

Summary
The innate immune system distinguishes low-level homeostatic microbial stimuli from those of invasive pathogens, yet we lack understanding of how qualitatively similar microbial products yield context-specific macrophage functional responses. Using quantitative approaches, we found that NF-κB and MAPK signaling were activated at different concentrations of a stimulatory TLR4 ligand in both mouse and human macrophages. Above a threshold of ligand, MAPK were activated in a switch-like manner, facilitating production of inflammatory mediators. At ligand concentrations below this threshold, NF-κB signaling occurred, promoting expression of a restricted set of genes and macrophage priming. Amongst TLR-induced genes, we observed an inverse correlation between MAPK dependence and ligand sensitivity, highlighting the role of this signaling dichotomy in partitioning innate responses downstream of a single receptor. Our study thus reveals an evolutionarily conserved innate immune response system, in which ‘danger discrimination’ is enforced by distinct thresholds for NF-κB and MAPK activation that provide sequential barriers to inflammatory mediator production.
Graphical abstract
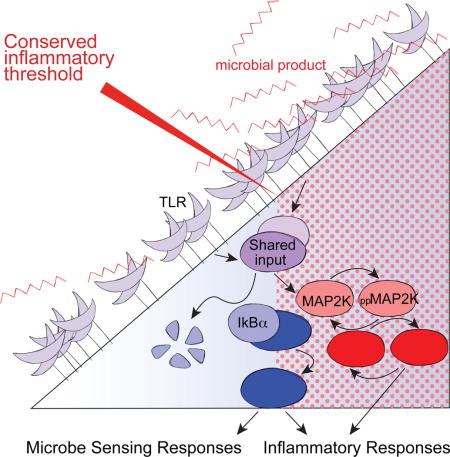
Introduction
Complex multi-cellular organisms employ a variety of immunological sensors to detect microbial invasion and muster host responses to clear or contain infections (Takeuchi and Akira, 2010). Among these, Toll Like Receptors (TLRs) are perhaps the best studied. Located on the cell surface and in endosomes, these trans-membrane proteins generate intracellular signals in response to engagement by conserved molecular patterns associated with microorganisms, such as the lipopolysaccharide (LPS) and peptidoglycan components of bacterial cell walls. At sites of infection, ligation of TLRs on innate immune cells induces rapid production of inflammatory mediators including tumor necrosis factor (TNF) and diverse chemokines. These factors facilitate host defense, but can also lead to inflammation-associated tissue destruction (Croft et al., 2013; Sedger and McDermott, 2014). Indeed, many chronic inflammatory conditions have been linked to dysregulated TLR signaling (Huang and Pope, 2010; Maloy and Powrie, 2011). Aberrant production of TNF stands out as a key driver of disease and blockade of this pathway is being used to effectively treat rheumatoid arthritis, psoriasis and inflammatory bowel disease (IBD) (Croft et al., 2013; Kaser et al., 2010; Melmed and Targan, 2010). Thus, strict regulation of TLR signaling pathways in cells such as macrophages is critical to permit efficient inflammatory cytokine responses toward pathogenic challenges while tolerating ongoing exposure to ligands from commensal microbes or minor tissue damage.
Recent evidence indicates that innate immune cells are not ignorant of microbial stimuli that typically do not induce robust inflammatory responses. Indeed, functionally relevant processing of commensal microbial products is important systemically for efficient immune responses (Abt et al., 2012; Clarke et al., 2010; Ganal et al., 2012; Iida et al., 2013). The innate immune system is thus capable of discriminating between non-dangerous TLR ligation and stimuli requiring potent inflammatory mediator production, reminiscent of antigen discrimination by T lymphocytes, whereby sensing of self-peptide-MHC supports homeostatic function while sufficiently strong T cell receptor (TCR) engagement activates effector function (Morris and Allen, 2012; Stefanova et al., 2002) In contrast to T cell activation, for which the consequences of TCR-ligand density and affinity on signaling and downstream functional responses are well appreciated (Corse et al., 2011), the signaling logic permitting context specific TLR-induced responses has not been delineated.
One variable that distinguishes inputs requiring strong inflammatory responses from those that should be tolerated to avoid unnecessary immunopathology is the higher concentration of TLR ligands sensed during invasive infection. Given the well-recognized requirements for both NF-κB and MAPK in TLR induced inflammatory responses (Figure 1A), a major challenge in understanding regulation of innate immunity is determining how these major signaling pathways translate quantitatively different TLR stimuli into qualitatively distinct responses. Computational modeling has been utilized to identify complex feedback mechanisms that shape the kinetics of robust TLR4-induced NF-κB activity (Basak et al., 2012; Lee et al., 2009; Sung et al., 2014; Werner et al., 2005) and to elucidate gene regulatory circuits allowing cells to differentiate between transient and persistent TLR4 signals (Litvak et al., 2009). However, much less work has so far addressed the quantitative linkage between TLR engagement, intracellular signaling, gene expression, and functional protein production, although it is apparent that such studies are required to better understand how the initiation of inflammatory responses is regulated on a background of homeostatic TLR stimuli.
Here we have conducted a quantitative analysis of concentration-dependent TLR-induced macrophage responses, seeking new insight into mechanisms yielding discrimination between non-dangerous ligand exposure and the presence of stimuli requiring a robust inflammatory response. This analysis revealed an evolutionarily conserved inflammatory threshold controlled by switch-like MAPK activation. In contrast, NF-κB signaling regulated non-inflammatory responses at TLR4 ligand concentrations below those required for MAPK-dependent inflammatory cytokine production. Thus, distinct TLR activation thresholds for NF-κB and MAPK partition concentration-dependent gene expression, minimizing undesirable inflammatory cytokine production during processing characterized by non-dangerous microbial stimuli, while robustly activating a protective response if stimuli exceed the MAPK switching threshold. While the dual-requirement for NF-κB and MAPK in inflammatory responses is well appreciated, these findings highlight a previously unappreciated divergence in the sensitivities of NF-κB and MAPK pathway activation downstream of TLR engagement, providing macrophages with discriminatory power not possible if the pathways behaved in lockstep. Considering the cooperative roles of these pathways in diverse biological systems, this may be a paradigm often used by cells to achieve context specific functional responses downstream of a common input stimulus.
Results
TLR4 stimulation yields a switch-like inflammatory threshold
Lipopolysaccharide (LPS) is present in the serum of healthy individuals (Kelly et al., 2012), but also plays a central role in pathologic inflammatory conditions (Brenchley et al., 2006). To examine dose-dependent induction of potentially tissue-damaging inflammatory mediators, we performed a detailed quantitative single-cell analysis of TLR4-stimulated mouse macrophages, first measuring the dose response characteristics for production of TNF and CCL3. Bone marrow-derived mouse macrophages (BMDM) were stimulated over a concentration range of five orders of magnitude (0.01 to 100 nM) with the TLR4 ligand Kdo2-Lipid A (KLA, the stimulatory portion of LPS) and BMDM expression of TNF and CCL3 protein produced during the first 4 hours of stimulation was measured by flow cytometry (Figure 1B). TLR4-induced production of inflammatory cytokines was switch-like with regard to the frequency of TNF+CCL3+ cells. The majority of cells were found to change from off to on within one order of magnitude of ligand concentration (Figure 1C–1E). This response can be described as ultrasensitive, based on an observed Hill coefficient greater than one (Figure 1E). A similar switch-like threshold for TLR-induced TNF production was observed in peritoneal macrophages (Figure S1). As seen in other immunological processes displaying ultrasensitivity (Altan-Bonnet and Germain, 2005), the sharp nature of this response suggests that mechanisms are in place to allow the system to filter out noise, minimizing inflammatory mediator production in the context of weak TLR4 stimulation.
Figure 1. TLR4 stimulation yields a switch-like inflammatory threshold.
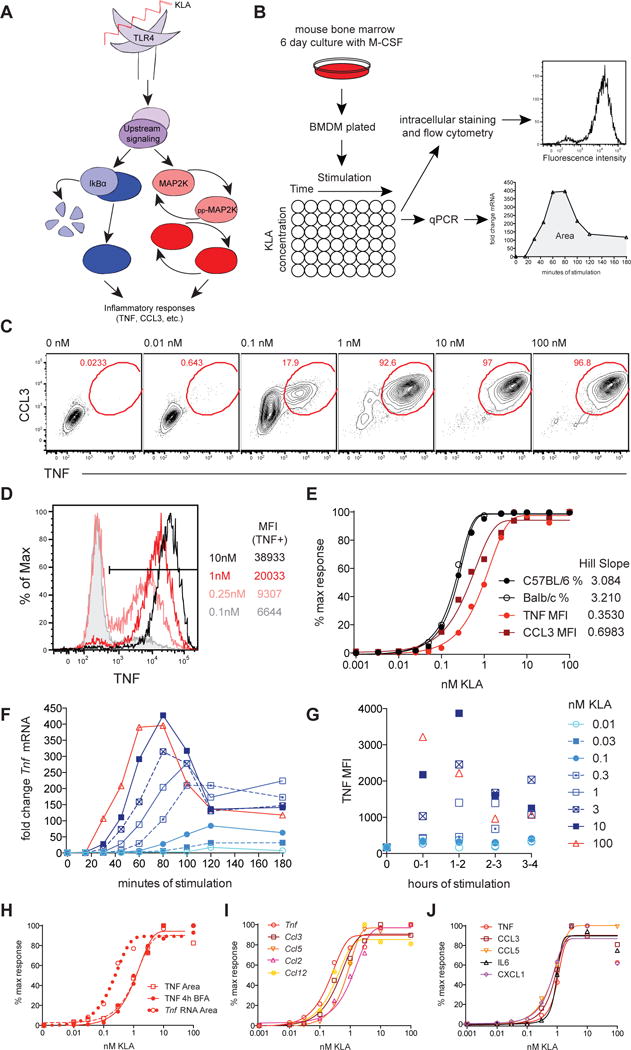
(A) TLR ligation induces inflammatory cytokine production via activation of NF-kB and MAPK (p38, Erk, and Jnk). (B) BMDM, derived from C57BL/6 unless otherwise noted, were stimulated with the indicated concentration of Kdo2-LipidA (KLA) and analyzed by flow cytometry or qPCR. (C–E) BMDM were stained intracellularly for TNF and CCL3 after 4 hours in the presence of Brefeldin A (BFA). (D) TNF response profiles for multiple KLA doses, with the TNF MFI for cells within the positive gate shown on the right. (E) The normalized frequency of TNF+CCL3+ cells (%) or cytokine MFI for the entire population. Timecourse data for Tnf mRNA and TNF protein are shown in (F) and (G) respectively and these dose responses are compared by plotting the area under the curve (Area), plotted with the 4 hour cumulative TNF protein MFI from two independent experiments (H). Dose responses for the indicated inflammatory mediators are were determined using mRNA Area (I) or protein from 4-hour stimulation supernatants (J). Data are representative of 3 or more independent experiments. See also Figure S1.
The mean level of intracellular cytokine and chemokine expressed by positive cells varied with supra-threshold inputs over a limited range of ligand (0.1 to 10nM of KLA, Figure 1C–1E), representing graded TNF and CCL3 production. Thus, while the decision of cells to make inflammatory cytokine is ultrasensitive, the quantity of cytokine produced is not digital, as would be defined by an all-or-none response. Time course analysis of Tnf mRNA and intracellular TNF protein revealed similar dose-dependent kinetics, with TLR ligand concentration impacting both the timing, duration, and amplitude of response peaks, while concentrations below 0.1nM KLA were found to be insufficient to induce robust TNF responses (Figure 1F–1G). Integration of these time course data by calculating the area under the curve (Area) for each KLA concentration identified parallel mRNA and protein dose responses, with mRNA detection being left-shifted (Figure 1H), consistent with additional layers of post-transcriptional regulation affecting protein production (Anderson, 2010; Caldwell et al., 2014). Population-wide TNF mean fluorescence intensity (MFI) measuring protein accumulated over 4 hours of stimulation provides a parameter that integrates both the frequency of responding cells and amount of TNF per cell and recapitulates dose responses calculated from time course data (Figure 1E and 1H). This readout is highly reproducible (closed red circles, Figure 1E and 1H) and will be used to represent the inflammatory threshold in subsequent figures. In addition to TNF and CCL3, we observed a comparable sharp dose response for Ccl2, Ccl5 and Ccl12 at the mRNA level, and for IL6, CCL5, and CXCL1, measured in culture supernatants (Figure 1I and 1J). These data strongly suggest the presence of a common, core inflammatory threshold.
Sub-threshold TLR stimulation primes macrophages without increasing ligand sensitivity
Recent reports have demonstrated that low-level exposure to TLR ligands enhances systemic myeloid cell inflammatory responses (Clarke et al., 2010; Ganal et al., 2012; Iida et al., 2013). Furthermore, priming macrophages with low doses of LPS in vitro results in enhanced inflammatory responses to subsequent stimulation (Deng et al., 2013; Hirohashi and Morrison, 1996). Considering the tightly regulated nature of switch-like TLR-induced inflammatory cytokine production, we sought to examine whether priming influences this threshold. Pre-stimulation of BMDM for two or more hours with 0.03nM KLA, a ligand concentration below the inflammatory cytokine production threshold (Figure 1H–1J), increased the per cell magnitude of TNF generation upon re-exposure to a wide range of KLA concentrations (Figure 2A and 2B). However, pre-stimulation had little influence on the threshold for the concentration-dependent TNF response (Figure 2C): while responding cells made more TNF if pre-stimulated, BMDM did not produce TNF until the ligand concentration reached the same switching point, regardless of priming. These data highlight the presence of a strict concentration-dependent barrier to TNF production, enforcing consistent switch-like inflammatory response thresholds, and suggest that the pathways or factors mediating this barrier are distinct from mechanisms facilitating priming.
Figure 2. Sub-threshold TLR stimulation primes macrophages without increasing ligand sensitivity.
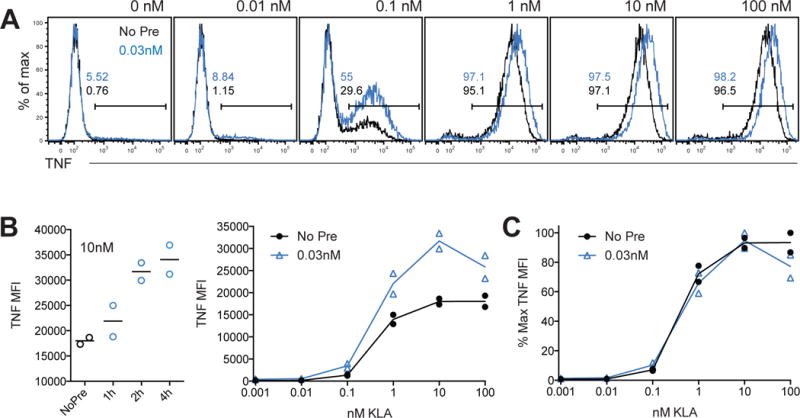
(A,B) BMDM were stimulated with the indicated concentration of KLA for 4 hours in the presence of BFA, with or without pre-stimulation with 0.03nM KLA, for 2h hours unless otherwise indicated (light blue histograms and lines). (C) Data from (B) were re-plotted as a percent of the maximum MFI measured across doses, for BMDM with or without pre-stimulation. Data are pooled from two independent experiments and representative of 3 experiments.
Restricted gene expression is induced below the inflammatory threshold
To gain insight into mechanisms that uncouple inflammatory responses from sensing of low-level microbial stimuli, we performed RNAseq analysis of BMDM stimulated with KLA concentrations above and below the inflammatory threshold. This analysis showed that transcripts of nearly a quarter of TLR4 response genes detected with the high 10nM dose of KLA were statistically significantly upregulated at the sub-threshold concentration of 0.03nM KLA, though generally with a reduced fold-change from baseline as compared to the high concentration of ligand (Figure 3A and 3B). To gain insight into genes with transcripts upregulated by 0.03nM KLA, we used DiRE to identify transcription factors that may be driving these responses. DiRE examines both proximal promoters and predicted distant regulatory elements and calculates both occurrence and importance, which is the product of occurrence and the specificity-based weight assigned to each motif (Gotea and Ovcharenko, 2008); the ten motifs with the highest importance or occurrence are displayed (Figure 3C and Figure S2). Considering the essential role for NF-κB in TLR-induced inflammatory responses, we were surprised to find that NF-κB-p65 was highlighted as the most important transcription factor for genes expressed below the inflammatory threshold, (Figure 3C). NF-κB motifs also stood out as the most highly significant in 0.03nM induced genes using the ToppGene Suite bioinformatics tool (Figure S2) (Chen et al., 2009). The lack of transcription factors highlighted as highly important in the 10nM only gene set, despite known critical roles for NF-κB, AP-1, and IRFs, may reflect the complexity of this transcriptional response. These data suggest that NF-κB contributes to information processing in response to low levels of microbial products, but that activation of this pathway is not sufficient for inflammatory cytokine production. These findings are consistent with in vivo studies demonstrating that constitutive intestinal NF-κB activation does not yield TNF protein production or associated tissue damage unless accompanied by a stimulus leading to generation of active MAPK (Guma et al., 2011).
Figure 3. Restricted NF-κB mediated gene expression occurs below inflammatory threshold.
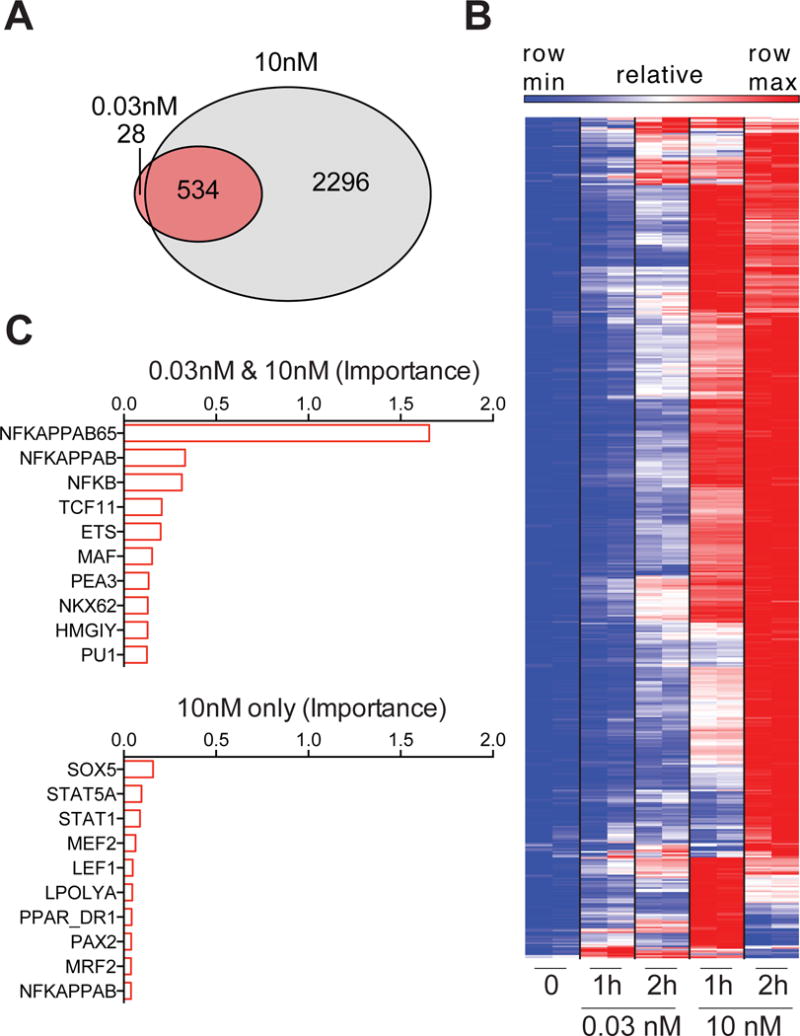
(A–D) BMDM were stimulated with KLA and gene expression was assessed using RNAseq. (A) Venn diagram shows number of genes significantly upregulated at 1 or 2 hours, relative to 0nM KLA stimulated cells (log2 fold change > 0.5 and 1% FDR). (B) DiRE transcription factor motif analysis for the indicated sets of genes, upregulated by both KLA concentrations or exclusively by 10nM (log2 fold change > 0.5 and 1% FDR); importance is a product of motif occurrence (fraction of regulatory elements containing motif) and weight score (motif prevalence compared to background gene list). (C) Relative expression for upregulated genes, significantly changed by both 0.03nM and 10nM KLA (log2 fold change > 0.5 and 1% FDR). RNAseq was performed on pooled RNA from 3 biological replicates, for 2 independent experiments. See also Figure S2.
Distinct NF-κB and MAPK signaling thresholds downstream of a common input
Given the enrichment of NF-κB motifs in genes induced by sub-inflammatory threshold TLR stimulation, we assessed concentration-dependent, TLR-induced NF-κB signaling by measuring levels of IκBα, an inhibitor whose degradation reports upstream IKK activity and results in the release of active NF-κB into the nucleus (Hayden and Ghosh, 2008). To allow for a consistent comparison of dynamic signaling events across ligand concentrations, the mean fluorescence intensity (MFI) for each response was plotted over time and the maximum change in peak height (Peak) or cumulative signal as determined by the area under the curve (Area) was extracted from the time course data (Figure 4A). Over a ligand titration ranging from 0.01 to 1000nM KLA and a dense two-hour time course analysis, we observed that the concentration of TLR4 ligand impacted both the timing and amplitude of the NF-κB response, with lower doses resulting in delayed and decreased IκBα degradation (Figure 4B). In contrast to the sharp dose curve for TLR4-induced inflammatory responses and the reported digital nature of NF-κB responses in non-hematopoietic cells (Tay et al., 2010), degradation of IκBα was more analog and was apparent at the lowest concentrations of ligand tested (Figure 4B and 4C), including those failing to evoke inflammatory cytokine production (Figure 4C, Figure 1). As expected, IkBα degradation at sub-threshold TLR4 ligand concentrations was coincident with increased nuclear localization of NF-κB p65 (Figure S3), consistent with previous evidence of analog NF-κB nuclear localization in macrophages (Sung et al., 2014). Together with our transcription factor motif analysis (Figure 3), these data suggest that NF-κB pathway activation occurs in response to weak TLR4 stimulation, but that this sensitive response does not induce inflammatory protein production to the same degree, pointing to the existence of additional concentration-dependent regulation for this functional output.
Figure 4. Distinct NF-κB and MAPK signaling thresholds downstream of a common input.
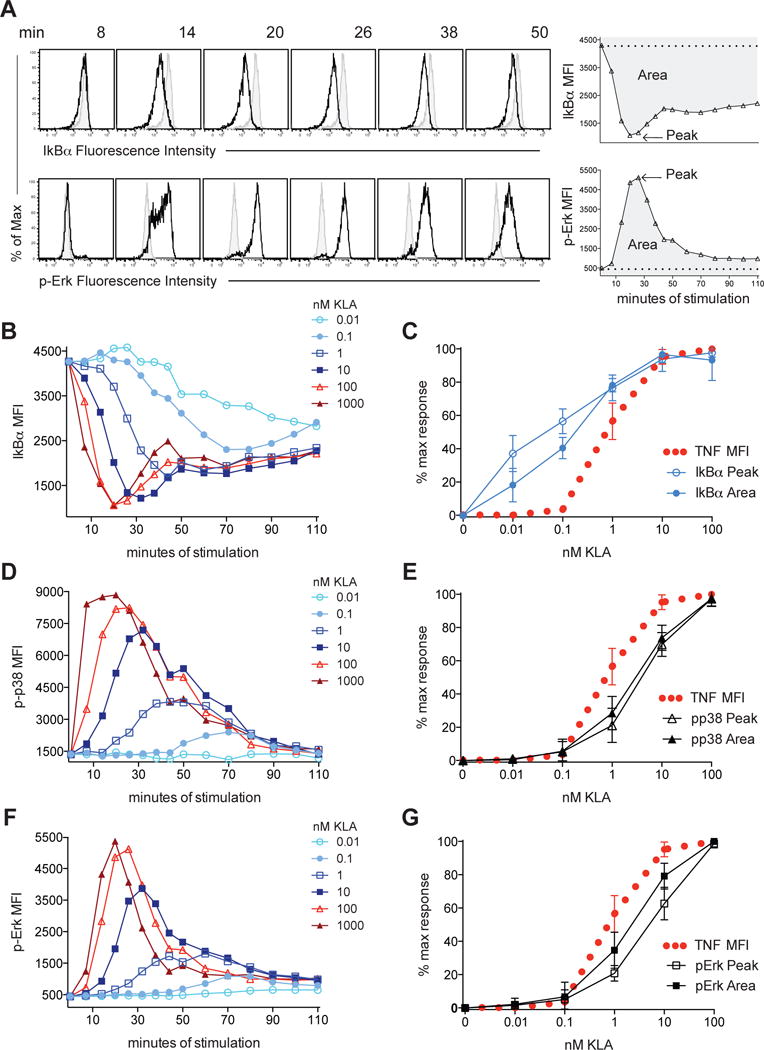
BMDM were stimulated with the indicated concentration of KLA and signaling intermediates were analyzed by flow cytometry. (A) Representative flow cytometry data (shaded histograms represent 0nM KLA stimulated cells) and schematic of data analysis. (B, D, F) MFI are plotted for each concentration of KLA over time and (C, E, G) the peak/maximum MFI (Peak) and area under the curve (Area) for each concentration of ligand was extracted from time course data and plotted over the dose response for TNF. Data in (A, B, D, F) are from one of 4 independent experiments pooled in (C, E, G), showing mean and standard deviation. See also Figure S3 and Figure S4.
NF-κB is necessary but not sufficient for TNF protein production, that, like other inflammatory mediators, requires activation of MAPK to regulate cytokine expression through a variety of transcriptional and post-transcriptional mechanisms (Anderson, 2010; Arthur and Ley, 2013; Caldwell et al., 2014; Guma et al., 2011). The MAPK cascade has been extensively studied in other signaling systems for its potential to set switch-like thresholds for cellular responses (Huang and Ferrell, 1996). In light of this, we hypothesized that distinct thresholds for NF-κB and MAPK activation could uncouple inflammatory responses from NF-κB activation. To examine MAPK responses to KLA, we measured phosphorylation of Erk and p38 on their activation loop residues (Thr202/Tyr204 and Thr180/Try182 respectively), and found that, like IκBα degradation, decreasing concentrations of TLR4 ligand resulted in delayed MAPK phosphorylation. However, compared to IκBα, these responses were more dampened at the lowest concentrations of KLA (Figure 4D and 4F). Comparable phospho-Erk dose responses were observed by Western blot analysis (Figure S3). Comparing the Peak and Area response measurements to mediator production at each TLR4 ligand concentration revealed that the MAPK dose response mirrored that of TNF production, with both increasing sharply above the same threshold (Figure 4E and 4G). Similar results were observed in BMDM responding to varying concentrations of heat-killed E. coli; IκBα degradation was detected a concentrations below the shared threshold for detectable MAPK activation and TNF production (Figure S4). Like inflammatory mediator expression, variable mean levels of MAPK activation were observed over a limited range of supra-threshold ligand concentrations, representing output gradation beyond an all-or-none, switch-like response. Together, these data demonstrate disparate sensitivities of the NF-κB and MAPK pathways to activation by TLR4 ligation and are consistent with MAPK regulation both providing a dose-dependent barrier to inflammatory mediator production and shaping the magnitude of the response above this threshold.
MAPK dependence enforces a two-tiered functional response to TLR ligation
The divergence of signaling thresholds downstream of a common ligand-receptor input provides a potential mechanism giving rise to selective responses to low ligand concentrations in the absence of potentially damaging inflammatory cytokine production. This model would predict that TLR-induced responses vary in their dependence on MAPK activity, and that this dependence would determine their relative expression below the MAPK threshold. To assess the relationship between ligand sensitivity and MAPK dependence, we first performed kinetic analysis of mRNA expression for 40 TLR-induced genes for eight ligand concentrations spanning five orders of magnitude using high-throughput qPCR (representative time course data shown in Figure 5A). From these time course data, ligand sensitivity was determined by plotting the area under the curve for each ligand concentration (Figure 5B). Example genes with high or low relative expression at the sub-threshold concentration of 0.1nM KLA are shown in the center and right panel, respectively.
Figure 5. Sub-threshold gene induction inversely correlates with MAPK dependence.
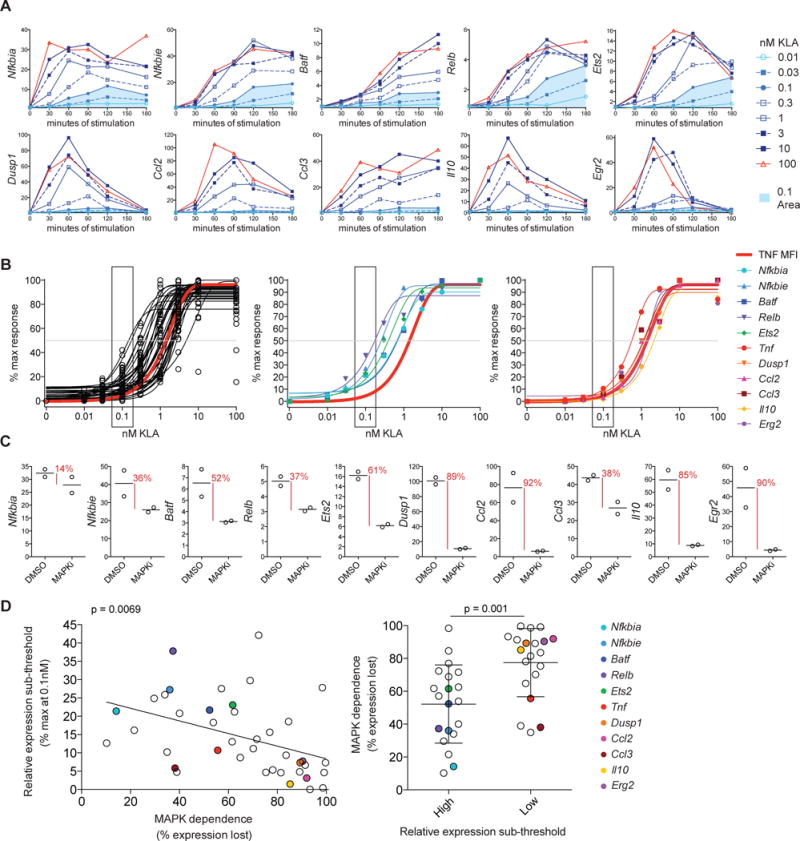
BMDM were stimulated with KLA and mRNA expression for 40 TLR-induced genes was measured using high-throughput qPCR. (A) Example timecourse data showing fold change in response to the indicated concentration of KLA, with area under the curve for 0.1nM shaded in blue. (B) Ligand sensitivities were determined using the area under the curve from timecourse experiments; plots show the distribution of forty genes (left plot) or responses for example genes with high or low relative expression at 0.1nM KLA (middle or right plot, respectively). (C) MAPK dependence was calculated based on the percentage of induction lost, comparing the 10nM fold change at peak timepoint (1h or 2h), with and without MAPK inhibition (MAPKi), MEK1/2 inhibitor U0126 and p38 inhibitor SB203580. (D) The MAPK dependence for the 40 genes assessed is shown as compared to relative induction at 0.1nM for each gene. Statistical significance for the slope of the regression line was determined using an F test and groups were compared using an unpaired t test, for the left and right plot respectively, with a statistical significance cutoff of p≤0.05. Data are representative of two independent experiments. See also Figure S5.
MAPK dependence was determined based on the percentage of maximum induction (seen at 10nM KLA) that was lost with MAPK inhibition, using a combination of MEK1/2 and p38 inhibitors that were previously titrated to determine optimal inhibitor concentration (MAPKi, Figure 5C and Figure S5A). While all TLR-induced genes we examined were influenced by MAPK inhibition (Figure 5C and 5D), genes varied with regard to their magnitude of MAPK dependence, with a subset losing over 80% of expression with MAPKi (Figure 5D). As predicted, these analyses revealed a statistically significant inverse relationship between MAPK dependence and relative expression at concentrations of TLR ligand below those required for robust MAPK activation; genes with lower induction at the sub-threshold concentration of 0.1nM KLA were more dependent on MAPK activity for TLR-induced expression (Figure 5D). Addition of a JNK inhibitor, thus dampening activity in all three MAPK branches, resulted in increased evidence for MAPK dependence, while yielding the same inverse relationship between gene MAPK dependence and relative expression at low ligand concentrations (Figure S5B and S5C). Together these findings are consistent with MAPK-dependent mechanisms shaping dose responses for a critical subset of inflammation-related TLR response genes and suggest that tight control of MAPK activation supports context-dependent NF-κB responses to TLR ligands, likely working in concert with mechanisms tuning NF-κB-mediated transcription in a target-specific manner (Lee et al., 2014).
Aberrant dose-dependent Erk activation can shift TLR ligand threshold for inflammatory cytokine production
The architecture underlying many MAPK cascades supports ultrasensitivity, whereby a graded input is translated into a switch-like output (Huang and Ferrell, 1996); the requirement for dual MAP2KK and MAPK phosphorylation, with concurrent de-phosphorylation by constitutive phosphatases, prevents low concentrations of ligand from activating downstream MAPK. Quantitative kinetic analysis of TLR4 ligand sensitivity in RAW264.7 (RAW) macrophages, a commonly studied mouse myeloid cell line established by Abelson leukemia virus transformation and used in a wide variety of TLR signaling studies (Raschke et al., 1978) (Kong et al., 2007; Sasai et al., 2010; Tang et al., 2014), revealed loss of this tight regulation of Erk activation. In contrast to BMDM, KLA stimulation of RAW macrophages resulted in prolonged Erk phosphorylation (Figure 6A). Interestingly, the slower Erk de-phosphorylation in RAW cells allowed for increased MAPK signal amplitude at low KLA concentrations compared to BMDM. The result was an Erk response lacking ultrasensitivity, and instead resembling the more graded dose response of IκBα degradation, which showed strikingly similar ligand sensitivity in both BMDM and RAW cells (Figure 6A). In contrast to Erk, p38 activity in RAW cells displayed dampened responses to weak stimuli, comparable to both Erk and p38 in BMDM (Figure S6A).
Figure 6. Dysregulated Erk activity is coincident with aberrant TNF production in RAW macrophages.
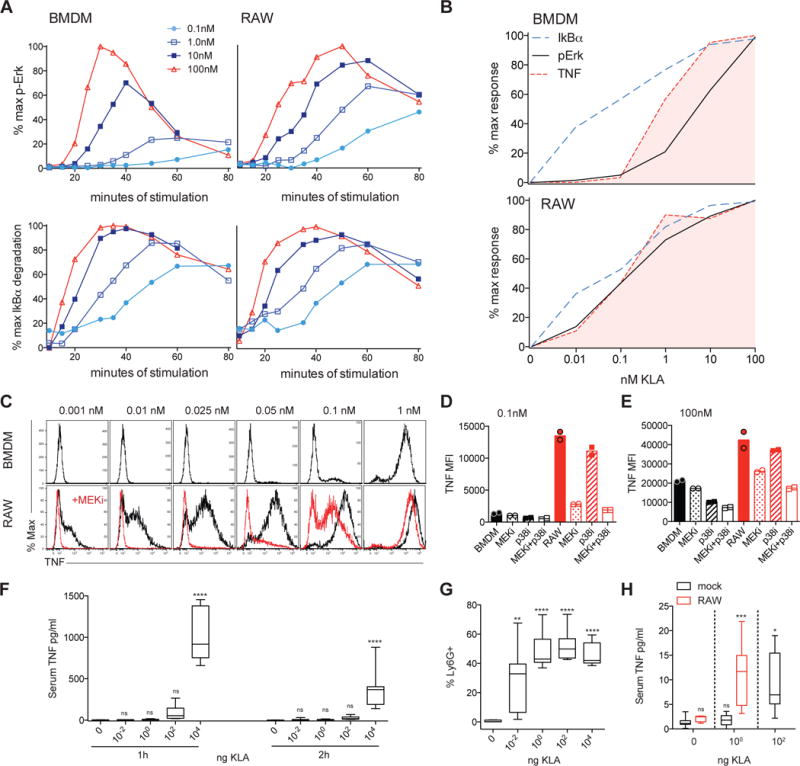
BMDM or RAW were stimulated with the indicated concentration of KLA and signaling intermediates or TNF expression were analyzed by flow cytometry. (A) Erk phosphorylation or IκBα degradation plotted as a percent of maximum response for each cell type. (B) The peak/maximum MFI for each concentration of ligand was extracted from IκBα degradation and Erk phosphorylation time course data, normalized, and plotted over the dose response for TNF production. Select BMDM data are re-plotted from Figure 4, with curves representing the mean of 4 or 3 independent experiments for BMDM or RAW, respectively. (C–E) Duplicate wells of BMDM or RAW were stimulated for 4 hours in the presence of BFA, with or without MEK1/2 inhibitor U0126 or p38 inhibitor SB203580. All in vitro data are representative of 3 or more independent experiments. (F,G) C57Bl/6 mice were injected i.p. with the indicated dose of KLA, data are pooled from two independent experiments (n=7). Serum TNF (F) was assessed at the indicated times and frequency of Ly6G+ neutrophils in peritoneal lavage was assessed at 2 hours (G). (H) Balb/c mice were injected i.p. with 2.5×106 CFSE labeled RAW cells just prior to administration of KLA, as above, and peritoneal macrophages and serum TNF were quantified 1h later; serum TNF levels pooled from 3 independent experiments (n=7, 4, 6, 9 and 6, from left to right). Box and whiskers represent 25th to 75th percentiles, and minimum to maximum values, respectively, and stars represent statistical significance based on ordinary one-way ANOVA, versus control (* p≤0.05; ** p≤0.01; *** p≤0.001, **** p≤0.0001). See also Figure S6.
Consistent with NF-κB and more stringent MAPK activation forming sequential concentration-dependent checkpoints for TNF production, simultaneous activation of both pathways resulted in atypical TNF production by RAW cells stimulated with low KLA concentrations (Figure 6B). To investigate whether this finding represented a causal relationship, with enhanced Erk activity being required for this hypersensitive TNF response, we treated RAW cells with a MEK1/2 inhibitor (MEKi). Treatment with this inhibitor blocked Erk phosphorylation, while IκBα degradation and p38 phosphorylation remained intact and unchanged (Figure S6B). The result was a loss of TNF production at low concentrations of KLA, largely restoring the ligand threshold seen in BMDM (Figure 6C). Inhibition of p38 (p38i) did not recapitulate this effect (Figure 6D), consistent with the abnormally high phosphorylation of Erk but not p38 we saw at low KLA concentrations (Figure S6). However, both MAPK pathways influence the magnitude of TNF expression at supra-threshold concentrations of KLA (Figure 6E). These data suggest that in the context of already-present NF-κB activity, heightened MAPK activity may be sufficient to allow for aberrant TNF production in response to otherwise sub-threshold TLR stimulation.
We sought to evaluate whether the level of dysregulation seen in vitro with RAW macrophages could influence systemic responses. As a framework of reference, we injected wild type mice intraperitoneally (i.p.) with doses of KLA ranging over 6 orders of magnitude. At doses equal to, or less than 1ng of KLA, we did not detect TNF in the serum, despite robust TLR4-induced neutrophil recruitment (Figure 6F and 6G). We then switched to an assay in which RAW cells were challenged with KLA in an in vivo setting, injecting CFSE-labeled RAW cells i.p. with numbers adjusted to achieve frequencies comparable to Tim4+ resident macrophages. Upon subsequent challenge, mice transferred with RAW cells showed a shifted dose response, whereby substantial amounts of serum TNF was present at doses otherwise not eliciting a detectable response in mock-injected mice (Figure 6H); responses in mock-injected mice were comparable to mice transferred with BMDM (Figure S6D). These data highlight multiple dose-dependent thresholds for in vivo inflammatory responses such as local neutrophil recruitment versus systemic TNF levels and suggest that the presence of cells with impaired control of TLR-induced MAPK activation could shift the threshold for aberrant inflammatory responses.
TLR4 inflammatory threshold in human macrophages
The role of MAPK in gating TNF production was seen with BMDM from two strains of mice differing in basal inflammatory propensity (Elson et al., 1996; Gueders et al., 2009), with a very well-conserved overall dose-response for both cell sources (Figure 1C). To examine whether the signaling control characteristics and tight regulation of the response found for mouse macrophages would also govern those of human macrophage inflammatory responses, we generated monocyte-derived macrophages from a set of 15 healthy donors, not pre-selected based on age, gender, or other factors, and stimulated the cells from each donor with a range of KLA concentrations ranging from 0.01 to 100nM KLA, as used in mouse BMDM experiments. The switch-like characteristics of TLR4-induced TNF production were similar in mouse and human macrophages (Figure 7A and 7B and Figure S7 compared to Figure 1C). Notably, the specific ligand threshold for TNF production showed little variability among individuals (Figure 7A and 7B and Figure S7); across donors, minimal TNF production was first detected at 0.1nM of ligand, with 50% of cells responding between the narrow range of 0.3nM and 1nM. As with mouse BMDM, Erk and p38 phosphorylation mirrored the TNF dose response, while IκBα degradation occurred at sub-threshold ligand concentrations (Figure 7C). The conserved nature of the TNF response to TLR4 ligand, across species and individuals, suggests the importance of strict MAPK ‘danger discrimination’, which may protect the host from undesirable and potentially tissue damaging inflammatory responses during processing of non-dangerous microbial stimuli, while robustly activating a protective response if stimuli exceed the MAPK-mediated inflammatory threshold.
Figure 7. Distinct TLR-induced NF-κB and MAPK signaling thresholds and tight dose-dependent TNF regulation in macrophages from diverse human subjects.
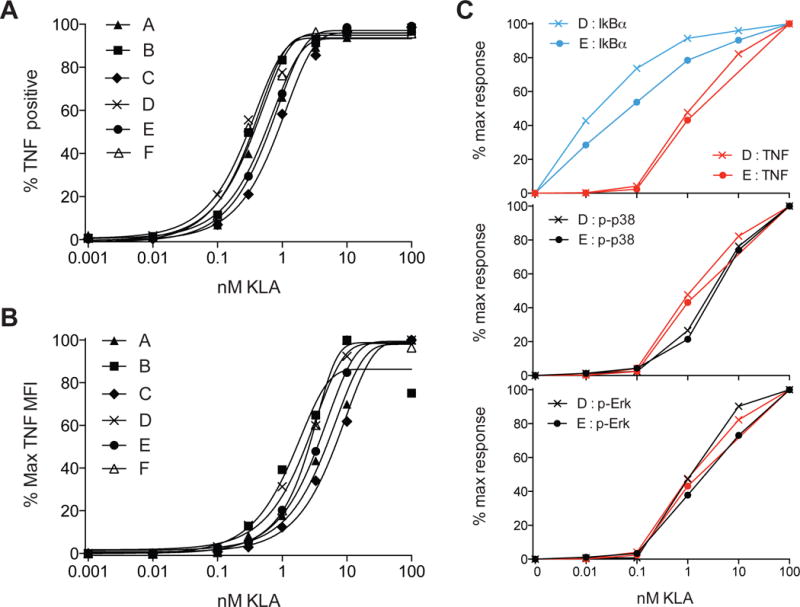
(A–B) Monocyte-derived macrophages from 6 independent healthy donors were stimulated with TLR4 ligand KLA for 4 hours in the presence of BFA. The frequency of TNF positive cells or % maximum MFI for TNF expression is plotted for each individual. (C) Macrophages from two healthy donors were stimulated with varying concentrations of KLA and signaling intermediates then analyzed by flow cytometry. The area under the curve for each concentration of ligand was extracted from time course data of IκBα degradation, p38 phosphorylation, or Erk phosphorylation and plotted over the dose response for TNF production. TNF dose responses are representative of 15 individual donors tested in 3 independent experiments. Signaling responses are representative of 4 individual donors tested in 2 independent experiments. See also Figure S7.
Discussion
Given the ubiquitous nature of TLR ligands and the variety of diseases associated with aberrant TNF production, cells of the innate immune system must exhibit stringent control of TLR-mediated induction of this and other highly inflammatory mediators. In this task, NF-κB and MAPK have essential complementary functions; NF-κB is indispensible for transcription of Tnf and other inflammatory genes, while MAPKs regulate production of these effector molecules through a variety of transcriptional and post-transcriptional mechanisms (Anderson, 2010; Arthur and Ley, 2013; Caldwell et al., 2014). Here, we report the existence of a previously unreported facet of this dual control operating through distinct ligand sensitivities for TLR-induced NF-κB and MAPK signaling that yield sequential barriers to inflammation, with MAPK ultimately shaping the inflammatory threshold. As a result, MAPK activation and inflammatory mediator expression share a combination of ultrasensitive and graded features, and TNF output is scalable by MAPK inhibition. Thus, in the context of licensing by NF-κB, MAPK regulation both thresholds initiation of inflammatory cytokine production and determines protein levels beyond this barrier. This ‘danger discrimination’ is reminiscent of antigen discrimination by T lymphocytes, whereby robust adaptive responses are thresholded by switch-like MAPK activation induced by TCR recognition of foreign ligand (Altan-Bonnet and Germain, 2005; Stefanova et al., 2003).
Our quantitative analysis has revealed the branching of TLR-proximal signals into distal outputs possessing distinct sensitivities to ligand input amount, thus facilitating signal-strength dependent partitioning of downstream functional responses. The TLR-proximal events that lead to activation of NF-κB and the multiple MAPK branches are shared until they diverge at the level of TAK1 (Banerjee and Gerondakis, 2007; Gantke et al., 2011). TAK1 is thought to directly phosphorylate MKK3 and MKK6, leading to activation of p38, and is also responsible for activating IKK, which subsequently phosphorylates IκBα and p105 and tags them for degradation (Banerjee and Gerondakis, 2007; Gantke et al., 2011). While IκBα degradation leads to release and nuclear translocation of NF-κB, the degradation of p105 permits activation of TPL-2 that acts as the MAP3K upstream of MEK1/2, and consequently Erk (Banerjee and Gerondakis, 2007; Gantke et al., 2011). Despite these differences in TAK1 control of MKK or MEK activation, we find that p38 and Erk showed comparably low responses to ligand concentrations eliciting substantial IKK-mediated IκBα degradation. This suggests that while IkBa degradation may reflect a more graded upstream input, this same input is converted into a more switch-like output by established architectural features of the MAPK cascade. Both MAP2K and downstream MAPK require two separate phosphorylation events to be activated. Combined with constitutive reversal of these events by phosphatases, this means that an increasing activating signal is required at each level of the cascade. It has been previously demonstrated these features allow MAPK cascades to filter out noise, responding less than a single-step enzyme to weak stimuli, and then switch from off to on within a narrow range of stimuli (Huang and Ferrell, 1996). This ‘digital’ behavior has been shown to be critical for cells making a ‘yes-no’ decision concerning their differentiation, such as in the case of oocyte activation (Huang and Ferrell, 1996), and applies here to the macrophage decision to make potentially damaging inflammatory mediators.
In contrast to the tight regulation of inflammatory mediator production through MAPK, more graded NF-κB responses to low concentrations of microbial products provide macrophages with an opportunity for ligand-induced changes in phenotype and function that may facilitate homeostatic “education” of myeloid cells by commensal TLR ligands. Indeed, in the absence of commensal stimuli, inflammatory responses upon pathogen infection or other challenge are impaired even at non-mucosal sites (Abt et al., 2012; Clarke et al., 2010; Ganal et al., 2012; Iida et al., 2013), consistent with steady-state systemic dispersion of low concentrations of soluble microbial products, such as LPS and peptidoglycan (Clarke et al., 2010; Kelly et al., 2012). We suggest that low-level TLR signaling involving NF-κB allows macrophages to process these weak stimuli and ready themselves at the level of locus accessibility. This idea is consistent with in vivo findings showing that sensing of commensals led to poised inflammatory genes ready for enhanced transcription factor binding (Ganal et al., 2012); such sensing in the steady-state also results in expression of transcription factors and other molecules less dependent on regulation by MAPK-mediated mechanisms. In support of this model, macrophage priming at low concentrations of TLR4 ligand was coincident with expression of genes containing NF-κB binding motifs. The inverse relationship between gene induction at low concentration and the percent of expression lost with MAPK inhibition, observed in the context of divergent signaling thresholds, supports the a model whereby varying MAPK dependence facilitates partitioning of genes into response categories with distinct ligand sensitivities. Thus, this sequential barrier signaling logic supports context-dependent innate immune responses during processing of ligand levels below or above the danger threshold. At the same time, various features of NF-κB signaling such as extent of nuclear occupancy, fold-change of active NF-κB, and oscillation dynamics in these two parameters, all previously reported in experiments using NF-κB reporter cell lines, likely work across a range of microbial ligand concentrations in concert with switch-like MAPK activation to facilitate context-specific gene expression (Basak et al., 2012; Lee et al., 2014; Sung et al., 2014; Werner et al., 2005).
Ultrasensitive MAPK activation provides the host with protection against undesirable inflammation in response to small degrees of TLR engagement, such as those that presumably occur on a regular basis from systemic dispersion of ligands from the intestinal microbiota (Clarke et al., 2010; Kelly et al., 2012). In response to TLR ligand concentrations sufficient to trigger switch-like MAPK activation, mRNA half-life for Tnf and other inflammatory mediators is increased due to MAPK-dependent mechanisms that block AU-rich motif (ARE)-mediated message destabilization (Deleault et al., 2008; Hao and Baltimore, 2009; Stoecklin et al., 2004). Mice engineered with myeloid-cell specific genetic deletion of Tnf AREs succumb to spontaneous inflammatory disease (Kontoyiannis et al., 2002; Kontoyiannis et al., 1999), in accord with the idea that NF-κB signaling and transcription of Tnf occur in the steady state and that post-transcriptional regulation by MAPK provides a critical second barrier to initiation of aberrant inflammation. GWAS have linked genes for MAPK-regulating phosphatases in the dual-specificity phosphatases (DUSP) family with IBD and colorectal cancer (Fernandez-Rozadilla et al., 2013; Houlston et al., 2010; Jostins et al., 2012; Yang et al., 2014), which are also associated with aberrant TNF production (Croft et al., 2013). It is interesting to speculate that impaired MAPK de-phosphorylation by DUSPs, due to quantitative trait loci–related changes in protein expression, may shift the MAPK dose-response towards the type of higher sensitivity we see in RAW macrophages. Such a dose-response shift may allow for enhanced TNF production in response to normally sub-threshold stimuli, thus contributing to human inflammatory disease.
Here we have used quantitative approaches to gain greater understanding of pathways known to be critical to inflammation but not previously placed in a context that explained a fundamental aspect of innate cell biology—namely, discrimination between dangerous and non-dangerous levels of microbial stimuli. Our findings point to a previously unappreciated divergence in the sensitivities of NF-κB and MAPK pathway activation downstream of single receptor engaging a single ligand. The highly conserved nature of these biochemical and functional responses across species and amongst disparate human subjects emphasizes the protective benefit offered by a signaling system generating multiple thresholds and the need for tight control mechanisms regulating inflammation. Furthermore, our observations offer a mechanism by which innate immune cells balance functionally relevant, homeostatic information processing and the potential risk associated with persistent TLR signaling. Considering the cooperative roles of NF-κB and MAPK in a variety of biological systems, this may be a common paradigm used by cells to achieve disparate functional response sensitivities to a single ligand.
From a broader systems perspective, our findings emphasize the importance of quantitative analysis of signaling in determining how common biochemical pathways are translated into specific physiologic outcomes. Qualitative identification of the relevant kinases, transcription factors, and biochemical interactions, while necessary to achieving such an understanding, is not sufficient for gaining insight into complex decision-making within cells. Such a quantitative view of biology is required to elucidate how potentially modest differences in the expression or activity of pathway components give rise to a shift from physiologic function to pathologic behavior, for example, in the case of disease-associated quantitative trait loci.
Experimental Procedures
Additional methods are described in Supplemental Experimental Procedures
Macrophages and in vitro treatments
Mice were maintained in specific-pathogen-free conditions and all procedures were approved by the NIAID Animal Care and Use Committee (National Institutes of Health, Bethesda, MD). Bone marrow progenitors were differentiated into BMDM during a 6-day culture in complete Dulbecco’s modified Eagle’s medium supplemented with 30ng/ml recombinant mouse M-CSF (R&D systems), added at days 0 and 3. RAW 264.7 cells purchased from ATCC were also maintained in complete DMEM. Human monocyte-derived macrophages were differentiated in X-vivo 15 media containing 100 ng/ml recombinant human M-CSF (R&D Systems). Kdo2-Lipid A (KLA, Avanti Polar Lipids) stimulation was performed in 48 well plates. In some experiments Brefeldin A (BD Golgi plug) was added 30 minutes after KLA, while MEK inhibitor U0126 (10uM Cell Signaling Technology) or p38 inhibitor SB203580 (10uM Cell Signaling Technology) was added 30 minutes prior to KLA.
RNA isolation and microfluidic qPCR
After stimulation, macrophage cells were lysed in Qiazol (Qiagen) and stored at −80 until RNA isolation. RNA isolation was performed using Direct-zol 96-well isolation column plates (Zymo Research). Approximately 10 ng of RNA was reverse transcribed using Superscript VILO cDNA synthesis system (Invitrogen). 1.25 uL of cDNA was combined with 0.5 uL of a pool of 96 primer sets (Deltagene assays, Fluidigm) at 500nM concentration each, 2.5 uL 2× Taqman Preamp Master Mix (Applied Biosystems), and made up to 5 uL total volume per reaction with water, in a low-profile 96-well PCR plate (Bio-rad). Specific Target Amplification was run on a CFX Connect thermal cycler (Bio-rad). Following pre-amplification, unincorporated primers were digested by the addition of Exonuclease 1 (New England Biolabs). Samples were analyzed by qPCR on the Fluidigm Biomark instrument using 96.96 chips according to manufacturer’s instructions (Fluidigm). Data were exported from Fluidigm Real-time PCR Analysis software version 3.1.3, using Linear (Derivative) Baseline method, a global threshold of 0.01, and a 0.65 quality threshold, parameters which were found to exclude non-specific amplification and reduce plate-plate variation.
Flow cytometry and qPCR data processing and statistics
Flow cytometry MFI and percentage values and qPCR fold change values were further processed in Excel (Microsoft) and in Prism (Graphpad). The percent maximum value was calculated in Excel, with subtraction of the baseline value. Area under the curve analysis, linear regression analysis, unpaired t tests, and ordinary one-way ANOVA were performed using Prism. Data were plotted in Prism; unless otherwise indicated, data points represent a single experimental value, with the exception of area under the curve data, for which each data point is an integration of the five or more time points for the given experiment.
Supplementary Material
Acknowledgments
We thank M. Narayanan, A. Nita-Lazar, V. Sjoelund, S.J. Vayttaden and members of the Germain laboratory and the Laboratory of Systems Biology for helpful discussions. This work was supported by the Intramural Research Program of National Institute of Allergy and Infectious Diseases, National Institutes of Health.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Author contributions
Conceptualization, R.A.G. and R.N.G.; Investigation, R.A.G., A.J.M., C.E.N. and S.U.; Formal Analysis and Visualization, R.A.G., A.J.M., B.R.A., and B.D.; Writing - Original Draft, R.A.G.; Writing – Review & Editing, A.J.M., B.R.A., I.D.C.F., M.M.S. and R.N.G; Supervision, J.S.T, I.D.C.F., M.M.S. and R.N.G.
References
- Abt MC, Osborne LC, Monticelli LA, Doering TA, Alenghat T, Sonnenberg GF, Paley MA, Antenus M, Williams KL, Erikson J, et al. Commensal bacteria calibrate the activation threshold of innate antiviral immunity. Immunity. 2012;37:158–170. doi: 10.1016/j.immuni.2012.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altan-Bonnet G, Germain RN. Modeling T cell antigen discrimination based on feedback control of digital ERK responses. PLoS biology. 2005;3:e356. doi: 10.1371/journal.pbio.0030356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson P. Post-transcriptional regulons coordinate the initiation and resolution of inflammation. Nature reviews Immunology. 2010;10:24–35. doi: 10.1038/nri2685. [DOI] [PubMed] [Google Scholar]
- Arthur JS, Ley SC. Mitogen-activated protein kinases in innate immunity. Nature reviews Immunology. 2013;13:679–692. doi: 10.1038/nri3495. [DOI] [PubMed] [Google Scholar]
- Banerjee A, Gerondakis S. Coordinating TLR-activated signaling pathways in cells of the immune system. Immunology and cell biology. 2007;85:420–424. doi: 10.1038/sj.icb.7100098. [DOI] [PubMed] [Google Scholar]
- Basak S, Behar M, Hoffmann A. Lessons from mathematically modeling the NF-kappaB pathway. Immunological reviews. 2012;246:221–238. doi: 10.1111/j.1600-065X.2011.01092.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brenchley JM, Price DA, Schacker TW, Asher TE, Silvestri G, Rao S, Kazzaz Z, Bornstein E, Lambotte O, Altmann D, et al. Microbial translocation is a cause of systemic immune activation in chronic HIV infection. Nature medicine. 2006;12:1365–1371. doi: 10.1038/nm1511. [DOI] [PubMed] [Google Scholar]
- Caldwell AB, Cheng Z, Vargas JD, Birnbaum HA, Hoffmann A. Network dynamics determine the autocrine and paracrine signaling functions of TNF. Genes & development. 2014;28:2120–2133. doi: 10.1101/gad.244749.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J, Bardes EE, Aronow BJ, Jegga AG. ToppGene Suite for gene list enrichment analysis and candidate gene prioritization. Nucleic acids research. 2009;37:W305–311. doi: 10.1093/nar/gkp427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clarke TB, Davis KM, Lysenko ES, Zhou AY, Yu Y, Weiser JN. Recognition of peptidoglycan from the microbiota by Nod1 enhances systemic innate immunity. Nature medicine. 2010;16:228–231. doi: 10.1038/nm.2087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corse E, Gottschalk RA, Allison JP. Strength of TCR-peptide/MHC interactions and in vivo T cell responses. Journal of immunology. 2011;186:5039–5045. doi: 10.4049/jimmunol.1003650. [DOI] [PubMed] [Google Scholar]
- Croft M, Benedict CA, Ware CF. Clinical targeting of the TNF and TNFR superfamilies. Nature reviews Drug discovery. 2013;12:147–168. doi: 10.1038/nrd3930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deleault KM, Skinner SJ, Brooks SA. Tristetraprolin regulates TNF TNF-alpha mRNA stability via a proteasome dependent mechanism involving the combined action of the ERK and p38 pathways. Molecular immunology. 2008;45:13–24. doi: 10.1016/j.molimm.2007.05.017. [DOI] [PubMed] [Google Scholar]
- Deng H, Maitra U, Morris M, Li L. Molecular mechanism responsible for the priming of macrophage activation. The Journal of biological chemistry. 2013;288:3897–3906. doi: 10.1074/jbc.M112.424390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elson CO, Beagley KW, Sharmanov AT, Fujihashi K, Kiyono H, Tennyson GS, Cong Y, Black CA, Ridwan BW, McGhee JR. Hapten-induced model of murine inflammatory bowel disease: mucosa immune responses and protection by tolerance. Journal of immunology. 1996;157:2174–2185. [PubMed] [Google Scholar]
- Fernandez-Rozadilla C, Cazier JB, Tomlinson IP, Carvajal-Carmona LG, Palles C, Lamas MJ, Baiget M, Lopez-Fernandez LA, Brea-Fernandez A, Abuli A, et al. A colorectal cancer genome-wide association study in a Spanish cohort identifies two variants associated with colorectal cancer risk at 1p33 and 8p12. BMC genomics. 2013;14:55. doi: 10.1186/1471-2164-14-55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ganal SC, Sanos SL, Kallfass C, Oberle K, Johner C, Kirschning C, Lienenklaus S, Weiss S, Staeheli P, Aichele P, et al. Priming of natural killer cells by nonmucosal mononuclear phagocytes requires instructive signals from commensal microbiota. Immunity. 2012;37:171–186. doi: 10.1016/j.immuni.2012.05.020. [DOI] [PubMed] [Google Scholar]
- Gantke T, Sriskantharajah S, Ley SC. Regulation and function of TPL-2, an IkappaB kinase-regulated MAP kinase kinase kinase. Cell research. 2011;21:131–145. doi: 10.1038/cr.2010.173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gotea V, Ovcharenko I. DiRE: identifying distant regulatory elements of co-expressed genes. Nucleic acids research. 2008;36:W133–139. doi: 10.1093/nar/gkn300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gueders MM, Paulissen G, Crahay C, Quesada-Calvo F, Hacha J, Van Hove C, Tournoy K, Louis R, Foidart JM, Noel A, et al. Mouse models of asthma: a comparison between C57BL/6 and BALB/c strains regarding bronchial responsiveness, inflammation, and cytokine production. Inflammation research: official journal of the European Histamine Research Society [et al] 2009;58:845–854. doi: 10.1007/s00011-009-0054-2. [DOI] [PubMed] [Google Scholar]
- Guma M, Stepniak D, Shaked H, Spehlmann ME, Shenouda S, Cheroutre H, Vicente-Suarez I, Eckmann L, Kagnoff MF, Karin M. Constitutive intestinal NF-kappaB does not trigger destructive inflammation unless accompanied by MAPK activation. The Journal of experimental medicine. 2011;208:1889–1900. doi: 10.1084/jem.20110242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hao S, Baltimore D. The stability of mRNA influences the temporal order of the induction of genes encoding inflammatory molecules. Nature immunology. 2009;10:281–288. doi: 10.1038/ni.1699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayden MS, Ghosh S. Shared principles in NF-kappaB signaling. Cell. 2008;132:344–362. doi: 10.1016/j.cell.2008.01.020. [DOI] [PubMed] [Google Scholar]
- Hirohashi N, Morrison DC. Low-dose lipopolysaccharide (LPS) pretreatment of mouse macrophages modulates LPS-dependent interleukin-6 production in vitro. Infection and immunity. 1996;64:1011–1015. doi: 10.1128/iai.64.3.1011-1015.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Houlston RS, Cheadle J, Dobbins SE, Tenesa A, Jones AM, Howarth K, Spain SL, Broderick P, Domingo E, Farrington S, et al. Meta-analysis of three genome-wide association studies identifies susceptibility loci for colorectal cancer at 1q41, 3q26.2, 12q13.13 and 20q13.33. Nature genetics. 2010;42:973–977. doi: 10.1038/ng.670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang CY, Ferrell JE., Jr Ultrasensitivity in the mitogen-activated protein kinase cascade. Proceedings of the National Academy of Sciences of the United States of America. 1996;93:10078–10083. doi: 10.1073/pnas.93.19.10078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang Q, Pope RM. Toll-like receptor signaling: a potential link among rheumatoid arthritis, systemic lupus, and atherosclerosis. Journal of leukocyte biology. 2010;88:253–262. doi: 10.1189/jlb.0310126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iida N, Dzutsev A, Stewart CA, Smith L, Bouladoux N, Weingarten RA, Molina DA, Salcedo R, Back T, Cramer S, et al. Commensal bacteria control cancer response to therapy by modulating the tumor microenvironment. Science. 2013;342:967–970. doi: 10.1126/science.1240527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jostins L, Ripke S, Weersma RK, Duerr RH, McGovern DP, Hui KY, Lee JC, Schumm LP, Sharma Y, Anderson CA, et al. Host-microbe interactions have shaped the genetic architecture of inflammatory bowel disease. Nature. 2012;491:119–124. doi: 10.1038/nature11582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaser A, Zeissig S, Blumberg RS. Inflammatory bowel disease. Annual review of immunology. 2010;28:573–621. doi: 10.1146/annurev-immunol-030409-101225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelly CJ, Colgan SP, Frank DN. Of microbes and meals: the health consequences of dietary endotoxemia. Nutrition in clinical practice: official publication of the American Society for Parenteral and Enteral Nutrition. 2012;27:215–225. doi: 10.1177/0884533611434934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kong XN, Yan HX, Chen L, Dong LW, Yang W, Liu Q, Yu LX, Huang DD, Liu SQ, Liu H, et al. LPS-induced down-regulation of signal regulatory protein {alpha} contributes to innate immune activation in macrophages. The Journal of experimental medicine. 2007;204:2719–2731. doi: 10.1084/jem.20062611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kontoyiannis D, Boulougouris G, Manoloukos M, Armaka M, Apostolaki M, Pizarro T, Kotlyarov A, Forster I, Flavell R, Gaestel M, et al. Genetic dissection of the cellular pathways and signaling mechanisms in modeled tumor necrosis factor-induced Crohn’s-like inflammatory bowel disease. The Journal of experimental medicine. 2002;196:1563–1574. doi: 10.1084/jem.20020281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kontoyiannis D, Pasparakis M, Pizarro TT, Cominelli F, Kollias G. Impaired on/off regulation of TNF biosynthesis in mice lacking TNF AU-rich elements: implications for joint and gut-associated immunopathologies. Immunity. 1999;10:387–398. doi: 10.1016/s1074-7613(00)80038-2. [DOI] [PubMed] [Google Scholar]
- Langmead B, Salzberg SL. Fast gapped-read alignment with Bowtie 2. Nature methods. 2012;9:357–359. doi: 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee RE, Walker SR, Savery K, Frank DA, Gaudet S. Fold change of nuclear NF-kappaB determines TNF-induced transcription in single cells. Molecular cell. 2014;53:867–879. doi: 10.1016/j.molcel.2014.01.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee TK, Denny EM, Sanghvi JC, Gaston JE, Maynard ND, Hughey JJ, Covert MW. A noisy paracrine signal determines the cellular NF-kappaB response to lipopolysaccharide. Science signaling. 2009;2:ra65. doi: 10.1126/scisignal.2000599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao Y, Smyth GK, Shi W. featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics. 2014;30:923–930. doi: 10.1093/bioinformatics/btt656. [DOI] [PubMed] [Google Scholar]
- Litvak V, Ramsey SA, Rust AG, Zak DE, Kennedy KA, Lampano AE, Nykter M, Shmulevich I, Aderem A. Function of C/EBPdelta in a regulatory circuit that discriminates between transient and persistent TLR4-induced signals. Nature immunology. 2009;10:437–443. doi: 10.1038/ni.1721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome biology. 2014;15:550. doi: 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maloy KJ, Powrie F. Intestinal homeostasis and its breakdown in inflammatory bowel disease. Nature. 2011;474:298–306. doi: 10.1038/nature10208. [DOI] [PubMed] [Google Scholar]
- Melmed GY, Targan SR. Future biologic targets for IBD: potentials and pitfalls. Nature reviews Gastroenterology & hepatology. 2010;7:110–117. doi: 10.1038/nrgastro.2009.218. [DOI] [PubMed] [Google Scholar]
- Morris GP, Allen PM. How the TCR balances sensitivity and specificity for the recognition of self and pathogens. Nature immunology. 2012;13:121–128. doi: 10.1038/ni.2190. [DOI] [PubMed] [Google Scholar]
- Raschke WC, Baird S, Ralph P, Nakoinz I. Functional macrophage cell lines transformed by Abelson leukemia virus. Cell. 1978;15:261–267. doi: 10.1016/0092-8674(78)90101-0. [DOI] [PubMed] [Google Scholar]
- Sasai M, Linehan MM, Iwasaki A. Bifurcation of Toll-like receptor 9 signaling by adaptor protein 3. Science. 2010;329:1530–1534. doi: 10.1126/science.1187029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sedger LM, McDermott MF. TNF and TNF-receptors: From mediators of cell death and inflammation to therapeutic giants - past, present and future. Cytokine & growth factor reviews. 2014;25:453–472. doi: 10.1016/j.cytogfr.2014.07.016. [DOI] [PubMed] [Google Scholar]
- Stefanova I, Dorfman JR, Germain RN. Self-recognition promotes the foreign antigen sensitivity of naive T lymphocytes. Nature. 2002;420:429–434. doi: 10.1038/nature01146. [DOI] [PubMed] [Google Scholar]
- Stefanova I, Hemmer B, Vergelli M, Martin R, Biddison WE, Germain RN. TCR ligand discrimination is enforced by competing ERK positive and SHP-1 negative feedback pathways. Nature immunology. 2003;4:248–254. doi: 10.1038/ni895. [DOI] [PubMed] [Google Scholar]
- Stoecklin G, Stubbs T, Kedersha N, Wax S, Rigby WF, Blackwell TK, Anderson P. MK2-induced tristetraprolin:14-3-3 complexes prevent stress granule association and ARE-mRNA decay. The EMBO journal. 2004;23:1313–1324. doi: 10.1038/sj.emboj.7600163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sung MH, Li N, Lao Q, Gottschalk RA, Hager GL, Fraser ID. Switching of the relative dominance between feedback mechanisms in lipopolysaccharide-induced NF-kappaB signaling. Science signaling. 2014;7:ra6. doi: 10.1126/scisignal.2004764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takeuchi O, Akira S. Pattern recognition receptors and inflammation. Cell. 2010;140:805–820. doi: 10.1016/j.cell.2010.01.022. [DOI] [PubMed] [Google Scholar]
- Tang S, Chen T, Yu Z, Zhu X, Yang M, Xie B, Li N, Cao X, Wang J. RasGRP3 limits Toll-like receptor-triggered inflammatory response in macrophages by activating Rap1 small GTPase. Nature communications. 2014;5:4657. doi: 10.1038/ncomms5657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tay S, Hughey JJ, Lee TK, Lipniacki T, Quake SR, Covert MW. Single-cell NF-kappaB dynamics reveal digital activation and analogue information processing. Nature. 2010;466:267–271. doi: 10.1038/nature09145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Werner SL, Barken D, Hoffmann A. Stimulus specificity of gene expression programs determined by temporal control of IKK activity. Science. 2005;309:1857–1861. doi: 10.1126/science.1113319. [DOI] [PubMed] [Google Scholar]
- Yang SK, Hong M, Zhao W, Jung Y, Baek J, Tayebi N, Kim KM, Ye BD, Kim KJ, Park SH, et al. Genome-wide association study of Crohn’s disease in Koreans revealed three new susceptibility loci and common attributes of genetic susceptibility across ethnic populations. Gut. 2014;63:80–87. doi: 10.1136/gutjnl-2013-305193. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.