

Abstract
Factor XIa is a plasma serine protease that contributes to thrombin generation primarily through proteolytic activation of factor IX. Traditionally considered part of the intrinsic pathway of coagulation, several lines of evidence now suggest that factor XIa serves as an interface between the vitamin-K dependent thrombin generation mechanism and the pro-inflammatory kallikrein-kinin system, allowing the two systems to influence each other. Work with animal models and results from epidemiologic surveys of human populations support a role for factor XIa in thromboembolic disease. These data, and the clinical observation that deficiency of factor XI, the zymogen of factor XIa, produces a relatively mild bleeding disorder suggest that drugs targeting factor XI or XIa could produce an antithrombotic effect while leaving hemostasis largely intact. Results of a recent trial comparing antisense-induced factor XI reduction to standard dose low molecular weight heparin as prophylaxis for venous thrombosis during knee replacement are encouraging in these regards. Here we discuss recent findings on the biochemistry, physiology and pathology of factor XI as they relate to thromboembolic disease.
Keywords: Factor XI, factor XII, thrombosis
Graphical abstract
Vitamin K antagonists such as warfarin have been mainstays of antithrombotic therapy for more than 50 years. While these drugs demonstrate efficacy across a spectrum of clinical settings, they increase the risk of major bleeding, and frequent monitoring is required to maintain the drug effect within a narrow therapeutic window [1]. In 2005, Hirsh and colleagues described features of an ideal anticoagulant that included a high efficacy-to-safety index and a predictable dose response that obviates the need for laboratory monitoring [2]. Direct oral anticoagulants (DOACs) that target thrombin or factor Xa are improvements on warfarin in these regards. They are at least as effective as warfarin for preventing stroke in patients with atrial fibrillation and treating venous thromboembolism (VTE), and are as effective as low molecular weight heparin for VTE prophylaxis in hip or knee arthroplasty [3]. They also appear to be safer than warfarin with lower rates of intracranial bleeding, and are given in fixed doses that do not require monitoring. However, the available DOACs, like warfarin, target proteins that are central to the body’s response to injury, and this places limits on how they can be used.
There is interest in targeting the plasma zymogen factor XI (fXI) and its protease form factor XIa (fXIa) for prevention or treatment of thrombosis. FXI appears to contribute to VTE and ischemic stroke in humans [4,5], and is required for formation of occlusive clots in animal thrombosis models [4,6]. As congenital absence of fXI is associated with a relatively mild bleeding disorder [7], it is anticipated that neutralizing fXI or fXIa would cause a smaller defect in hemostasis than would warfarin or a DOAC. Results from a recent clinical trial involving patients undergoing knee replacement support the premise that an antithrombotic effect can be achieved by targeting fXI without precipitating severe bleeding [8]. Here we discuss recent findings on the biochemistry and physiology of fXI, the pre-clinical and clinical evidence supporting a role for this protein in thrombosis, and the mechanisms by which it may contribute to thromboembolism.
The Factor XI Molecule
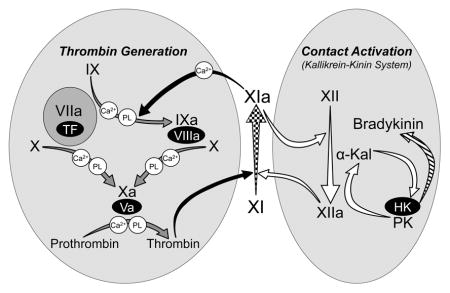
FXI arose from a duplication of the gene for prekallikrein (PK), the zymogen of α-kallikrein [9,10]. PK, factor XII (fXII) and high molecular weight-kininogen (HK) comprise the plasma kallikrein-kinin system (KKS, Figure 1) [11,12]. Among its functions, the KKS may contribute to the host-response to infection by assembling on microorganisms, and generating inflammatory kinins and antimicrobial peptides. A similar process, contact activation, leads to coagulation when blood is exposed to artificial surfaces, such as medical devices used in extracorporeal blood oxygenation [11,12]. During contact activation, reciprocal conversion of PK and fXII to α-kallikrein and factor XIIa (fXIIa) occurs on a surface (Figure 1). FXI, like its homolog PK, is activated by fXIIa and, similar to α-kallikrein, fXIa has some capacity to activate fXII [4,13]. However, fXI has features distinguishing it from PK that facilitate interactions with the thrombin generation mechanism (Figure 1) [10]. PK and fXI polypeptides each have four apple domains and a trypsin-like catalytic domain. The fXI apple 3 domain contains a factor IX-binding exosite not present on PK. The amino acid sequence adjacent to the fXI activation site (Arg369-Ile370) also differs from corresponding PK sequence, permitting fXI to be activated by thrombin, as well as by fXIIa. The combination of PK-like and novel features permit fXI to promote thrombin generation through fXIIa-dependent and fXIIa-independent processes (Figure 1) [4].
Figure 1. The Relationship of Factor XI to Thrombin Generation and Contact Activation.

Proteolytic reactions required for thrombin generation at an injury site are shown in the gray oval on the left, with each reaction indicated by a yellow arrow. The factor VIIa/tissue factor (TF) complex initiates thrombin generation by activating factors X and IX. Activated factor X (factor Xa) is responsible for cleaving prothrombin to form thrombin. Protease zymogens are indicated in black, and their active forms are indicated by a lower case “a”. Cofactors are shown as red ovals. Calcium (Ca2+) and phospholipid (PL) dependent reactions are indicated. Thrombin generated early in coagulation converts fXI to fXIa, which sustains thrombin production through factor IX activation (green arrows). Note that fXI activation does not require fXIIa, explaining why fXII deficiency does not cause bleeding. Proteolytic reactions involved in contact activation are shown in the gray oval on the right, with each reaction indicated by a black arrow. Artificial or abnormal surfaces facilitate fXII autoactivation. FXIIa converts prekallikrein (PK) to α-kallikrein, which activates additional fXII and cleaves high-molecular-weight kininogen (HK), liberating bradykinin (BK) and antimicrobial peptides (AMPs). Contact activation can promote thrombin generation through fXIIa-mediated activation of FXI [11,12]. There is evidence that fXIa, in turn, can activate fXII [13], although this is not a standard part of contact activation models. In plasma, PK and fXI circulate as complexes with HK, which may serve as a cofactor for PK and fXI activation. Activation of fXI by fXIIa is not required for hemostasis, but contributes to thrombosis in animal models. FXI is considered a component of contact activation (kallikrein-kinin) and thrombin generation in the scheme shown here, functioning as a bidirectional interface between the two systems. Hypothetically, activation of either system could activate the other through fXI conversion to fXIa. Image adapted from references 4 and 13.
Factor XI in Hemostasis
The phenotype associated with congenital fXI deficiency indicates fXI has a role in limiting trauma induced-bleeding. In humans severe deficiency (≤15% normal level) may exacerbate post-traumatic bleeding, particularly in areas with high fibrinolytic activity (urinary tract, nose, mouth) [7]. Hemorrhage in other tissues is less frequent, and procedures such as appendectomy and cholecystectomy may be well tolerated without factor replacement [7,14]. As deficiencies of fXII, PK or HK are not associated with abnormal bleeding, fXI is probably activated by fXIIa-independent processes during hemostasis. In the model in Figure 1, fXI is activated by thrombin after the VIIa/tissue factor complex initiates coagulation, with fXIa sustaining thrombin generation through factor IX activation [15]. In addition to promoting fibrin formation, fXI-dependent thrombin generation may promote activation of TAFI (thrombin-activatable fibrinolysis inhibitor), a metalloproteinase that modifies fibrin by removing binding sites for fibrinolytic proteins, rendering it resistant to fibrinolytic degradation [16]. While severe fXI deficiency delays clot formation in surface-dependent assays such as the activated partial thromboplastin time (aPTT), the magnitude of the abnormality correlates poorly with symptoms, and some patients with severe deficiency may not bleed abnormally, even with trauma. It is conceivable that some individuals have relatively robust factor VIIa/tissue factor activity or weak fibrinolytic activity that tips the balance in favor of clot stability, rendering fXIa unnecessary. The clinical experience suggests that inhibitors targeting fXI/fXIa would leave some patients more prone to trauma-induced bleeding. However, “spontaneous” soft tissue bleeding is not part of the phenotype of fXI deficiency, and such drugs would not be expected to precipitate severe bleeding as frequently as would warfarin or DOACs.
Factor XI and Thrombosis in Humans
Despite its modest role in hemostasis, there is substantial evidence supporting a role for fXI in thrombosis. Plasma fXI levels at the upper end of the normal range are linked to modest increases in VTE and ischemic stroke risk [4,5]. The 10% of subjects with the highest fXI levels in the Leiden Thrombophilia Study had a two-fold higher risk of VTE than the lower 90% [17]. This result is supported by data from the Longitudinal Investigation of Thromboembolism Etiology (LITE) cohort [18], and the observation that fXI deficiency reduces the incidence of VTE [19]. High fXI or fXIa levels were associated with increased risk for ischemic stroke in several studies, including the Atherosclerosis Risk in Communities (ARIC) study [20] and the Risk of Arterial Thrombosis In relation to Oral contraceptives (RATIO) study [21]. Severe fXI deficiency reduces incidence of stroke [22]. A role for fXI in myocardial infarction (MI) is less clear. FXI levels correlated with MI risk in the Study of Myocardial Infarction Leiden (SMILE) [23], but not in the ARIC or RATIO studies [21,22], and the incidence of MI in fXI-deficient persons is similar to the expected incidence in age-matched controls [24]. These data indicate that fXI participates in thrombosis in humans, but suggest the contribution varies between vascular beds.
Factor XI and Thrombosis – Animal Models
Mice lacking coagulation factors have been compared for resistance to thrombosis by a variety of techniques [4,6]. Our data for ferric chloride-induced carotid artery occlusion are summarized in Figure 2. Factor IX-deficient mice have a significant bleeding disorder (hemophilia B), and high FeCl3 concentrations are required to induce thrombosis in these animals [25]. Mice lacking fXI or fXII do not have obvious hemostatic abnormalities. Despite this they are at least as resistant to FeCl3-induced thrombosis as factor IX-deficient mice [25,26], showing that a bleeding phenotype is not a prerequisite for resistance to thrombus formation. Mice lacking PK or HK, proteins required for optimal fXII, are also resistant to FeCl3-induced thrombosis [27], suggesting a contact activation-like process drives thrombosis in mice.
Figure 2. Ferric chloride-induced carotid artery occlusion.

Carotid artery occlusion was induced in wild type (WT) C57Bl/6 mice, and in mice lacking factor IX (IX−/−), factor XI (fXI−/−), fXII (fXII−/−), prekallikrein (PK−/−), or high molecular weight kininogen (HK−/−) with varying concentrations of FeCl3 as indicated at the bottom of the graph. The percent of animals with patent arteries 30 minutes after FeCl3 exposure is shown (n = 10 for each bar). HK−/− mice are homozygous null for deletions of the Kng1 gene. Mice, unlike humans, have two kininogen genes (Kng1 and Kng2). Kng1 is though to be responsible for most, if not all, of the HK in plasma. Image from data in references 25–27.
In a primate model fXI and fXII contributed to thrombosis, but in contrast to mice, fXI inhibition had a greater effect than fXII inhibition [26,28–30]. Perhaps this reflects stronger thrombin-mediated fXI activation in primates. When tissue factor- or collagen-coated vascular grafts are inserted into arteriovenous shunts in baboons, platelets and fibrin deposit within the coated graft segment, followed by clot extension downstream into the uncoated graft. Polyclonal anti-fXI IgG [28] or a monoclonal IgG that blocks fXIa activation of factor IX (O1A6) [29] reduce platelet and fibrin accumulation within coated graft portions, and block downstream growth. Monoclonal IgG inhibiting fXI activation by fXIIa (14E11/3G3) [26] or fXII activation (15H8) [30] have little effect on platelet accumulation, and a modest effect on fibrin deposition, in coated portions of grafts. (Figure 3), but do limit clot extension. The results support a larger role for fXI than fXII in the primate model, in agreement with data indicating that fXI makes a greater contribution to VTE and stroke than does fXII in humans [5].
Figure 3. Acute vascular graft thrombosis.

Thrombosis is induced by deployment of vascular grafts with collagen- or TF-coated segments into chronic arteriovenous shunts in baboons. Graft platelet content during blood perfusion and fibrin content after graft removal were measured in naïve and antibody-treated animals. Meta-analysis of experiments with antibodies that target specific reactions involving fXI and fXII indicate that inhibition of fXIa and inhibition of fXI activation by fXIIa have different effects on thrombus formation, with the most pronounced antithrombotic effect obtained with antibodies that inhibit factor IX activation by fXIa. Left Panel - values for total fibrin content and platelet deposition over the 60 minutes of each study, and maximum, average and final (last 20 minutes of the study) platelet deposition rates are shown as percentages of baseline values in the same study subject prior to antibody treatment. Red bars show mean values for animals treated with polyclonal anti-fXI antibody or the monoclonal IgG O1A6, both of which block factor IX activation by fXIa. The black bars show mean values for animals treated with monoclonal IgG 14E11 (or its humanized counterpart 3G3) which binds to fXI and specifically blocks its activation by fXIIa without interfering with fXIa activity, or with the anti-fXII monoclonal antibody 15H8, which blocks fXII activation. Error bars represent +/− one SD. Right Panel – platelet thrombus growth over time in animals treated with anti-fXI or anti-fXII antibodies (red curves) compared to untreated animals (black curves). The value for platelet deposition 60 minutes after the start of the experiment in untreated animals was arbitrarily assigned a value of 100%. Images derived from data in references 26 and 28–30.
In the mouse and primate models, lack of fXI activity interferes with thrombus growth. While this could reflect a decreased rate of clot formation, increased clot breakdown may also be a factor. Fibrinolytic degradation of plasma thrombi introduced into the jugular veins of rabbits was ~two-fold greater in the presence of neutralizing anti-fXI antibody than in controls [31], consistent with in vitro results demonstrating increased clot sensitivity to fibrinolysis in the absence of fXI [32]. A role for fXI in enhanced clot resistance to fibrinolysis is consistent with the observation that fXI-deficient individuals bleed most frequently from tissues with high intrinsic fibrinolytic activity.
Factor XI and Inflammation – Animal Models
While the contributions of fXI to coagulation have received considerable study, data from mouse models suggest fXI influences inflammatory processes in a manner that impacts blood vessel biology [6]. Absence of protein C (PC), the zymogen of a protease that regulates coagulation and inflammation, results in perinatal death with intravascular thrombus formation and tissue inflammation prominently featured. FXI deficiency reduces the severity of this phenotype, with some PC−/−/fXI−/− mice living to adulthood [33]. FXI deficiency reduces ischemia-reperfusion injury after temporary middle cerebral artery occlusion [34]. While reduced fibrin deposition may have a role in the tissue sparing effect, alteration of the inflammatory response could be involved. Shnerb-Ganor et al. reported that fXI deficiency reduces atherosclerotic plaque growth and plaque infiltration by macrophages in ApoE deficient mice [35]. van Montfoort et al. also noted reduced macrophage infiltration and an absence of neutrophils in arteries in ApoE-null mice after knockdown of fXI expression [36]. FXI-deficient mice have a survival advantage over wild type mice after sepsis induced by cecal ligation and puncture (CLP) [13,37]. While initial studies pointed to reduced consumptive coagulation in fXI-deficient animals, we noted that a coagulopathy is not a consistent feature after CLP and that fXI deficiency improves survival in the absence of a coagulopathy. The early cytokine response and rise in markers of systemic inflammation after CLP are reduced in fXI-deficient mice [13,37]. The mechanisms by which fXI promotes inflammation are just starting to be investigated. Certainly, a contribution based on its role in thrombin and fibrin formation seems likely. However, in the CLP model fXI deficiency also decreased fXII and PK turnover, suggesting it contributes to activation of these proteins (Figure 1) [13]. If this is the case, fXI deficiency could reduce α-kallikrein generation and subsequent bradykinin production. In addition, fXI modulates neutrophil migration in vitro, and its absence could alter neutrophil behavior during inflammatory processes [37,38].
Therapeutic Targeting of Factor XI and Factor XIa
Several therapeutic strategies to target fXI and fXIa are under development [reviewed in 4,39]. Antisense oligonucleotide (ASO) knockdown of fXI expression has undergone phase II testing. 2′-methoxethyl DNA ASOs are avidly taken up by hepatocytes and bind to mRNAs through complementary base-pairing, followed by RNase H-dependent degradation of the ASO-mRNA complex [40]. The anti-fXI ASO IONIS-FXIRX (formerly ISIS-416858) was compared to standard dose enoxaparin for prevention of VTE in patients undergoing knee replacement [8]. IONIS-FXIRX was given in 200 or 300 mg doses at specific intervals over 36 days before surgery. On the day of surgery, average plasma fXI levels were 38% and 20% of normal in patients on 200 or 300 mg ASO, respectively. There were few symptomatic clots in any treatment group in this study. Venography performed 8 to 12 days post-surgery detected lower extremity thrombi in 30% of patients on enoxaparin, 27% on 200 mg ASO, but only 4% on 300 mg ASO. Interestingly, thrombi were not only rarer, but smaller in the 300 mg ASO group, suggesting thrombus growth is compromised when fXI is reduced below a threshold. Alternatively, as venography was performed more than a week after surgery, the results could reflect greater fibrinolytic degradation of clot formed intraoperatively in the absence of fXI. While clinically relevant bleeding was not statistically different in ASO- and enoxaparin-treated patients the study was not powered to show a difference in bleeding. It is worth noting that patients started ASO treatment five weeks before surgery, and were under the full drug effect during surgery [8]. Despite this, abnormal intra-operative hemostasis was not observed, and post-operative bleeding was rare, even with fXI levels <10% of normal.
Because of the slow onset of action, ASO therapy cannot be applied in situations where an antithrombotic effect is required rapidly. Anti-fXI antibodies have shown promise in pre-clinical studies [26,28,29], and an antibody specifically targeting fXIa is entering clinical evaluation. Antibody-based therapies are highly specific and provide rapid onset of action. Their half-lives make them better suited for long-term inhibition than for situations requiring brief treatment. Small molecule active site inhibitors of fXIa are efficacious in rodent and rabbit thrombosis models [reviewed in 4 and 39], and some are entering phase I testing. Most have relatively short half-lives, facilitating dose adjustment and more rapid dissipation of effect after discontinuation. Specificity is an issue with these agents, as the fXIa active site is structurally similar to those of other proteases. Some anti-fXIa compounds demonstrate activity toward the fXIa homolog α-kallikrein. This might be beneficial, as it may produce anti-inflammatory and antithrombotic effects distinct from those due to fXIa inhibition.
Conclusions, Conjecture and Future Considerations
FXI was identified as a plasma constituent missing in patients with abnormal surface-dependent clotting and a mild bleeding disorder [4,7]. As part of the intrinsic pathway, the protein provides a link between contact activation and factor IX, driving thrombin generation in aPTT assays [11,12]. Subsequent work identified fXIIa-independent mechanisms for fXI activation, explaining the phenotypic differences between fXI and fXII deficiencies [4,15]. Genomic studies have shed additional light on the relationship between fXI and the traditional coagulation cascade [9]. Most terrestrial vertebrates have a vitamin K-dependent mechanism for thrombin generation that is required for hemostasis, and a KKS (Figure 1). However, in most lineages these two ancient systems lack the connection provided by fXI. The duplication of the PK gene that led to fXI appears to have occurred during mammalian evolution [9]. FXI retains features of PK that allow it to interact with fXII/fXIIa, while possessing adaptations that facilitate interactions with factor IX and thrombin [4,10]. Based on the structural, biochemical and genomic data, it seems reasonable to classify fXI as a component of contact activation and as a coagulation factor. The dual role may be central to its ability to promote thrombosis, as it allows the KKS and coagulation mechanisms to activate each other (Figure 1) [4,13]. This strategic location indicates that fXI may be an excellent target for controlling thrombo-inflammatory processes initiated by either system.
The prominent roles for fXI and fXII in thrombosis models stand in stark contrast to their limited importance for hemostasis [4,6,7], indicating fundamental differences in processes that form hemostatic and thrombotic clots. In mice, fXI or fXII deficiency do not prevent clot formation on surfaces of injured vessels, but do cause an impressive defect in clot propagation into the vessel lumen [6,11]. Platelet deposition in thrombogenic grafts in baboons treated with anti-fXI IgG (Figure 3) is also primarily on the graft surface [28,29]. Intraluminal thrombi fragment in the absence of fXI or fXII in flowing blood. In this environment, these proteins may support thrombin generation to maintain thrombus stability and growth. Hemostatic clots may not require fXII, and in most cases fXI, because they form largely outside of damaged vessels and within vessel walls, away from the higher shear environment in the lumen. Perhaps unrecognized features of thrombotic clots allow them to recruit fXI and fXII more efficiently than do hemostatic clots. We need to identify and characterize factors that promote fXI and fXII activation during thrombosis, as they may turn out to be targets for antithrombotic therapy in their own right. Polyanions that facilitate fXI and fXII activation such as polyphosphate, DNA-containing neutrophil extracellular traps, and RNA, are candidates for cofactors that enhance fXI and fXII activation and may contribute to thrombus formation in vivo (reviewed in 4).
The recent ASO trial provided proof-of-concept that targeting fXI can produce a useful therapeutic effect in patients at risk of thrombosis [8]. Future work will be directed at determining if these promising results are relevant to other clinical settings. It seems prudent to first test fXIa inhibitors for prophylaxis, such as in primary or secondary prevention of VTE or prevention of stroke in patients with atrial fibrillation. At this point, we do not have pre-clinical data to indicate that fXI/fXIa inhibition will be useful in treating active thrombosis. FXIa inhibition may be useful in patients who are poor candidates for warfarin or DOACs, or as short-term prophylaxis after procedures where even modest bleeding is problematic (e.g. neurosurgery). Inhibitors of fXI/fXIa should also be considered in situations where blood is exposed to artificial surfaces (dialysis, extracorporeal membrane oxygenation, cardiopulmonary bypass, artificial heart valves) [11,41].
The advantage of strategies targeting fXI over current therapies likely lies in the area of safety. Pre-clinical and epidemiologic data indicate that fXI/fXIa inhibitors will not promote bleeding after injury to the same extent as warfarin or DOAC therapy and, therefore, may not need to be discontinued prior to certain invasive procedures. That patients successfully underwent knee replacement with reduced fXI levels is encouraging, but does not establish the safety profile for drugs targeting this protease for all hemostatic challenges. Considering the phenotype of fXI deficiency, we should anticipate that fXIa inhibitors will exacerbate bleeding in some individuals undergoing surgery, particularly when the oropharynx or urinary tract is involved, and in some women with a propensity for menorrhagia or who are postpartum [7]. An ideal antithrombotic agent could be defined as a drug that, at its effective dose, completely and specifically blocks a prothrombotic activity while leaving hemostasis intact, eliminating concerns with overdose or monitoring. Along this line of thinking, fXIIa seems to be an ideal target. Pre-clinical studies in rodents and rabbits support a role for fXII in thrombus formation [4,11,26] and selective blockade of fXI activation by fXIIa without inhibiting other functions of fXIIa or fXI demonstrated antithrombotic efficacy in mice and primates [26]. However, enthusiasm must be tempered by observations that fXII does not appear to contribute to VTE, stroke or MI in humans [5], and that fXII inhibition is less effective than fXI inhibition in the primate model [29,30]. Also, fXII likely contributes to several homeostatic and host-defense processes [11,12], and the repercussions of long-term inhibition of fXII are uncertain. Still, brief treatment with fXII inhibitors may be effective for procedures in which blood is exposed to artificial surfaces [11]. If inhibitors of fXI/fXIa and fXII/fXIIa prove to be effective in the clinic, their safety profiles should broaden the spectrum of clinical situations in which antithrombotic therapy can be applied.
HIGHLIGHTS SECTION.
The plasma protease factor XIa contributes to thrombin generation by activating factor IX. Traditionally considered part of the intrinsic pathway of coagulation, factor XIa may function as an interface between thrombin generation and the pro-inflammatory kallikrein-kinin system.
Data from animal models and epidemiologic surveys indicate factor XI contributes to thromboembolic disease.
Therapeutic strategies designed to neutralize factor XIa (antibodies, RNA aptamers, small molecule active site inhibitors) or its zymogen form factor XI (antisense oligonucleotides, antibodies) are being developed, with the goal of testing them in prevention or treatment of thromboembolic disease.
Congenital factor XI deficiency is associated with a mild to moderate bleeding disorder, and it is anticipated that therapies targeting this protein will be associated with a relatively low risk of serious bleeding compared to currently available anticoagulants.
Results of a recent trial of antisense oligonucleotide-mediates factor XI reduction for venous thrombosis prophylaxis in knee replacement surgery indicates that factor XI is an important contributor to thrombosis in this setting, and that targeting factor XI can produce a useful therapeutic effect.
Acknowledgments
The authors thank Dr. Michael Wallisch (Aronora) for his contributions to this review.
SOURCES OF FUNDING
The authors acknowledge support from awards HL81326 and HL58837 (D. Gailani) and HL101972, GM116184, and AI088937 (A. Gruber) from the National Institutes of Health.
Footnotes
DISCLOSURE
D. Gailani is a consultant for several pharmaceutical companies (Bayer, Bristol-Myers Squibb, Dyax, Instrument Laboratory, Ionis, and Ono) and receives consultant fees or research support. A. Gruber is an employee of Aronora, Inc.
BIBLIOGRPAHY
- 1.Crowther MA, Warkentin TE. Bleeding risk and the management of bleeding complications in patients undergoing anticoagulant therapy: focus on new anticoagulant agents. Blood. 2008;111:4871–4879. doi: 10.1182/blood-2007-10-120543. [DOI] [PubMed] [Google Scholar]
- 2.Hirsh J, O’Donnell M, Weitz JI. New Anticoagulants. Blood. 2005;105:453–463. doi: 10.1182/blood-2003-12-4195. [DOI] [PubMed] [Google Scholar]
- 3.Yeh CH, Hogg K, Weitz JI. Overview of the new oral anticoagulants: opportunities and challenges. Arterioscler Thromb Vasc Biol. 2015;35:1056–1065. doi: 10.1161/ATVBAHA.115.303397. [DOI] [PubMed] [Google Scholar]
- 4.Gailani D, Bane CE, Gruber A. Factor XI and contact activation as targets for antithrombotic therapy. J Thromb Haemost. 2015;13:1383–1395. doi: 10.1111/jth.13005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Key NS. Epidemiologic and clinical data linking factors XI and XII to thrombosis. Hematology Am Soc Hematol Educ Program. 2014;2014:66–70. doi: 10.1182/asheducation-2014.1.66. [DOI] [PubMed] [Google Scholar]
- 6.van Montfoort ML, Meijers JC. Recent insights into the role of the contact pathway in thrombo-inflammatory disorders. Hematology Am Soc Hematol Educ Program. 2014;2014:60–65. doi: 10.1182/asheducation-2014.1.60. [DOI] [PubMed] [Google Scholar]
- 7.Duga S, Salomon O. Congenital factor XI deficiency: an update. Semin Thromb Hemost. 2013;39:621–631. doi: 10.1055/s-0033-1353420. [DOI] [PubMed] [Google Scholar]
- 8.Büller HR, Bethune C, Bhanot S, Gailani D, Monia BP, Raskob GE, Segers A, Verhamme P, Weitz JI FXI-ASO TKA Investigators. Factor XI antisense oligonucleotide for prevention of venous thrombosis. N Engl J Med. 2015;372:232–240. doi: 10.1056/NEJMoa1405760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Doolittle RF. Step-by-step evolution of vertebrate blood coagulation. Cold Spring Harb Symp Quant Biol. 2009;74:35–40. doi: 10.1101/sqb.2009.74.001. [DOI] [PubMed] [Google Scholar]
- 10.Emsley J, McEwan PA, Gailani D. Structure and function of factor XI. Blood. 2010;115:2569–2577. doi: 10.1182/blood-2009-09-199182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Long AT, Kenne E, Jung R, Fuchs TA, Renné T. Contact system revisited: an interface between inflammation, coagulation, and innate immunity. J Thromb Haemost. 2016;14:427–437. doi: 10.1111/jth.13235. [DOI] [PubMed] [Google Scholar]
- 12.Schmaier AH. The contact activation and kallikrein/kinin systems: pathophysiologic and physiologic activities. J Thromb Haemost. 2016;14:28–39. doi: 10.1111/jth.13194. [DOI] [PubMed] [Google Scholar]
- 13.Bane CE, Ivanov I, Matafonov A, Boyd KL, Cheng Q, Sherwood ER, Tucker EI, Smiley ST, McCarty OJT, Gruber A, Gailani D. Factor XI deficiency alters the cytokine response and activation of contact proteases during polymicrobial sepsis in mice. PLOS. 2016;11:e0152968. doi: 10.1371/journal.pone.0152968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Salomon O, Steinberg DM, Seligshon U. Variable bleeding manifestations characterize different types of surgery in patients with severe factor XI deficiency enabling parsimonious use of replacement therapy. Haemophilia. 2006;12:490–493. doi: 10.1111/j.1365-2516.2006.01304.x. [DOI] [PubMed] [Google Scholar]
- 15.Broze GJ., Jr The role of tissue factor pathway inhibitor in a revised coagulation cascade. Semin Hematol. 1992;29:159–169. [PubMed] [Google Scholar]
- 16.Plug T, Meijers JC. Structure-function relationships in Thrombin-activatable fibrinolysis Inhibitor. J Thromb Haemost. 2016;14:633–644. doi: 10.1111/jth.13261. [DOI] [PubMed] [Google Scholar]
- 17.Meijers JC, Tekelenburg WL, Bouma BN, Bertina RM, Rosendaal FR. High levels of coagulation factor XI as a risk factor for venous thrombosis. N Engl J Med. 2000;342:696–701. doi: 10.1056/NEJM200003093421004. [DOI] [PubMed] [Google Scholar]
- 18.Cushman M, O’Meara ES, Folsom AR, Heckbert SR. Coagulation factors IX through XIII and the risk of future venous thrombosis: the longitudinal investigation of thromboembolism etiology. Blood. 2009;114:2878–2883. doi: 10.1182/blood-2009-05-219915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Salomon O, Steinberg DM, Zucker M, Varon D, Zivelin A, Seligsohn U. Patients with severe factor XI deficiency have a reduced incidence of deep-vein thrombosis. Thromb Haemost. 2011;105:269–273. doi: 10.1160/TH10-05-0307. [DOI] [PubMed] [Google Scholar]
- 20.Suri MF, Yamagishi K, Aleksic N, Hannan PJ, Folsom AR. Novel hemostatic factor levels and risk of ischemic stroke: the Atherosclerosis Risk in Communities (ARIC) Study. Cerebrovasc Dis. 2010;29:497–502. doi: 10.1159/000297966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Siegerink B, Govers-Riemslag JW, Rosendaal FR, Ten Cate H, Algra A. Intrinsic coagulation activation and the risk of arterial thrombosis in young women: results from the Risk of Arterial Thrombosis in relation to Oral contraceptives (RATIO) case-control study. Circulation. 2010;122:1854–1861. doi: 10.1161/CIRCULATIONAHA.110.943738. [DOI] [PubMed] [Google Scholar]
- 22.Salomon O, Steinberg DM, Koren-Morag N, Tanne D, Seligsohn U. Reduced incidence of ischemic stroke in patients with severe factor XI deficiency. Blood. 2008;111:4113–4117. doi: 10.1182/blood-2007-10-120139. [DOI] [PubMed] [Google Scholar]
- 23.Doggen CJ, Rosendaal FR, Meijers JC. Levels of intrinsic coagulation factors and the risk of myocardial infarction among men: Opposite and synergistic effects of factors XI and XII. Blood. 2006;108:4045–4051. doi: 10.1182/blood-2005-12-023697. [DOI] [PubMed] [Google Scholar]
- 24.Salomon O, Steinberg DM, Dardik R, Rosenberg N, Zivelin A, Tamarin I, Ravid B, Berliner S, Seligsohn U. Inherited factor XI deficiency confers no protection against acute myocardial infarction. J Thromb Haemost. 2003;1:658–661. doi: 10.1046/j.1538-7836.2003.00195.x. [DOI] [PubMed] [Google Scholar]
- 25.Wang X, Cheng Q, Xu L, Feuerstein GZ, Hsu MY, Smith PL, Seiffert DA, Schumacher WA, Ogletree ML, Gailani D. Effects of factor IX or factor XI deficiency on ferric chloride-induced carotid artery occlusion in mice. J Thromb Haemost. 2005;3:695–702. doi: 10.1111/j.1538-7836.2005.01236.x. [DOI] [PubMed] [Google Scholar]
- 26.Cheng Q, Tucker EI, Pine MS, Sisler I, Matafonov A, Smith SA, Sun M-F, Renné T, Gruber A, Gailani D. A role for factor XIIa-mediated factor XI activation in thrombus formation in vivo. Blood. 2010;116:3981–3989. doi: 10.1182/blood-2010-02-270918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kokoye Y, Ivanov I, Cheng Q, Matafonov A, Dickeson SK, Mason M, Sexton DJ, Renné T, McCrae K, Feener EP, Gailani D. A comparison of the effects of factor XII deficiency and prekallikrein deficiency on thrombus formation. Thromb Res. 2016;140:118–124. doi: 10.1016/j.thromres.2016.02.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gruber A, Hanson SR. Factor XI-dependence of surface- and tissue factor-initiated thrombus propagation in primates. Blood. 2003;102:953–955. doi: 10.1182/blood-2003-01-0324. [DOI] [PubMed] [Google Scholar]
- 29.Tucker EI, Marzec UM, Hurst S, McCarty OJT, Rugonyi S, Gailani D, Gruber A, Hanson SR. Prevention of vascular graft occlusion and thrombus-associated thrombin generation by inhibition of factor XI. Blood. 2009;113:936–944. doi: 10.1182/blood-2008-06-163675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Matafonov A, Leung PY, Gailani AE, Grach SL, Puy CG, Cheng Q, Sun M-f, McCarty OJT, Tucker EI, Kataoka H, Renné T, Morrissey JH, Gruber A, Gailani D. Factor XII inhibition reduces thrombus formation in a primate thrombosis model. Blood. 2014;123:1739–1746. doi: 10.1182/blood-2013-04-499111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Minnema MC, Friederich PW, Levi M, von dem Borne PA, Mosnier LO, Meijers JC, Biemond BJ, Hack CE, Bouma BN, ten Cate H. Enhancement of rabbit jugular vein thrombolysis by neutralization of factor XI. In vivo evidence for a role of factor XI as an anti-fibrinolytic factor. J Clin Invest. 1998;101:10–14. doi: 10.1172/JCI781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Dai L, Mitchell M, Savidge G, Alhaq A. The profibrinolytic effect of plasma thrombomodulin in factor XI deficiency and its implications in hemostasis. J Thromb Haemost. 2004;2:2200–2204. doi: 10.1111/j.1538-7836.2004.01034.x. [DOI] [PubMed] [Google Scholar]
- 33.Chan JCY, Ganopolsky JG, Cornelissen I, Suckow MA, Sandoval-Cooper MJ, Brown EC, Noria F, Gailani D, Rosen ED, Ploplis VA, Castellino F. The characterization of mice with a targeted combined deficiency of protein C and factor XI. Am J Path. 2001;158:469–479. doi: 10.1016/S0002-9440(10)63989-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kleinschnitz C, Stoll G, Bendszus M, Schuh K, Ulrich P, Burfeind P, Gailani D, Nieswandt B, Renné T. Targeting coagulation factor XII provides protection from pathological thrombosis in cerebral ischemia without interfering with hemostasis. J Exp Med. 2006;203:513–518. doi: 10.1084/jem.20052458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Shnerb Ganor R, Harats D, Schiby G, Gailani D, Levkovitz H, Tamarin I, Shaish A, Salomon O. Factor XI deficiency protects against atherogenesis in apolipoprotein E/factor XI double knockout mice. Atheroscler Thromb Vasc Biol. 2016;36:475–481. doi: 10.1161/ATVBAHA.115.306954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.van Montfoort ML, Kuijpers MJ, Knaup VL, Bhanot S, Monia BP, Roelofs JJ, Heemskerk JW, Meijers JC. Factor XI regulates pathologic thrombus formation on acutely ruptured atherosclerotic plaques. Arterioscler Thromb Vasc Biol. 2014;34:1668–73. doi: 10.1161/ATVBAHA.114.303209. [DOI] [PubMed] [Google Scholar]
- 37.Tucker EI, Verbout NG, Leung PY, Hurst S, McCarty OJT, Gailani D, Gruber A. Inhibition of factor XI activation attenuates inflammation and coagulopathy while improving the survival of mouse polymicrobial sepsis. Blood. 2012;119:4762–4768. doi: 10.1182/blood-2011-10-386185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Itakura A, Verbout NG, Phillips KG, Insall RH, Gailani D, Tucker EI, Gruber A, McCarty OJ. Activated factor XI inhibits chemotaxis of polymorphonuclear leukocytes. J Leukoc Biol. 2011;90:923–927. doi: 10.1189/jlb.0411182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gailani D. Future prospects for contact factors as therapeutic targets. Hematology Am Soc Hematol Educ Program. 2014;2014:52–59. doi: 10.1182/asheducation-2014.1.52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zhang H, Löwenberg EC, Crosby JR, MacLeod AR, Zhao C, Gao D, Black C, Revenko AS, Meijers JC, Stroes ES, Levi M, Monia BP. Inhibition of the intrinsic coagulation pathway factor XI by antisense oligonucleotides: a novel antithrombotic strategy with lowered bleeding risk. Blood. 2010;116:4684–4692. doi: 10.1182/blood-2010-04-277798. [DOI] [PubMed] [Google Scholar]
- 41.Jaffer IH, Fredenburgh JC, Hirsh J, Weitz JI. Medical device-induced thrombosis: what causes it and how can we prevent it? J Thromb Haemost. 2015 Jun;13(Suppl 1):S72–81. doi: 10.1111/jth.12961. [DOI] [PubMed] [Google Scholar]