

Abstract
The manzamines represent a class of marine natural products that show considerable promise in the control of malaria but generate GI distress in rodents when administered orally in high doses. In an effort to generate manzamine prodrugs with improved antimalarial activity and reduced GI toxicity, we prepared acetylated 8-hydroxymanzamine A analogues including 8-acetoxymanzamine A (3) and 8,12-diacetoxymanzamine A (4), and 8-methoxymanzamine A (5) beginning with 8-hydroxymanzamine A (2). The semisynthetic analogues were assayed for antimalarial and antimicrobial activities, cytotoxicity, and biological and chemical stability. Due to gradual hydrolysis of the ester group, application of monoacetate 3 as an antimalarial prodrug was investigated. The in vitro and in vivo bioassays show that acetylated analogues exhibit significant antimalarial activity (IC50(3) 9.6–30 ng/mL), which are comparable to the parent molecule; however the monoaceate 3 was shown to actually produce higher toxicity at 30 mg/kg when administered orally.
Malaria is a major tropical infectious disease and remains one of the most serious global health challenges. There are four species of Plasmodium that infect humans: P. falciparum, P. vivax, P. ovale, and P. malariae. Among these four species most deaths are caused by P. falciparum, which yields complications such as cerebral malaria, severe anemia, and renal and pulmonary failure. Worldwide, malaria is prevalent in 100 countries and is responsible for 273 million clinical cases and 1.12 million deaths annually. About 2.1 billion people are estimated to be at risk. An estimated 76% of productive life years are lost due to malaria.1,2
Numerous attempts to control malaria have been made, including exploitation of the P. falciparum, Anophles gambiae, and human genome for the development of new vaccines and drugs.3–5 The challenge in malaria management is to overcome the spread of drug-resistant P. falciparum strains. Global concerns regarding malaria continue to increase as a result of the widespread development of P. falciparum resistance to the antimalarial drug chloroquine. Resistance to all antimalarial drugs has been reported at varied rates for each treatment. Indeed, in some parts of Asia resistance to all antimalarial drugs generates serious clinical complications.5–7 The emergence of drug resistance in most malaria-endemic regions has fueled the need for aggressive controls through either management of the insect vector or the development of new antimalarial drugs. Artemisinin and its derivates rapidly reduce parasite levels but suffer from chemical, biopharmaceutical, and treatment compliance issues, leaving an unmet need for the discovery of new and unique antimalarial drug classes. Antimalarial drugs must demonstrate efficacy and safety and also have additional properties such as oral availability and effective single dose or short dosing schedules.8
Since Higa’s first report in the mid 1980s of manzamine A (1) from the Okinawan sponge of the genus Haliclona,9 more than 60 manzamine and related alkaloids have been isolated from 16 species of sponges belonging to nine different genera.10,11 Manzamine A is an important new class of antimalarial leads with potent in vitro activity against chloroquine-, quinine-, and pyrimethamine-resistant strains of P. falciparum. In addition 2 is one of the common manzamine alkaloids that has been isolated from sponges of the genera Amphimedon, Xestospongia, Acanthostrongylophora, and Pachypellina.12–14 A reported single intraperitoneal and oral administration of manzamine alkaloid 1 or 2 prolonged the survival of P. bergei-infected mice more than 10 days, far exceeding the chloroquine and artemisin survival times.15
Despite highly promising antimalarial activity, toxicity is the major problem for utilizing alkaloids 1 and 2 as drugs, and this issue was the driving force for this study. Whereas comprehensive structure–activity relationship (SAR) studies of both natural products are being investigated, it is noteworthy to explore manzamine prodrugs for potentially less toxic drug leads, which are currently limited by GI distress. As a part of this strategy, we selected 2 as a starting material for modification at the 8-position in an attempt to yield comparable activity and reduced toxicity relative to manzamine A. Our hypothesis, based on numerous examples from the pharmacopoeia, was that the protection of the phenoxy group of the β-carboline ring may reduce toxicity of analogue 2 and yield the antimalarial metabolite after hydrolysis in vivo. Previous attempts for preparation of 3 using common acetylation methods in the presence of acetic anhydride and moderate bases such as potassium carbonate failed. Therefore, it appeared that utilization of a Lewis acid catalyst (BF3·OEt2) for acetylation would secure the irreversible generation of acetate, provided that we apply a proper solvent in order to avoid solvolysis of the unstable acetoxy group.
We anticipated that generating acetylated manzamine analogues may create less toxic prodrugs that could be converted into active drugs in the GI tract. Due to inherent hydrolytic instability of analogue 3 to an esterase or a nonenzymatic hydrolysis, we assayed the in vivo antimalarial activity and toxicity of 3 with rats at 0.1 N HCl.
In 1994, Scheuer et al. reported the semisynthesis of analogue 5 from the alkaloid 2 (free base) in almost quantitative yield for structure elucidation purposes.13 Interestingly, they discovered that replacement of a hydroxyl group with methoxy at the 8-position reduced the toxicity of compound 5 in a KB carcinoma cell line. Furthermore, the cytotoxity increased against LoVo and exhibited no activity against HSV-II. To date, there were no reports regarding the activity against malaria, bacteria, or Vero toxicity. In comparison to acetylated analogues 3 and 4, we believe that semisynthetic alkaloid 5 is a good candidate for further biological evaluation and pharmacokinetic studies.
The use of HPLC-HRMS permitted identification, quantification, evaluation, and stability analysis of the natural and semisynthetic products. High-resolution micro LC-TOF-ESI-MS was used to report the stability and provide data for a kinetic study of structurally modified 8- and 12-substituted manzamine analogues.
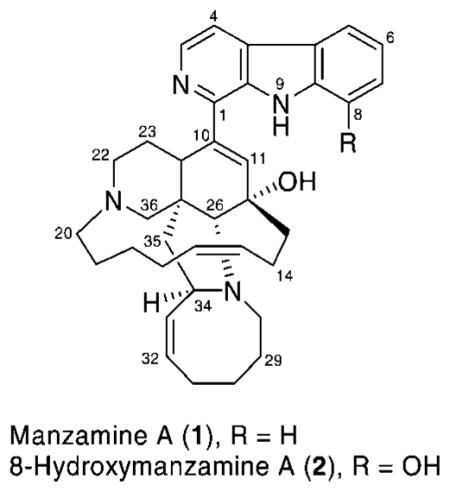
We recently reported glycogen synthase kinase-3 (GSK-3) and cyclin-dependent kinase (CDK5) as primary targets for the manzamine class of alkaloids.16 GSK-3β functions as a mediator for phosphorylation of glycogen synthase, an enzyme that stores excess glucose residues in a polymeric chains (glycogen). Inhibition of GSK-3 has also been reported as a possible treatment for malaria.17 To date, three isoforms of GSK-3 including GSK-3α, GSK-3β, and GSK-3β2 have been determined in mammals. In vitro assay of manzamine A exhibited activity against GSK-3 kinases with IC50 values of 10 and 25 μM for GSK-3β and GSK-3α, respectively. This result suggests that a possible target for manzamine against malaria is its inhibition of parasite kinases.
Results and Discussion
Semisynthesis
Ramirez et al. reported that the acetylation of secondary and tertiary alcohols could be readily achieved by treatment with acetic anhydride and BF3·OEt2 at room temperature in just a few seconds.18 We found this procedure to be both clean and straightforward without major side reactions. These conditions were compatible with the sensitive manzamine molecule bearing several functional groups, whereas commonly utilized basic media were expected to decompose the alkaloid. In addition, while acetylation of individual phenoxy groups has been reported in the literature, acetylation of 8-hydroxy on the β-carboline moiety in the vicinity of other functional groups of the manzamine alkaloids, and in particular acetylation of a tertiary alcohol at the C-12 position, may emerge with more challenges. Indeed, in practice, we encountered unexpected difficulties, especially in the control of the reaction time and purification of unstable acetates.
Treatment of 2 with acetylating reagent BF3·OEt2 and acetic anhydride in dichloromethane resulted in the formation of 3 in good yield. No trace of acetylated 1 under the same reaction conditions validates the high specificity of this reaction (Scheme 1).
Scheme 1.

Stirring of the reaction mixture for 12 h under the same conditions led to the formation of the diacetate product 4. All attempts to purify analogue 4 with flash column chromatography and preparative silica gel TLC decomposed the product. Finally, we succeeded in purification of the impure mixture by using gradient reversed-phase HPLC on a C8 column, eluting with a mixture of water/acetonitrile. Diacetate 4 was obtained as a fluffy white solid in moderate yield and was stored at −20 °C.
Following a literature method,13 the O-methylation of 2 as the hydrochloride salt was carried out with freshly prepared diazomethane in CH2Cl2 at ambient temperature for 12 h to yield analogue 5 in near quantitative yield as a pale yellow solid.
The structure of analogue 3 was confirmed with spectroscopic and MS techniques. The high-resolution ESIMS of this analogue showed the [M+H]+ ion peak at m/z 607.3648 amu in positive mode, in accordance with its molecular formula (C38H47N4O3).
The 1H NMR spectrum of 3 in CD2Cl2 in 400 MHz clearly indicated the presence of a new singlet methyl group at 2.58 (s, 3H, CH3CO) ppm, while the NH group of the β-carboline moiety and tertiary alcohol still remained distinct at 11.43 (s, 1H, NH) and 6.01 (s, 1H, OH) ppm, respectively. This finding was unambiguously confirmed by a new carbonyl signal at 170.0 ppm and a methyl singlet at 21.9 ppm in the 13C NMR spectrum. The HMBC spectrum of 3 supported the acetylation of the 8-OH group, as shown by a correlation between a CH3 singlet at δ 2.58 and a carboxyl signal at δ 170.0 (Supporting Information). The HMBC correlation between the free NH of the β-carboline and the adjacent aromatic carbons at 124.2 (C-4b), 129.6 (C-4a, C-8a), and 133.5 (C-9a) ppm, along with correlation between the tertiary alcohol and carbons at 39.0 (C-13), 70.4 (C-12), and 135.9 (C-11) ppm, clearly revealed that O-acetylation had occurred only on the phenolic alcohol. The tertiary alcohol (O-acetylation) and free pyrrolic NH (N-acetylation) were not involved in these Lewis acid-type catalyzed acetylation reactions.
High-resolution TOF-ESIMS of analogue 4 provided a molecular mass of [M + H]+ at m/z 649.3751, corresponding to the addition of two acetyl groups relative to the parent molecule. Spectroscopic studies of compound 4 were carried out at 600 MHz in C6D6 as an inert solvent in order to exclude any possible solvent-catalyzed degradation. The 1H NMR of compound 4 contained two methyl proton signals at δ 2.19 (s, 3H) and 1.78 (s, 3H), which were clearly correlated to the corresponding carbon signals at δ 167.9 and 170.9, respectively, in the HMBC experiment. Closer inspection of 1H NMR and 13C NMR data for 4 revealed the presence of olefinic methine protons including H-11 (δ 6.26 (s)), H-15 (δ 5.64 (m)), H-16 (δ 5.57 (s)), H-32 (δ 5.12 (t)), and H-33 (δ 5.71 (m)) along with corresponding carbons at C-11 (δ 134.7), C-15 (δ 133.3), C-16 (δ 127.6), C-32 (δ 131.4), and C-33 (δ 132.5), respectively, in agreement with structure 2, and these data revealed that diacetate 4 remains stable in benzene. Significant differences in 1H NMR chemical shifts, comprised of considerable shielding for the protons of the phenyl ring (Δδ = 0.11–0.31) with a concomitant upfield shift of H-11 (Δδ = 0.70), were observed in conjunction with the anisotropic effect of the ester groups. Furthermore, due to the inductive effect of the ester, the chemical shifts of C-8 and C-12 in compound 4 now resonated upfield at δ 137.1 and 84.3. Unambiguous confirmation of O-acetylation rather than N-acetylation was secured by the observation of HMBC correlations between H-9 (NH) and C-9a (δ 134.2), C-8a (δ 133.7), C-4a (δ 129.0), and C-4b (δ 124.7) (Supporting Information).
Stability Study
LC-MS analysis of monoacetate 3 in MeOH as a function of time showed rapid conversion to starting material 2 over a period of 15 h (Supporting Information). The proton and carbon NMR spectra, as well as high-resolution ESIMS analysis of the resulting starting material, were identical with those of an authentic sample of alkaloid 2.
We also completed further investigations regarding the stability of 3 in various solvents. Subsequent experiments revealed that analogue 3 hydrolyzed fastest in MeOH (15 h, 98%) as compared to other solvents such as EtOH, MeCN/H2O, and MeCN/HCl (0.1 N). Hydrolysis of analogue 3 in DMSO and IPA was negligible, and particularly in MeCN and DMSO we found no detectable trace of hydroxyl moiety 2 (Figure 1). As a result, we were confident regarding the stability of the samples during various in vitro biological assays in DMSO.
Figure 1.

Stability of monoacetate 3 vs time in various solvents.
The diacetate product 4 was unstable and easily degraded to a mixture of unknown species in the presence of several tested solvents with the exception of benzene, in which no changes were observed even after a few days of 1H NMR acquisition (Supporting Information). Particularly, 1H NMR studies unambiguously confirmed decomposition of compound 4 in CDCl3 and CD2Cl2 accompanied by a change in the appearance of the solution from colorless to brownish-yellow after 24 h.
The stability of analogue 5 was confirmed in different solvents over a period of two months with LC-MS; no expected hydrolysis to compound 2 was observed.
Biological Activity
To compare the activity and toxicity of these possible prodrugs with the parent compound, all synthetic analogues (3–5) were evaluated for the in vitro antimalarial and antimicrobial activity in addition to cytotoxicity using mammalian cells.
The antimalarial activity of each analogue is shown in Table 1. Analogues 3–5 exhibited antimalarial activity against the chloroquine-sensitive (D6) and the chloroquine-resistant (W2) strains of P. falciparum with values ranging from 9 to 1300 ng/mL. Compound 3, with an IC50 of 9.6 and 30 ng/mL against the D6 and W2 strains, respectively, displayed the most significant in vitro antimalarial activity of about 0.5-fold less than the parent molecule and artemisinin against the D6 clone. Analogue 4, which contains acetoxy groups at both the 8- and 12 -positions, was the least potent compound, with IC50 values of 1300 and 1200 ng/mL against the D6 and W2 clones, respectively. Analogues 3 and 5 exhibited toxicity to the normal Vero cell line (monkey kidney fibroblast cells), while the diacetate 4 did not show cytotoxicity at the highest tested concentration of 4700 ng/mL. These data indicated that modification of the β-carboline phenoxy group resulted in retained antimalarial efficacy, as well as the toxicity in the case of compound 3. However, further acetylation of 3 at the C-12 hydroxy group (to yield 4) led to the loss of antimalarial activity, indicating that this hydroxyl group is an essential pharmacophore for antimalarial activity.
Table 1.
In Vitro Data against Malaria and Leishmania
| compound |
P. falciparum (D6 clone)
|
P. falciparum (W2 clone)
|
cytotoxicity (Vero)
|
L. donovani
|
|
|---|---|---|---|---|---|
| IC50 (ng/mL) | IC50 (ng/mL) | TC50 (μg/mL) | IC50 (μg/mL) | IC90 (μg/mL) | |
| 1 | 4.2 | 5.0 | 680 | 0.9 | 1.8 |
| 2 | 6.0 | 8.0 | 1100 | 6.2 | 11 |
| 3 | 9.6 | 30 | 370 | 3.4 | 7.0 |
| 4 | 1300 | 1200 | NCa | 16 | 35 |
| 5 | 37 | 47 | 980 | 5 | 24 |
| chloroquine | 14 | 155 | –b | – | – |
| artemisinin | 4.3 | 4.4 | – | – | – |
| pentamidine | – | – | – | 2.6 | 6.9 |
| amphotericin B | – | – | – | 0.4 | 0.36 |
NC, no cytotoxicity (concentration: 4.76 μg/mL).
–, not tested.
Antileishmanial activity against Leishmania donovani promastigotes was determined using an Almar Blue assay. Analogues 3–5 displayed potent in vitro antileishmanial activity, as shown in Table 1, with IC50 values of 3.4, 16, and 5 μg/mL, respectively, and were comparable to the activity of the standard drug pentamidine (IC50 = 2.6 μg/mL) used as a positive control. Their IC90 values ranged from 7 to 35 μg/mL as compared to an IC90 of 6.9 μg/mL for pentamidine. However, they were less potent than amphotericin B (IC50 = 0.40 and IC90 = 0.36 μg/mL). It is noteworthy to point out that analogue 3 displayed improved antileishmanial potency compared to parent molecule 2.
Analogue 3 alongside natural products 1 and 2 was also evaluated for in vivo antimalarial activity against P. berghei (sensitive strain) in a rodent malaria model (Table 2). Assays were conducted at two concentrations of 30 and 10 mg/kg/day × 3 (oral). Treatment started on day 0, i.e., 2 h after the infection, and the compounds were administered as three doses once daily on days 0–2 post-infection. The results, parasitemia on days 7, 10, and 14 post-infection, as well as cure (the mice without parasitemia up to day 28 post-infection), were compared to a positive control group of mice treated with chloroquine at the suppressive dose of 50 mg/ kg/day × 3 (orally). These data revealed that parent molecule 2 is comparatively less active than 1, but is not toxic up to 30 mg/kg/ day × 3 doses (manzamine A shows toxicity at this dose). Analogue 3 reveals significant antimalarial activity and is comparable to 2, but is toxic at 30 mg/kg/day × 3 dose after 14 days of oral administration.
Table 2.
In Vivo Antimalarial Dataa
| compound/drug | dose mg/kg × days | % suppression in parasitemia (% activity)
|
cure | ||
|---|---|---|---|---|---|
| day 7 | day 10 | day 14 | |||
| chloroquine | 50mg×3 | 100 | 100 | 100 | 3/4 |
| 1 | 10mg×3 | 100 | 100 | 90 | 1/4 |
| 2 | 30mg×3 | 100 | 83 | 67 | 0/4 |
| 10mg×3 | 57 | 35 | 32 | 0/4 | |
| 3 | 30mg×3 | 93 | 84 | 48 | 0/3 |
| 10mg×3 | 61 | 51 | 8 | 0/4 | |
Oral treatment, vehicle = 0.1 N HCl.
The antifungal and antibacterial activities of analogues 3–5 against Candida albicans, Cryptococcus neoformans, methicillin-resistant S. aureus (MRSA), and Mycobacterium intracellulare are reported as IC50, MIC, MFC, and MBC in Table 3. Amphotericin B and ciprofloxacin were included as positive controls for comparison. Analogues 3 and 5 showed a significant improvement in activity with IC50 values in the range 0.1–3.0 μg/mL and MIC, MFC, and MBC in the range 0.16–20 μg/mL against all pathogenic bacteria and fungi. In contrast, analogue 4 was inactive against C. albicans and C. neoformans but displayed moderate activity against MRSA and M. intracellulare. These results indicated that the tertiary alcohol group at the 12-position is also crucial for antimicrobial activity.
Table 3.
In Vitro Antimicrobial Activitiesa
| compound |
C. albicans (μg/mL)
|
C. neoformans (μg/mL)
|
MRSA (μg/mL)
|
M. intracellulare (μg/mL)
|
||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| IC50 | MIC | MFC | IC50 | MIC | MFC | IC50 | MIC | MFC | IC50 | MIC | MBC | |
| 1 | 2.0 | –b | – | 3.0 | 10 | – | 0.7 | – | – | 0.35 | 0.63 | – |
| 2 | 10 | 20 | – | 3.0 | 5.0 | – | 6.5 | – | – | 0.25 | 0.31 | – |
| 3 | 1.5 | 2.5 | 5.0 | 1.5 | 2.5 | 5.0 | 0.45 | 0.5 | 20 | 0.1 | 0.16 | >20 |
| 4 | >20 | – | – | >20 | – | – | 2.0 | 5.0 | >20 | 4.5 | 10 | >20 |
| 5 | 3.0 | 10 | >20 | 2.0 | 5.0 | 5.0 | 0.55 | >20 | >20 | 0.15 | 0.63 | 20 |
| amphotericin B | 0.1 | 0.63 | 1.25 | 1.00 | 2.5 | 2.5 | – | – | – | – | – | – |
| ciprofloxacin | – | – | – | – | – | – | 0.06 | 0.25 | 1.0 | 0.25 | 0.5 | >20 |
Amphotericin B and ciprofloxacin are used as positive antifungal and antibacterial controls, respectively. The IC50 is the concentration that affords 50% inhibition of growth. MIC (minimum inhibitory concentration) is the lowest test concentration that yields detectable growth. MFC/MBC (minimum fungicidal/bactericidal concentration) is the lowest test concentration that kills 100% of the organism.
– = not tested.
In summary, due to the instability of ester groups during isolation and structure elucidation of natural products, it is clear that the application of MeOH as a solvent for extraction and purification can lead to significant hydrolysis relative to EtOH. On the basis of in vitro and in vivo studies, derivatization of the 8-OH position of the β-carboline moiety improved the antimalarial and antimicrobial activities, while acetylation of the C-12 hydroxy group drastically decreased in vitro activity.
Supplementary Material
Acknowledgments
We are grateful to F. T. Wiggers for assistance with spectroscopic data and M. Jacob and S. Khan for testing the antimicrobial, antimalarial, and antileishmania activities at the National Center for Natural Products Research. This work was funded by NIH grants NCRR P20 RR021929, NIAID 5RO1AI1036596, facilities improvement grant C06 RR-14503-01, and Center for Disease Control, Cooperative Agreement No. U50/CCU418839.
Footnotes
Supporting Information Available: Experimental details and spectroscopic data for compounds 3 and 4, including 1H, 13C, DEPT 135 NMR, HMQC, and HMBC spectra. This material is available free of charge via the Internet at http://pubs.acs.org.
References and Notes
- 1.Toure YT, Oduola A. Nat Microbiol Rev. 2004;2:276–277. [Google Scholar]
- 2.Heddini A. Int J Parasitol. 2002;32:1587–1598. doi: 10.1016/s0020-7519(02)00187-x. [DOI] [PubMed] [Google Scholar]
- 3.Osta MA, George CK, Fotis KC. Science. 2004;303:2030–2033. [Google Scholar]
- 4.Hemingway J, Craig A. Science. 2004;303:1984–1985. doi: 10.1126/science.1096548. [DOI] [PubMed] [Google Scholar]
- 5.Phillips RS. Clin Microbiol Rev. 2001;14:208–226. doi: 10.1128/CMR.14.1.208-226.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bjorkman A. Int J Parasitol. 2002;32:1637–1643. doi: 10.1016/s0020-7519(02)00192-3. [DOI] [PubMed] [Google Scholar]
- 7.Rosenthal PJ. J Exp Biol. 2003;206:3735–3744. doi: 10.1242/jeb.00589. [DOI] [PubMed] [Google Scholar]
- 8.Enserink M. Science. 2000;287:1956–1958. doi: 10.1126/science.287.5460.1956. [DOI] [PubMed] [Google Scholar]
- 9.Sakai R, Higa T, Jefford CW, Bernardinelli G. J Am Chem Soc. 1986;108:6404–6405. [Google Scholar]
- 10.Hu JF, Hamann MT, Hill RT, Kelly M. The Alkaloids. Vol. 60. Elsevier Science; New York: 2003. pp. 207–285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yousaf M, Hammond NL, Peng J, Wahyuono S, McIntosh KA, Charman WN, Mayer AMS, Hamann MT. J Med Chem. 2004;47:3512–3517. doi: 10.1021/jm030475b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tsuda M, Kobayashi J. Heterocycles. 1997;46:765–794. [Google Scholar]
- 13.Ichiba T, Corgiat JM, Scheuer PJ, Kelly-Borges M. J Nat Prod. 1994;57:168–170. doi: 10.1021/np50103a027. [DOI] [PubMed] [Google Scholar]
- 14.Rao KV, Santarsiero BD, Mesecar AD, Schinazi RF, Tekwani BL, Hamann MT. J Nat Prod. 2003;66:823–828. doi: 10.1021/np020592u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ang KK, Holmes MJ, Higa T, Hamann MT, Kara UA. Antimicrob Agents Chemother. 2000;44:1645–1649. doi: 10.1128/aac.44.6.1645-1649.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hamann MT, Alonso D, Martín-Aparicio E, Fuertes A, Pérez-Puerto MJ, Castro A, Morales S, Navarro ML, Monte-Millán M, Medina M, Pennaka H, Balaiah A, Peng J, Cook J, Wahyuono S, Martínez A. J Nat Prod. 2007;70:1397–1405. doi: 10.1021/np060092r. [DOI] [PubMed] [Google Scholar]
- 17.(a) Doerig C, Billker O, Pratt D, Endicott J. Biochim Biophys Acta. 2005;1754:132–150. doi: 10.1016/j.bbapap.2005.08.027. [DOI] [PubMed] [Google Scholar]; (b) Droucheau E, Primot A, Thomas V, Mattei D, Knockaert M, Richardson C, Sallicandro P, Alano P, Jafarshad A, Baratte B, Kunick C, Parzy D, Pearl L, Doerig C, Meijer L. Biochim Biophys Acta. 2004;1697:181–196. doi: 10.1016/j.bbapap.2003.11.023. [DOI] [PubMed] [Google Scholar]
- 18.Martinez-Pascual R, Vinas-Bravo O, Mezareyes S, Iglesias-Arteaga MA, Sandoval-Ramirez J. Synth Commun. 2004;34:4591–4596. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.