targeting of bone in living subjects increases with the number of appended iminodiacetate groups as the bone targeting ligands. An interesting conceptual question is whether the difference in probe affinity is simply due to the different number of bone targeting ligands or whether there is a multivalent cooperative binding effect. In the literature, these types of studies are conducted with purified synthetic or model in vitro binding systems,5,12 and they typically involve probe avidity measurements over a range of different concentrations. But the complexity of in vivo systems makes it much harder to design meaningful experiments that can delineate the different structural factors that control probe targeting performance. This is the case with bone probes 6B ⊃S and 2(6B) ⊃S3S because they have significant structural differences beyond the number of bone targeting ligands. Most importantly, the structure of 6B ⊃S is flanked by two long PEG44 chains, whereas the structure of 2(6B) ⊃S3S has two short PEG3 chains. Such a large difference in PEG chain lengths may produce concentration-dependent differences in probe pharmacokinetics or the kinetics of probe association to the bone target site,11,31 thus making it impossible to unambiguously assign any observed difference in the degree of in vivo bone targeting to a single parameter. This is one of the major reasons why we are pursuing a probe library fabrication and screening approach as an efficient method to discover high performance molecular probes for in vivo applications.
Our results imply that in vivo targeting of multivalent probe to bone can be fine-tuned by altering the number of iminodiacetate groups. Inspection of the bone imaging literature indicates increasing community interest in using fluorescent probes for preclinical studies,44–46 and there are also ongoing efforts to produce multimodality probes for clinical imaging.47,48 Furthermore, researchers are beginning to develop bone-targeting systems for drug delivery and bone tissue engineering.49–53 Most of these studies employ bisphosphonates as the bone-targeting ligands.54,55 While bisphosphonates have very high bone affinity, they also have potent pharmaceutical activities that can potentially disturb the physiological processes they are supposed to target.49 More benign types of bone-targeting ligands are receiving increased attention,56–60 and a probe design based on multiple iminodiacetate groups is a promising new paradigm.
Conclusions
The results of this study show that the Synthavidin method for macrocycle threading by a PEGylated squaraine scaffold (Figure 2) can be utilized for programmed pre-assembly of multivalent near-infrared fluorescent probes for biological imaging. The absorption/emission bands are narrow and correspond to the common Cy5.5 filter set on microscopes, microarrays, and in vivo imaging stations. The two N-propyl units attached to each end of the squaraine docking station are key structural elements that endow the pre-assembly process with a very favorable mix of kinetic and thermodynamic properties. Thus, a simple process that mixes a squaraine scaffold with one or two docking stations, and a macrocycle decorated with multiple copies of a targeting ligand at high micromolar concentration in water, leads to essentially quantitative formation of a threaded complex that is stable enough in biological conditions to act as an effective multivalent molecular probe for fluorescence imaging. Structure and purity of the threaded complexes were readily determined by diagnostic features in the absorption and fluorescence spectra and also by straightforward analysis using gel electrophoresis. The practical utility of the pre-assembly method was demonstrated by preparing two multivalent probes for targeting bone in a living mouse. A pre-assembled probe with twelve bone-targeting iminodiacetate ligands produced more bone accumulation than an analogous pre-assembled probe with six iminodiacetate ligands. Most notably there was no loss in probe fluorescence intensity at the bone target site after 24 hours in the living animal, proving that the pre-assembled probe maintained very high mechanical and chemical stability. The results suggest that Synthavidin technology can be exploited as a versatile pre-assembly method for producing libraries of fluorescent molecular probes that differ in the number of targeting groups, linker lengths, linker flexibility, and degree of PEGylation. The libraries can then be screened to find the library members with the best targeting properties.
The preparation of additional probe libraries with a new targeting ligand should be relatively easy since the same sub-library of PEG-squaraine scaffolds can be used repeatedly. Furthermore, common macrocycle building blocks such as the hexa-alkyne precursor used to prepare 6B, can be conjugated with multiple copies of a new targeting ligand to create a sub-library of ligand appended macrocycles. In some disease areas, such as cancer, many low molecular weight targeting ligands are known,61,62 and there is good evidence that multivalent presentation will enhance affinity and target selectivity.63 Parallel combinatorial mixing of the PEG-squaraine and macrocycle sub-libraries will instantly create a new library of pre-assembled fluorescent multivalent probes for immediate screening. Each pre-assembled molecular probe in the library should exhibit a slightly different multivalent targeting performance in living subjects,11 and thus the library can be screened to find the library member that exhibits the best targeting performance for any specific biological sample or disease state. Not only can this technology be used to produce new and effective molecular probes for imaging and diagnostics, it can be exploited as a discovery platform for new drug delivery systems and targeted therapeutics.
Experimental
Synthesis, Structure, and Biocompatibility
The synthetic methods used to make the two squaraine scaffolds and two macrocycles are described in the Supporting Information, along with the structure characterization data. Also provided are the absorption, fluorescence, 1H NMR, MALDI-TOF MS, and gel electrophoresis data and analyses that proved the structure and high purity of the four separate pre-assembled complexes. In addition, the probe biocompatibility assays and related data are included.
Kinetic Studies of Step-Wise Threading of Macrocycle onto Two-Station Scaffold
Separate stock solutions (1.0 mM) were prepared of the two macrocycles (6C and 6B) in water, and S3S in 1:3 DMSO:H2O. Aliquots were combined to produce a 1:1 mixture of squaraine scaffold (S3S) and one of the two macrocycles, each at final concentration of 250 μM in a solution containing 6 % DMSO in H2O at 25 °C. Macrocycle threading was monitored over time by periodically removing a 2 μL aliquot, diluting it with 1 mL of H2O, and measuring the fluorescence emission intensity at 715 nm (ex: 670 nm). After equilibrium had been reached (indicating complete formation of singly threaded two-station complex), a second molar equivalent of macrocycle (250 μM) was added, and threading of the second macrocycle onto the scaffold was monitored over time by periodically removing a 2 μL aliquot, diluting it with 1 mL of H2O, and measuring the fluorescence emission intensity at 715 nm. Control experiments measured the threading of macrocyle 6C by one-station scaffold S to form 6C ⊃ S and found that the presence of 6 % DMSO lowered kon by a factor of three. Thus, the presence of 6 % DMSO cannot be the sole reason why kon with S3S is 20–50 times lower than with S.
In Vitro Bone Targeting Studies
Targeting Bone Powder
Tibia and femur bones were excised from SKH1 mice and ground up to make bone powder. A solution of 6C ⊃ S, 6B ⊃ S, or 2(6B) ⊃ S3S (4 μM) in water was combined with varying amounts of dried bone powder (0 to 200 mg) in separate centrifuge tubes and the tubes were shaken for 1 h at 37 °C. To separate the probe not bound to the bone powder, the tubes were centrifuged at 2000g for 5 min and the pellet of bone powder was discarded. An absorbance spectrum of the unbound probe remaining in the supernatant was acquired and absorbance maxima were normalized to a dye solution containing no bone powder.
Targeting Bone Histology Sections
Tibia tissue from an SKH1 hairless mouse was excised and flash frozen in OCT media. The bone tissue was sliced (12 μm thickness) at −17 °C, and the slices adhered to Unifrost microscope slides (Azer Scientific, USA), then fixed with chilled acetone for 10 min, and air-dried for an additional 20 min. The tissue slices were submerged in either 6C ⊃ S, 6B ⊃ S, or 2(6B) ⊃ S3S (10 μM) in water for 1 h, then (a) washed with phosphate buffered saline buffer (pH = 7.4) in triplicate for 5 min intervals, (b) incubated with Image-IT FX Signal Enhancer (Invitrogen, Eugene, USA) for 15 min and washed three additional times, and (c) submerged in ProLong Gold Antifade Reagent (Invitrogen, Eugene, USA). Finally, a coverslip was adhered, and the slide was allowed to dry for at least 1 h. Bright field and deep-red fluorescence images of the slices were acquired using a Nikon TE-2000U epifluorescence microscope equipped with the appropriate Cy5.5 filter set (ex: 620/60, em: 700/75). Images were captured using NIS-Elements software (Nikon, USA) and analyzed using ImageJ 1.40g.
In Vivo Bone Targeting Studies
All animal experiments used protocols that were approved by the Notre Dame Institutional Animal Care and Use Committee. Female SKH1 hairless mice (N =3) were anesthetized with 2–3% isoflurane with an oxygen low rate of 2 L min−1 and given retro-orbital injections of 6C ⊃ S, 6B ⊃ S, or 2(6B) ⊃ S3S (10 nmol) in water (50 μL). At the experimental endpoint, the mice were anesthetized and euthanized via cervical dislocation. The skin and organs were immediately removed to expose the skeleton. Fluorescence images were acquired using the IVIS Lumina Imaging system with the following parameters: Cy5.5 fluorescence (ex: 615–665 nm, em: 695–770 nm, acquisition time: 3 seconds, binning: 4 × 4, F-stop: 2, field-of-view: 10 cm × 10 cm). Prior to imaging, syringes containing aqueous solutions of the fluorescent probes were imaged using the following parameters: Cy5.5 fluorescence (ex: 615–665 nm, em: 695–770 nm, acquisition time: 0.5 seconds, binning: 4 × 4, F-stop: 4, field-of-view: 10 cm × 10 cm). All images were analyzed using ImageJ 1.40g. The 16-bit images were imported, opened, and converted to an image stack. Background subtraction was applied to the images using the rolling ball algorithm (radius = 500 pixels or 1000 pixels). The stack was then converted to a montage and pseudocolored as “Fire” (under the “Lookup Tables” menu) which color-codes the fluorescence counts contained in each pixel. Probe biodistribution was measured by Region of Interest (ROI) analysis of the fluorescent images of the dissected tissues. Using ImageJ 1.40g software, a free-hand ROI was drawn around each tissue image and the mean pixel intensity was measured. The biodistribution analyses assume that probe fluorescence from a specific organ suffers the same amount of signal attenuation and thus, the mean pixel intensities reflect relative probe concentration in the organ. The ROI values were plotted using GraphPad Prism 5 (Graphpad Software Inc., San Diego, CA).
Supplementary Material
Acknowledgments
This work was supported by grants from the NSF (CHE1401783 to B.D.S.) and the NIH (R01GM059078 to B.D.S. and T32GM075762 to D.R.R. and F.M.R).
Footnotes
Synthetic procedures, compound characterization and spectral data, experimental methods, and additional imaging data is available free of charge on the ACS Publications website at DOI: 10.1021/?
References
- 1.Srinivasarao M, Galliford CV, Low PS. Principles in the design of ligand-targeted cancer therapeutics and imaging agents. Nat Rev Drug Discov. 2015;14:203–19. doi: 10.1038/nrd4519. [DOI] [PubMed] [Google Scholar]
- 2.Sim N, Parker D. Critical design issues in the targeted molecular imaging of cell surface receptors. Chem Soc Rev. 2015;44:2122–34. doi: 10.1039/c4cs00364k. [DOI] [PubMed] [Google Scholar]
- 3.Warram JM, de Boer E, Sorace AG, Chung TK, Kim H, Pleijhuis RG, van Dam GM, Rosenthal EL. Antibody-based imaging strategies for cancer. Cancer Metastasis Rev. 2014;33:809–22. doi: 10.1007/s10555-014-9505-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Xu S, Olenyuk BZ, Okamoto CT, Hamm-Alvarez SF. Targeting receptor-mediated endocytotic pathways with nanoparticles: rationale and advances. Adv Drug Deliv Rev. 2013;65:121–38. doi: 10.1016/j.addr.2012.09.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kiessling LL, Gestwicki JE, Strong LE. Synthetic multivalent ligands in the exploration of cell-surface interactions. Curr Opin Chem Biol. 2000;4:696–703. doi: 10.1016/s1367-5931(00)00153-8. [DOI] [PubMed] [Google Scholar]
- 6.Hlavacek WS, Percus JK, Percus OE, Perelson AS, Wofsy C. Retention of antigen on follicular dendritic cells and B lymphocytes through complement-mediated multivalent ligand-receptor interactions: theory and application to HIV treatment. Math Biosci. 2002;176:185–202. doi: 10.1016/s0025-5564(02)00091-3. [DOI] [PubMed] [Google Scholar]
- 7.Kitov PI, Bundle DR. On the nature of the multivalency effect: a thermodynamic model. J Am Chem Soc. 2003;125:16271–84. doi: 10.1021/ja038223n. [DOI] [PubMed] [Google Scholar]
- 8.Vauquelin G, Charlton SJ. Exploring avidity: understanding the potential gains in functional affinity and target residence time of bivalent and heterobivalent ligands. Br J Pharmacol. 2013;168:1771–85. doi: 10.1111/bph.12106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Fischer G, Lindner S, Litau S, Schirrmacher R, Wangler B, Wangler C. Next step toward optimization of GRP receptor avidities: determination of the minimal distance between BBN(7-14) units in peptide homodimers. Bioconjug Chem. 2015;26:1479–83. doi: 10.1021/acs.bioconjchem.5b00362. [DOI] [PubMed] [Google Scholar]
- 10.Guo Z, He B, Jin H, Zhang H, Dai W, Zhang L, Zhang H, Wang X, Wang J, Zhang X, et al. Targeting efficiency of RGD-modified nanocarriers with different ligand intervals in response to integrin alphavbeta3 clustering. Biomaterials. 2014;35:6106–17. doi: 10.1016/j.biomaterials.2014.04.031. [DOI] [PubMed] [Google Scholar]
- 11.Hennig R, Pollinger K, Veser A, Breunig M, Goepferich A. Nanoparticle multivalency counterbalances the ligand affinity loss upon PEGylation. J Control Release. 2014;194:20–7. doi: 10.1016/j.jconrel.2014.07.062. [DOI] [PubMed] [Google Scholar]
- 12.Lundquist JJ, Toone EJ. The cluster glycoside effect. Chem Rev. 2002;102:555–78. doi: 10.1021/cr000418f. [DOI] [PubMed] [Google Scholar]
- 13.Care BR, Soula HA. Impact of receptor clustering on ligand binding. BMC Syst Biol. 2011;5:48. doi: 10.1186/1752-0509-5-48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Martinez A, Ortiz Mellet C, Garcia Fernandez JM. Cyclodextrin-based multivalent glycodisplays: covalent and supramolecular conjugates to assess carbohydrate-protein interactions. Chem Soc Rev. 2013;42:4746–73. doi: 10.1039/c2cs35424a. [DOI] [PubMed] [Google Scholar]
- 15.Appelhans D, Klajnert-Maculewicz B, Janaszewska A, Lazniewska J, Voit B. Dendritic glycopolymers based on dendritic polyamine scaffolds: view on their synthetic approaches, characteristics and potential for biomedical applications. Chem Soc Rev. 2015;44:3968–96. doi: 10.1039/c4cs00339j. [DOI] [PubMed] [Google Scholar]
- 16.Tassa C, Shaw SY, Weissleder R. Dextran-coated iron oxide nanoparticles: a versatile platform for targeted molecular imaging, molecular diagnostics, and therapy. Acc Chem Res. 2011;44:842–52. doi: 10.1021/ar200084x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Montet X, Funovics M, Montet-Abou K, Weissleder R, Josephson L. Multivalent effects of RGD peptides obtained by nanoparticle display. J Med Chem. 2006;49:6087–93. doi: 10.1021/jm060515m. [DOI] [PubMed] [Google Scholar]
- 18.Cheng Z, Al Zaki A, Hui JZ, Muzykantov VR, Tsourkas A. Multifunctional nanoparticles: cost versus benefit of adding targeting and imaging capabilities. Science. 2012;338:903–10. doi: 10.1126/science.1226338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lammers T, Kiessling F, Hennink WE, Storm G. Drug targeting to tumors: principles, pitfalls and (pre-) clinical progress. J Control Release. 2012;161:175–87. doi: 10.1016/j.jconrel.2011.09.063. [DOI] [PubMed] [Google Scholar]
- 20.Bae YH, Park K. Targeted drug delivery to tumors: myths, reality and possibility. J Control Release. 2011;153:198–205. doi: 10.1016/j.jconrel.2011.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Fox ME, Szoka FC, Frechet JM. Soluble polymer carriers for the treatment of cancer: the importance of molecular architecture. Acc Chem Res. 2009;42:1141–51. doi: 10.1021/ar900035f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Dix AV, Moss SM, Phan K, Hoppe T, Paoletta S, Kozma E, Gao ZG, Durell SR, Jacobson KA, Appella DH. Programmable nanoscaffolds that control ligand display to a G-protein-coupled receptor in membranes to allow dissection of multivalent effects. J Am Chem Soc. 2014;136:12296–303. doi: 10.1021/ja504288s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Dix AV, Conroy JL, George Rosenker KM, Sibley DR, Appella DH. PNA-based multivalent scaffolds activate the dopamine D2 receptor. ACS Med Chem Lett. 2015;6:425–9. doi: 10.1021/ml500478m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Selden NS, Todhunter ME, Jee NY, Liu JS, Broaders KE, Gartner ZJ. Chemically programmed cell adhesion with membrane-anchored oligonucleotides. J Am Chem Soc. 2012;134:765–8. doi: 10.1021/ja2080949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Grunstein D, Maglinao M, Kikkeri R, Collot M, Barylyuk K, Lepenies B, Kamena F, Zenobi R, Seeberger PH. Hexameric supramolecular scaffold orients carbohydrates to sense bacteria. J Am Chem Soc. 2011;133:13957–66. doi: 10.1021/ja2036767. [DOI] [PubMed] [Google Scholar]
- 26.Ornelas-Megiatto C, Becher TB, Megiatto JD. Interlocked systems in nanomedicine. Curr Top Med Chem. 2015;15:1236–56. doi: 10.2174/1568026615666150330111614. [DOI] [PubMed] [Google Scholar]
- 27.Kobayashi H, Longmire MR, Ogawa M, Choyke PL. Rational chemical design of the next generation of molecular imaging probes based on physics and biology: mixing modalities, colors and signals. Chem Soc Rev. 2011;40:4626–48. doi: 10.1039/c1cs15077d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Nguyen QT, Tsien RY. Fluorescence-guided surgery with live molecular navigation--a new cutting edge. Nat Rev Cancer. 2013;13:653–62. doi: 10.1038/nrc3566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chi C, Du Y, Ye J, Kou D, Qiu J, Wang J, Tian J, Chen X. Intraoperative imaging-guided cancer surgery: from current fluorescence molecular imaging methods to future multi-modality imaging technology. Theranostics. 2014;4:1072–84. doi: 10.7150/thno.9899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhang X, Wang H, Ma Z, Wu B. Effects of pharmaceutical PEGylation on drug metabolism and its clinical concerns. Expert Opin Drug Metab Toxicol. 2014;10:1691–702. doi: 10.1517/17425255.2014.967679. [DOI] [PubMed] [Google Scholar]
- 31.Guo Y, Yuan H, Rice WL, Kumar AT, Goergen CJ, Jokivarsi K, Josephson L. The PEG-fluorochrome shielding approach for targeted probe design. J Am Chem Soc. 2012;134:19338–41. doi: 10.1021/ja309085b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Smith BD. Smart molecules for imaging, sensing and health (SMITH) Beilstein J Org Chem. 2015;11:2540–8. doi: 10.3762/bjoc.11.274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Peck EM, Liu W, Spence GT, Shaw SK, Davis AP, Destecroix H, Smith BD. Rapid macrocycle threading by a fluorescent dye-polymer conjugate in water with nanomolar affinity. J Am Chem Soc. 2015;137:8668–71. doi: 10.1021/jacs.5b03573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Liu W, Peck EM, Hendzel KD, Smith BD. Sensitive structural control of macrocycle threading by a fluorescent squaraine dye flanked by polymer chains. Org Lett. 2015;17:5268–71. doi: 10.1021/acs.orglett.5b02633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Liu W, Peck EM, Smith BD. High affinity macrocycle threading by a near-infrared croconaine dye with flanking polymer chains. J Phys Chem B. 2016;120:995–1001. doi: 10.1021/acs.jpcb.5b11961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.White AG, Fu N, Leevy WM, Lee JJ, Blasco MA, Smith BD. Optical imaging of bacterial infection in living mice using deep-red fluorescent squaraine rotaxane probes. Bioconjug Chem. 2010;21:1297–304. doi: 10.1021/bc1000998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Rice DR, White AG, Leevy WM, Smith BD. Fluorescence imaging of interscapular brown adipose tissue in living mice. J Mater Chem B Mater Biol Med. 2015;3:1979–1989. doi: 10.1039/C4TB01914H. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Harris JM, Chess RB. Effect of pegylation on pharmaceuticals. Nat Rev Drug Discov. 2003;2:214–21. doi: 10.1038/nrd1033. [DOI] [PubMed] [Google Scholar]
- 39.Harmatys KM, Cole EL, Smith BD. In vivo imaging of bone using a deep-red fluorescent molecular probe bearing multiple iminodiacetate groups. Mol Pharm. 2013;10:4263–71. doi: 10.1021/mp400357v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Parkesh R, Gowin W, Lee TC, Gunnlaugsson T. Synthesis and evaluation of potential CT (computer tomography) contrast agents for bone structure and microdamage analysis. Org Biomol Chem. 2006;4:3611–7. doi: 10.1039/b606976b. [DOI] [PubMed] [Google Scholar]
- 41.Jackson SN, Woods AS. Imaging of noncovalent complexes by MALDI-MS. J Am Soc Mass Spectrom. 2013;24:1950–6. doi: 10.1007/s13361-013-0745-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Gassensmith JJ, Arunkumar E, Barr L, Baumes JM, DiVittorio KM, Johnson JR, Noll BC, Smith BD. Self-assembly of fluorescent inclusion complexes in competitive media including the interior of living cells. J Am Chem Soc. 2007;129:15054–9. doi: 10.1021/ja075567v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Cole EL, Arunkumar E, Xiao S, Smith BA, Smith BD. Water-soluble, deep-red fluorescent squaraine rotaxanes. Org Biomol Chem. 2012;10:5769–73. doi: 10.1039/c2ob06783h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hyun H, Wada H, Bao K, Gravier J, Yadav Y, Laramie M, Henary M, Frangioni JV, Choi HS. Phosphonated near-infrared fluorophores for biomedical imaging of bone. Angew Chem Int. 2014;53:10668–72. doi: 10.1002/anie.201404930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Bao K, Nasr KA, Hyun H, Lee JH, Gravier J, Gibbs SL, Choi HS. Charge and hydrophobicity effects of NIR fluorophores on bone-specific imaging. Theranostics. 2015;5:609–17. doi: 10.7150/thno.11222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sun S, Blazewska KM, Kadina AP, Kashemirov BA, Duan X, Triffitt JT, Dunford JE, Russell RG, Ebetino FH, Roelofs AJ, et al. Fluorescent bisphosphonate and carboxyphosphonate probes: a versatile imaging toolkit for applications in bone biology and biomedicine. Bioconjug Chem. 2016;27:329–40. doi: 10.1021/acs.bioconjchem.5b00369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Bhushan KR, Tanaka E, Frangioni JV. Synthesis of conjugatable bisphosphonates for molecular imaging of large animals. Angew Chem Int Ed. 2007;46:7969–71. doi: 10.1002/anie.200701216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ventura M, Boerman OC, de Korte C, Rijpkema M, Heerschap A, Oosterwijk E, Jansen JA, Walboomers XF. Preclinical imaging in bone tissue engineering. Tissue Eng Part B Rev. 2014;20:578–95. doi: 10.1089/ten.TEB.2013.0635. [DOI] [PubMed] [Google Scholar]
- 49.Anada T, Takeda Y, Honda Y, Sakurai K, Suzuki O. Synthesis of calcium phosphate-binding liposome for drug delivery. Bioorg Med Chem Lett. 2009;19:4148–50. doi: 10.1016/j.bmcl.2009.05.117. [DOI] [PubMed] [Google Scholar]
- 50.Singh T, Kaur V, Kumar M, Kaur P, Murthy RS, Rawal RK. The critical role of bisphosphonates to target bone cancer metastasis: an overview. J Drug Target. 2015;23:1–15. doi: 10.3109/1061186X.2014.950668. [DOI] [PubMed] [Google Scholar]
- 51.Liu XM, Zhang Y, Chen F, Khutsishvili I, Fehringer EV, Marky LA, Bayles KW, Wang D. Prevention of orthopedic device-associated osteomyelitis using oxacillin-containing biomineral-binding liposomes. Pharm Res. 2012;29:3169–79. doi: 10.1007/s11095-012-0812-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Santos-Ferreira I, Bettencourt A, Almeida AJ. Nanoparticulate platforms for targeting bone infections: meeting a major therapeutic challenge. Nanomedicine. 2015;10:3131–45. doi: 10.2217/nnm.15.134. [DOI] [PubMed] [Google Scholar]
- 53.Schott S, Vallet S, Tower RJ, Noor S, Tiwari S, Schem C, Busch C. In vitro and in vivo toxicity of 5-FdU-alendronate, a novel cytotoxic bone-seeking duplex drug against bone metastasis. Invest New Drugs. 2015;33:816–26. doi: 10.1007/s10637-015-0253-3. [DOI] [PubMed] [Google Scholar]
- 54.Kootala S, Tokunaga M, Hilborn J, Iwasaki Y. Anti-resorptive functions of poly(ethylene sodium phosphate) on human osteoclasts. Macromol Biosci. 2015;15:1634–40. doi: 10.1002/mabi.201500166. [DOI] [PubMed] [Google Scholar]
- 55.Ossipov DA. Bisphosphonate-modified biomaterials for drug delivery and bone tissue engineering. Expert Opin Drug Deliv. 2015;12:1443–58. doi: 10.1517/17425247.2015.1021679. [DOI] [PubMed] [Google Scholar]
- 56.Groger D, Kerschnitzki M, Weinhart M, Reimann S, Schneider T, Kohl B, Wagermaier W, Schulze-Tanzil G, Fratzl P, Haag R. Selectivity in bone targeting with multivalent dendritic polyanion dye conjugates. Adv Healthc Mater. 2014;3:375–85. doi: 10.1002/adhm.201300205. [DOI] [PubMed] [Google Scholar]
- 57.Mizrahi DM, Ziv-Polat O, Perlstein B, Gluz E, Margel S. Synthesis, fluorescence and biodistribution of a bone-targeted near-infrared conjugate. Eur J Med Chem. 2011;46:5175–83. doi: 10.1016/j.ejmech.2011.08.040. [DOI] [PubMed] [Google Scholar]
- 58.Kubicek V, Lukes I. Bone-seeking probes for optical and magnetic resonance imaging. Future Med Chem. 2010;2:521–31. doi: 10.4155/fmc.09.162. [DOI] [PubMed] [Google Scholar]
- 59.Brounts SH, Lee JS, Weinberg S, Lan Levengood SK, Smith EL, Murphy WL. High affinity binding of an engineered, modular peptide to bone tissue. Mol Pharm. 2013;10:2086–90. doi: 10.1021/mp300662r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Bain JL, Bonvallet PP, Abou-Arraj RV, Schupbach P, Reddy MS, Bellis SL. Enhancement of the regenerative potential of anorganic bovine bone graft utilizing a polyglutamate-modified BMP2 peptide with improved binding to calcium-containing materials. Tissue Eng Part A. 2015;21:2426–36. doi: 10.1089/ten.tea.2015.0160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Gray BP, Li S, Brown KC. From phage display to nanoparticle delivery: functionalizing liposomes with multivalent peptides improves targeting to a cancer biomarker. Bioconjug Chem. 2013;24:85–96. doi: 10.1021/bc300498d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Gray BP, Brown KC. Combinatorial peptide libraries: mining for cell-binding peptides. Chem Rev. 2014;114:1020–81. doi: 10.1021/cr400166n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Ruzza P, Marchiani A, Antolini N, Calderan A. Peptide-receptor ligands and multivalent approach. Anticancer Agents Med Chem. 2012;12:416–27. doi: 10.2174/187152012800617849. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.