

Abstract
Satellite cells are adult myogenic stem cells that function to repair damaged muscle. The enduring capacity for muscle regeneration requires efficient satellite cell expansion after injury, differentiation to produce myoblasts that can reconstitute damaged fibers, and self-renewal to replenish the muscle stem cell pool for subsequent rounds of injury and repair. Emerging studies indicate that misregulations of satellite cell fate and function contribute to age-associated muscle dysfunction and influence the severity of muscle diseases, including Duchenne Muscular Dystrophy (DMD). It has also become apparent that satellite cell fate during muscle regeneration, aging, and in the context of DMD is governed by an intricate network of intrinsic and extrinsic regulators. Targeted manipulation of this network may offer unique opportunities for muscle regenerative medicine.
Introduction
Skeletal muscle is a contractile, post-mitotic tissue composed of multinucleated muscle cells, known as myofibers, which are formed by fusion of differentiated mononuclear muscle cells. Skeletal muscle accounts for 30–50% of body mass in humans, and is one of the few organs that retains a highly adaptive and robust capacity to regenerate throughout most of life. Muscle regeneration depends upon resident muscle stem cells known as satellite cells. These mesoderm-derived cells comprise a heterogeneous population of adult stem cells (Box 1), capable of both self-renewal and myogenic differentiation, which reside in a specialized niche between the muscle sarcolemma and the basal lamina of individual myofibers1(Fig. 1). The satellite cell niche is comprised of both acellular and cellular components, including extracellular matrix proteins and growth factors, myofibers, and muscle-resident non-myogenic cells such as fibro-adipogenic progenitors (FAPs), macrophages, and regulatory T-cells (Tregs) 2–9. Collectively, components of the satellite cell niche create a complex microenvironment that plays a crucial role in maintaining satellite cell identity and ensuring robust regenerative responses to muscle insult2, 4–9.
Box 1. Origin and heterogeneity of satellite cells.
Most satellite cells in postnatal muscle originate from a population of embryonic precursors that expresses PAX7 and/or the related Paired box protein, PAX3. These embryonic precursors of adult muscle are of mesodermal origin and arise from a dorsal structure of the developing somite (known as the dermomyotome) 136, 137. In the mouse, by embryonic day 16.5 to 18.5, a subset of myogenic progenitors in the dermomyotome migrates to its prospective niche (analogous to the niche of satellite cells in postnatal skeletal muscle), which is positioned between a primitive basal lamina structure and the myotome136. Shortly after birth, postnatal satellite cells expand extensively to accommodate organismal growth, and begin acquiring characteristic molecular features, including expression of specific surface markers, and the emergence of distinct high- and low- cycling sub-populations90, an aspect of satellite cell heterogeneity in adult muscle that is discussed in more depth below.
We define muscle satellite cells as bona fide muscle stem cells, capable of self-renewal and differentiation to produce myoblasts, which can then fuse (with each other as well as with existing fibers) to generate myofibers. Yet, several lines of evidence indicate that satellite cells in postnatal muscle exhibit notable molecular and phenotypic heterogeneity that can influence the fate and function of individual cells within the satellite cell pool. Mouse molecular genetic tools have been particularly useful in delineating subsets of muscle satellite cells, suggesting the coexistence in this compartment of a population of committed progenitors ready for myogenic differentiation and a distinct, self-renewing population that is capable of reconstituting the satellite cell niche45, 82, 83, 90. In one of the studies Cre recombinase-mediated lineage tracing was used to distinguish a minority of adult muscle satellite cells (~10% of the total pool) that were not marked by Myf5-Cre, suggesting that they had never expressed Myf5. This study further showed that this subset of cells repopulates the satellite cell compartment more readily (as compared to Myf5-expressing satellite cells) when transplanted into PAX7-null animals, indicating that these cells may retain more robust self-renewal capabilities83. On the other hand, satellite cells that did experience Myf5 expression were more prone to myogenic differentiation in these in vivo engraftment assays83. In another study, satellite cells that expressed higher levels of PAX7 RNA (Pax7hi cells) as determined by flow cytometry using a Pax7-GFP reporter mouse82, displayed slower cycling, lower metabolic activity, and the exclusive capacity to replenish the entire complement of Pax7hi and Pax7low satellite cells upon transplantation. Studies to determine whether satellite cells that have never expressed Myf5 are enriched in the Pax7hi subset, or vice versa, have yet to be reported. Satellite cells have also been functionally segregated based solely on their proliferative history, with several studies indicating that low-cycling satellite cells exhibit a higher engraftment potential than high-cycling satellite cells when both populations are transplanted into injured animals45, 90, 138. These data clearly document phenotypic and functional heterogeneity within the satellite cell pool; yet, how cells toggle between these potentially interchangeable states and how each subset is maintained and regulated during homeostasis and throughout regeneration remains an area of very active exploration.
Figure 1. Classical view of muscle myogenesis.

(A) During muscle homeostasis, satellite cells are maintained in a quiescent state (red) beneath the basal lamina and above the muscle sarcolemma of the muscle fiber. Quiescent satellite cells express Pax7 and lack MyoD. Upon muscle insult (ruptured fiber), satellite cells become activated (blue) and proliferate. Proliferating satellite cells maintain expression of Pax7 and induced expression of MyoD. A subset of these dividing satellite cells commit to differentiation (green), through the expression of Myogenin (MyoG) and downregulation of Pax7, ultimately producing myoblasts that exit the cell cycle and fuse to repair muscle. Alternatively, another subset of activated satellite cells will self-renew (purple) by inhibiting myogenic determination and re-instating quiescence. This ensures that the muscle retains a sufficient number of functional muscle stem cells to support future rounds of muscle repair. (B) Cell-intrinsic factors discussed in this review having roles in satellite cell quiescence, activation, and self-renewal.
Previous studies have established the central importance of a regulated cascade of transcription factors that mediate satellite cell maintenance, activation, and differentiation3. Satellite cells are distinguished from other mononuclear cells in muscle by expression of the canonical satellite cell regulator gene, Paired box protein 7 (PAX7)10, 11. Satellite cells exist in a quiescent (or non-dividing) state in uninjured muscle, a state often referred to as muscle homeostasis (Fig. 1). But, these cells become activated upon muscle trauma, which promotes their subsequent proliferation and, for a subset of activated satellite cells, differentiation into fusion-competent myoblasts. Satellite cell activation is mediated in part by the induced expression of Myogenic determination protein (MYOD) and Myogenic Factor 5 (MYF5) 12–17. The differentiation of committed myoblasts involves downregulation of PAX7 and de novo expression of myogenin (also known as MYOG), which is followed by fusion of the differentiated myoblasts with each other and with any remaining myofibers to repair damaged muscle18. Activated satellite cells that do not commit to differentiation can downregulate MYOD and MYF5 and undergo self-renewing proliferation that replenishes the pool of muscle satellite cells, thereby ensuring that the capacity to respond to future injuries is maintained in the muscle3, 19–21 (Fig. 1).
Extensive evidence supports the notion that adult satellite cells are required for healthy muscle repair, and severe muscular and regenerative deficits result in their absence22, 23. Although there has been debate regarding the requirements for PAX7 in satellite cell maintenance and muscle repair24, recent efforts have affirmed an absolute requirement for PAX7 in satellite cell proliferation and self-renewal, and hence, for efficient repair of muscle after injury25–27. Functional loss of the satellite cell pool has been observed in association with age-related muscle dysfunction and other muscle degenerative pathologies28, 29, highlighting the critical need to maintain this unique cell population in order to sustain effective regenerative function over a lifetime.
The development of efficient methods to prospectively isolate satellite cells from skeletal muscle has fuelled progress in further delineating their regenerative functions in vivo. Satellite cells can be purified from muscle using protocols that combine chemical and mechanical dissociation of muscle and separation of mononuclear cells from fibers with Fluorescence Activating Cell Sorting (FACS) using positive and negative selection markers to enrich for myogenic populations19, 30–32. Various surface marker combinations have been reported to positively enrich for highly functional satellite cells including, CD34/α7-integrin19, VCAM130, 33–35, CXCR4/β1-integrin36, 37, NCAM (CD56)38, 39, EGFR40, and Syndecan3/441. Whether these different surface marker sorting schemes yield identical myogenic populations has yet to be fully addressed; however, cells enriched by any of these methods can efficiently engraft skeletal muscle when transplanted into either injured or diseased muscle, and can improve muscle strength and function19, 37, 42. Satellite cells transplanted into pre-injured, diseased muscle also repopulate the satellite cell niche, and can contribute to future rounds of muscle repair 19, 37, 42. These observations have raised enthusiasm for the possibility of utilizing satellite cells as a cellular source for regenerative medicine (Box 2). New insights into how these cells regulate self-renewal and differentiation decisions in vivo, and how ex vivo culture systems impact satellite cell fate will certainly be required to achieve this goal.
Box 2. Translational applications for modulating satellite cell fate decisions.
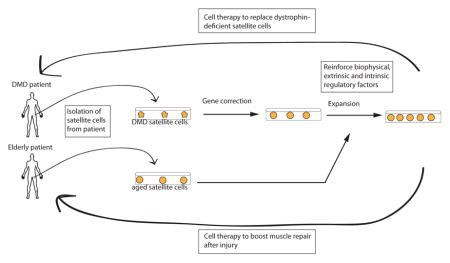
There has been tremendous excitement over the years in using satellite cells as a transplantable cell population that could restore the functional stem cell pool in aged and diseased muscle, for instance in patients with Duchenne Muscular Dystrophy (DMD). One could envision a scenario in which satellite cells isolated from the patient are genetically corrected ex vivo139, expanded, and transplanted back to promote therapeutic muscle repair (Figure). However, a major obstacle to this approach is that these cells are very rare, encompassing only ~1% of the mononuclear cells associated with myofibers37. In addition, removal of satellite cells from their endogenous niche rapidly alters their cellular state and, ultimately, their functional capacity42, 140–142.
New culture systems that more precisely mimic the in vivo environment will likely prove essential for generating sufficient numbers of engraftable satellite cells. For example, it was shown that culturing satellite cells on soft hydrogels to mimic in vivo forces and re-create niche signals, promotes enhanced cell survival and self-renewal. Although this approach is a definite improvement over standard culturing conditions, 3-fold more hydrogel-cultured satellite cells are needed to obtain as much as 50% of the engraftment achieved by freshly isolated cells 97, 140. Reestablishing in culture the full functional potential of satellite cells will likely require a combination of extrinsic and intrinsic signals57, 58, 143.
Cell delivery represents a second major impediment to the realization of cell therapeutic approaches in skeletal muscle. Two delivery strategies have been utilized so far: systemic and intramuscular delivery. An ideal stem cell therapy to combat disseminated muscle diseases such as DMD or sarcopenia would be compatible with systemic delivery. However, prior work shows that satellite cells delivered systemically into mice get trapped in other organs (lungs, kidney, spleen). 144. Therefore, methods to achieve more efficient muscle “homing” of satellite cells, similar to the homing properties of circulating immune cells7, as well as to improve their mobility within the tissue145, will be critical in overcoming this technical hurdle.
In this review, we discuss recent progress in our understanding of the intrinsic molecular mechanisms regulating satellite cell fate during muscle regeneration, aging, and in the context of Duchenne Muscular Dystrophy (DMD), the most common and severe muscle pathology diagnosed in childhood. As the details of myogenic differentiation and function of satellite cells during embryonic development have been recently addressed elsewhere3, we focus here on adult satellite cells and the molecular networks governing their quiescence, activation, and self-renewal as key stages of the satellite cell life cycle. We also highlight the critical influence of extrinsic factors that can modulate satellite cell intrinsic signalling to direct satellite cell fate decisions during muscle regeneration and aging. We anticipate that further elucidation of the gene regulatory circuits that determine satellite cell fate will provide a molecular blueprint that can be used to establish novel therapies to treat the increasing numbers of individuals suffering from age-associated muscle dysfunction and muscular diseases that currently have no cure.
Satellite cell fate decisions
Careful regulation of satellite cell identity and myogenic commitment are essential for maintaining the muscle satellite cell pool and appropriately activating repair mechanisms in response to muscle damage. We discuss here recent advances that have defined critical new effectors of satellite cell regulation that govern the maintenance of satellite cell quiescence, activation of proliferation, and capacity for self-renewal.
Satellite cell quiescence
Satellite cell division is restrained in uninjured muscle through mechanisms that appear to enforce satellite cell quiescence during homeostasis. In this section we discuss recent advances in the transcriptional, epigenetic, and signalling pathways controlling the quiescent state.
Quiescent satellite cells (QSCs) express PAX7 but lack MYOD and other markers associated with cellular proliferation20, 43 (Fig. 1). Satellite cells isolated from fresh muscle by FACS also express PAX7 and have low to undetectable levels of MYOD19, 37, 42. Given these molecular similarities, and keeping with convention in the field19, 33, 37, 42, this review refers to freshly isolated satellite cells as QSCs, although we note the likelihood that the process of satellite cell isolation from muscle may trigger some initiating events in satellite cell activation. Future studies to provide a better molecular understanding of the very early molecular changes that signal satellite cell activation will help to clarify this issue.
Recent genome-wide RNA expression maps have revealed that QSCs possess a unique transcriptional profile that distinguishes them from their more activated progeny. QSCs express roughly 500 uniquely enriched genes involved in transcriptional control, negative regulation of the cell cycle (including several members of the Sprouty family44), and cell-cell adhesion, extracellular matrix, and lipid transporter activity33, 34. Although the functional importance of many of these genes is yet to be understood, it seems likely that this transcriptional program acts to inhibit QSC proliferation44, 45, to anchor satellite cells in their anatomical niche46, and to provide mechanisms for efficient transport and processing of lipids that are required for metabolic reactions characteristic of quiescent cells47. At the epigenetic level, nearly half of all protein coding genes are marked by active chromatin, specifically acquiring the histone 3 modification, H3K4me3 33, 48. Interestingly, genes that are expressed at low levels in quiescent satellite cells, but induced upon satellite cell activation, exhibit this active histone mark, a likely indicator that these gene regions are in a “poised” state, anticipating release from quiescence33. These results emphasize the notion that satellite cell quiescence should not be mistaken as a form of cellular dormancy or inactivity. Rather, it represents an actively maintained state typified by a quiescence-specific gene expression program that is poised to execute the transcriptional and epigenetic transformations that enable a rapid switch to activation in the presence of muscle trauma.
Given the central role of developmental signalling pathways in modulating stem cell behaviours, it is perhaps not surprising that Notch signalling plays a key role in promoting satellite cell quiescence49–51. The Notch gene family consists of 4 transmembrane proteins (NOTCH1, 2, 3, and 4) that bind 5 distinct ligands52. Upon receptor-ligand binding, Notch undergoes a series of proteolytic cleavages that leads to nuclear translocation of the Notch Intracellular Domain (NICD). In the nucleus, NICD interacts with downstream effector proteins, such as Recombining Binding Protein Jκ (RBP-Jκ), to regulate transcription of Notch target genes52. Supporting a role for Notch signalling in QSCs, Notch 3 mRNA and protein levels decline rapidly upon satellite cell activation49, and genetic deletion of RBP-Jκ causes loss of PAX7-expressing satellite cells in muscle sections, even in the absence of muscle injury. This loss of RBP-Jκ deficient satellite cells has been attributed to entry of these cells into the cell cycle, and subsequent differentiation 50. Conversely, ectopic expression of NICD in cultured primary muscle myoblasts slowed cycling kinetics, upregulated PAX7, and suppressed MYOD, indicating that active Notch signalling is sufficient to program at least some of the molecular and phenotypic aspects of quiescence51. The molecular mechanisms that link activated Notch with preserved quiescence are far from complete, but insights have been gained from studies indicating that Notch target genes, like the Hes and Hey family of transcription factors, can reduce MyoD mRNA levels, and thereby suppress myogenic commitment. In addition, RBP-Jκ can bind directly to the Pax7 promoter leading to its transcriptional activation51. This enables PAX7 to further reduce MYOD protein stability via transcription-independent mechanisms53, 54 (Fig 2a).
Figure 2. Emerging regulators of satellite cell quiescence, activation, and self-renewal.

(A)During homeostasis most satellite cells are quiescent. (Aa) The Notch signalling pathway plays a key role in enforcing satellite cell quiescence. Upon ligand/receptor binding, the Notch Intracellular Domain (NICD, pink oval) undergoes several proteolytic cleavages (not shown), and then translocates to the nucleus where it interacts with the Recombining Binding Protein J-κ effector protein (RBPJ-κ, blue circle). The NICD:RBPJ-κ complex activates expression of target genes, including the Hes and Hey family of transcription factors, which function to inhibit MYOD. RBPJ-κ also binds the promoter of Pax7 and induces its expression. PAX7 induction further downregulates MYOD via transcription-independent mechanisms (not shown). The Forkhead box 3 (FOXO3, green circle) transcription factor can induce expression of NOTCH1 and NOTCH3 trans-membrane proteins, which facilitates Notch signalling by ensuring an adequate number of receptors are produced and shuttled to the cell membrane where they can interact with available ligands in the local environment. (Ab) Satellite cell entry into the cell cycle is inhibited through the activities of p27Kip1, which inhibits cyclin-dependant kinases (CDKs). (B) After muscle injury, activated satellite cells detect pro-myogenic stimuli from the local environment and begin proliferation. (Ba) Exposure to FGF2 induces p38α/β mitogen-activated protein kinase (MAPK) activity, which inhibits the RNA-binding destabilizing protein, Tristetraprolin (TTP), leading to stabilization of the MyoD mRNA. This allows MYOD protein to accumulate (purple circle) and induce expression of Cdc6, a DNA replication gene that promotes cell cycle entry and thus proliferation. (Bb) Another activation regulator, MYF5, is post-transcriptionally repressed by miRNA-31 (miR-31), which sequesters Myf5 mRNA into RNA granules in quiescent satellite cells (QSCs) and prevents activation of MYF5 targets. Upon muscle trauma, the RNA granules dissociate, miRNA-31 levels decrease, and MYF5 protein is produced and can activate target genes. (Bc) The Myf5 gene is also transcriptionally regulated through the activities of CARM1 (pink circle), an arginine methyl-transferase that methylates PAX7 (small purple circles on the light blue circle) leading to PAX7 association with the Histone Acetyl-Transferase complex (HAT, purple complex), and the induced expression of target genes (such as MYF5) in activated satellite cells (ASCs). (C) Satellite cells are capable of self renewal. (Ca) A balance between self-renewal and differentiation of satellite cells is acheived by the mode of cell division. Apico-basal divisions generate one self-renewing (red, Myf5-) and one differentiating cell (blue, Myf5+). On the other hand, planar divisions (parallel to the muscle fiber) are symmetric and generate two daughters that retain self-renewal capabilities. (Cb) Self-renewal of satellite cells can be followed by their return to quiescence. FOXO3 induces expression of Notch signalling genes, enabling the cascade of molecular events leading to the inhibition of MYOD-mediated differentiation and blockade of cell cycle progression. (Cc) The return to quiescence also requires Sprouty1 (Spry1), a tyrosine kinase inhibitor that inhibits FGF signalling, thereby restricting cellular proliferation.
Mechanisms to regulate Notch signalling include modulating expression of the ligand or the receptor. Recent data indicate that satellite cells lacking the Forkhead Box protein 3 (FOXO3) transcription factor exhibit reduced levels of the Notch1 and Notch3 receptor RNAs. FOXO3-deficient satellite cells also proliferate faster and differentiate more readily in culture, consistent with a failure to preserve cellular quiescence. Mice carrying an inactivating Foxo3 deletion in satellite cells display markedly impaired muscle regeneration after two consecutive muscle injuries, perhaps due to a failure of satellite cells to self-renew after the primary injury. These studies also raise the possibility that FOXO3 might promote either proliferation and/or survival of satellite cells during the transient amplification phase, since fewer PAX7-positive satellite cells were associated with centrally nucleated fibers one week after injury in these mice. Further supporting a potentially direct link between FOXO3 and Notch signalling in QSCs, ectopic expression of NICD in FOXO3-deficient satellite cells partially recovered QSC number in single fiber assays55. Together, these results suggest that a key function of FOXO3 in satellite cells is to maintain Notch receptor expression, which is in turn essential for maintaining the quiescent state (Fig 2a). Future work should build upon these and other findings discussed elsewhere56–58 to fully characterize critical transcription factors, non-coding RNAs, epigenetic regulators, and the translational regulators that are required to enforce satellite cell quiescence.
Satellite cell activation
Proper satellite cell activation is crucial to ensuring an adequate regenerative response after muscle injury. Upon muscle insult, QSCs are exposed to extrinsic, pro-myogenic stimuli, including Fibroblast Growth Factor (FGF) proteins59, 60, Hepatocyte Growth factor (HGF)61, 62, insulin-like growth Factor (IGF-1) factors63, and Tumor Necrosis Factor-α (TNF-α )64, 65, which activate intrinsic pathways that stimulate proliferation. It has been reported that the first cell division is the rate-limiting step in satellite cell conversion to a highly proliferative state66, which likely explains why the peak of satellite cell expansion occurs roughly 3–4 days after muscle injury in vivo44. In addition, the long delay in initiation of satellite cell division may hint at the possibility that QSCs may transition through one or more intermediate cell states on the path to full activation (Box 3).
Box 3. GAlert state of satellite cells.
Recently, a new satellite cell state – the GAlert state was revealed when QSCs were exposed to injury at a distance66. In this study, animals were subjected to muscle injury in one limb, and then Activated Satellite Cells (ASCs) were isolated from the injured limb muscle and Contralateral Satellite cells (CSCs) were isolated from the contralateral, uninjured limb muscle. Interestingly, as compared to QSCs, CSCs exhibited a shortened time until their first division, were slightly larger in size, had higher quantities of functional mitochondria and increased mtDNA content. CSCs also exhibited a higher propensity to differentiate and fuse with damaged fibers in vivo when compared to QSCs66. However, CSCs were still less activated, according to most of the phenotypic criteria described above, than ASCs that were proximal to muscle injury66. Thus, it was proposed that CSCs had entered an “alert” state, representing a discrete stage of the cell cycle. This state is characterized by an intermediate cellular program that can be reversibly adopted by satellite cells as they anticipate a pending need for a rapid transition from quiescence to activation to enable efficient muscle repair66. Interestingly, entry into the GAlert state appears to require mTORC1, since genetic deletion of one of its inhibitors, TSC1 (tuberous sclerosis complex 1), induced QSCs in uninjured animals to adopt a GAlert phenotype. Conversely, genetic deletion of RAPTOR (Regulatory-Associated Protein of mTOR), an adaptor protein required for mTORC1 activity149, impaired the GAlert phenotype in CSCs. Abrogation of HGF signalling inhibited the GAlert phenotype, revealing that secreted HGF in the local microenvironment62 may be required for this alternative satellite cell state. Together, these data suggest that QSCs are highly responsive to extracellular damage cues and can execute somewhat preemptive alterations to their cellular phenotype that prime them for activation, enabling them to respond more rapidly to subsequent perturbations of muscle homeostasis in their vicinity. It will be interesting in future experiments to test whether the GAlert state is also required as an intermediate step for satellite cells responding directly to local muscle injury.
From an epigenetic perspective, the transition of satellite cells to full activation requires the implementation of a highly specialized, epigenetic and regulatory gene expression program33, 34. For example, freshly isolated ASCs exhibit enriched expression of gene pathways involved in metabolic processes (see Box 4), cell cycle progression, chemotaxis, and immune system regulation33, 34, reflecting the need for satellite cells to grow in size, proliferate, migrate, and interact with muscle-resident immune cells in the context of muscle repair. In addition, the chromatin of ASCs is uniquely altered from QSCs, with ASCs having more genes associated with the repressive H3K27me3 chromatin mark67. H3K27me3 is deposited by EZH2, the enzymatic unit of the Polycomb repressive complex 2 (PRC2), which functions during development to restrict alternative cell fates during lineage commitment68. Interestingly, pro-inflammatory cytokines secreted during muscle injury and repair can stimulate EZH2, which (in complex with the transcriptional regulator, YYI) acts to repress PAX7 expression in cultured satellite cells that have already begun downregulating PAX7, suggesting that EZH2 can play a role in mediating myogenic differentiation69. However, a subsequent study from the same group showed that genetic deletion of Ezh2 in PAX7-expressing satellite cells prior to injury hampers their proliferation and predisposes them to differentiate, ultimately impairing muscle repair after injury70, 71. These studies highlight the complex activities of epigenetic regulators at multiple stages in the satellite cell life cycle, and the need to confirm findings from ex vivo systems using in vivo animal models. Nonetheless, these findings clearly support the importance of chromatin reorganization and repression of key target genes in satellite cells during the transition from a QSC to ASC.
Box 4. Metabolic sensing and control of satellite cell quiescence and activation.
Muscle satellite cells undergo extreme changes in size and metabolic activity as they transition from a quiescent to a proliferating state47. In fact, even during homeostasis, heterogeneities in the metabolic status of quiescent satellite cells exist. For example, freshly isolated muscle satellite cells expressing lower levels of Pax7 transcripts (suggestive of a more activated state) have higher levels of active mitochondria, ATP, and mitochondrial transcription factor RNAs, indicating that an increase in oxidative phosphorylation (OXPHOS) may be required for satellite cell activation and proliferation47, 82. Increased mitochondrial activity is also found in satellite cells from mice subjected to short-term calorie restriction, a dietary regimen shown to enhance muscle regenerative activity146.
A direct functional link between metabolic state and the transition from satellite cell quiescence to activation has been unclear. However, it was recently reported that autophagic flux in quiescent satellite cells provides key bioenergetic resources needed for cell growth and proliferation during activation147. Interestingly, deletion of the NAD-dependent deacetylase Sirt1 in satellite cells led to a reduction in autophagy and consequently less active mitochondria, leading to impairments in satellite cell proliferation147. The activities of SIRT1 in satellite cells are likely complicated, since it has also been reported that proliferating satellite cell-derived myoblasts in culture, exhibit reduced NAD+ levels rendering SIRT1 inactive, and that Sirt1 deletion in vivo led to precocious satellite cell differentiation 148. As these two studies utilized different genetic systems for Sirt1 ablation (inducible versus constitutive), it may be that the timing of expression and cellular distribution of SIRT1, as well as potential compensatory mechanisms, underlie these seemingly contradictory observations.
In addition to modulating satellite cell entry and exit from the cell cycle, metabolic status may also directly influence the ability of these cells to support recovery from muscle damage as well as the effectiveness of their transplantation. As noted above, satellite cells from calorie-restricted mice displayed enhanced oxygen consumption and mitochondrial activity. They were also more abundant, and engrafted host muscle more efficiently upon transplantation37. These functional alterations suggest that metabolic reprogramming of quiescent satellite cells may promote their survival as well as induce their proliferation and/or differentiation – all properties that would increase the capacity of these cells to contribute to new and existing fibers. These data hint at exciting possibilities for enhancing muscle regenerative function through metabolic modulation. However, further investigations are needed to clarify the precise mechanisms through which metabolic shifts alter satellite cell function and to delineate additional key regulators of this process.
MyoD and MYF5 are “master” transcription factors that are critical for enforcing the unique regulatory gene expression program ascribed to ASCs72, 73. MYOD and MYF5 are induced in satellite cells in vivo within 3 hours after cardiotoxin-mediated muscle injury, implicating them in the early stages of satellite cell activation16. Indeed, ex vivo knockdown of MYOD in satellite cells associated with muscle fibers reduced the expression of Cdc6, a regulator of DNA replication, and blocked satellite cell entry into S-phase74. The activities of MYOD and MYF5 may only partially overlap, since MYOD-null animals have a severe myogenic differentiation defect14, while in MYF5-null animals, proliferation is primarily impaired15, 17. Future studies should focus on elucidating the mechanisms by which MYOD can enable both proliferative expansion and myogenic differentiation in ASCs, and whether co-expression of MYF5 influences these cell fate decisions.
There has been great interest in identifying the relevant upstream regulators of MYOD and MYF5. Although MYOD protein is undetectable in QSCs, PAX7 and FOXO3 induce transcriptional activation at the MyoD locus in these cells75, further highlighting that satellite cells are primed for activation, but also hinting towards post-transcriptional mechanisms to regulate MYOD protein levels in QSCs. Insight into such regulation was revealed when it was shown that p38 α and β MAPK (p38α/β-MAPK) is crucial for awakening QSCs into the proliferative state76. p38α/β-MAPK are increased in proliferating satellite cells, where they induce MYOD protein expression by inhibiting the RNA destabilizing protein, Tristetraprolin (TTP) 76, 77 (Fig. 2b). The activities of TTP in QSCs ensure that MyoD mRNA is stabilized for satellite cell activation. Like MYOD, MYF5 also can be regulated both transcriptionally and post-transcriptionally, and its regulation is critical for a rapid transition to the full ASC state78–80. For example, CARM1, an arginine methyltransferase enzyme, methylates PAX7 protein in satellite cells committed to myogenesis leading to association of PAX7 with the Histone Methyl-Transferase (HMT) complex (which is comprised of Absent, Small, or Homeotic – like (ASH2L); Mixed-lineage leukemia 1 or 2 (MLL-1 or -2);WD Repeat Domain 5 (WDR5); Retinoblastoma Binding Protein 5 (RBBP5)) and subsequent gene activation of Myf579. The HMT complex, which functions as a chromatin-interacting unit that recruits elements required for active transcription, has previously been shown to collaborate with PAX7 to induce target gene expression81 (Fig. 2b). Another study showed that Myf5 mRNA is post-transcriptionally repressed by miRNA-31 in QSCs, via a mechanism involving mRNA sequestration in cytoplasmic RNA granules80. These RNA granules disassemble upon satellite cell activation leading to Myf5 mRNA release and subsequent robust protein expression80(Fig 2b).
Altogether, these studies highlight the complex regulation involving signalling pathways, transcription factors, epigenetic regulators, RNA-binding proteins, and non-coding RNAs that are required for the transition from quiescence to the full ASC program. Future studies should focus on a more detailed understanding of the earliest stages of satellite cell activation, to define more completely the complexity of how QSCs awaken and prepare for cell division. New investigations into these unexplored areas may reveal insight into mechanisms that can be exploited to prime satellite cells for enhanced muscle repair in vivo, such as the newly described poised (GAlert) state of these cells (Box 4)
Satellite cell self renewal
The satellite cell population must self-renew to ensure that a sufficient number of reserve cells can seed future rounds of muscle repair. Current data indicates that most resident satellite cells enter the cell cycle in response to muscle injury, as the majority Pax7-positive satellite cells in an injured muscle incorporate BrdU within the first 7-days of muscle regeneration44. Thus, to achieve self-renewal, a sub-population of proliferating satellite cells must first avoid myogenic differentiation and then subsequently return to G0, or the QSC state, to replenish the available pool of stem cells. In this section, we discuss the various modes of cellular division that have recently been shown to influence these crucial cell fate decisions, and then highlight recent progress in identifying novel regulators that drive the self-renewal process during muscle regeneration.
Adult muscle stem cells can undergo either symmetric or asymmetric cell divisions (Figure 2c). The mode of division depends on the distribution of cellular components, signalling molecules, and genetic material and is paramount in balancing the maintenance and differentiation of the muscle stem cell pool76, 82–86. For example, upon satellite cell activation, the template DNA strand used for DNA replication, the asymmetric determinant regulator, NUMB, and p38α/β-MAPK signalling molecules can segregate asymmetrically to daughter cells, with those daughters that receive p38α/β-MAPK acquiring a more committed/differentiated state (Figure 3a); those daughters that acquire NUMB and the template DNA strand (and not p38α/β-MAPK) are destined to self-renew to replenish the QSC pool76, 82, 84, 87. The evolutionarily conserved PAR (Partitioning-defective) complex and the Scrib (Scribbled) cell polarity complex77, 88 are likely drivers of the asymmetric distribution pattern of p38α/β-MAPK. It has also been suggested that satellite cells that have never expressed Myf5, as determined by Cre-based lineage tracing, represent a satellite stem cell population that divides asymmetrically, with the daughter that maintains contact with the basal lamina eventually acquiring a self-renewed state (remaining Myf5-negative), and the apical daughter becoming Myf5-positive and committing to differentiation83. On the other hand, satellite cells can also self-renew using symmetric cell division through the activities of Wnt7a signalling83, 89, or when the JAK-STAT signalling pathway is genetically or pharmacologically impaired85, 86. Further studies will be needed to decipher the molecular determinants that regulate asymmetric versus symmetric satellite cell division, to determine whether these modes of cellular division occur concurrently in vivo, and to identify additional intrinsic gene regulatory factors that must be distributed selectively to self-renewing satellite cells to enable their stalled differentiation and return to quiescence.
Figure 3. Intrinsic cell fate regulators are misregulated in aged and dystrophic satellite cells.

(A) In youth and adulthood (age 2–6 months in mice), quiescent satellite cells (red circles) asymmetrically distribute p38 to daughter cells, such that one daughter receives p38 and is competent for myogenic commitment and differentiation (blue circle with arrow pointing forwards to denote lineage commitment), while the other daughter self-renews and returns to the quiescent state (red circle with arrow pointing backwards to denote their self-renewal and return to quiescence). Asymmetric satellite cell divisions in young animals yield a healthy balance between satellite cells that are competent for myogenic differentiation and cells that self-renew during the expansion phase, which ensures the efficient repair of muscle and the return of a sufficient number of cells to the satellite cell niche to support future demands for muscle regeneration (homeostasis). However, in aged animals (18–24 months in mice), p38 is symmetrically partitioned to daughter cells and JAK-STAT signalling is elevated in proliferating cells, leading to an overall increase in myogenic commitment within the satellite cell population, and reducing the number of quiescent satellite cells at homeostasis. In “geriatric” muscle (28–32 months of age in mice), p16Ink4a is additionally elevated in the satellite cell population, leading to a block in satellite cell activation and directing these cells to an irreversible state of cellular senescence (grey circles). The intrinsic deficits in aged and “geriatric” satellite cells deplete the self-renewing satellite cell population at homeostasis, and thus impair the regenerative response in skeletal muscle as animals age (Black triangle). (B) Satellite cells from 2-month old animals (adult) carrying a loss-of-function mutation in the Dystrophin gene (mdx) display defects in asymmetric division and a stark reduction in apico-basally oriented divisions due to reduced levels of polarity kinase MARK2 (also known as Par-1). This alters the localization of PARD3 (shown in green) and impairs mitosis, leading to a failure to properly expand the lineage committed satellite cells, and a consequent inefficiency of muscle repair in response to injury. In mice carrying an alternative mdx allele (mdx5cv ), dystrophic satellite cells (either QSC or ASC) can undergo a cell type transition towards a fibrogenic state (brown oval) via overactivation of Wnt and TGFβ signalling. This may over time and in a context-dependant manner lead to depletion of highly functional muscle stem cells.
Some insight into the crucial factors that may promote satellite cell self-renewal comes from recent studies of a slow-dividing sub-population of satellite cells that is established after birth and requires the cell-cycle inhibitor, CDKN1B (p27kip1), for maintenance90. Sprouty1 (SPRY1), a membrane-associated inhibitor of receptor tyrosine kinase signalling91, is expressed specifically in this slow cycling sub-population of satellite cells45, 90, and its genetic deletion in PAX7-expressing satellite cells led to a 50% reduction in the quiescent PAX7+MYOD-population present after muscle regeneration44. Thus, Sprouty1 function in satellite cells appears to be required for efficient return of a subset of ASCs to quiescence after muscle repair is complete (Fig. 2c). Sprouty1 likely acts to slow cell division through inhibition of FGF signalling, which normally stimulates proliferation45, 60. FOXO3 also may play a role in the suppression of myogenic differentiation and the return to quiescence (Fig. 2c), since FOXO3-deficient satellite cells were more prone to spontaneous myogenic differentiation, and mice carrying a Foxo3 deletion displayed reduced satellite cell numbers after two rounds of muscle regeneration55. Collectively, these data suggest a model whereby both FOXO3 and Sprouty1, independently or in concert, act to suppress myogenic differentiation in self-renewing satellite cells and return these cells to the G0, QSC state (Fig. 2). It is now important to identify upstream regulators of FOXO3 and Sprouty1 and define the mechanism by which these factors are induced in only self-renewing satellite cells during muscle regeneration. It will also be beneficial to pinpoint exactly when during the regeneration process these cell fate decisions, to self-renew or to differentiate, are made.
Satellite cell fate in aging muscle
Muscle aging is characterized by a decline in muscle repair and progressive loss of muscle mass and function due to muscular atrophy, a process commonly referred to as sarcopenia. As elderly individuals represent the fastest growing demographic in many national populations, there is an urgent need to identify new therapeutics to boost muscle repair and prevent muscle wasting and dysfunction. Although satellite cell dysfunction alone may be insufficient to drive the emergence of sarcopenia, misregulation of this cell population may promote increased muscle fibrosis and certainly impedes recovery after injury in older individuals92–94. It is increasingly apparent that intrinsic misregulation of gene pathways in aging satellite cells leads to alterations in their proliferation and differentiation in vivo, impeding muscle regeneration. Likewise, extrinsic signals from the local niche and even circulating, systemic factors can greatly influence muscle regenerative function and contribute to the decline of satellite cell regenerative function in aged muscle.
Intrinsic changes in aging satellite cells
Recent studies have revealed critical changes in the transcriptional and epigenetic landscape of QSCs as they age33, 86, 95. These analyses generally indicate that aged QSCs feature an increase in repressed chromatin states, with widespread H3K27me3 across the genome33, so it would not be surprising if these chromatin re-organization events contribute to the misregulation of key signalling pathways that are required for a robust regenerative response. Work in this area recently converged to highlight the critical influence of overactive p38α/β-MAPK signalling in aged satellite cells, which leads to a drastic decline in satellite cell self-renewal and impairs satellite cell function96, 97.
A recent study showed that when as few as 10 freshly isolated satellite cells from aged or young mice were intramuscularly transplanted into immuno-compromised recipient mice, aged satellite cells engrafted (as defined by bioluminescence and histology) skeletal muscle at two-thirds the rate of younger cells97. This difference was lost when higher numbers of muscle satellite cells were transplanted (between100 and 300), indicating that a sub-population of aged satellite cells may retain high functionality98. Interestingly, aged satellite cells exhibited a reduced proliferative output and were associated with elevated levels of p38α/β-MAPK and senescence regulator genes, which could be reversed by pharmacological inhibition of p38α/β-MAPK97. A parallel study demonstrated that the normal asymmetric partitioning of p38α/β-MAPK signalling to satellite cell daughters is disrupted in aged cells and that this disruption causes symmetric inheritance of p38α/β-MAPK and impedes satellite cell self-renewal (Fig. 3a), which could be recovered by treatment with a small molecule inhibitor of p38 signalling96.
Another recent study found elevated levels of p16Ink4a, an inhibitor of cyclin-dependent kinases (CDKs)99, in “geriatric” satellite cells from 28–32 month old mice. The elevation of p16Ink4a in “geriatric” satellite cells led to their conversion from a reversibly quiescent to an irreversibly senescent state95. Paradoxically, in addition to its known role in satellite cell activation76, p38 α/β-MAPK signalling can also activate p16Ink4a to promote cellular senescence99. A genetic link between p38α/β-MAPK and p16Ink4a was recently demonstrated in skeletal muscle, when it was found that p16Ink4a protein levels were reduced in the gastrocnemius muscle of animals expressing a dominant-negative p38α allele, although future studies will need to verify this connection in satellite cells100. Cumulatively, these data suggest that an age-regulated intrinsic p38 signalling-axis exists in the satellite cell pool. Under this model, while p38 α/β-MAPK is asymmetrically distributed to daughter cells in young satellite cells, the switch to symmetric p38 α/β-MAPK inheritance leads to a depletion of self-renewed satellite cells and through unknown mechanisms activates p16Ink4a, driving cellular senescence (Fig 3a).
Finally, in two parallel studies, it was shown that JAK-STAT signaling and effector target genes play a pivotal role in myogenic commitment and differentiation, and that this pathway is misregulated in satellite cells of aged muscle85, 86. JAK-STAT target genes are elevated in 18-month old mice, relative to 3-month old animals, and genetic perturbation of JAK-STAT members in freshly isolated satellite cells from aged animals improved their functional engraftment when transplanted into host skeletal muscle. SiRNA knockdown of JAK2 and STAT3 in satellite cells still associated with aged single fibers led to increases in the symmetric expansion of the Pax7+Myf5-satellite stem cell population86. STAT3 is required for MYOD-mediated myogenic commitment ex vivo, and a transient pharmacological inhibition of STAT3 in aged animals in vivo increased the population of proliferating satellite cells and improved muscle regeneration85. Altogether, these data support a model in which elevated JAK-STAT signaling in aged satellite cells leads to their rapid differentiation and failure to undergo efficient symmetric expansion in response to muscle injury (Fig. 3a).
In future studies, it will be useful to determine whether senescence is a state that all satellite cells will eventually reach, or rather, a condition that only a subset of satellite cells will experience during muscle aging. It will also be helpful to clarify whether p38α/β-MAPK directly influences the senescent phenotype in aging satellite cells. Furthermore, the role of JAK-STAT signaling in regulating cell fate decisions of muscle stem cells is probably context dependent and likely varies for different JAK-STAT family members, since JAK-STAT proteins have been reported to promote satellite cell proliferation and prevent myogenic differentiation101, in addition to their role in promoting myogenic differentiation (discussed above)85, 86. Future studies to generate genomic binding profiles and identify interacting partners of the various STAT transcription factors in young and aged satellite cells should shed light on their important functions during muscle regeneration and aging.
Regulation by local factors
Satellite cells are greatly influenced by their local microenvironment, as muscle fibers and other non-myogenic cells secrete growth factors and cytokines that influence their behaviour during regeneration3. As satellite cells age, so does the niche in which they reside, and these structural, and molecular alterations can impede satellite cell function. One example comes from a recent study showing that aged muscle secretes higher levels of FGF2 (Fibroblast growth factor 2) into the satellite cell niche, leading to a considerable depletion of the low-cycling satellite cell population. This study also reported that inhibition of FGF receptor 1 (FGFR1) can rescue age-related defects in satellite cell cycling and apoptosis45. Importantly, FGF2 was shown to inhibit the activities of Sprouty1, a seminal regulator of satellite cell quiescence, explaining the shift towards more high-cycling satellite cells. Curiously, a different study found that the activity of FGFR1 was attenuated in aged satellite cells, and that ectopic expression of a constitutively active FGFR1 recombinant protein enhanced the asymmetric inheritance of p38α/β-MAPK102. As many signalling pathways exhibit bimodal or multimodal effects, which can be influenced by cellular context, timing, and dosage, it is likely that these seemingly contradictory results reflect a critical need to maintain FGF signalling in satellite cells within a tight, physiological window, as overactivation or attenuation of this pathway can perturb muscle homeostasis and contribute to satellite cell dysfunction.
Altogether, these data provide an intriguing example of how aging-related misregulation of extrinsic factors in the local niche can influence critical intrinsic pathways that modify satellite cell fate.
Regulation by systemic signals
Evaluation of potential systemic influences on muscle regenerative function has been undertaken by a number of research groups using a 150 year-old technique known as parabiosis103, which involves the surgical joining of the circulatory systems of two mice. Remarkably, heterochronic parabiotic pairings of young and aged mice led to the restoration of satellite cell activation and a more robust regenerative response after injury in the aged partner of the pair104, 105. These data provided initial support for the notion that young blood may contain “rejuvenation” factors capable of regulating muscle repair in older animals. Importantly, the physiological impacts of heterochronic parabiosis are not limited to the aged partner, and exposure of a young animal to an aged circulation through such pairing has been shown to slow regeneration in young muscle 92, 106, 107. Thus, in addition to loss of pro-regenerative factors, aged blood also appears to accumulate suppressive factors that can actively impair healthy function in the muscle and other organs.
Molecular identification of circulating molecules that regulate cell function in aged animals has been a major emphasis in recent years. Published reports implicate increases in TGFβ1108, 109 and Wnt92, 110 signalling as key contributors to the active suppression of satellite cell function in aged muscle, and these factors could potentially contribute to rare, age-related cell fate conversion events, in which satellite cells transition from a myogenic to a fibrogenic state92, 111. A few “rejuvenating” molecules have also been identified. For example, it has been recently reported that Oxytocin, a hormone produced by the hypothalamus with a demonstrated role in social behavior112, is reduced 3-fold in the blood of aged mice, and that its receptor is down-regulated by nearly 50% in aged satellite cells113. Administration of Oxytocin to aged mice relieved the proliferation and differentiation defects of old satellite cells, and enhanced the regenerative response to muscle injury113. In an intriguing and still unexplained link to the studies of cell intrinsic alterations in aged satellite cells discussed above, Oxytocin induced the activation of the MAPK/ERK pathway in satellite cells and improved their proliferation potential. Similar beneficial effects on muscle repair following cryoinjury in aging mice have been reported for the circulating cytokine Growth Differentiation Factor-11 (GDF11), a member of the TGFβ superfamily that has significant structural and functional homology to Myostatin (MSTN)114. Several studies suggest that expression of Gdf11 declines with age in key GDF11-producing organs, and that the combined pool of GDF11 and MSTN is diminished in the blood of aged animals and humans115–117. Moreover, supplementation of recombinant GDF11 protein in older mice yielded improvements in muscle ultrastructure, satellite cell function and muscle regeneration118, supporting the notion that this protein, in particular, promotes healthier function in aged muscle. It is important to note, however, that these results were recently challenged by another group, who reported that GDF11 levels may increase with age in some species and that GDF11 supplementation in old mice has no effect and in young mice may impede muscle regeneration in response to cardiotoxin injury119. These apparently discrepant results may relate to cross-reactivity of reagents used to track GDF11 protein levels, which also react with GDF8119–121 and immunoglobulin light chain116, as well as to differences in experimental design, including differences in the modes of muscle injury employed, in the doses of GDF11 injected, in the strategies for analysis of muscle regeneration employed, and even in the sources of GDF11 protein and aged animals used by the different groups. While further studies have clarified some of the reported discrepancies116, additional experiments will be needed to fully reconcile these observations and determine how the levels of circulating GDF11 protein change throughout life as well as how such changes influence satellite cells.
Satellite cells in muscular dystrophy
Duchenne muscular dystrophy (DMD) is a severe, genetic disorder that causes progressive muscle wasting in affected individuals and currently has no cure. Dystrophin, the defective gene product that causes DMD, plays a pivotal structural role in anchoring the muscle fiber to the extracellular matrix in the muscle niche. Numerous studies in mice have shown that dystrophic fibers rupture and die after repeated muscle contractions, and are eventually replaced with fibrotic tissue122. Fibrosis arises due to an excess of connective tissue deposited in muscle during regenerative processes, and is thought to be mediated in part by muscle-resident fibroblasts123. Most of the physiological and functional deficits exhibited by DMD patients have been attributed to abnormalities in mature muscle fibers, where a known role for Dystrophin has been documented. However, recent work has shown that dystrophic satellite cells are impaired in various aspects of the satellite cell life cycle, which impact their muscle regenerative functions37, 111, 124–129. We discuss here emerging data suggesting that intrinsic misregulation of satellite cell fate in dystrophic muscle may impair muscle regenerative function and contribute directly to the DMD pathology.
Dystrophin functions near the cell membrane and acts as a structural component of the muscle cell that regulates the flow of signalling molecules. Therefore, it would not be surprising if lack of dystrophin, which normally is expressed by satellite cells, would directly influence their downstream cell-intrinsic, signalling pathways to alter their fate, and ultimately, function. Initial evidence that functional impairments in the satellite cell pool contribute to the DMD phenotype derives from a recent study that created a telomerase and dystrophin double knockout animal, mainly as a tool to impair the functional muscle stem cell pool130. These mice exhibit severe molecular and physiological defects in skeletal muscles, including the diaphragm130, similar to human DMD patients. Interestingly, satellite cells from these mice showed reduced proliferation both in vivo and during ex vivo culturing, and they were functionally impaired when transplanted into host animals130. These data suggested, for the first time, that the severity of DMD can be influenced by the regenerative functions of endogenous muscle stem cells.
Evidence for a direct role of Dystrophin in satellite cells remained elusive, however, until a recent study showed that the Dystrophin protein has a highly specialized function in regulating cell division mechanisms in muscle stem cells129. Specifically, it was shown that the presence of Dystrophin enhances MARK2(Par1b) protein levels and that MARK2 then mediates the asymmetric localization of the PARD3 polarity protein, which functions to properly guide asymmetric cell division in proliferating satellite cells. Accordingly, in response to injury, dystrophic satellite cells undergo a prolonged mitosis and their ability to undergo apico-basal cell divisions is perturbed, leading to impaired asymmetric division and a smaller population of progenitors capable of contributing to muscle repair129(Fig. 3b). In line with this, after culturing dystrophic muscle fibers for 72h an increase in the subset of Pax7+ satellite cells that have never expressed Myf5 (discussed above83) and a reduction in the number of MYOG-expressing progenitors was observed, further suggesting that dystrophic satellite cells may display enhanced self-renewal and that some may be blocked with respect to myogenic differentiation129. These findings may explain why dystrophic muscle or single fibers contain more PAX7-positive satellite cells than control muscle or wildtype fibers128, 131. Interestingly, a prior report suggested that dystrophin-deficient mice exhibit defects in satellite cell self-renewal, with reduced numbers of Pax7+Myf5-satellite cells associated with single fibers from pre-injured animals128. As this effect was more pronounced in 6 month old, as compared to 2 month old mice (the approximate age used in the study discussed above), these data may suggest that the impact of dystrophin deficiency on satellite cell self-renewal can vary with age. Alternatively, it is possible that these differences may be explained by the different activation models used - isolation of single fibers from pre-injured animals128 versus single fibers from uninjured animals cultured for 72 hrs129. Regardless, further studies are needed to harmonize these results.
Impaired self-renewal of the Pax7+Myf5-sub-population of dystrophic satellite cells have also been associated with reduced Notch signalling, and enforced activation of this pathway restored satellite cell self-renewal128. Given the role that Notch plays in promoting satellite cell quiescence (discussed above), reduced Notch signaling in dystrophin deficient satellite cells may also contribute to the reported increase in the high-cycling satellite cell subset in these animals90, 132. The notion that modulation of Notch signalling may improve the dystrophic phenotype has gained further support from a very recent analysis of two dystrophin-deficient dogs presenting a milder DMD phenotype133, 134. Intriguingly, these “escaper” animals were found to possess a single mutation in the promoter of the Notch ligand Jagged-1, which creates a novel Myogenin binding site and causes upregulation of Jagged1 expression134. Myogenic cells from escaper dogs overexpressing Jagged-1 also showed enhanced proliferation, and further studies in dystrophic zebrafish confirmed that overactivation of Jagged-1 ameliorates the dystrophic phenotype134. These results demonstrate that studies aimed at revealing the mechanisms underlying the effects of Jagged-1 in dystrophic muscles, as well as to delineate additional genetic suppressors of dystrophic phenotypes, may open novel treatment avenues for DMD.
Finally, it has been recently demonstrated that, apart from changes regarding their self-renewal and differentiation potential, a subset of dystrophic satellite cells may initiate a Wnt3a and TGFβ-dependant fibrogenic-like cellular program (Fig. 3b). This, over time, may serve to deplete functional muscle stem cells and impede effective muscle repair. It also may help to explain the increase in fibrotic tissue seen in DMD muscle111.
Collectively, the data discussed above strongly suggest that the loss of satellite cell regenerative function exacerbates DMD symptoms, and that dystrophin deficiency directly influences satellite cell fate. More studies will be required to fully understand the cellular and molecular mechanisms driving these apparently context-dependant effects135. It will also be important to decipher the transcriptional and epigenetic changes that may underwrite functional misregulation in dystrophin-deficient satellite cells and to determine the contribution that extrinsic signals may play in modulating these intrinsic changes to the muscle stem cell fate.
Conclusion/perspective
An emerging theme from the work discussed here is that the intrinsic gene regulatory circuits that control adult muscle satellite cell behaviour in healthy muscle are misregulated in aging and disease. Elucidating the transcriptional and epigenetic “drivers”, and their targets, that act downstream of dystrophin-deficiency or become altered in aged satellite cells, will continue to reveal a molecular roadmap of potentially drug-able targets that could be manipulated with small-molecule intervention to enable the modulation of stem cell activity. Furthermore, reinstating self-renewal pathways in cultured satellite cells may also enable their efficient expansion in culture, a barrier that currently impedes their use in the clinic (see Box 2). There is no better time to be studying gene regulation, muscle stem cell fate, and translational medicine, and we anticipate that novel discoveries in basic science will improve the prognosis and treatment of age associated muscle dysfunction and currently incurable muscle pathologies.
Online summary.
Satellite cell quiescence, activation, and self-renewal are coordinated by extrinsic and intrinsic regulators.
Quiescent satellite cells are molecularly and functionally heterogeneous and have a unique transcriptional and epigenetic profile that distinguishes them from activated satellite cells.
Aged satellite cells misregulate intrinsic factors like p38-α/β-MAPK, JAK-STAT, and p16Ink4a, leading to impairments in self-renewal and eventually, conversion to a senescent state.
Local and systemic factors influence satellite cell fate decisions, and systemic delivery of youthful blood-borne factors can ameliorate some aspects of the aging phenotype in skeletal muscle.
Dystrophin-deficiency directly misregulates the satellite cell state and function, which further exacerbates DMD symptoms.
Acknowledgments
We wish to thank all reviewers of this manuscript for their critical evaluation and their insightful feedback in preparing this manuscript. This work was supported in part by a Burroughs Wellcome Fund Postdoctoral fellowship Award and a T32 NIH training grant (5T32DK007260-38) to A.E.A., and National Institutes of Health (NIH) RO1 ES024935 and R56 AG048917 to A.J.W. Content is solely the responsibility of the authors and does not necessarily represent the official views of the NIH.
Glossary
- Satellite cell niche
A structural and growth-factor rich environment above the muscle sarcolemma but beneath the basal lamina, consisting of cellular and acelluar components that support and regulate muscle satellite cells.
- Muscle sarcolemma
Outer cell membrane of a myofiber.
- Basal lamina
Inner layer of the basement membrane, composed of extracellular matrix proteins, that is adjacent to the muscle sarcolemma.
- Fibro-adipogenic progenitors (FAPs)
A bipotent progenitor population that can adopt both fibrogenic and adopgenic cell fates, located in the interstitial space of muscle fibers.
- Cellular senescence
A cellular state induced by extensive proliferation, induction of oncogene expression, inflammatory signals, or other stimuli, in which cells irreversibly exit the cell cycle.
- Cyclin-dependent kinases (CDKs)
A family of kinases that regulate cell cycle transitions.
- Gastrocnemius muscle
A skeletal muscle located at the back of the lower legs.
- Somite
A division in the developing embryo consisting of paired blocks of paraxial mesoderm.
- Myotome
The dorsal part of the somite in the developing embryo that gives rise to the skeletal muscle.
- Cre recombinase-mediated lineage tracing
A molecular genetic technique used to trace each cell that has ever expressed a gene-of-interest, through gene-specific Cre-mediated recombination that leads to expression of a fluorescent reporter gene.
- Oxidative phosphorylation (OXPHOS)
A metabolic pathway in which mitrochondria oxidize nutrients to form ATP.
- Autophagic flux
A measurement to quantify cellular autophagic activity.
- NAD+
The oxidized form of Nicotinamide Adenine Dinucleotide (NAD) that can accept an electron becoming reduced to its NADH form.
- Quiescence
A reversible state of dormancy characterized by withdrawal from the cell cycle, or lack of proliferation.
- Activation
A reversible state characterized by entry into the cell-cycle, or transition to a proliferative state.
- Symmetric cell division
The partitioning of cellular components during cell division that leads to two daughter cells being molecularly and functionally identical.
- Asymmetric cell division
The partitioning of cellular components during cell division that leads to two daughter cells being molecularly and functionally distinct.
- Pax3
A paired box protein family member expressed in satellite cells in a few adult muscle groups with a prominent role in embryonic myogenesis.
- Sprouty1
A receptor tyrosine kinase inhibitor that restricts entry to the cell cycle by abrogating cell signalling pathways, like the Fibroblast Growth Factor Receptor (FGFR).
- mTORC1
A gene product encoding the mammalian Target of Rapamycin Complex 1 that is part of a protein complex that controls protein synthesis by sensing nutrients, energy, and metabolic products in the cell.
- TTP
Tristetroprolin is an RNA-binding protein that binds the 3′ untranslated region of target RNAs, leading to their rapid decay through the recruitment of cytoplasmic RNA degradation machinery.
- Active chromatin
Regions of the genome that are actively transcribed and are flanked by histone H3 tri-methylation at lysine 4 (H3K4me3).
- p38α/β MAPK
Two isoforms of the p38 family of MAPKs, which plays a seminal role in detecting extracellular signals from the outside of the cell and directing a cascade of phosphorylation events that activates downstream effectors and ultimately target gene expression.
- Parabiosis
The surgical joining of the circulatory systems between two animals.
- Heterochronic parabiosis
The surgical joining of the circulatory systems between a young and old animal.
Biographies
Albert E. Almada is a Burroughs Wellcome Fund postdoctoral fellow in the laboratory of Amy J. Wagers at the department of Stem Cell and Regenerative Biology at Harvard University, where he is investigating the transcriptional and epigenetic mechanisms that program muscle stem cell identity and function. He obtained his Ph.D. in Biology at the Massachusetts Institute of Technology (MIT) under the tutelage of Dr. Phillip A. Sharp, where he studied the biogenesis and transcriptional regulation of a novel class of promoter-associated non-coding RNAs in embryonic stem cells.
Amy J. Wagers is the Forst Family Professor of Stem Cell and Regenerative Biology at Harvard University, a Senior Investigator in the Section on Islet Cell and Regenerative Biology at the Joslin Diabetes Center and a member of the Harvard Stem Cell Institute and Paul F. Glenn Center for the Biology of Aging at Harvard University and Harvard Medical School. She received her Ph.D. in Immunology and Microbial Pathogenesis from Northwestern University, and completed postdoctoral training in stem cell biology at Stanford University. Dr. Wagers has studied muscle stem cell biology for more than 15 years and her research seeks to understand how changes in stem cell activity impact tissue homeostasis and repair throughout life and how these cells may be harnessed for regenerative medicine.
References
- 1.Mauro A. Satellite cell of skeletal muscle fibers. J Biophys Biochem Cytol. 1961;9:493–5. doi: 10.1083/jcb.9.2.493. The first reported observation of the satellite cell in skeletal muscle in frog. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bentzinger CF, et al. Fibronectin regulates Wnt7a signaling and satellite cell expansion. Cell Stem Cell. 2013;12:75–87. doi: 10.1016/j.stem.2012.09.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Yin H, Price F, Rudnicki MA. Satellite cells and the muscle stem cell niche. Physiol Rev. 2013;93:23–67. doi: 10.1152/physrev.00043.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Urciuolo A, et al. Collagen VI regulates satellite cell self-renewal and muscle regeneration. Nat Commun. 2013;4:1964. doi: 10.1038/ncomms2964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Brohl D, et al. Colonization of the satellite cell niche by skeletal muscle progenitor cells depends on Notch signals. Dev Cell. 2012;23:469–81. doi: 10.1016/j.devcel.2012.07.014. [DOI] [PubMed] [Google Scholar]
- 6.Castiglioni A, et al. FOXP3+ T Cells Recruited to Sites of Sterile Skeletal Muscle Injury Regulate the Fate of Satellite Cells and Guide Effective Tissue Regeneration. PLoS One. 2015;10:e0128094. doi: 10.1371/journal.pone.0128094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Burzyn D, et al. A special population of regulatory T cells potentiates muscle repair. Cell. 2013;155:1282–95. doi: 10.1016/j.cell.2013.10.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Murphy MM, Lawson JA, Mathew SJ, Hutcheson DA, Kardon G. Satellite cells, connective tissue fibroblasts and their interactions are crucial for muscle regeneration. Development. 2011;138:3625–37. doi: 10.1242/dev.064162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Joe AW, et al. Muscle injury activates resident fibro/adipogenic progenitors that facilitate myogenesis. Nat Cell Biol. 2010;12:153–63. doi: 10.1038/ncb2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Seale P, Ishibashi J, Scime A, Rudnicki MA. Pax7 is necessary and sufficient for the myogenic specification of CD45+:Sca1+ stem cells from injured muscle. PLoS Biol. 2004;2:E130. doi: 10.1371/journal.pbio.0020130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Seale P, et al. Pax7 is required for the specification of myogenic satellite cells. Cell. 2000;102:777–86. doi: 10.1016/s0092-8674(00)00066-0. [DOI] [PubMed] [Google Scholar]
- 12.Yablonka-Reuveni Z, et al. The transition from proliferation to differentiation is delayed in satellite cells from mice lacking MyoD. Dev Biol. 1999;210:440–55. doi: 10.1006/dbio.1999.9284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.White JD, et al. Myotube formation is delayed but not prevented in MyoD-deficient skeletal muscle: studies in regenerating whole muscle grafts of adult mice. J Histochem Cytochem. 2000;48:1531–44. doi: 10.1177/002215540004801110. [DOI] [PubMed] [Google Scholar]
- 14.Megeney LA, Kablar B, Garrett K, Anderson JE, Rudnicki MA. MyoD is required for myogenic stem cell function in adult skeletal muscle. Genes Dev. 1996;10:1173–83. doi: 10.1101/gad.10.10.1173. [DOI] [PubMed] [Google Scholar]
- 15.Gayraud-Morel B, et al. A role for the myogenic determination gene Myf5 in adult regenerative myogenesis. Dev Biol. 2007;312:13–28. doi: 10.1016/j.ydbio.2007.08.059. [DOI] [PubMed] [Google Scholar]
- 16.Cooper RN, et al. In vivo satellite cell activation via Myf5 and MyoD in regenerating mouse skeletal muscle. J Cell Sci. 1999;112( Pt 17):2895–901. doi: 10.1242/jcs.112.17.2895. [DOI] [PubMed] [Google Scholar]
- 17.Ustanina S, Carvajal J, Rigby P, Braun T. The myogenic factor Myf5 supports efficient skeletal muscle regeneration by enabling transient myoblast amplification. Stem Cells. 2007;25:2006–16. doi: 10.1634/stemcells.2006-0736. [DOI] [PubMed] [Google Scholar]
- 18.Faralli H, Dilworth FJ. Turning on myogenin in muscle: a paradigm for understanding mechanisms of tissue-specific gene expression. Comp Funct Genomics. 2012;2012:836374. doi: 10.1155/2012/836374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sacco A, Doyonnas R, Kraft P, Vitorovic S, Blau HM. Self-renewal and expansion of single transplanted muscle stem cells. Nature. 2008;456:502–6. doi: 10.1038/nature07384. Report demonstrating that a single satellite cell is sufficient to restore a functional satellite cell pool. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zammit PS, et al. Muscle satellite cells adopt divergent fates: a mechanism for self-renewal? J Cell Biol. 2004;166:347–57. doi: 10.1083/jcb.200312007. A report to suggest that satellite cells adopt two fates after activation: downregulation of Pax7 to differente or retain Pax7 and suppress MyoD to return to a more quiescent state. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Collins CA, et al. Stem cell function, self-renewal, and behavioral heterogeneity of cells from the adult muscle satellite cell niche. Cell. 2005;122:289–301. doi: 10.1016/j.cell.2005.05.010. [DOI] [PubMed] [Google Scholar]
- 22.Sambasivan R, et al. Pax7-expressing satellite cells are indispensable for adult skeletal muscle regeneration. Development. 2011;138:3647–56. doi: 10.1242/dev.067587. [DOI] [PubMed] [Google Scholar]
- 23.Lepper C, Partridge TA, Fan CM. An absolute requirement for Pax7-positive satellite cells in acute injury-induced skeletal muscle regeneration. Development. 2011;138:3639–46. doi: 10.1242/dev.067595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lepper C, Conway SJ, Fan CM. Adult satellite cells and embryonic muscle progenitors have distinct genetic requirements. Nature. 2009;460:627–31. doi: 10.1038/nature08209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Brack AS. Pax7 is back. Skelet Muscle. 2014;4:24. doi: 10.1186/s13395-014-0024-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.von Maltzahn J, Jones AE, Parks RJ, Rudnicki MA. Pax7 is critical for the normal function of satellite cells in adult skeletal muscle. Proc Natl Acad Sci U S A. 2013;110:16474–9. doi: 10.1073/pnas.1307680110. Report demonstrating the absolute requirement for Pax7-expressing satellite cells in muscle regeneration. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gunther S, et al. Myf5-positive satellite cells contribute to Pax7-dependent long-term maintenance of adult muscle stem cells. Cell Stem Cell. 2013;13:590–601. doi: 10.1016/j.stem.2013.07.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Blau HM, Cosgrove BD, Ho AT. The central role of muscle stem cells in regenerative failure with aging. Nat Med. 2015;21:854–62. doi: 10.1038/nm.3918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sousa-Victor P, Garcia-Prat L, Serrano AL, Perdiguero E, Munoz-Canoves P. Muscle stem cell aging: regulation and rejuvenation. Trends Endocrinol Metab. 2015 doi: 10.1016/j.tem.2015.03.006. [DOI] [PubMed] [Google Scholar]
- 30.Liu L, Cheung TH, Charville GW, Rando TA. Isolation of skeletal muscle stem cells by fluorescence-activated cell sorting. Nat Protoc. 2015;10:1612–24. doi: 10.1038/nprot.2015.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Conboy MJ, Cerletti M, Wagers AJ, Conboy IM. Immuno-analysis and FACS sorting of adult muscle fiber-associated stem/precursor cells. Methods Mol Biol. 2010;621:165–73. doi: 10.1007/978-1-60761-063-2_11. [DOI] [PubMed] [Google Scholar]
- 32.Pasut A, Oleynik P, Rudnicki MA. Isolation of muscle stem cells by fluorescence activated cell sorting cytometry. Methods Mol Biol. 2012;798:53–64. doi: 10.1007/978-1-61779-343-1_3. [DOI] [PubMed] [Google Scholar]
- 33.Liu L, et al. Chromatin modifications as determinants of muscle stem cell quiescence and chronological aging. Cell Rep. 2013;4:189–204. doi: 10.1016/j.celrep.2013.05.043. A study demonstrating the the epigenetic landscape of quiescent satellite cells is distinct from activated satellite cells, and that widespread chromatin alterations occur during satellite cell activation. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Fukada S, et al. Molecular signature of quiescent satellite cells in adult skeletal muscle. Stem Cells. 2007;25:2448–59. doi: 10.1634/stemcells.2007-0019. [DOI] [PubMed] [Google Scholar]
- 35.Fukada S, et al. Purification and cell-surface marker characterization of quiescent satellite cells from murine skeletal muscle by a novel monoclonal antibody. Exp Cell Res. 2004;296:245–55. doi: 10.1016/j.yexcr.2004.02.018. [DOI] [PubMed] [Google Scholar]
- 36.Sherwood RI, et al. Isolation of adult mouse myogenic progenitors: functional heterogeneity of cells within and engrafting skeletal muscle. Cell. 2004;119:543–54. doi: 10.1016/j.cell.2004.10.021. [DOI] [PubMed] [Google Scholar]
- 37.Cerletti M, et al. Highly efficient, functional engraftment of skeletal muscle stem cells in dystrophic muscles. Cell. 2008;134:37–47. doi: 10.1016/j.cell.2008.05.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Castiglioni A, et al. Isolation of Progenitors that Exhibit Myogenic/Osteogenic Bipotency In Vitro by Fluorescence-Activated Cell Sorting from Human Fetal Muscle. Stem Cell Reports. 2014;2:92–106. doi: 10.1016/j.stemcr.2013.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Mackey AL, et al. Assessment of satellite cell number and activity status in human skeletal muscle biopsies. Muscle Nerve. 2009;40:455–65. doi: 10.1002/mus.21369. [DOI] [PubMed] [Google Scholar]
- 40.Charville GW, et al. Ex Vivo Expansion and In Vivo Self-Renewal of Human Muscle Stem Cells. Stem Cell Reports. 2015;5:621–32. doi: 10.1016/j.stemcr.2015.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Cornelison DD, Filla MS, Stanley HM, Rapraeger AC, Olwin BB. Syndecan-3 and syndecan-4 specifically mark skeletal muscle satellite cells and are implicated in satellite cell maintenance and muscle regeneration. Dev Biol. 2001;239:79–94. doi: 10.1006/dbio.2001.0416. [DOI] [PubMed] [Google Scholar]
- 42.Montarras D, et al. Direct isolation of satellite cells for skeletal muscle regeneration. Science. 2005;309:2064–7. doi: 10.1126/science.1114758. A study revealing that FACS-isolated satellite cells can engraft muscle in an mdx model, and showed that cultured satellite cells loose regenerative potential after several days in culture. [DOI] [PubMed] [Google Scholar]
- 43.Yoshida N, Yoshida S, Koishi K, Masuda K, Nabeshima Y. Cell heterogeneity upon myogenic differentiation: down-regulation of MyoD and Myf-5 generates ‘reserve cells’. J Cell Sci. 1998;111( Pt 6):769–79. doi: 10.1242/jcs.111.6.769. [DOI] [PubMed] [Google Scholar]
- 44.Shea KL, et al. Sprouty1 regulates reversible quiescence of a self-renewing adult muscle stem cell pool during regeneration. Cell Stem Cell. 2010;6:117–29. doi: 10.1016/j.stem.2009.12.015. Identification of Sprouty1 as a critical cell signalling regulator that is required for the return to satellite cell quiescence during regeneration. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Chakkalakal JV, Jones KM, Basson MA, Brack AS. The aged niche disrupts muscle stem cell quiescence. Nature. 2012;490:355–60. doi: 10.1038/nature11438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Chen S, Lewallen M, Xie T. Adhesion in the stem cell niche: biological roles and regulation. Development. 2013;140:255–65. doi: 10.1242/dev.083139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Koopman R, Ly CH, Ryall JG. A metabolic link to skeletal muscle wasting and regeneration. Front Physiol. 2014;5:32. doi: 10.3389/fphys.2014.00032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Guenther MG, Levine SS, Boyer LA, Jaenisch R, Young RA. A chromatin landmark and transcription initiation at most promoters in human cells. Cell. 2007;130:77–88. doi: 10.1016/j.cell.2007.05.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Mourikis P, et al. A critical requirement for notch signaling in maintenance of the quiescent skeletal muscle stem cell state. Stem Cells. 2012;30:243–52. doi: 10.1002/stem.775. [DOI] [PubMed] [Google Scholar]
- 50.Bjornson CR, et al. Notch signaling is necessary to maintain quiescence in adult muscle stem cells. Stem Cells. 2012;30:232–42. doi: 10.1002/stem.773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wen Y, et al. Constitutive Notch activation upregulates Pax7 and promotes the self-renewal of skeletal muscle satellite cells. Mol Cell Biol. 2012;32:2300–11. doi: 10.1128/MCB.06753-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kopan R, Ilagan MX. The canonical Notch signaling pathway: unfolding the activation mechanism. Cell. 2009;137:216–33. doi: 10.1016/j.cell.2009.03.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Olguin HC, Olwin BB. Pax-7 up-regulation inhibits myogenesis and cell cycle progression in satellite cells: a potential mechanism for self-renewal. Dev Biol. 2004;275:375–88. doi: 10.1016/j.ydbio.2004.08.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Olguin HC, Yang Z, Tapscott SJ, Olwin BB. Reciprocal inhibition between Pax7 and muscle regulatory factors modulates myogenic cell fate determination. J Cell Biol. 2007;177:769–79. doi: 10.1083/jcb.200608122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Gopinath SD, Webb AE, Brunet A, Rando TA. FOXO3 promotes quiescence in adult muscle stem cells during the process of self-renewal. Stem Cell Reports. 2014;2:414–26. doi: 10.1016/j.stemcr.2014.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Cheung TH, et al. Maintenance of muscle stem-cell quiescence by microRNA-489. Nature. 2012;482:524–8. doi: 10.1038/nature10834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Sato T, Yamamoto T, Sehara-Fujisawa A. miR-195/497 induce postnatal quiescence of skeletal muscle stem cells. Nat Commun. 2014;5:4597. doi: 10.1038/ncomms5597. [DOI] [PubMed] [Google Scholar]
- 58.Zismanov V, et al. Phosphorylation of eIF2alpha Is a Translational Control Mechanism Regulating Muscle Stem Cell Quiescence and Self-Renewal. Cell Stem Cell. 2015 doi: 10.1016/j.stem.2015.09.020. [DOI] [PubMed] [Google Scholar]
- 59.Allen RE, Dodson MV, Luiten LS. Regulation of skeletal muscle satellite cell proliferation by bovine pituitary fibroblast growth factor. Exp Cell Res. 1984;152:154–60. doi: 10.1016/0014-4827(84)90239-8. [DOI] [PubMed] [Google Scholar]
- 60.Gospodarowicz D, Weseman J, Moran J. Presence in brain of a mitogenic agent promoting proliferation of myoblasts in low density culture. Nature. 1975;256:216–9. doi: 10.1038/256216a0. [DOI] [PubMed] [Google Scholar]
- 61.Tatsumi R, Hattori A, Ikeuchi Y, Anderson JE, Allen RE. Release of hepatocyte growth factor from mechanically stretched skeletal muscle satellite cells and role of pH and nitric oxide. Mol Biol Cell. 2002;13:2909–18. doi: 10.1091/mbc.E02-01-0062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Sheehan SM, Tatsumi R, Temm-Grove CJ, Allen RE. HGF is an autocrine growth factor for skeletal muscle satellite cells in vitro. Muscle Nerve. 2000;23:239–45. doi: 10.1002/(sici)1097-4598(200002)23:2<239::aid-mus15>3.0.co;2-u. [DOI] [PubMed] [Google Scholar]
- 63.Schiaffino S, Mammucari C. Regulation of skeletal muscle growth by the IGF1-Akt/PKB pathway: insights from genetic models. Skelet Muscle. 2011;1:4. doi: 10.1186/2044-5040-1-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Chen SE, Jin B, Li YP. TNF-alpha regulates myogenesis and muscle regeneration by activating p38 MAPK. Am J Physiol Cell Physiol. 2007;292:C1660–71. doi: 10.1152/ajpcell.00486.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Li YP. TNF-alpha is a mitogen in skeletal muscle. Am J Physiol Cell Physiol. 2003;285:C370–6. doi: 10.1152/ajpcell.00453.2002. [DOI] [PubMed] [Google Scholar]
- 66.Rodgers JT, et al. mTORC1 controls the adaptive transition of quiescent stem cells from G0 to G(Alert) Nature. 2014;510:393–6. doi: 10.1038/nature13255. A study reporting two states of satellite cell quiescence, a G0 and the GAlert state. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Kouzarides T. Chromatin modifications and their function. Cell. 2007;128:693–705. doi: 10.1016/j.cell.2007.02.005. [DOI] [PubMed] [Google Scholar]
- 68.Bracken AP, Helin K. Polycomb group proteins: navigators of lineage pathways led astray in cancer. Nat Rev Cancer. 2009;9:773–84. doi: 10.1038/nrc2736. [DOI] [PubMed] [Google Scholar]
- 69.Palacios D, et al. TNF/p38alpha/polycomb signaling to Pax7 locus in satellite cells links inflammation to the epigenetic control of muscle regeneration. Cell Stem Cell. 2010;7:455–69. doi: 10.1016/j.stem.2010.08.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Juan AH, et al. Polycomb EZH2 controls self-renewal and safeguards the transcriptional identity of skeletal muscle stem cells. Genes Dev. 2011;25:789–94. doi: 10.1101/gad.2027911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Woodhouse S, Pugazhendhi D, Brien P, Pell JM. Ezh2 maintains a key phase of muscle satellite cell expansion but does not regulate terminal differentiation. J Cell Sci. 2013;126:565–79. doi: 10.1242/jcs.114843. [DOI] [PubMed] [Google Scholar]
- 72.Blum R, Vethantham V, Bowman C, Rudnicki M, Dynlacht BD. Genome-wide identification of enhancers in skeletal muscle: the role of MyoD1. Genes Dev. 2012;26:2763–79. doi: 10.1101/gad.200113.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Cao Y, et al. Genome-wide MyoD binding in skeletal muscle cells: a potential for broad cellular reprogramming. Dev Cell. 2010;18:662–74. doi: 10.1016/j.devcel.2010.02.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Zhang K, Sha J, Harter ML. Activation of Cdc6 by MyoD is associated with the expansion of quiescent myogenic satellite cells. J Cell Biol. 2010;188:39–48. doi: 10.1083/jcb.200904144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Hu P, Geles KG, Paik JH, DePinho RA, Tjian R. Codependent activators direct myoblast-specific MyoD transcription. Dev Cell. 2008;15:534–46. doi: 10.1016/j.devcel.2008.08.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Troy A, et al. Coordination of satellite cell activation and self-renewal by Par-complex-dependent asymmetric activation of p38alpha/beta MAPK. Cell Stem Cell. 2012;11:541–53. doi: 10.1016/j.stem.2012.05.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Hausburg MA, et al. Post-transcriptional regulation of satellite cell quiescence by TTP-mediated mRNA decay. Elife. 2015;4 doi: 10.7554/eLife.03390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Diao Y, et al. Pax3/7BP is a Pax7- and Pax3-binding protein that regulates the proliferation of muscle precursor cells by an epigenetic mechanism. Cell Stem Cell. 2012;11:231–41. doi: 10.1016/j.stem.2012.05.022. [DOI] [PubMed] [Google Scholar]
- 79.Kawabe Y, Wang YX, McKinnell IW, Bedford MT, Rudnicki MA. Carm1 regulates Pax7 transcriptional activity through MLL1/2 recruitment during asymmetric satellite stem cell divisions. Cell Stem Cell. 2012;11:333–45. doi: 10.1016/j.stem.2012.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Crist CG, Montarras D, Buckingham M. Muscle satellite cells are primed for myogenesis but maintain quiescence with sequestration of Myf5 mRNA targeted by microRNA-31 in mRNP granules. Cell Stem Cell. 2012;11:118–26. doi: 10.1016/j.stem.2012.03.011. [DOI] [PubMed] [Google Scholar]
- 81.McKinnell IW, et al. Pax7 activates myogenic genes by recruitment of a histone methyltransferase complex. Nat Cell Biol. 2008;10:77–84. doi: 10.1038/ncb1671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Rocheteau P, Gayraud-Morel B, Siegl-Cachedenier I, Blasco MA, Tajbakhsh S. A subpopulation of adult skeletal muscle stem cells retains all template DNA strands after cell division. Cell. 2012;148:112–25. doi: 10.1016/j.cell.2011.11.049. [DOI] [PubMed] [Google Scholar]
- 83.Kuang S, Kuroda K, Le Grand F, Rudnicki MA. Asymmetric self-renewal and commitment of satellite stem cells in muscle. Cell. 2007;129:999–1010. doi: 10.1016/j.cell.2007.03.044. Work demonstrating that satellite cells are heterogeneous for Myf5 expression, and that satellite cells that have never experienced Myf5 are capable of reconsituting the satellite cell niche. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Shinin V, Gayraud-Morel B, Gomes D, Tajbakhsh S. Asymmetric division and cosegregation of template DNA strands in adult muscle satellite cells. Nat Cell Biol. 2006;8:677–87. doi: 10.1038/ncb1425. [DOI] [PubMed] [Google Scholar]
- 85.Tierney MT, et al. STAT3 signaling controls satellite cell expansion and skeletal muscle repair. Nat Med. 2014;20:1182–6. doi: 10.1038/nm.3656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Price FD, et al. Inhibition of JAK-STAT signaling stimulates adult satellite cell function. Nat Med. 2014;20:1174–81. doi: 10.1038/nm.3655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Yennek S, Burute M, Thery M, Tajbakhsh S. Cell adhesion geometry regulates non-random DNA segregation and asymmetric cell fates in mouse skeletal muscle stem cells. Cell Rep. 2014;7:961–70. doi: 10.1016/j.celrep.2014.04.016. [DOI] [PubMed] [Google Scholar]
- 88.Ono Y, et al. Muscle stem cell fate is controlled by the cell-polarity protein Scrib. Cell Rep. 2015;10:1135–48. doi: 10.1016/j.celrep.2015.01.045. [DOI] [PubMed] [Google Scholar]
- 89.Le Grand F, Jones AE, Seale V, Scime A, Rudnicki MA. Wnt7a activates the planar cell polarity pathway to drive the symmetric expansion of satellite stem cells. Cell Stem Cell. 2009;4:535–47. doi: 10.1016/j.stem.2009.03.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Chakkalakal JV, et al. Early forming label-retaining muscle stem cells require p27kip1 for maintenance of the primitive state. Development. 2014;141:1649–59. doi: 10.1242/dev.100842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Kim HJ, Bar-Sagi D. Modulation of signalling by Sprouty: a developing story. Nat Rev Mol Cell Biol. 2004;5:441–50. doi: 10.1038/nrm1400. [DOI] [PubMed] [Google Scholar]
- 92.Brack AS, et al. Increased Wnt signaling during aging alters muscle stem cell fate and increases fibrosis. Science. 2007;317:807–10. doi: 10.1126/science.1144090. [DOI] [PubMed] [Google Scholar]
- 93.Fry CS, et al. Inducible depletion of satellite cells in adult, sedentary mice impairs muscle regenerative capacity without affecting sarcopenia. Nat Med. 2015;21:76–80. doi: 10.1038/nm.3710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Jackson JR, et al. Reduced voluntary running performance is associated with impaired coordination as a result of muscle satellite cell depletion in adult mice. Skelet Muscle. 2015;5:41. doi: 10.1186/s13395-015-0065-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Sousa-Victor P, et al. Geriatric muscle stem cells switch reversible quiescence into senescence. Nature. 2014;506:316–21. doi: 10.1038/nature13013. Study showing that induction of p16Ink4a directs a fraction of geriatric satellite cells towards a reversible, scenescent state. [DOI] [PubMed] [Google Scholar]
- 96.Bernet JD, et al. p38 MAPK signaling underlies a cell-autonomous loss of stem cell self-renewal in skeletal muscle of aged mice. Nat Med. 2014;20:265–71. doi: 10.1038/nm.3465. A parallel study demonstrating the aged satellite cells failed to properly self-renew by symmetically distributing p38, leading to two daugher cells committed to myogenic differentiation. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Cosgrove BD, et al. Rejuvenation of the muscle stem cell population restores strength to injured aged muscles. Nat Med. 2014;20:255–64. doi: 10.1038/nm.3464. A key study demonstrating that p38 inhibition in conjunction with culturing on soft-hydrogels restores the robust functional capacity of aged satellite cells. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Collins CA, Zammit PS, Ruiz AP, Morgan JE, Partridge TA. A population of myogenic stem cells that survives skeletal muscle aging. Stem Cells. 2007;25:885–94. doi: 10.1634/stemcells.2006-0372. [DOI] [PubMed] [Google Scholar]
- 99.Munoz-Espin D, Serrano M. Cellular senescence: from physiology to pathology. Nat Rev Mol Cell Biol. 2014;15:482–96. doi: 10.1038/nrm3823. [DOI] [PubMed] [Google Scholar]
- 100.Papaconstantinou J, et al. Attenuation of p38alpha MAPK stress response signaling delays the in vivo aging of skeletal muscle myofibers and progenitor cells. Aging (Albany NY) 2015;7:718–33. doi: 10.18632/aging.100802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Sun L, et al. JAK1-STAT1-STAT3, a key pathway promoting proliferation and preventing premature differentiation of myoblasts. J Cell Biol. 2007;179:129–38. doi: 10.1083/jcb.200703184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Sheehan SM, Allen RE. Skeletal muscle satellite cell proliferation in response to members of the fibroblast growth factor family and hepatocyte growth factor. J Cell Physiol. 1999;181:499–506. doi: 10.1002/(SICI)1097-4652(199912)181:3<499::AID-JCP14>3.0.CO;2-1. [DOI] [PubMed] [Google Scholar]
- 103.Conboy MJ, Conboy IM, Rando TA. Heterochronic parabiosis: historical perspective and methodological considerations for studies of aging and longevity. Aging Cell. 2013;12:525–30. doi: 10.1111/acel.12065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Conboy IM, Conboy MJ, Smythe GM, Rando TA. Notch-mediated restoration of regenerative potential to aged muscle. Science. 2003;302:1575–7. doi: 10.1126/science.1087573. [DOI] [PubMed] [Google Scholar]
- 105.Conboy IM, et al. Rejuvenation of aged progenitor cells by exposure to a young systemic environment. Nature. 2005;433:760–4. doi: 10.1038/nature03260. [DOI] [PubMed] [Google Scholar]
- 106.Villeda SA, et al. The ageing systemic milieu negatively regulates neurogenesis and cognitive function. Nature. 2011;477:90–4. doi: 10.1038/nature10357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Salpeter SJ, et al. Systemic regulation of the age-related decline of pancreatic beta-cell replication. Diabetes. 2013;62:2843–8. doi: 10.2337/db13-0160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Carlson ME, et al. Relative roles of TGF-beta1 and Wnt in the systemic regulation and aging of satellite cell responses. Aging Cell. 2009;8:676–89. doi: 10.1111/j.1474-9726.2009.00517.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Carlson ME, Hsu M, Conboy IM. Imbalance between pSmad3 and Notch induces CDK inhibitors in old muscle stem cells. Nature. 2008;454:528–32. doi: 10.1038/nature07034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Liu H, et al. Augmented Wnt signaling in a mammalian model of accelerated aging. Science. 2007;317:803–6. doi: 10.1126/science.1143578. [DOI] [PubMed] [Google Scholar]
- 111.Biressi S, Miyabara EH, Gopinath SD, Carlig PM, Rando TA. A Wnt-TGFbeta2 axis induces a fibrogenic program in muscle stem cells from dystrophic mice. Sci Transl Med. 2014;6:267ra176. doi: 10.1126/scitranslmed.3008411. An interesting report showing that Wnt and Tgfb1 program a portion of satellite cells to a fibrogenic state in animals harboring a loss of function allele of dystropin (mdx) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Carter CS. Oxytocin pathways and the evolution of human behavior. Annu Rev Psychol. 2014;65:17–39. doi: 10.1146/annurev-psych-010213-115110. [DOI] [PubMed] [Google Scholar]
- 113.Elabd C, et al. Oxytocin is an age-specific circulating hormone that is necessary for muscle maintenance and regeneration. Nat Commun. 2014;5:4082. doi: 10.1038/ncomms5082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.McPherron AC, Huynh TV, Lee SJ. Redundancy of myostatin and growth/differentiation factor 11 function. BMC Dev Biol. 2009;9:24. doi: 10.1186/1471-213X-9-24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Loffredo FS, et al. Growth differentiation factor 11 is a circulating factor that reverses age-related cardiac hypertrophy. Cell. 2013;153:828–39. doi: 10.1016/j.cell.2013.04.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Poggioli T, et al. Circulating Growth Differentiation Factor 11/8 Levels Decline with Age. Circ Res. 2015 doi: 10.1161/CIRCRESAHA.115.307521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Olson KA, et al. Association of growth differentiation factor 11/8, putative anti-ageing factor, with cardiovascular outcomes and overall mortality in humans: analysis of the Heart and Soul and HUNT3 cohorts. Eur Heart J. 2015 doi: 10.1093/eurheartj/ehv385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Sinha M, et al. Restoring systemic GDF11 levels reverses age-related dysfunction in mouse skeletal muscle. Science. 2014;344:649–52. doi: 10.1126/science.1251152. Report describing that young blood containes factors that can rejuvenate some aspects of the muscle defects exhibited by aged animals. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Egerman MA, et al. GDF11 Increases with Age and Inhibits Skeletal Muscle Regeneration. Cell Metab. 2015 doi: 10.1016/j.cmet.2015.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Hathout Y, et al. Large-scale serum protein biomarker discovery in Duchenne muscular dystrophy. Proc Natl Acad Sci U S A. 2015;112:7153–8. doi: 10.1073/pnas.1507719112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Smith SC, et al. GDF11 Does Not Rescue Aging-Related Pathological Hypertrophy. Circ Res. 2015;117:926–32. doi: 10.1161/CIRCRESAHA.115.307527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Rahimov F, Kunkel LM. The cell biology of disease: cellular and molecular mechanisms underlying muscular dystrophy. J Cell Biol. 2013;201:499–510. doi: 10.1083/jcb.201212142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Klingler W, Jurkat-Rott K, Lehmann-Horn F, Schleip R. The role of fibrosis in Duchenne muscular dystrophy. Acta Myol. 2012;31:184–95. [PMC free article] [PubMed] [Google Scholar]
- 124.Blau HM, Webster C, Pavlath GK. Defective myoblasts identified in Duchenne muscular dystrophy. Proc Natl Acad Sci U S A. 1983;80:4856–60. doi: 10.1073/pnas.80.15.4856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Yablonka-Reuveni Z, Anderson JE. Satellite cells from dystrophic (mdx) mice display accelerated differentiation in primary cultures and in isolated myofibers. Dev Dyn. 2006;235:203–12. doi: 10.1002/dvdy.20602. [DOI] [PubMed] [Google Scholar]
- 126.Luz MA, Marques MJ, Santo Neto H. Impaired regeneration of dystrophin-deficient muscle fibers is caused by exhaustion of myogenic cells. Braz J Med Biol Res. 2002;35:691–5. doi: 10.1590/s0100-879x2002000600009. [DOI] [PubMed] [Google Scholar]
- 127.Heslop L, Morgan JE, Partridge TA. Evidence for a myogenic stem cell that is exhausted in dystrophic muscle. J Cell Sci. 2000;113( Pt 12):2299–308. doi: 10.1242/jcs.113.12.2299. [DOI] [PubMed] [Google Scholar]
- 128.Jiang C, et al. Notch signaling deficiency underlies age-dependent depletion of satellite cells in muscular dystrophy. Dis Model Mech. 2014;7:997–1004. doi: 10.1242/dmm.015917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Dumont NA, et al. Dystrophin expression in muscle stem cells regulates their polarity and asymmetric division. Nat Med. 2015 doi: 10.1038/nm.3990. Evidence that dystrophin functions in muscle satellite cells by enforcing asymmetric cellular division, which is required for the production of a sufficient numbers of progenitors for muscle repair. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Sacco A, et al. Short telomeres and stem cell exhaustion model Duchenne muscular dystrophy in mdx/mTR mice. Cell. 2010;143:1059–71. doi: 10.1016/j.cell.2010.11.039. Report introducing the dystrophin/tert1 deficient mouse as a better model that more closely recapitulates the human disorder, and providing evidence that stem cell depletion exacerbates DMD symptoms. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Kottlors M, Kirschner J. Elevated satellite cell number in Duchenne muscular dystrophy. Cell Tissue Res. 2010;340:541–8. doi: 10.1007/s00441-010-0976-6. [DOI] [PubMed] [Google Scholar]
- 132.Kudryashova E, Kramerova I, Spencer MJ. Satellite cell senescence underlies myopathy in a mouse model of limb-girdle muscular dystrophy 2H. J Clin Invest. 2012;122:1764–76. doi: 10.1172/JCI59581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Vieira NM, et al. Muscular dystrophy in a family of Labrador Retrievers with no muscle dystrophin and a mild phenotype. Neuromuscul Disord. 2015;25:363–70. doi: 10.1016/j.nmd.2015.02.012. [DOI] [PubMed] [Google Scholar]
- 134.Vieira NM, et al. Jagged 1 Rescues the Duchenne Muscular Dystrophy Phenotype. Cell. 2015;163:1204–13. doi: 10.1016/j.cell.2015.10.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Boldrin L, Zammit PS, Morgan JE. Satellite cells from dystrophic muscle retain regenerative capacity. Stem Cell Res. 2015;14:20–9. doi: 10.1016/j.scr.2014.10.007. A report demonstrating that escaper Golden Retriever Dogs, which contain inactivating dystrophin mutations, acquired a mutation in the Jagged-1 promoter leading to its activation and enhanced muscle function. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Relaix F, Rocancourt D, Mansouri A, Buckingham M. A Pax3/Pax7-dependent population of skeletal muscle progenitor cells. Nature. 2005;435:948–53. doi: 10.1038/nature03594. [DOI] [PubMed] [Google Scholar]
- 137.Kassar-Duchossoy L, et al. Pax3/Pax7 mark a novel population of primitive myogenic cells during development. Genes Dev. 2005;19:1426–31. doi: 10.1101/gad.345505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Ono Y, et al. Slow-dividing satellite cells retain long-term self-renewal ability in adult muscle. J Cell Sci. 2012;125:1309–17. doi: 10.1242/jcs.096198. [DOI] [PubMed] [Google Scholar]
- 139.Sander JD, Joung JK. CRISPR-Cas systems for editing, regulating and targeting genomes. Nat Biotechnol. 2014;32:347–55. doi: 10.1038/nbt.2842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Gilbert PM, et al. Substrate elasticity regulates skeletal muscle stem cell self-renewal in culture. Science. 2010;329:1078–81. doi: 10.1126/science.1191035. A compelling report showing that satellite cells grown on soft-hydrogels, that mimic in vivo biophyscial properties, improve their survival and self-renewal during ex vivo culturing. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Briggs D, Morgan JE. Recent progress in satellite cell/myoblast engraftment -- relevance for therapy. FEBS J. 2013;280:4281–93. doi: 10.1111/febs.12273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Asakura A, et al. Increased survival of muscle stem cells lacking the MyoD gene after transplantation into regenerating skeletal muscle. Proc Natl Acad Sci U S A. 2007;104:16552–7. doi: 10.1073/pnas.0708145104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Fu X, et al. Combination of inflammation-related cytokines promotes long-term muscle stem cell expansion. Cell Res. 2015;25:655–73. doi: 10.1038/cr.2015.58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Elster JL, et al. Skeletal muscle satellite cell migration to injured tissue measured with 111In-oxine and high-resolution SPECT imaging. J Muscle Res Cell Motil. 2013;34:417–27. doi: 10.1007/s10974-013-9368-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Bentzinger CF, et al. Wnt7a stimulates myogenic stem cell motility and engraftment resulting in improved muscle strength. J Cell Biol. 2014;205:97–111. doi: 10.1083/jcb.201310035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Cerletti M, Jang YC, Finley LW, Haigis MC, Wagers AJ. Short-term calorie restriction enhances skeletal muscle stem cell function. Cell Stem Cell. 2012;10:515–9. doi: 10.1016/j.stem.2012.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Tang AH, Rando TA. Induction of autophagy supports the bioenergetic demands of quiescent muscle stem cell activation. EMBO J. 2014;33:2782–97. doi: 10.15252/embj.201488278. A study suggesting that the transition from quiescence to activation requires Sirt1-dependant autophagy. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Ryall JG, et al. The NAD(+)-dependent SIRT1 deacetylase translates a metabolic switch into regulatory epigenetics in skeletal muscle stem cells. Cell Stem Cell. 2015;16:171–83. doi: 10.1016/j.stem.2014.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Laplante M, Sabatini DM. mTOR signaling in growth control and disease. Cell. 2012;149:274–93. doi: 10.1016/j.cell.2012.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]