

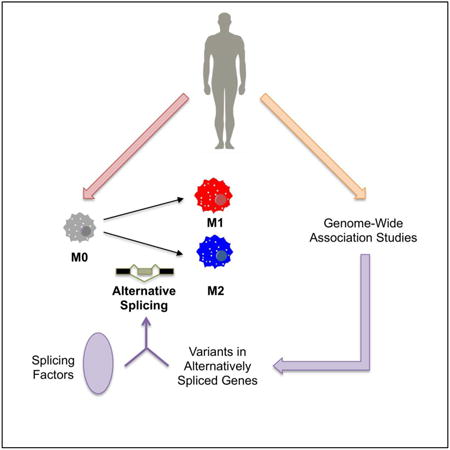
Abstract
Objective
Human macrophages can shift phenotype across the inflammatory M1 and reparative M2 spectrum in response to environmental challenges, but the mechanisms promoting inflammatory and cardiometabolic disease-associated M1 phenotypes remain incompletely understood. Alternative splicing (AS) is emerging as an important regulator of cellular function, yet its role in macrophage activation is largely unknown. We investigated the extent to which AS occurs in M1 activation within the cardiometabolic disease context and validated a functional-genomic cell model for studying human macrophage-related AS events.
Approach and Results
From deep RNA-seq of resting, M1, and M2 primary human monocyte derived macrophages, we found 3860 differentially expressed genes in M1 activation and detected 233 M1-induced AS events; the majority of AS events were cell- and M1-specific with enrichment for pathways relevant to macrophage inflammation. Using genetic variant data for 10 cardiometabolic traits, we identified in M1 activation 28 trait-associated variants within the genomic loci of 21 alternatively spliced genes and 15 variants within seven differentially expressed regulatory splicing factors. Knockdown of one such splicing factor, CELF1, in primary human macrophages led to increased inflammatory response to M1 stimulation, demonstrating CELF1's potential modulation of the M1 phenotype. Finally, we demonstrated that an induced pluripotent stem cell derived macrophage system recapitulates M1-associated AS events and provides a high-fidelity macrophage AS model.
Conclusions
AS plays a role in defining macrophage phenotype in a cell- and stimulus-specific fashion. Alternatively spliced genes and splicing factors with trait-associated variants may reveal novel pathways and targets in cardiometabolic diseases.
Keywords: alternative splicing, RNA, genetics, macrophages, inflammation
Graphical abstract
Introduction
As the phagocytic cells of the innate immune system, macrophages reside in almost every organ of the human body and mediate homeostasis through surveillance for tissue injury and invading pathogens.1-3 The most plastic cells of the hematopoietic system, they can rapidly evolve function in response to their microenvironment,1,4-8 adopting phenotypes across a spectrum that consists of pro-inflammatory M1 and reparative M2 states at the extremes. Emerging evidence has demonstrated that systemic chronic inflammation seen in cardiometabolic diseases can induce macrophages to shift toward the M1 phenotype in disease relevant tissues, e.g., the arterial wall in atherosclerosis and adipose in obesity,5,8-14 thereby exacerbating the disease-promoting inflammatory milieu and highlighting the potential of therapeutic strategies in mitigating M1 macrophage activation.
Although a few transcription factors have been implicated in the divergent gene expression profiles seen in M1 vs. M2 macrophages,1,2,8,15,16 the mechanisms underlying the promotion of these specific phenotypes remain incompletely understood. A missing link is whether post-transcriptional regulation also contributes to the function of key proteins that modulate macrophage activation. As such, the study of alternative splicing (AS), or differential inclusion of exons into multiple mature mRNA transcripts,17-19 may provide new insights into this process given the potential for functional differences in the translated protein products. Such study may be of particular importance in the human context given the rapid evolution of AS in primate species.20,21
AS occurs in >95% of human multi-exon genes, enhancing the proteomic diversity encoded by the genome. Often, AS profiles are tissue- or cell type-specific22-26 with variable interspecies conservation,20,21,27 and these patterns are coordinated by a balance of the expression or activity of splicing factors, which are RNA binding proteins that bind to cis-regulatory elements near alternative exons. This binding affects recruitment of the spliceosome to pre-mRNA splice sites and influences whether an alternative exon is incorporated into the final transcript.28 In addition, AS patterns can be disrupted by genetic variants affecting the “splicing code” of these cis-regulatory elements as well as the actual splice sites recognized during exon inclusion.26,29,30
As a regulatory process, differential AS on a transcriptome-wide scale has been shown to occur during cellular phenotypic changes, e.g., during epithelial to mesenchymal transition, somatic cell reprogramming for induced pluripotent stem cell (iPSC) generation, neuronal and cardiomyocyte development, and T-cell activation.31-35 Therefore, we hypothesized that aberrant splicing of a network of genes may contribute to pathogenic M1 activation in cardiometabolic diseases. Although a few studies have used RNA-sequencing (RNA-seq) to profile the transcriptomes of M1 and M2 macrophages,36-38 differential AS during macrophage activation and its relationship to cardiometabolic diseases have not yet been explored.
To address this gap in knowledge, we sought to determine the extent to which AS and its regulation modulate human macrophage phenotype, with a focus on the cardiometabolic disease-associated M1 macrophage activation paradigm. The aims of our study were three-fold: (1) to characterize the AS landscapes of macrophage activation in primary human monocyte derived macrophages (HMDMs), (2) to identify splicing targets and splicing factors as novel targets for cardiometabolic traits using genetic variant data, and (3) to validate iPSC-derived macrophages (iPSDMs) as a cellular model system for investigating macrophage-relevant AS events. Here, we report the first comprehensive assessment of AS in human macrophage activation with translational context for the study of human cardiometabolic diseases.
Materials and Methods
Materials and Methods are available in the online-only Data Supplement.
Results
Macrophage Activation Leads to Distinct Transcriptomic Landscapes
To study the full spectrum of transcriptomic changes occurring during macrophage activation, we performed deep RNA-seq (∼130 million reads per sample) of resting M0-HMDMs, M1-HMDMs (LPS / IFN-γ), and M2-HMDMs (IL-4) from six healthy human participants as previously described, analyzing differences between resting and activated HMDMs (Figure 1A, GSE55536).16 We characterized elsewhere the divergent transcript-level profiles of M1-HMDM and M2-HMDM activation, including comprehensive differential gene expression of M1 and M2 mRNA markers as well as enrichment of different gene ontology pathways for each condition (pathways are summarized in Supplemental Table I).16 In this previous analysis, we had found 3860 genes to be differentially expressed in M1 activation while only 357 genes were differentially expressed in M2 activation.16 To explore whether macrophage activation also involved changes in AS patterns, we analyzed our RNA-seq data using the PennDiff, DEXseq, and IUTA39-41 software programs to detect differential AS between M0-HMDMs and M1-HMDMs, our primary analysis, and between M0-HMDMs and M2-HMDMs in our secondary analysis. Three different software programs were used to maximize the number of detected AS events and to analyze AS on both the exon-level (DEXseq and PennDiff) and whole-gene level (degree of AS occurring at a locus, IUTA and PennDiff). We extended this approach to RNA-seq of PBMCs collected before and after intravenous bolus of 1 ng LPS per kg body mass exposure in eight healthy participants in a human experimental endotoxemia trial (GSE50792).42-44 The PBMC transcriptomic profiles served as a basis to probe whether M1-HMDM AS changes occurred uniquely within the macrophage system or more broadly in other inflamed hematopoietic cells and contexts. Given that M1-HMDM activation induced differential expression of genes enriched for pathways distinct from those seen in M2-HMDM activation and in post-LPS PBMCs (Supplemental Table I)16, we hypothesized that AS patterns would also manifest in a cell-specific and stimulation-specific manner.
Figure 1. RNA-seq differential AS analysis of HMDMs, iPSDMs, and PBMCs.

A, Overview of cell types studied. PBMCs were isolated from human donors and were differentiated into primary M0-HMDMs, reprogrammed into iPSCs with subsequent macrophage differentiation, or stimulated with LPS in vivo in an evoked endotoxemia experiment. M0-HMDMs and M0-iPSDMs were then activated to M1 or M2 states. B, Distribution of differential AS and alternative promoter events for each experimental condition and cell type. C, Venn diagrams of single cassette exon events for each type of cell stimulation shows distinct differential AS patterns for each condition. HMDMs = human monocyte derived macrophages; iPSDMs = induced pluripotent stem cell derived macrophages; PBMCs = peripheral blood mononuclear cells; AS = alternative splicing.
Differential AS events in each experimental condition (M0- vs. M1-HMDMs, M0- vs. M2-HMDMs, or pre- vs. post-LPS PBMCs; Figure 1A) were considered significant if they were called by either DEXseq or PennDiff to have > 5% Δ exon inclusion level at a false discovery rate (FDR) P-value < 0.05 as determined by the Benjamini-Hochberg method.39,40 We found that out of the three experimental conditions, M1-HMDM activation was accompanied by the most dynamic landscape for AS differences: the 233 AS events for M1-HMDM activation, which were predominately single cassette exon events, amounted to (1) more than 20 times the number of events occurring in M2-HMDM activation and (2) more 2 times the number of events occurring in LPS-stimulation of PBMCs (Figure 1B, Supplemental Tables II-IV). Although on a different scale, this pattern echoes what we had seen in differential expression of genes for M1 vs. M2 activation.
The computational programs we used also allowed us to detect alternative promoter usage in addition to AS events, and we found that the majority of isoform-switching across all experimental contexts involved alternative promoter sites (Figure 1B, Supplemental Tables II-IV). Previous studies have reported widespread usage of alternative promoters in the human genome, including differential alternative promoter events in M1-HMDMs,36,45 and within genes using alternative promoters, concurrent AS events have been seen.46,47 Indeed, for M1-HMDM activation one-sided Fisher's exact tests revealed robust enrichment of alternative promoter events not only in genes that were differentially expressed (P = 2.8 × 10-6) but even more so in genes undergoing differential AS (P < 2.2 × 10-16). Enrichment of AS events in differentially expressed genes was less robust (P = 0.02, Supplemental Figure I). Alternative promoter sites in M1-HMDM activation, however, had significant overlap with post-LPS PBMCs (Supplemental Table VI), suggesting that unlike AS (see below), alternative promoter events may not be driving most of the macrophage-specific changes in M1-HMDM activation.
Differential Alternative Splicing Patterns Are Specific to Cell Type and Stimulus
Differentially Spliced Genes in M1-HMDM Activation Are Enriched for Inflammatory Pathways
After establishing that M1-HMDM activation resulted in its own pattern of transcriptomic changes, we evaluated whether differential AS occurred in genes enriched for particular pathways specific to M1-HMDM activation. We interrogated differentially spliced genes for each type of cell stimulation using Ingenuity Pathway Analysis and found distinct pathway enrichment patterns for each experimental condition. Top pathways in M1-HMDM activation were consistent with intracellular responses to inflammation and included apoptosis as well as signaling in the AKT, mTOR, CDK5, ERK / MAPK, and NF-κB pathways (Table 1). These pathways contrasted with those involved in PBMC inflammation and M2-HMDM activation, which both included metabolic pathways such as lipoate modification and glycogen synthesis, respectively (Table 1). Top pathways in genes undergoing differential AS for each condition also differed from those for genes differentially expressed in the corresponding condition; as an example, in M1-HMDM activation the top pathways for alternatively spliced genes including apoptosis and mTOR signaling differ from top pathways for differentially expressed genes (Table 1, Supplemental Table I). Furthermore, the top functions enriched in networks of differentially spliced genes for each condition also differed. For example, angiogenesis and atherosclerosis predominated networks in M1-HMDM activation, while fatty acid metabolism and wound healing predominated M2-HMDM networks, and cellular migration and adhesion were top functions of post-LPS PBMC networks (Supplemental Figures II A-C).
Table 1. Top Enriched Pathways Associated with Differential Alternative Splicing Events.
| Pathway | -log(P-value) |
|---|---|
| M0-HMDM vs M1-HMDM | |
| Apoptosis Signaling | 6.03 |
| PI3K/AKT Signaling | 4.15 |
| mTOR Signaling | 4.08 |
| CDK5 Signaling | 4.02 |
| ERK/MAPK Signaling | 3.94 |
| NF-κB Signaling | 2.04 |
| M0-HMDM vs M2-HMDM | |
| TWEAK Signaling | 3.47 |
| TNFR1 Signaling | 3.03 |
| PEDF / PPARγ Signaling | 2.54 |
| Interferon Signaling | 2.11 |
| PKR in inflammation | 1.95 |
| Glycogen Biosynthesis | 1.70 |
| Pre- vs Post-LPS PBMCs | |
| IGF-1 Signaling | 2.10 |
| Lipoate Salvage and Modification | 1.74 |
| Cell Cycle Checkpoint Regulation | 1.57 |
| Hypoxia Signaling in the Cardiovascular System | 1.57 |
| Insulin Receptor Signaling | 1.51 |
| Nitric Oxide Signaling in the Cardiovascular System | 1.51 |
Pathways identified by Ingenuity Pathway Analysis for genes that are differentially spliced in each experimental condition. LPS = lipopolysaccharide, PBMC = peripheral blood mononuclear cells, HMDM =human monocyte derived macrophage.
Cassette Exon Events Occur in Cell- and Stimulus-Specific Manner
To determine whether specific differential AS patterns are unique to cell type and condition, we focused on single cassette exons, the most common type of differential AS we found in our experiments (Figure 1B). M1-HMDM activation had only two out of its 123 cassette exon events overlapping with those occurring in M2-HMDM activation and only 14 overlapping with those in LPS-stimulated PBMCs (Figure 1C). We selected a subset of cassette exon events observed in M1-HMDM activation for RT-PCR validation prioritized based on biological relevance (e.g., PLD1 and GSK3B for macrophage metabolism; ARRB1, CD74, and TLR4 for immune response). This confirmed that these splicing changes were specifically seen in M1-HMDMs but not in M2-HMDMs (Figure 2A-B, Figure 3A). Differential AS of the alternative exons for these genes may have biological relevance as they encode a portion of the functional domains of the corresponding proteins such as the protein kinase domain of GSK3B and the TRAF6 interacting domain of ARRB1 (Supplemental Figures IV, V). These AS patterns differed not only from those for reparative M2-HMDMs but also from patterns observed during inflammatory stimulation of several other cells and tissues; for example, differential AS events seen in M1-HMDMs were not seen in visualization of our RNA-seq data for inflamed PBMCs, adipose, or renal epithelial cells (Supplemental Figure III).
Figure 2. RT-PCR validation of identified differential AS events.

A, Genome browser view of M0-HMDM vs. M1-HMDM RNA-seq wiggle tracks showing differential exon 16 inclusion in PLD1 (gray shading with arrow) and RT-PCR validation of AS of exon 16 in PLD1 in M1-HMDMs in 6 human donors independent of the RNA-seq experiments. % exon inclusion and SD calculated from gel densitometry of (inclusion PCR product) / (total PCR product). L denotes the DNA ladder in bp. B, Genome browser view and RT-PCR validation of AS of GSK3B's exon 9. SD = standard deviation; AS = alternative splicing; bp = base pairs.
Figure 3. iPSDMs recapitulate AS events in M1 activation.

A, RT-PCR validation of bioinformatically identified differential AS events in HMDMs and iPSDMs. % exon inclusion and SD calculated from gel densitometry of (inclusion PCR product) / (total PCR product). B, Correlation plot of AS events predicted for each gene (calculated to be value θ) in M1-HMDM vs. M1-iPSDM. Spearman's rho = 0.77, P < 0.001. C, Correlation plot of differential expression of splicing factors on the log(2) scale. Spearman's rho = 0.78, P < 0.001. iPSDMs = induced pluripotent stem cell derived macrophages; RT-PCR = reverse transcriptase polymerase chain reaction; SD = standard deviation; AS = alternative splicing; HMDM = human monocyte derived macrophage.
Because these differential AS events occur specifically in M1-HMDM activation, we hypothesized that they play a functional role during this phenotypic transition. CD74 has macrophage-relevant roles in antigen presentation,48 and increased inclusion of CD74's exon 7, which encodes the proteolytic thyroglobulin domain of the CD74 protein (Supplemental Figure V), is observed during M1-HMDM (Figure 3A, Supplemental Table II). To test if exon 7 is involved in the inflammatory M1 response, we used anti-sense locked nucleic acid oligonucleotides to target exon 7 and found that compared to non-targeting control, knockdown of the exon 7-containing isoform induced even higher IL6 and CCL5 mRNA expression after 6 hours of 100 ng/mL LPS with 20 ng/mL IFN-γ stimulation of HMDMs (Supplemental Figure VI B) and almost complete macrophage detachment by 18 hours of stimulation (data not shown), suggesting that the inclusion isoform plays a role in modulating the M1-HMDM response.
Splicing Factors in M1-HMDM Activation
After determining that M1-HMDMs have a distinct AS pattern, we focused on potential splicing factors, which regulate tissue-specific splicing,23,26,49 that might contribute to these differences. Performing RNA-binding motif enrichment analysis for simple cassette exon events in M1-HMDM activation, we found that regions within 300 base-pairs (bp) of a differentially spliced exon were enriched for binding sites of 12 splicing factors listed in Table 2. Enrichment for these splicing factor binding sites was not found near differentially spliced exons in M2-HMDM activation and LPS-stimulation of PBMCs. Because splicing factors that regulate smaller subsets of transcripts may not be identified through enrichment testing for all differential AS events, we examined the RNA-seq data of M0-HMDMs vs. M1-HMDMs for splicing factor gene expression patterns. Out of 220 known and putative splicing factors,50 37 were differentially expressed (>1.5 fold change, FDR < 0.05), with 14 upregulated in M1-HMDM activation (Supplemental Table VII) whereas no splicing factors were differentially expressed in M2-HMDM activation, consistent with the more dynamic and diverse changes in AS in M1-HMDMs. Although splicing factors do not necessarily need to be differentially expressed to have differential functionality in one condition over another, we used differential expression as a criterion for selecting M1-HMDM modulated splicing factor CELF1 for further study, as described below.
Table 2. Splicing Factor Binding Motif Enrichment Flanking Alternatively Spliced Exons in M1-HMDM Activation.
| Splicing Factor | IUPAC for Motif | Unadjusted P | FDR –Adjusted P | Bonferroni-Adjusted P |
|---|---|---|---|---|
| CELF1 | UGUUUGUUUGU | 4.54E-04 | 4.5E-04 | 4.90E-02 |
| HNRNPC | HUUUUUK | 2.11E-04 | 2.5E-04 | 2.13E-02 |
| HuR | UUDUUUU | 9.91E-07 | 4.4E-06 | 1.01E-04 |
| PABPC1 | ARAAAAM | 2.77E-06 | 5.5E-06 | 2.83E-04 |
| PTBP1 | HYUUUYU | 1.10E-06 | 4.4E-06 | 1.12E-04 |
| RBM3 | RADACKA | 3.68E-04 | 4.0E-04 | 3.69E-02 |
| SART3 | ARAAAAM | 2.73E-06 | 5.5E-06 | 2.78E-04 |
| SRSF10 | AGAGAVM | 2.30E-08 | 2.8E-07 | 2.34E-06 |
| SRSF2 | GGAGWD | 1.93E-04 | 2.5E-04 | 1.95E-02 |
| SRSF9 | AKGAVMR | 3.89E-05 | 6.7E-05 | 3.95E-03 |
| TIA1 | WUUUUUB | 7.03E-05 | 1.1E-04 | 7.14E-03 |
| U2AF2 | UUUUUYC | 1.51E-06 | 4.5E-06 | 1.54E-04 |
RNA-binding motif enrichment identified for splicing factors in the 300 bp regions flanking differentially spliced exons using the Analysis for Motif Enrichment tool of MEME-Suite.79 Adjusted P-values reflect false discovery rate (FDR) and Bonferroni corrections for multiple testing. FDR was determined using the Benjamini-Hochberg method,80 and Bonferroni correction entailed dividing the unadjusted P-value for the number of tests performed.
Common Genetic Variants Associated with Cardiometabolic Traits Highlight Potential Modulators of M1 Macrophage Activation Through Alternative Splicing
Because previous studies have shown that common genetic variants associated with cardiometabolic traits can have functional effects in disease pathophysiology,51,52 we probed the relationship between trait-associated variants and differential AS in each cell condition.
Variants in Differentially Spliced Genes during M1-HMDM Activation
To determine which of the AS events occurring in macrophage activation and PBMC stimulation might be implicated in cardiometabolic diseases, we interrogated the genomic loci of these differentially spliced genes for cardiometabolic trait-associated SNPs in publicly available GWAS databases.53-56 Our primary hypothesis for this analysis was that (1) trait-associated SNPs may identify genes differentially spliced in M1-HMDM activation that contribute to pathogenic M1-activation, and (2) a subset of these SNPs may directly cause disease-promoting splicing events by altering the splicing code. As a complementary study, we also interrogated SNPs in genes differentially spliced in M2-HMDM activation and in post-LPS PBMCs. After Bonferroni correction for the number of SNPs tested, we identified 28 tag SNPs that fall within the loci of genes differentially spliced in M1-HMDM activation and are associated with cardiometabolic traits including lipid phenotypes, obesity, and coronary artery disease (Table 3); the tag SNPs also fall within loci of genes with alternative promoter usage. These SNPs, which are mostly intronic and more than 300 bp away from splice sites, have very low LD with each other and are located throughout the genome at metabolism-related genes such as PLD1 and GSK3B or inflammation-related genes such as TAX1BP1, FBXL19, and ALOX5. Interestingly, trait-associated SNPs were not found in genes differentially spliced in M2-HMDM activation, and although GSK3B, GUCY1A3, and SYNE1 have tag SNPs for both M1-HMDM AS and post-LPS PBMC AS, the tag SNPs for genes alternatively spliced in both inflamed cell populations mostly differed (Table 3 and Supplemental Table X).
Table 3. Cardiometabolic Trait-Associated Genetic Variants in Differentially Spliced M1-HMDM Genes.
| SNP | Chr | Position | Gene | Effect Allele | Trait | Consortium | Unadjusted P | FDR –Adjusted P | Bonferroni-Adjusted P |
|---|---|---|---|---|---|---|---|---|---|
| rs12765320 | 10 | 45930805 | ALOX5 | T | HDL | GLGC | 2.8E-07 | 1.7E-04 | 2.8E-04 |
| rs12325278 | 16 | 28848818 | ATXN2L | G | BMI | GIANT | 3.5E-10 | 8.1E-07 | 2.8E-07 |
| rs12448482 | 16 | 28838073 | ATXN2L | G | BMI | GIANT | 8.8E-08 | 7.3E-05 | 7.1E-05 |
| rs10838663 | 11 | 47184971 | C11orf49 | T | HDL | GLGC | 2.8E-16 | 2.1E-12 | 2.8E-13 |
| rs2291443 | 11 | 47179861 | C11orf49 | A | HDL | GLGC | 3.1E-07 | 1.7E-04 | 3.0E-04 |
| rs7117404 | 11 | 47127153 | C11orf49 | A | HDL | GLGC | 3.5E-05 | 2.9E-03 | 3.4E-02 |
| rs2156499 | 18 | 47007234 | C18orf32 | A | HDL | GLGC | 2.5E-08 | 2.0E-05 | 2.5E-05 |
| rs3094379 | 9 | 136334910 | CACFD1 | T | CAD | CARDIoGRAM | 2.6E-05 | 1.5E-02 | 2.2E-02 |
| rs739468 | 9 | 136326248 | CACFD1 | T | LDL, TC | GLGC | 5.5E-06 | 8.8E-03 | 5.5E-03 |
| rs11086985 | 20 | 44532255 | CTSA | A | HDL | GLGC | 6.6E-08 | 4.4E-05 | 6.5E-05 |
| rs3817731 | 20 | 44518843 | CTSA | A | HDL | GLGC | 4.8E-06 | 7.8E-03 | 4.7E-03 |
| rs10982200 | 9 | 117168551 | DFNB31 | A | HDL | GLGC | 4.8E-05 | 3.7E-03 | 4.7E-02 |
| rs9319588 | 16 | 30930983 | FBXL19 | T | TG | GLGC | 7.9E-06 | 1.7E-02 | 7.7E-03 |
| rs6781942* | 3 | 119758049 | GSK3B | A | TC | GLGC | 3.8E-05 | 2.2E-03 | 2.4E-02 |
| rs2306556* | 4 | 156638573 | GUCY1A3 | A | CAD | CARDIoGRAM | 3.4E-06 | 9.2E-04 | 2.2E-03 |
| rs5763662 | 22 | 30378703 | MTMR3 | T | TC | GLGC | 3.6E-05 | 1.5E-02 | 3.5E-02 |
| rs881803 | 16 | 15802334 | MYH11 | T | CAD | CARDIoGRAM | 5.5E-05 | 2.1E-02 | 4.7E-02 |
| rs4687614 | 3 | 52492085 | NISCH | A | WHR_BMI | GIANT | 2.0E-07 | 3.3E-04 | 1.5E-04 |
| rs360399 | 3 | 171362977 | PLD1 | A | HbA1c | MAGIC | 8.2E-06 | 2.2E-02 | 6.8E-03 |
| rs1265151 | 8 | 103213460 | RRM2B | A | CAD | CARDIoGRAM | 3.2E-05 | 1.5E-02 | 2.6E-02 |
| rs13332660 | 16 | 29905882 | SEZ6L2 | T | TC | GLGC | 1.5E-05 | 9.4E-03 | 1.4E-02 |
| rs13107325 | 4 | 103188709 | SLC39A8 | T | BMI | GIANT | 1.4E-07 | 5.4E-04 | 7.8E-05 |
| rs13107325 | 4 | 103188709 | SLC39A8 | T | HDL | GLGC | 7.2E-11 | 6.6E-08 | 7.1E-08 |
| rs9939417 | 16 | 31053467 | STX4 | T | TG | GLGC | 2.7E-07 | 1.5E-03 | 2.7E-04 |
| rs594522* | 6 | 152581079 | SYNE1 | A | TG | GLGC | 4.3E-05 | 4.0E-02 | 2.8E-02 |
| rs17611937 | 7 | 27775894 | TAX1BP1 | T | CAD | CARDIoGRAM | 3.0E-05 | 1.E-02 | 2.5E-02 |
| rs2655260 | 3 | 12563640 | TSEN2 | T | LDL | GLGC | 7.3E-06 | 8.8E-03 | 7.3E-03 |
| rs1618545 | 3 | 12560763 | TSEN2 | A | TC | GLGC | 1.3E-06 | 9.4E-03 | 1.3E-03 |
Adjusted P values are corrected for multiple testing. HDL = high density lipoprotein, Fasting Glu = fasting glucose, T2DM = type 2 diabetes mellitus, BMI = body mass index, CAD = coronary artery disease, TG = triglycerides, FDR = false discovery rate.
Also present in post-LPS PBMCs
The effects of these SNPs on protein abundance, isoform expression levels, and function remain unknown, but they may exert effects on AS patterns. For example, ALOX5 is identified by the PennDiff software to have 7% more inclusion of exon 13 in M1-HMDM activation (P = 5.2 × 10-4, Supplemental Table II, wiggle file in Figure 4A) and hosts rs12765320, a SNP associated with circulating high-density lipoprotein (HDL-C). Because the vast majority of tag SNPs in our analyses do not fall within the splicing regulatory regions of 300 bp flanking an alternative splice site, we performed additional LD analysis using 1000 Genomes SNP data to identify potential SNPs that could affect the splicing code. The intronic tag SNP rs12765320 is in high LD (R2 ≥ 0.8) with rs56145077, an intronic SNP falling within 300 bp of alternative exon 13 which encodes a portion of the enzyme's lipoxygenase domain (Supplemental Figure V). rs56145077 lies within bioinformatically identified binding sites of splicing factors SRSF1 (P = 1.8 × 10-3), RBM45 (P = 3.6 × 10-2), and RBM5 (4.4 × 10-2), thus potentially affecting the splicing code as the minor allele is predicted to decrease exon 13 inclusion by 32% (Figure 4A). Additional tag SNPs in LD with 1000 Genomes SNPs (European data) are listed in Supplemental Table XI.
Figure 4. Cardiometabolic trait-associated variants within genomic loci of alternatively spliced genes and splicing factors.

A, Genome browser view of ALOX5 RNA-seq wiggle tracks showing more exon 13 (purple) inclusion in M1-HMDMs, with a SNP, in LD with a tag SNP for HDL-C, predicted to disrupt the splicing code to decrease exon inclusion by 32%. The LD SNP falls within the binding sites for splicing factors SRSF1, RBM45, and RBM5. B, Genome browser view of splicing factor gene CELF1: RefSeq annotation in blue, C/EBPβ ChIP-seq peaks,58 and histone tail modification marks on ChIP-seq.81 Yellow highlights indicate positions of SNPs in LD with tag SNPs for T2DM (green diamond) and fasting glucose (teal circle), notably overlapping C/EBPβ binding sites. HMDMs = human monocyte derived macrophages; SNP = single nucleotide polymorphism; LD = linkage disequilibrium; ChIP-seq = chromatin immunoprecipitation sequencing; T2DM = type 2 diabetes mellitus.
Variants in Differentially Expressed Splicing Factor Genes during M1-HMDM Activation
In addition to exerting effects on the splicing code of cis-regulatory elements, cardiometabolic trait-associated genetic variants may also affect AS if they are located within the loci encoding relevant splicing factors. Among the 37 differentially expressed splicing factor genes during M1-HMDM activation, seven have SNPs associated with cardiometabolic traits after Bonferroni correction (Table 4). CELF1, a splicing factor implicated in other human diseases,34,57 has the broadest and most statistically significant associations with cardiometabolic traits including HDL-C, body mass index, fasting glucose, and type 2 diabetes mellitus (T2DM) (Table 4). We chose to study CELF1 further for the following reasons: (1) its robust associations with multiple cardiometabolic traits, (2) the enrichment of its RNA-binding motif near differentially spliced exons in M1-HMDM activation (Table 2), and (3) its differential expression on both the mRNA and protein levels during M1 activation (Figure 5A and 5B).
Table 4. Cardiometabolic Trait-Associated Genetic Variants in M1-HMDM Regulated Splicing Factors.
| SNP | Chr | Position | Gene | Effect Allele | Trait | Consortium | Unadjusted P | FDR –Adjusted P | Bonferroni-Adjusted P |
|---|---|---|---|---|---|---|---|---|---|
| rs1044269* | 11 | 47487740 | CELF1 | A | HDL | GLGC | 4.9E-12 | 2.8E-10 | 1.3E-10 |
| rs1317149† | 11 | 47486885 | CELF1 | T | HDL | GLGC | 4.1E-10 | 1.3E-08 | 1.1E-08 |
| rs7933019‡ | 11 | 47509137 | CELF1 | C | Fasting Glu | MAGIC | 4.4E-05 | 7.5E-03 | 1.8E-03 |
| rs11607518‡ | 11 | 47504935 | CELF1 | A | T2DM | DIAGRAM | 1.2E-03 | 1.7E-02 | 2.6E-02 |
| rs2242081‡ | 11 | 47500267 | CELF1 | T | BMI | GIANT | 1.5E-09 | 1.7E-07 | 4.3E-08 |
| rs2232965‡ | 19 | 18426950 | LSM4 | T | CAD | CARDIoGRAM | 7.9E-04 | 1.9E-02 | 2.2E-02 |
| rs3891584‡ | 11 | 114269266 | RBM7 | C | T2DM | DIAGRAM | 1.1E-01 | 1.7E-02 | 2.4E-02 |
| rs7304944‡ | 12 | 56958447 | RBMS2 | T | TG | GLGC | 1.7E-04 | 1.2E-02 | 5.0E-03 |
| rs2657888‡ | 12 | 56938383 | RBMS2 | T | TG | GLGC | 2.9E-04 | 1.2E-02 | 8.7E-03 |
| rs11171894‡ | 12 | 56951219 | RBMS2 | A | Fasting Glu | MAGIC | 2.7E-04 | 1.2E-02 | 1.1E-02 |
| rs12232026‡ | 12 | 56960766 | RBMS2 | A | Fasting Glu | MAGIC | 7.0E-04 | 1.2E-02 | 2.8E-02 |
| rs17465651† | 1 | 38418496 | SF3A3 | T | CAD | CARDIoGRAM | 4.7E-04 | 1.9E-02 | 1.3E-02 |
| rs17465826* | 1 | 38423521 | SF3A3 | T | TG | GLGC | 1.0E-03 | 1.2E-02 | 3.1E-02 |
| rs3766287‡ | 1 | 31765685 | SNRNP40 | T | LDL | GLGC | 9.9E-05 | 1.5E-02 | 7.7E-03 |
| rs2302593‡ | 19 | 46196634 | SNRPD2 | C | Fasting Glu | MAGIC | 4.8E-04 | 1.2E-02 | 1.9E-02 |
Adjusted P-values are corrected for multiple testing. HDL = high-density lipoprotein, Fasting Glu = fasting glucose, T2DM = type 2 diabetes mellitus, BMI = body mass index, CAD = coronary artery disease, TG = triglycerides, FDR = false discovery rate.
3′ UTR,
intergenic,
intronic
Figure 5. Functional studies for CELF1 reveal role in modulating M1-HMDM activation.

A, Relative CELF1 mRNA expression in M1-HMDMs vs. M0-HMDMs by QPCR, normalized to ACTB. N=6. P < 0.01. B, CELF1 protein expression in M1-HMDMs vs. M0-HMDMs by Western blot. N=1. C, siRNA knockdown efficiency of CELF1 compared to non-targeting siRNA control, data normalized to ACTB. N = 3. D, Measured differences on dot blot cytokine array of secreted proteins in the cell supernatant of HMDMs with CELF1 siRNA knockdown vs. non-targeting siRNA control. PAI-1 (P < 0.05) and IL-1β (P < 0.01) were quantitated on densitometry normalized to reference spots. N = 3. N represents number of individuals and does not include triplicate replication. HMDMs = human monocyte derived macrophages.
LD analysis of CELF1's trait-associated tag SNPs revealed that rs11607518 (T2DM, adjusted P = 2.6 × 10-2) and rs7933019 (fasting glucose, adjusted P = 1.8 × 10-3) are in high LD (R2> 0.8) with rs77204316, rs11604233, and rs11039290—SNPs that fall within ChIP-seq binding sites for macrophage-relevant transcription factor C/EBPβ (GSE31621).58 The location of these trait-associated SNPs at the CELF1 locus within binding sites for an essential macrophage transcription factor as well as evidence that M1 macrophages are involved in T2DM59,60 prompted us to ask whether CELF1 plays a role in modulating M1 activation. First, we confirmed in samples of independent healthy human donors, that CELF1 mRNA (Figure 5A) and protein (Figure 5B) expression increased during M1-HMDM activation. Next, we performed siRNA-mediated knockdown of CELF1 (Figure 5C) and evaluated inflammatory response after HMDM stimulation with LPS and IFN-γ. CELF1 knockdown led to increased expression of a subset of inflammatory markers, including secreted IL-1β on cytokine array (Figure 5D, P < 0.01; Supplemental Figure VII B); mRNA expression of IL6 (P = 0.04), IRF1 (P = 0.02), and CXCL10 (P < 0.01) (Supplemental Figure VII A); and secreted IL-6 and CCL5 (Supplemental Figure VII C, P = 0.04 and P = 0.03, respectively). These results suggest that CELF1 may modulate a portion of the anti-inflammatory response during M1 activation.
Induced Pluripotent Stem Cell Derived Macrophages Demonstrate High Fidelity of Differential Alternative Splicing Events
With genetic variation emerging as a significant basis of complex cardiometabolic diseases,54,56,61,62 the use of differentiated iPSCs, including iPSDMs, to model human diseases has become increasingly important in understanding functional impact and translational implications.16,36,63-65 Recent studies have demonstrated that genetic variants may be causal in human diseases by inducing aberrant splicing through altering the splicing code (see above),29,30,66 so we evaluated whether the iPSDM system can be a model for studying AS events. We analyzed RNA-seq data for M1-iPSDM and M2-iPSDM activation (derived from the same donors contributing to HMDM RNA-seq, Figure 1A) to determine if iPSDM cellular models recapitulated the AS profiles for M1-HMDMs and M2-HMDMs. On a transcriptome-wide scale, a strong correlation in gene-level differential AS events (defined as degree of differential isoform expression θ, using identical PennDiff and IUTA criteria as for HMDMs) is seen between M1-HMDMs and M1-iPSDMs (ρ = 0.77, P < 0.001; Figure 3B). RT-PCR assays also confirmed fidelity of predicted differences in AS patterns for both M1-HMDMs and M1-iPSDMs (examples from three sequenced donors shown in Figure 3A). Furthermore, a strong correlation in differential expression of splicing factor genes was seen between the two cell systems (ρ = 0.78, P < 0.001; Figure 3C), indicating that splicing regulation is largely similar between iPSDMs and HMDMs. These results demonstrate that the iPSDM system can be a useful tool in functional genomic interrogation AS events.
Discussion
To our knowledge, this the first study to characterize comprehensively the AS landscape of human macrophage activation, particularly with respect to cell- and phenotypic specificity and cardiometabolic diseases. Although one previous study demonstrated that a common genetic variant associated with low density lipoprotein cholesterol can alter AS,66 ours is the first to integrate summary-level data from multiple GWAS consortia to explore the relationship between cardiometabolic variants and AS in M1 activation. We present novel evidence that the splicing factor CELF1, mostly studied in the context of muscular dystrophy and cardiomyopathy, plays a role in modulating M1 activation of human macrophages and also harbors genetic variation for multiple cardiometabolic traits. Finally, our iPSDM system provides a reliable cellular model for functional interrogation of human macrophage splicing events facilitating translational study of disease associated genetic variation in both the splicing code and splicing factors.
Transcriptional programs have been recognized as regulators of macrophage activation,1,2,8,15,16 yet little is known about whether isoform-level expression profiles are also distinct for M1 and M2 macrophages. Because knockdown of whole genes may lead to unanticipated off-target effects, identifying disease-relevant isoform-level differences in a tissue- or cell-specific manner may open the door for more selective therapeutic targeting in re-programming macrophage phenotype, a focus of increasing interest in clinical translation.8,67,68 Here we demonstrated that M1-HMDM activation was indeed associated with many changes on the isoform level, including more than 200 differential AS events. Although the number of AS events was far fewer than the number of differentially expressed genes in M1 activation, these AS events were enriched for a network of biologically relevant genes involved in macrophage functions such as autophagy (e.g., PLD1)69 as well as immune processes (e.g., CD74 and TLR4),70-73 all of which are linked to the modulation of macrophage inflammation. M2 activation also confers a recognized pattern of differential gene expression,16 but it induces relatively few differential AS events, suggesting that the inflammatory stress response of M1 activation may create more transcriptomic diversity through AS than in M2 activation. Differential AS events in inflamed PBMCs overlapped minimally with those in M1-HMDMs, reinforcing the concept that stress-induced splicing changes are mostly cell- and context-specific.
After establishing that AS contributes to the transcriptomic response during macrophage activation, the next mechanistic steps to advance clinical translation are to study (1) specific splicing targets and (2) the larger regulatory mechanisms underlying differential AS in macrophage activation. Here we showed through our proof-of-principle experiment with CD74 isoform-specific knockdown that transcript variants can have major effects on cellular function and survival. Whether transcriptome-wide differential AS events in M1-HMDM activation have systematic functional and pathophysiological significance requires further investigation. It is more likely that genes differentially spliced in M1-HMDM activation can modulate cardiometabolic pathophysiology if they house SNPs associated with relevant traits. The combination of structural mRNA changes in response to environmental challenges and the existence of trait-associated variants within a locus make a case for the potential importance of a gene in macrophage phenotype. We found 28 such tag SNPs within the loci of 21 genes differentially spliced in macrophage M1 activation. A subset of these SNPs are in LD (identifiable via 1000 Genomes74 data, Supplemental Table XI) with specific variants that fall within regulation sites of alternative splicing. They may alter the splicing code and subsequently induce aberrant splicing to generate specific isoforms favoring a disease phenotype, a phenomenon documented for rare and common genetic variants for a variety of human diseases.29,66 For example, here we showed that ALOX5, a gene involved in pro-inflammatory functions and leukotriene synthesis,75 is predicted to have a decrease in its alternative exon inclusion level in the presence of a minor allele SNP associated with HDL-C. Further functional validation of this variant in terms of its effect on ALOX5's alternative splicing and downstream pathways is warranted. Because the majority of genes alternatively spliced in macrophage activation do not house cardiometabolic variants at their loci, while only a subset of those with trait-associated variants have sites where SNPs may affect splicing efficiency, trait-associated variants likely are not the primary driver of alternative splicing in this process. However, tag SNPs, serving as guideposts to identify genes functionally relevant in cardiometabolic traits, may still help identify and prioritize novel targets in studies of macrophage activation within the human disease context.
Beyond specific AS events and specific spliced genes, the expression and function of splicing factors determining isoform switch patterns in a tissue- and cell-specific manner holds promise for novel mechanistic insights and therapeutic translation. We found that M1-associated glucometabolic traits76 are statistically associated with SNPs at the CELF1 locus overlying binding sites for the macrophage-relevant transcription factor C/EBPβ, suggesting that regulation of CELF1 gene expression in macrophages may impact human cardiometabolic diseases. Indeed, knockdown of CELF1 levels modulated expression of a subset of M1 markers, suggesting that CELF1 splicing targets are involved in M1 activation. Because AS is a critical process during development,31,34,57 the increased splicing switches that occur in M1 activation may indicate that inflammatory stress induces a dedifferentiation profile. This concept is consistent with our observation of increased M1-HMDM expression of CELF1, which is more highly expressed in the embryonic state.50,57 Remarkably, 36% of our bioinformatically identified AS events in M1-HMDM activation overlap with transcripts found to interact with CELF1 in CLIP-seq of murine hearts (GSE61892-61893),57 although CLIP-seq within the human macrophage system will need to be performed in order to confirm experimentally that CELF1 is a regulator of AS in M1 activation.
Beyond splicing factors, additional regulatory mechanisms could be at play in influencing splicing patterns in macrophage biology. As expected, M1-HMDM activation was accompanied by a wealth of alternative promoter usage given that transcriptional regulation has been established as a driving force in determining macrophage phenotype. However, our data show that alternative promoter usage was highly correlated with AS events in the same gene, a poorly studied and understood phenomenon.46 Future studies are needed to identify whether specific pro-inflammatory transcription factors interact with splicing factors to orchestrate M1-HMDM isoform switches and whether these switches can be explained by chromatin changes during M1 activation. In this respect, future studies on epigenetics, which have also been implicated in AS,77 may be informative.
This study has several strengths. First, we took an unbiased approach to discover M1-HMDM differential AS events not previously identified in the literature. Second, our use of primary human macrophages for our primary analysis and for RT-PCR validation strengthens the applicability of our findings to human physiology and thus future clinical translational potential given the species-specific nature of many AS events.18,20,21,78 Third, we leverage GWAS data for cardiometabolic traits to identify potentially novel modulators of M1 activation in complex human diseases. Fourth, our exploration of whether differential AS events can be recapitulated in the iPSDM system validates the use of this cutting-edge genomic technology for future disease variant-oriented studies of AS in macrophages.
This study also has limitations that suggest further avenues of research. First, while we identified many differential AS events and validated a few for M1-HMDM activation, demonstrating that specific isoforms are necessary for M1 activation was beyond the scope of this work. Second, although we were able to validate splicing changes on the mRNA level, we did not present protein-level data as our attempts to resolve predicted small molecular weight changes and modest isoform shifts using Western blot methods did not yield detectable differences. These differences would be more accurately detected and quantitated by mass spectrometry, which will be employed in future targeted follow-up. Also, our use of HMDMs from healthy individuals as a model for M1 and M2 activation may not capture the full spectrum of macrophage-specific transcriptomic changes in chronic cardiometabolic diseases (e.g., atherosclerosis) that develop and progress from factors including but not limited to age, genetic background, and other co-morbidities. In addition, our analysis of splicing factors lacked human macrophage CLIP-seq data, which can help prove that CELF1's effects on the M1 phenotype are via AS rather than other RNA binding regulatory mechanisms. Finally, our GWAS translation can be considered a starting point to generate experimental data to demonstrate whether the disease-associated SNPs are truly causal in altering splicing patterns and in modulating M1 activation. Future studies on the molecular mechanisms, RNA-binding protein interactions, causal splicing variants, and novel pathways of alternative splicing changes in macrophage activation are warranted.
In summary, we show that isoform-level differences help define macrophage phenotype and that the splicing mechanisms for producing isoform switches occur in a cell- and stimulus-specific fashion. Aberrant splicing that leads to deleterious traits may occur in the presence of trait-associated genetic variants that provide a causal link between splicing event and disease-related phenotype. These findings lay the groundwork for future macrophage isoform-specific studies and for evaluating novel causal mechanisms and therapeutic targets in cardiometabolic diseases.
Supplementary Material
Highlights.
Alternative splicing (AS), the generation of multiple isoforms from a single gene, is emerging as a key mechanism for proteomic diversity that contributes to cellular reprogramming and cellular phenotypes. For complex cardiometabolic diseases, AS is an understudied cell-specific process that may contribute to inflammatory macrophage activation and disease pathophysiology. This is the first comprehensive assessment of AS and its regulation in macrophage activation, with a translational focus on the cardiometabolic disease-associated, pro-inflammatory M1 phenotype.
We characterized in primary human macrophages cell-specific, phenotype-specific, and context-specific AS landscapes of macrophage activation.
Using human genetic data, we identified alternatively spliced genes and splicing factors relevant to M1 activation as novel candidates and targets for cardiometabolic pathophysiologies.
iPSDMs were validated as a functional genomic cell model system for investigating human AS events.
This work lays the foundation for further isoform-specific functional studies of potential novel causal mechanisms and therapeutic targets for cardiometabolic diseases and advances the study of AS in the context of human genetics toward future translation in precision medicine.
Acknowledgments
We would like to thank the participants of the study, the staff at the University of Pennsylvania Clinical Translational Research Center, and the University of Pennsylvania iPS Core.
Sources of Funding: This work was supported by R01-HL-113147 (M.P.R. and M.L.), K24-HL-107643 (to M.P.R.), R01-GM-108600 (to M.L.), R01-HL-107196 (to A.S.F.), and U01-HG-006398 (to D.J.R.). J.L. is supported by KL2-TR-000139.
Nonstandard Abbreviations and Acronyms
- AS
Alternative splicing
- BMI
Body mass index
- CAD
Coronary artery disease
- CLIP-seq
Cross-linking and immunoprecipitation sequencing
- GWAS
Genome-wide association study
- HDL
High density lipoprotein
- HMDM
Human monocyte derived macrophages
- iPSC
Induced pluripotent stem cell
- iPSDM
Induced pluripotent stem cell derived macrophages
- LD
Linkage disequilibrium
- LDL
Low density lipoprotein
- LPS
Lipopolysaccharide
- PBMC
Peripheral blood mononuclear cell
- RNA-seq
RNA sequencing
- RT-PCR
Reverse transcriptase polymerase chain reaction
- SNP
Single nucleotide polymorphism
- T2DM
Type 2 diabetes mellitus
- TG
Triglyceride
Footnotes
Contributions to this work: Study design and concept (J.L., M.P.R., M.L.), data analysis (Y.H., S.N., A.S.F., C.X., J.L.), data acquisition (J.L., H.Z., M.G., W.L.), editing of manuscript (J.L., M.P.R., M.L., A.S.F., Y.H., K.M., D.J.R.), final approval of manuscript (J.L., M.P.R.).
Disclosures: None
Contributor Information
Jennie Lin, Email: linjenn@mail.med.upenn.edu, Renal, Electrolyte, and Hypertension Division; Department of Medicine, Perelman School of Medicine at the University of Pennsylvania, 1 Founders, 3400 Spruce Street, Philadelphia, PA 19104, Tel: 215-898-6388; Fax: 215-615-0349.
Yu Hu, Department of Biostatistics and Epidemiology, University of Pennsylvania, Philadelphia, PA.
Sara Nunez, Department of Mathematics and Statistics, Mount Holyoke College, South Hadley, MA.
Andrea S. Foulkes, Department of Mathematics and Statistics, Mount Holyoke College, South Hadley, MA
Benjamin Cieply, Department of Genetics, Perelman School of Medicine at the University of Pennsylvania, Philadelphia, PA.
Chenyi Xue, Division of Cardiology; Department of Medicine, Columbia University Medical Center, New York, NY.
Mark Gerelus, Cardiovascular Institute; Department of Medicine, Perelman School of Medicine at the University of Pennsylvania, Philadelphia, PA; Cardiovascular Institute; Department of Medicine, Perelman School of Medicine at the University of Pennsylvania, Philadelphia, PA.
Wenjun Li, Cardiovascular Institute; Department of Medicine, Perelman School of Medicine at the University of Pennsylvania, Philadelphia, PA; Cardiovascular Institute; Department of Medicine, Perelman School of Medicine at the University of Pennsylvania, Philadelphia, PA.
Hanrui Zhang, Division of Cardiology; Department of Medicine, Columbia University Medical Center, New York, NY.
Daniel J. Rader, Department of Genetics, Perelman School of Medicine at the University of Pennsylvania, Philadelphia, PA
Kiran Musunuru, Department of Genetics, Perelman School of Medicine at the University of Pennsylvania, Philadelphia, PA; Cardiovascular Institute; Department of Medicine, Perelman School of Medicine at the University of Pennsylvania, Philadelphia, PA.
Mingyao Li, Department of Biostatistics and Epidemiology, University of Pennsylvania, Philadelphia, PA.
Muredach P. Reilly, Email: mpr2144@cumc.columbia.edu, Division of Cardiology; Department of Medicine, Columbia University Medical Center, New York, NY; Irving Institute for Clinical and Translational Research, Columbia University Medical Center, 622 West 168th Street, PH10-305, New York, NY 10032, Tel: 212-305-9453; Fax: 212-305-3213.
References
- 1.Gosselin D, Link VM, Romanoski CE, Fonseca GJ, Eichenfield DZ, Spann NJ, Stender JD, Chun HB, Garner H, Geissmann F, Glass CK. Environment Drives Selection and Function of Enhancers Controlling Tissue-Specific Macrophage Identities. Cell. 2014;159:1327–1340. doi: 10.1016/j.cell.2014.11.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Murray PJ, Wynn TA. Protective and pathogenic functions of macrophage subsets. Nature reviews: Immunology. 2011;11:723–737. doi: 10.1038/nri3073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Colin S, Chinetti-Gbaguidi G, Staels B. Macrophage phenotypes in atherosclerosis. Immunological Reviews. 2014;262:153–166. doi: 10.1111/imr.12218. [DOI] [PubMed] [Google Scholar]
- 4.Auton A, Abecasis GR, Altshuler DM, Durbin RM, Abecasis GR, Bentley DR, Chakravarti A, Clark AG, Donnelly P, Eichler EE, Flicek P, Gabriel SB, Gibbs RA, Green ED, Hurles ME, et al. A global reference for human genetic variation. Nature. 2015;526:68–74. doi: 10.1038/nature15393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Stöger JL, Gijbels MJJ, van der Velden S, Manca M, van der Loos CM, Biessen EAL, Daemen MJAP, Lutgens E, de Winther MPJ. Distribution of macrophage polarization markers in human atherosclerosis. Atherosclerosis. 2012;225:461–468. doi: 10.1016/j.atherosclerosis.2012.09.013. [DOI] [PubMed] [Google Scholar]
- 6.Calvano SE, Xiao W, Richards DR, Felciano RM, Baker HV, Cho RJ, Chen RO, Brownstein BH, Cobb JP, Tschoeke SK, Miller-Graziano C, Moldawer LL, Mindrinos MN, Davis RW, Tompkins RG, et al. A network-based analysis of systemic inflammation in humans. Nature. 2005;437:1032–1037. doi: 10.1038/nature03985. [DOI] [PubMed] [Google Scholar]
- 7.Rees AJ. Monocyte and Macrophage Biology: An Overview. YSNEP. 2010;30:216–233. doi: 10.1016/j.semnephrol.2010.03.002. [DOI] [PubMed] [Google Scholar]
- 8.Moore KJ, Sheedy FJ, Fisher EA. Macrophages in atherosclerosis: a dynamic balance. Nature reviews Immunology. 2013;13:709–721. doi: 10.1038/nri3520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Fadini GP, Simoni F, Cappellari R, Vitturi N, Galasso S, Vigili de Kreutzenberg S, Previato L, Avogaro A. Pro-inflammatory monocyte-macrophage polarization imbalance in human hypercholesterolemia and atherosclerosis. Atherosclerosis. 2014;237:805–808. doi: 10.1016/j.atherosclerosis.2014.10.106. [DOI] [PubMed] [Google Scholar]
- 10.Bories G, Caiazzo R, Derudas B, Copin C, Raverdy V, Pigeyre M, Pattou F, Staels B, Chinetti-Gbaguidi G. Impaired alternative macrophage differentiation of peripheral blood mononuclear cells from obese subjects. Diabetes and Vascular Disease Research. 2012;9:189–195. doi: 10.1177/1479164111430242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Peled M, Fisher EA. Dynamic Aspects of Macrophage Polarization during Atherosclerosis Progression and Regression. Frontiers in immunology. 2014;5:579. doi: 10.3389/fimmu.2014.00579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lee CW, Hwang I, Park CS, Lee H, Park DW, Kang SJ, Lee SW, Kim YH, Park SW, Park SJ. Macrophage Heterogeneity of Culprit Coronary Plaques in Patients With Acute Myocardial Infarction or Stable Angina. American Journal of Clinical Pathology. 2013;139:317–322. doi: 10.1309/AJCP7KEYGN3OBGQX. [DOI] [PubMed] [Google Scholar]
- 13.Ley K, Miller YI, Hedrick CC. Monocyte and Macrophage Dynamics During Atherogenesis. Arteriosclerosis, Thrombosis, and Vascular Biology. 2011;31:1506–1516. doi: 10.1161/ATVBAHA.110.221127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wynn TA, Chawla A, Pollard JW. Macrophage biology in development, homeostasis and disease. Nature. 2013;496:445–455. doi: 10.1038/nature12034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hanna RN, Shaked I, Hubbeling HG, Punt JA, Wu R, Herrley E, Zaugg C, Pei H, Geissmann F, Ley K, Hedrick CC. NR4A1 (Nur77) Deletion Polarizes Macrophages Toward an Inflammatory Phenotype and Increases Atherosclerosis. Circulation Research. 2012;110:416–427. doi: 10.1161/CIRCRESAHA.111.253377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhang H, Xue C, Shah R, Bermingham K, Hinkle CC, Li W, Rodrigues A, Tabita-Martinez J, Millar JS, Cuchel M, Pashos EE, Liu Y, Yan R, Yang W, Gosai SJ, et al. Functional analysis and transcriptomic profiling of iPSC-derived macrophages and their application in modeling Mendelian disease. Circulation Research. 2015;117:17–28. doi: 10.1161/CIRCRESAHA.117.305860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fu XD, Ares M. Context-dependent control of alternative splicing by RNA-binding proteins. Nature Reviews Genetics. 2014;15:689–701. doi: 10.1038/nrg3778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Modrek B, Lee CJ. Alternative splicing in the human, mouse and rat genomes is associated with an increased frequency of exon creation and/or loss. Nature genetics. 2003;34:177–180. doi: 10.1038/ng1159. [DOI] [PubMed] [Google Scholar]
- 19.Chen M, Manley JL. Mechanisms of alternative splicing regulation: insights from molecular and genomics approaches. Nature reviews Molecular cell biology. 2009;10:741–754. doi: 10.1038/nrm2777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Takeda JI, Suzuki Y, Sakate R, Sato Y, Seki M, Irie T, Takeuchi N, Ueda T, Nakao M, Sugano S, Gojobori T, Imanishi T. Low conservation and species-specific evolution of alternative splicing in humans and mice: comparative genomics analysis using well-annotated full-length cDNAs. Nucleic Acids Research. 2008;36:6386–6395. doi: 10.1093/nar/gkn677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gueroussov S, Gonatopoulos-Pournatzis T, Irimia M, Raj B, Lin ZY, Gingras AC, Blencowe BJ. RNA Splicing. An alternative splicing event amplifies evolutionary differences between vertebrates. Science. 2015;349:868–873. doi: 10.1126/science.aaa8381. [DOI] [PubMed] [Google Scholar]
- 22.Heinzen EL, Ge D, Cronin KD, Maia JM, Shianna KV, Gabriel WN, Welsh-Bohmer KA, Hulette CM, Denny TN, Goldstein DB. Tissue-Specific Genetic Control of Splicing: Implications for the Study of Complex Traits. In: Liu E, editor. PLoS biology. Vol. 6. 2008. p. e1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ellis JD, Barrios-Rodiles M, Çolak R, Irimia M, Kim T, Calarco JA, Wang X, Pan Q, O'Hanlon D, Kim PM, Wrana JL, Blencowe BJ. Tissue-specific alternative splicing remodels protein-protein interaction networks. Molecular cell. 2012;46:884–892. doi: 10.1016/j.molcel.2012.05.037. [DOI] [PubMed] [Google Scholar]
- 24.Wang ET, Sandberg R, Luo S, Khrebtukova I, Zhang L, Mayr C, Kingsmore SF, Schroth GP, Burge CB. Alternative isoform regulation in human tissue transcriptomes. Nature. 2008;456:470–476. doi: 10.1038/nature07509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Johnson JM, Castle J, Garrett-Engele P, Kan Z, Loerch PM, Armour CD, Santos R, Schadt EE, Stoughton R, Shoemaker DD. Genome-wide survey of human alternative pre-mRNA splicing with exon junction microarrays. Science. 2003;302:2141–2144. doi: 10.1126/science.1090100. [DOI] [PubMed] [Google Scholar]
- 26.Barash Y, Calarco JA, Gao W, Pan Q, Wang X, Shai O, Blencowe BJ, Frey BJ. Deciphering the splicing code. Nature. 2010;465:53–59. doi: 10.1038/nature09000. [DOI] [PubMed] [Google Scholar]
- 27.Zhang XO, Yin QF, Wang HB, Zhang Y, Chen T, Zheng P, Lu X, Chen LL, Yang L. Species-specific alternative splicing leads to unique expression of sno-lncRNAs. BMC genomics. 2014;15:1–15. doi: 10.1186/1471-2164-15-287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cieply B, Carstens RP. Functional roles of alternative splicing factors in human disease. Wiley Interdisciplinary Reviews: RNA. 2015;6:311–326. doi: 10.1002/wrna.1276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Xiong HY, Alipanahi B, Lee LJ, Bretschneider H, Merico D, Yuen RKC, Hua Y, Gueroussov S, Najafabadi HS, Hughes TR, Morris Q, Barash Y, Krainer AR, Jojic N, Scherer SW, et al. RNA splicing. The human splicing code reveals new insights into the genetic determinants of disease. Science. 2015;347:1254806. doi: 10.1126/science.1254806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Rosenberg AB, Patwardhan RP, Shendure J, Seelig G. Learning the Sequence Determinants of Alternative Splicing from Millions of Random Sequences. Cell. 2015;163:698–711. doi: 10.1016/j.cell.2015.09.054. [DOI] [PubMed] [Google Scholar]
- 31.Warzecha CC, Jiang P, Amirikian K, Dittmar KA, Lu H, Shen S, Guo W, Xing Y, Carstens RP. An ESRP-regulated splicing programme is abrogated during the epithelial–mesenchymal transition. The EMBO Journal. 2010;29:3286–3300. doi: 10.1038/emboj.2010.195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ohta S, Nishida E, Yamanaka S, Yamamoto T. Global Splicing Pattern Reversion during Somatic Cell Reprogramming. CellReports. 2013;5:357–366. doi: 10.1016/j.celrep.2013.09.016. [DOI] [PubMed] [Google Scholar]
- 33.Irimia M, Weatheritt RJ, Ellis JD, Parikshak NN, Gonatopoulos-Pournatzis T, Babor M, Quesnel-Vallières M, Tapial J, Raj B, O'Hanlon D, Barrios-Rodiles M, Sternberg MJE, Cordes SP, Roth FP, Wrana JL, et al. A Highly Conserved Program of Neuronal Microexons Is Misregulated in Autistic Brains. Cell. 2014;159:1511–1523. doi: 10.1016/j.cell.2014.11.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Verma SK, Deshmukh V, Liu P, Nutter CA, Espejo R, Hung ML, Wang GS, Yeo GW, Kuyumcu-Martinez MN. Reactivation of fetal splicing programs in diabetic hearts is mediated by protein kinase C signaling. Journal of Biological Chemistry. 2013;288:35372–35386. doi: 10.1074/jbc.M113.507426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ip JY, Tong A, Pan Q, Topp JD, Blencowe BJ, Lynch KW. Global analysis of alternative splicing during T-cell activation. RNA. 2007;13:563–572. doi: 10.1261/rna.457207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Alasoo K, Martinez FO, Hale C, Gordon S, Powrie F, Dougan G, Mukhopadhyay S, Gaffney DJ. Transcriptional profiling of macrophages derived from monocytes and iPS cells identifies a conserved response to LPS and novel alternative transcription. Scientific reports. 2015;5:12524. doi: 10.1038/srep12524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Beyer M, Mallmann MR, Xue J, Staratschek-Jox A, Vorholt D, Krebs W, Sommer D, Sander J, Mertens C, Nino-Castro A, Schmidt SV, Schultze JL. High-Resolution Transcriptome of Human Macrophages. In: Zirlik A, editor. PLoS ONE. Vol. 7. 2012. p. e45466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Xue J, Schmidt SV, Sander J, Draffehn A, Krebs W, Quester I, De Nardo D, Gohel TD, Emde M, Schmidleithner L, Ganesan H, Nino-Castro A, Mallmann MR, Labzin L, Theis H, et al. Transcriptome-Based Network Analysis Revealsa Spectrum Model of Human Macrophage Activation. Immunity. 2014;40:274–288. doi: 10.1016/j.immuni.2014.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hu Y, Lin J, Hu G, Wang K, Zhang H, Stambolian D, Reilly MP, Li M. PennDiff: Detecting Differential Alternative Splicing from RNA-Seq Data. doi: 10.1093/bioinformatics/bty097. Submitted. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Anders S, Reyes A, Huber W. Detecting differential usage of exons from RNA-seq data. Genome Research. 2012;22:2008–2017. doi: 10.1101/gr.133744.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Niu L, Huang W, Umbach DM, Li L. IUTA: a tool for effectively detecting differential isoform usage from RNA-Seq data. BMC genomics. 2014;15:862. doi: 10.1186/1471-2164-15-862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ferguson JF, Patel PN, Shah RY, Mulvey CK, Gadi R, Nijjar PS, Usman HM, Mehta NN, Shah R, Master SR, Propert KJ, Reilly MP. Race and gender variation in response to evoked inflammation. Journal of Translational Medicine. 2013;11:63. doi: 10.1186/1479-5876-11-63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Liu Y, Ferguson JF, Xue C, Ballantyne RL, Silverman IM, Gosai SJ, Serfecz J, Morley MP, Gregory BD, Li M, Reilly MP. Tissue-Specific RNA-Seq in Human Evoked Inflammation Identifies Blood and Adipose LincRNA Signatures of Cardiometabolic Diseases. Arteriosclerosis, Thrombosis, and Vascular Biology. 2014;34:902–912. doi: 10.1161/ATVBAHA.113.303123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Mehta NNN, Heffron SPS, Patel PNP, Ferguson JJ, Shah RDR, Hinkle CCC, Krishnamoorthy PP, Shah RR, Tabita-Martinez JJ, Terembula KK, Master SRS, Rickels MRM, Reilly MPM. A human model of inflammatory cardio-metabolic dysfunction; a double blind placebo-controlled crossover trial. Journal of Translational Medicine. 2012;10:124–124. doi: 10.1186/1479-5876-10-124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Singer GA, Wu J, Yan P, Plass C, Huang TH, Davuluri RV. Genome-wide analysis of alternative promoters of human genes using a custom promoter tiling array. BMC genomics. 2008;9:349. doi: 10.1186/1471-2164-9-349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Xin D, Hu L, Kong X. Alternative promoters influence alternative splicing at the genomic level. PLoS ONE. 2008;3:e2377. doi: 10.1371/journal.pone.0002377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kornblihtt AR. Promoter usage and alternative splicing. Current Opinion in Cell Biology. 2005;17:262–268. doi: 10.1016/j.ceb.2005.04.014. [DOI] [PubMed] [Google Scholar]
- 48.Takahashi K, Koga K, Linge HM, Zhang Y, Lin X, Metz CN, Al-Abed Y, Ojamaa K, Miller EJ. Macrophage CD74 contributes to MIF-induced pulmonary inflammation. Respiratory Research. 2009;10:33. doi: 10.1186/1465-9921-10-33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Carstens RP. Networking in an Alternative Splicing World. Molecular cell. 2014;54:903–904. doi: 10.1016/j.molcel.2014.06.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Han H, Irimia M, Ross PJ, Sung HK, Alipanahi B, David L, Golipour A, Gabut M, Michael IP, Nachman EN, Wang E, Trcka D, Thompson T, O'Hanlon D, Slobodeniuc V, et al. MBNL proteins repress ES-cell-specific alternative splicing and reprogramming. Nature. 2013;498:241–245. doi: 10.1038/nature12270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Musunuru K, Strong A, Frank-Kamenetsky M, Lee NE, Ahfeldt T, Sachs KV, Li X, Li H, Kuperwasser N, Ruda VM, Pirruccello JP, Muchmore B, Prokunina-Olsson L, Hall JL, Schadt EE, et al. From noncoding variant to phenotype via SORT1 at the 1p13 cholesterol locus. Nature. 2010;466:714–719. doi: 10.1038/nature09266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Soccio RE, Chen ER, Rajapurkar SR, Safabakhsh P, Marinis JM, Dispirito JR, Emmett MJ, Briggs ER, Bin Fang, Everett LJ, Lim HW, Won KJ, Steger DJ, Wu Y, Civelek M, et al. Genetic Variation Determines PPARg Function and Anti-diabetic Drug Response In Vivo. Cell. 2015;162:33–44. doi: 10.1016/j.cell.2015.06.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.CARDIoGRAMplusC4D Consortium. Deloukas P, Kanoni S, Willenborg C, Farrall M, Assimes TL, Thompson JR, Ingelsson E, Saleheen D, Erdmann J, Goldstein BA, Stirrups K, König IR, Cazier JB, Johansson A, et al. Large-scale association analysis identifies new risk loci for coronary artery disease. Nature genetics. 2013;45:25–33. doi: 10.1038/ng.2480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Gaulton KJ, Ferreira T, Lee Y, Raimondo A, Mägi R, Reschen ME, Mahajan A, Locke A, Rayner NW, Robertson N, Scott RA, Prokopenko I, Scott LJ, Green T, Sparso T, et al. ng.3437. Nature genetics. 2015;47:1415–1425. doi: 10.1038/ng.3437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.DIAbetes Genetics Replication And Meta-analysis (DIAGRAM) Consortium, Asian Genetic Epidemiology Network Type 2 Diabetes (AGEN-T2D) Consortium, South Asian Type 2 Diabetes (SAT2D) Consortium, Mexican American Type 2 Diabetes (MAT2D) Consortium, Type 2 Diabetes Genetic Exploration by Nex-generation sequencing in muylti-Ethnic Samples (T2D-GENES) Consortium. Mahajan A, Go MJ, Zhang W, Below JE, Gaulton KJ, Ferreira T, Horikoshi M, Johnson AD, Ng MCY, Prokopenko I, et al. Genome-wide trans-ancestry meta-analysis provides insight into the genetic architecture of type 2 diabetes susceptibility. Nature genetics. 2014;46:234–244. doi: 10.1038/ng.2897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Do R, Willer CJ, Schmidt EM, Sengupta S, Gao C, Peloso GM, Gustafsson S, Kanoni S, Ganna A, Chen J, Buchkovich ML, Mora S, Beckmann JS, Bragg-Gresham JL, Chang HY, et al. Common variants associated with plasma triglycerides and risk for coronary artery disease. Nature genetics. 2013;45:1345–1352. doi: 10.1038/ng.2795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Wang ET, Ward AJ, Cherone JM, Giudice J, Wang TT, Treacy DJ, Lambert NJ, Freese P, Saxena T, Cooper TA, Burge CB. Antagonistic regulation of mRNA expression and splicing by CELF and MBNL proteins. Genome Research. 2015;25:858–871. doi: 10.1101/gr.184390.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Pham TH, Benner C, Lichtinger M, Schwarzfischer L, Hu Y, Andreesen R, Chen W, Rehli M. Dynamic epigenetic enhancer signatures reveal key transcription factors associated with monocytic differentiation states. Blood. 2012;119:e161–e171. doi: 10.1182/blood-2012-01-402453. [DOI] [PubMed] [Google Scholar]
- 59.Reddy MA, Chen Z, Park JT, Wang M, Lanting L, Zhang Q, Bhatt K, Leung A, Wu X, Putta S, Saetrom P, Devaraj S, Natarajan R. Regulation of Inflammatory Phenotype in Macrophages by a Diabetes-Induced Long Noncoding RNA. Diabetes. 2014;63:4249–4261. doi: 10.2337/db14-0298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Murray PJ, Allen JE, Biswas SK, Fisher EA, Gilroy DW, Goerdt S, Gordon S, Hamilton JA, Ivashkiv LB, Lawrence T, Locati M, Mantovani A, Martinez FO, Mege JL, Mosser DM, et al. Macrophage activation and polarization: nomenclature and experimental guidelines. Immunity. 2014;41:14–20. doi: 10.1016/j.immuni.2014.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Edwards SL, Beesley J, French JD, Dunning AM. Beyond GWASs: Illuminatingthe Dark Road from Association to Function. The American Journal of Human Genetics. 2013;93:779–797. doi: 10.1016/j.ajhg.2013.10.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.MB DMPR, PhD ML, PhD JH, PhD JFF, PhD IMS, MD NNM, PhD MSB, PhD JMD, MD CWK, PhD PJRT, PhD BDH, PhD AFS, MD TLA, MD PSW, PhD HA, et al. ArticlesIdentification of. Lancet. 2011;377:383–392.
- 63.Fox CS, Hall JL, Arnett DK, Ashley EA, Delles C, Engler MB, Freeman MW, Johnson JA, Lanfear DE, Liggett SB, Lusis AJ, Loscalzo J, MacRae CA, Musunuru K, Newby LK, et al. Future translational applications from the contemporary genomics era: a scientific statement from the American Heart Association. Circulation. 2015;131:1715–1736. doi: 10.1161/CIR.0000000000000211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Musunuru K. Genome editing of human pluripotent stem cells to generate human cellular disease models. Disease Models & Mechanisms. 2013;6:896–904. doi: 10.1242/dmm.012054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Gupta RM, Meissner TB, Cowan CA, Musunuru K. Genome-Edited Human Pluripotent Stem Cell-Derived Macrophages as a Model of Reverse Cholesterol Transport-Brief Report. Arteriosclerosis, Thrombosis, and Vascular Biology. 2016;36:15–18. doi: 10.1161/ATVBAHA.115.305956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Burkhardt R, Kenny EE, Lowe JK, Birkeland A, Josowitz R, Noel M, Salit J, Maller JB, Pe'er I, Daly MJ, Altshuler D, Stoffel M, Friedman JM, Breslow JL. Common SNPs in HMGCR in Micronesians and Whites Associated With LDL-Cholesterol Levels Affect Alternative Splicing of Exon13. Arteriosclerosis, Thrombosis, and Vascular Biology. 2008;28:2078–2084. doi: 10.1161/ATVBAHA.108.172288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Li X, Yao W, Yuan Y, Chen P, Li B, Li J, Chu R, Song H, Xie D, Jiang X, Wang H. Targeting of tumour-infiltrating macrophages via CCL2/CCR2 signalling as a therapeutic strategy against hepatocellular carcinoma. Gut. 2015;0:1–11. doi: 10.1136/gutjnl-2015-310514. [DOI] [PubMed] [Google Scholar]
- 68.Schultze JL. Reprogramming of macrophages — new opportunities for therapeutic targeting. Current Opinion in Pharmacology. 2016;26:10–15. doi: 10.1016/j.coph.2015.09.007. [DOI] [PubMed] [Google Scholar]
- 69.Dall'Armi C, Hurtado-Lorenzo AES, Tian H, Morel E, Nezu A, Chan RB, Yu WH, Robinson KS, Yeku O, Small SA, Duff K, Frohman MA, Wenk MR, Yamamoto A, Di Paolo G. The phospholipase D1 pathway modulates macroautophagy. Nature Communications. 2010;1:142–11. doi: 10.1038/ncomms1144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Martin-Ventura JL, Madrigal-Matute J, Munoz-Garcia B, Blanco-Colio LM, Van Oostrom M, Zalba G, Fortuno A, Gomez-Guerrero C, Ortega L, Ortiz A, Diez J, Egido J. Increased CD74 expression in human atherosclerotic plaques: contribution to inflammatory responses in vascular cells. Cardiovascular Research. 2009;83:586–594. doi: 10.1093/cvr/cvp141. [DOI] [PubMed] [Google Scholar]
- 71.Starlets D, Gore Y, Binsky I, Haran M, Harpaz N, Shvidel L, Becker-Herman S, Berrebi A, Shachar I. Cell-surface CD74 initiates a signaling cascade leading to cell proliferation and survival. Blood. 2006;107:4807–4816. doi: 10.1182/blood-2005-11-4334. [DOI] [PubMed] [Google Scholar]
- 72.Waltz P, Carchman EH, Young AC, Rao J, Rosengart MR, Kaczorowski D, Zuckerbraun BS. Lipopolysaccaride induces autophagic signaling in macrophages via a TLR4, heme oxygenase-1 dependent pathway. Autophagy. 2011;7:315–320. doi: 10.4161/auto.7.3.14044. [DOI] [PubMed] [Google Scholar]
- 73.Xu YY, Jagannath CC, Liu XDX, Sharafkhaneh AA, Kolodziejska KEK, Eissa NTN. Toll-like Receptor 4 Is a Sensor for Autophagy Associated with Innate Immunity. Immunity. 2007;27:10–10. doi: 10.1016/j.immuni.2007.05.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Lappalainen T, Sammeth M, Friedländer MR, 't Hoen PAC, Monlong J, Rivas MA, Gonzàlez-Porta M, Kurbatova N, Griebel T, Ferreira PG, Barann M, Wieland T, Greger L, van Iterson M, Almlöf J, et al. Transcriptome and genome sequencing uncovers functional variation in humans. Nature. 2013;501:506–511. doi: 10.1038/nature12531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Demetz E, Schroll A, Auer K, Heim C, Patsch JR, Eller P, Theurl M, Theurl I, Theurl M, Seifert M, Lener D, Stanzl U, Haschka D, Asshoff M, Dichtl S, et al. The Arachidonic Acid Metabolome Servesas a Conserved Regulator of Cholesterol Metabolism. Cell Metabolism. 2014;20:787–798. doi: 10.1016/j.cmet.2014.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Kitade H, Sawamoto K, Nagashimada M, Inoue H, Yamamoto Y, Sai Y, Takamura T, Yamamoto H, Miyamoto KI, Ginsberg HN. CCR5 plays a critical role in obesity-induced adipose tissue inflammation and insulin resistance by regulating both macrophage recruitment and M1/M2 status. Diabetes. 2012;61:1680–1690. doi: 10.2337/db11-1506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Luco RF, Allo M, Schor IE, Kornblihtt AR, Misteli T. Epigenetics in Alternative Pre-mRNA Splicing. Cell. 2011;144:16–26. doi: 10.1016/j.cell.2010.11.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Mudge JM, Frankish A, Fernandez-Banet J, Alioto T, Derrien T, Howald C, Reymond A, Guigo R, Hubbard T, Harrow J. The Origins, Evolution, and Functional Potential of Alternative Splicing in Vertebrates. Molecular Biology and Evolution. 2011;28:2949–2959. doi: 10.1093/molbev/msr127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Bailey TL, Bodén M, Whitington T, Machanick P. The value of position-specific priors in motif discovery using MEME. BMC Bioinformatics. 2010;11:179. doi: 10.1186/1471-2105-11-179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Benjamini Y, Hochberg Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. Journal of the Royal Statistical Society Series B. 1995;57:289–300. [Google Scholar]
- 81.Saeed S, Quintin J, Kerstens HHD, Rao NA, Aghajanirefah A, Matarese F, Cheng SC, Ratter J, Berentsen K, van der Ent MA, Sharifi N, Janssen-Megens EM, Huurne Ter M, Mandoli A, van Schaik T, et al. Epigenetic programming of monocyte-to-macrophage differentiation and trained innate immunity. Science. 2014;345:1251086–1251086. doi: 10.1126/science.1251086. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.