

Abstract
Objectives
We present the rationale for and design of a randomized controlled superiority trial comparing two vaginal surgical approaches for the treatment of uterovaginal prolapse. The Study of Uterine Prolapse Procedures-Randomized Trial (SUPeR) trial compares the efficacy and safety of native tissue repair with vaginal hysterectomy and suture apical suspension versus uterine conservation with mesh hysteropexy through 36 to 60 months postoperatively for primary repair of uterovaginal prolapse.
Methods
The selection of the primary outcome measure, timing of randomization, patient and evaluator masking to surgical intervention, collection and adjudication of adverse events, cost effectiveness evaluations, partnering with industry, and surgeon certification of competency to perform the two procedures is described. A composite primary outcome of success defined as no prolapse symptoms, no objective prolapse beyond the hymen, and no retreatment of prolapse, with a minimum of 36 months post-surgery follow-up using survival analyses is planned. Secondary outcomes measured at baseline and every 6 months post-surgery include validated condition-specific and general quality of life assessments, global impression of improvement, body image, and sexual function measures. Unique challenges during the trial design include maintenance of patient masking to the intervention with routine gynecologic health maintenance and maintenance of evaluator masking.
Results
Recruitment and randomization of 180 participants is complete and participants are currently in the follow-up phase.
Conclusions
This trial will provide information to help surgeons counsel patients and contribute evidence-based information regarding risks and benefits of two approaches for the treatment of uterovaginal prolapse.
Introduction
Uterovaginal prolapse is common and makes up a substantial proportion of the 300,000 surgeries for pelvic organ prolapse performed in the U.S. each year.1 While uterovaginal prolapse surgery can be performed by either a vaginal or abdominal route, most of the surgeries for this condition are performed using the vaginal approach.2, 3 Traditionally, surgical correction of uterovaginal prolapse has included hysterectomy, despite beliefs that the uterus is a passive structure in the disease process.4 Increased interest has developed in prolapse procedures that preserve the uterus while suspending the vaginal apex and uterus (hysteropexy). Uterine preservation at the time of prolapse repair has evolved from simply a desire to maintain fertility to avoidance of the added surgical risk and costs of hysterectomy, and a common perception among patients that hysterectomy may negatively impact sexual function or body image4–6 despite much evidence that hysterectomy does not negatively impact sexual function.
Vaginal hysteropexies can be performed with suture techniques (usually sacrospinous hysteropexy) or with mesh techniques but there is limited comparative data and the results are not definitive.4 A small randomized controlled trial comparing sacrospinous hysteropexy demonstrated a higher prolapse recurrence rate with sacrospinous hysteropexy especially with advanced prolapse, but shorter hospitalizations and quicker recovery.7, Transvaginal mesh systems were introduced with the goal of improving long-term success rates over native tissue repair with suture fixation. In studies with trocar mesh kits for hysteropexy, which were mostly retrospective and non-comparative, 86% average success rates were obtained with 9% average erosion rates. 4 Newer trocarless transvaginal mesh kits with lightweight mesh are being promoted for hysteropexy and limited preliminary non-comparative data suggest high success rates with mesh exposure rates of approximately 2–7% at 1 year.8, 9
The relative risks and benefits of hysterectomy versus uterine preservation at time of vaginal prolapse repair for uterovaginal prolapse are currently ill-defined. High quality clinical trials are needed that compare mesh hysteropexy to native tissue repairs with long-term follow-up in order to better clarify the benefits and risks of uterine preservation including risk of subsequent uterine pathology and recurrent prolapse. The primary aim of the Study of Uterine Prolapse Procedures-Randomized Trial (SUPeR Trial) is to compare the effectiveness and safety of two transvaginal apical suspension strategies for women with symptomatic uterovaginal prolapse beyond the introitus. The purpose of this manuscript is to describe trial design, including selection and measurement of the primary and secondary outcomes, the plans to evaluate cost effectiveness, unusual challenges with patient and evaluator masking, and study conduct.
Methods
A. Study design overview
SUPeR is conducted by the NICHD sponsored Pelvic Floor Disorders Network, a team of clinical researchers who work in conjunction with a data coordinating center, representatives from the NICHD sponsor, and a steering committee chair to design and conduct high impact randomized clinical trials at 8 clinical sites. The primary aim of SUPeR is to determine whether treatment success in women with symptomatic uterovaginal prolapse beyond the introitus undergoing transvaginal mesh augmented hysteropexy (using the Uphold® LITE transvaginal mesh kit) differs from women undergoing vaginal hysterectomy and native tissue cuff suspension with suture at time points for a minimum of 3 years post-surgery. The study will test the null hypothesis that risk of treatment failure is not different between the two groups. Our primary outcome, treatment failure, includes anatomic and patient reported symptoms.
The secondary aims of the study address detailed anatomic and comprehensive functional outcomes such as prolapse, urinary, bowel and health related quality of life (HRQOL); safety including adverse events (such as mesh erosion and exposure), pain, and complications requiring subsequent procedures for the two treatments. POPQ measures are recorded every 6 months for the duration of the study and POPQ points Ba, Bp, and C will be compared in both groups. In addition, the study is designed to evaluate whether advanced prolapse, age, obesity, smoking, menopausal status, exogenous estrogen use, previous prolapse surgery, and physical activity levels, alone or in combination, predict higher treatment failure rates. The study will provide data to compare the cost effectiveness of the two surgical approaches and assess the relationship between the cost of care and health utilities and health quality of life. Finally, we also aim to determine if the surgeries result in significant changes in body image and if these changes are related to altered sexual function. SUPeR is registered with Clinicaltrials.gov: NCT01802281.
B. Study Population
The study population comprises adult women (≥21 years of age) with symptomatic uterovaginal prolapse beyond the hymen with uterine prolapse at least into the lower half of the vagina who desire vaginal surgical management. Table 1 describes the inclusion and exclusion criteria. This study was intended for women who have completed child-bearing and have a uterus with inactive endometrium, defined as amenorrhea for 1 year. Therefore, participants are postmenopausal or have undergone an endometrial ablation and have amenorrhea. Amenorrhea caused from exogenous steroids, or hypothalamic disorders are excluded. This protocol adheres to the CONSORT guidelines for performing and reporting randomized controlled trials.10 Women who are eligible but decline enrollment will be characterized in a manner consistent with the CONSORT recommendations.
Table 1.
Inclusion and Exclusion criteria in the SUPeR study.
Inclusion Criteria
|
Exclusion Criteria
|
C. Baseline Assessments and Certification
Once eligibility was confirmed, baseline information was obtained which included age, race/ethnicity, marital status, education, obstetric history, prior pelvic surgeries, menopause/estrogen status, medical history, prior treatment of pelvic organ prolapse or urinary incontinence, smoking, diabetes, urinary tract infection history and current medication use. A detailed physical examination included height and weight, and pelvic organ prolapse quantification (POPQ)11 measurements. Since hysteropexy and mesh procedures could be associated with postoperative cervical elongation, the study group developed a novel measurement to capture possible cervical elongation. At each POPQ exam, the evaluators recorded the posterior cervical vaginal junction (PCVJ which was described as the point where the cervix merges with the vagina posteriorly. We would then define cervical length as point C minus PCVJ.
To reduce bias related to surgical experience, surgeons were required to have adequate experience with both procedures being performed in this study. Because vaginal hysterectomies and uterosacral ligament suspension (TVH/ULS) procedures have been commonly performed for decades, while the Uphold® mesh hysteropexy procedures are less than 10 years old and not universally adopted, it was acknowledged that most surgeons in this study had more experience with the TVH/ULS arm. All participating surgeons in this study are Board-eligible or board- certified Female Pelvic Medicine and Reconstructive Surgery specialists. The primary skills required to do the Uphold® procedure (e.g. anterior vaginal dissection to the sacrospinous ligament and the use of a Capio® device) were familiar to urogynecologic surgeons participating in the trial and experience with these techniques was felt to be transferrable to the Uphold® LITE procedure. Certification criteria for the Uphold® LITE procedure required that all surgeons had previously performed a minimum of 20 sacrospinous ligament dissections with performance of at least 10 anterior vaginal dissections to the sacrospinous ligament, at least 10 Capio® suture applications, and five Uphold Lite procedures independently. Surgeons who had not met these criteria were required to have a certified surgeon perform the apical suspension. We felt that these criteria ensured that all study surgeries would be performed by surgeons who were well trained on procedures for both arms to assure study validity, while at the same time establishing standards for the trial that would allow results to be generalized to the population of urogynecologic surgeons likely to perform these surgeries in the future. Surgeon certification required an attestation by the surgeon, affirmed by the site Principal Investigator (PI). A more detailed description of the certification is described in Table 2.
Table 2.
Surgeon Certification Processes for the Two Surgical Procedures in the SUPeR study
FOR TVH/USLS CERTIFICATION
|
FOR UPHOLD® CERTIFICATION
|
D. Randomization
After eligibility was determined and consent was obtained, surgery was scheduled and performed within 4 months of enrollment. The participant was randomized to one of the two treatment arms in the operating room (after anesthesia induction) by the surgeon telephoning an automated randomization center at the Data Coordinating Center (DCC). The 1:1 randomization system used permuted blocks, with a block size that was known only to the DCC, stratified by site. Additional surgeries that were allowed in the study were native tissue vaginal wall prolapse repairs (e.g. anterior colporrhaphy, posterior colporrhaphy, perineorrhaphy) and anti-incontinence procedures.
E. Standardization of the Surgical Procedures
Uterosacral Ligament Suspension (ULS)
The ULS procedure used in this protocol (Table 3) is a modification of the technique described by Shull 12 and was used in a previous surgical trial performed by the PFDN.13 With considerations of mesh load, future availability of devices, familiarity with the delivery system, and recently published favorable results,8 the SUPeR protocol committee decided by consensus to standardize the mesh hysteropexy procedure to a single standardized mesh kit (Boston Scientific, Uphold® LITE). The Uphold® LITE procedure used in this protocol is a modification of the technique described by Vu and Goldberg.8 The procedures are illustrated in Figure 1 and the specific steps of the procedure that were standardized across sites are described in Table 3.
Table 3.
Standardized Procedures for the Two Surgeries in the SUPeR study
TVH/ULS
|
Uphold® LITE Procedure
|
Figure 1.
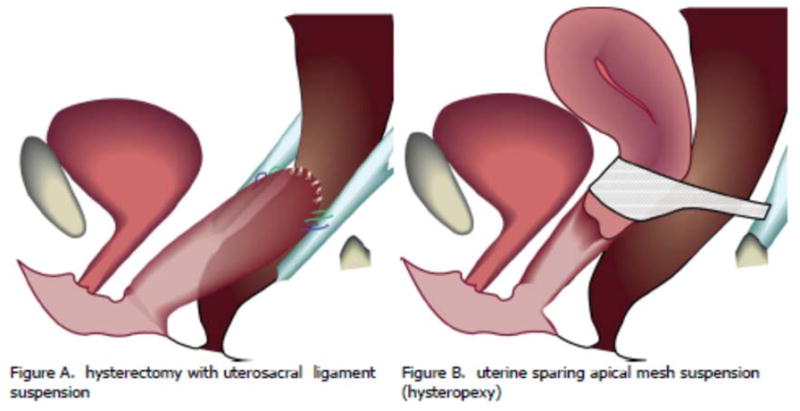
A drawing depicting the 2 surgical procedures being compared in the SUPeR randomized trial.
F. Other Measures
After surgery, participants are seen at 6 weeks and then every 6 months for the duration of the study (36 to 60 months with follow-up for each participant ending when the last participant randomized reaches 36 months). Every 6 months, participants will undergo a POPQ exam, an exam for mesh exposure, an adverse event survey, and a review of bulge symptoms (PFDI14- Question 3) Other study measures are reported in Table 4. Adverse event categorization and standardization is performed by a blinded adjudication committee.
Table 4.
Other Study Measures Captured in the SUPeR Study
|
G. Masking of the Randomized Intervention
Although the primary anatomic outcome does not depend on masked patients, and anatomic evaluators cannot be masked, several important patient-reported secondary aims were subject to patient reporting bias if the participant was aware of her group assignment. For example, in this current era of multi-district litigation against mesh manufacturers, many urinary, bowel, and sexual complaints are being attributed to transvaginal mesh, raising concerns participants’ knowledge about mesh-augmented repair might lead to biased adverse events reporting. On the other hand, internet testimonials attest that hysterectomy worsens sexual function and a patient’s knowledge of having or not having a uterus could bias her reports on sexual function and body image. For these reasons, the ideal study design encourages masking to treatment assignment. Other trials have achieved patient and evaluator masking of supracervical and total hysterectomies.15, 16.
Since the study surgeon provides clinical care to enrolled participants; masking the surgeon to treatment allocation or participant symptoms is not practical or feasible, other than the allocation concealment prior to surgical randomization. To improve objectivity in the primary outcome and eliminate potential ascertainment bias, the study surgeon is not involved in any postoperative anatomic outcome assessments. The anatomic outcome evaluator cannot be masked because they will need to know if the participant has a cervix or not to properly record POPQ values, but the study is designed so that this evaluator was not involved in the surgery in order to limit bias. To minimize biases, subjective and all objective outcomes (except for the POPQ exam) were obtained by study nurses or coordinators masked to the procedure. Thus, when feasible and ethical, all outcomes assessors (except for the POPQ exam) and the participant are masked to treatment allocation.
At the time of enrollment, participants were asked to remain masked to their treatment group for the duration of the study, although we recognize that unintentional unmasking may occur. In order to minimize unmasking due to details of their operation disclosed on a pathology payment bill, the study covered the costs for pathologic assessment of the uterus in those women randomized to native tissue repair.
Consistent with other regulatory bodies, current American Congress of Obstetrics and Gynecology (ACOG) guidelines have recommended women age 30 years or older with known recent negative cervical cytology and negative HPV testing be screened no sooner than every 3 years.17 Current US Preventative Services Task Force, American Cancer Society, American Society for Colposcopy and Cervical Pathology, and American Society for Clinical Pathology recommendations allow no screening for women age <21, every 3 year screening for ages 21–29, every 5 year screening ages 30–65, and no screening for women older than 65. 18 Therefore, based on age, all participants in this study would likely at least be able to space screening to 3 years and the overwhelming majority would qualify for screening intervals every 5 years. Unmasking did occur for an eligible amenorrheic 21–30 year old woman with a uterus who in the opinion of the study surgeon needed screening during the study. We recognize that women may still have pelvic exams for reasons other than cervical screening. We requested that the patient undergoing an exam remind her provider not to tell her of the absence or presence of her uterus. Participants have been encouraged to see the study team for any gynecologic problems or evaluations during the course of the study. It is possible that participants may be unblinded if they examine themselves and feel a cervix. In addition, there may be health situations where it is necessary for them to know if they have a cervix and if so, they will be informed by their study surgeon.
At every 6 month visit the participants are queried about whether they have remained masked, and if not, what caused the unmasking and which randomized surgical treatment they believe they have received. This is done in a manner that reaffirms that masking is preferred for the duration of the study. If a patient has an anatomic failure requiring reoperation and reoperation counselling requires unmasking, then unmasking is allowed. After the study is completed (maximum 60 months), participants will be queried as to what randomization arm they think they received and will then be notified of their uterine and mesh status
H. Primary outcome measure
A participant is considered a treatment failure if any ONE of the following criteria is met:
Report of bothersome vaginal bulge symptoms (see definition below), or
Re-treatment for prolapse (surgery or pessary), or
Any prolapse measure (POPQ measures Ba, C, Bp) beyond the hymen (i.e. > 0 cm) 19
Bothersome vaginal bulge symptoms is measured by a positive response to Question 3 of the Pelvic Floor Distress Inventory short form (PFDI-20)14: “Do you usually have a bulge or something falling out that you can see or feel in your vaginal area?” and patient report of any degree of bother associated with this symptom. An affirmative answer to this question was 96% sensitive (95% CI 92–100) and 79% specific (95% CI 77–92) for prolapse beyond the hymen. The 1-week test-retest reliability was good (kappa 0.84).20 Participants not meeting criteria for a treatment failure for the primary outcome are considered a treatment success.
I. Statistical Design
Sample Size/Power Calculations
Power and sample size calculations were generated to determine the sample size needed to test for treatment difference favoring the mesh augmented strategy (i.e., a superiority trial) across the study arms for a variety of assumptions about effect size and study follow-up time. For all power analyses, we assumed that failure in both arms follows an exponential survival model and that the native tissue repair has a success rate of 80% at 24 months. This 80% calculation is based on an assumed 85% anatomic success rate for uterosacral ligament suspension and an additional 5% failure rate based on symptom failure.13 In the only published Uphold® series a 98 % 12 month anatomic success rate was reported.8 Although no 2-year outcome data are available for the Uphold® procedure, under an assumption of a constant hazard, the 1-year success rate of 98% yields an estimated 2-year anatomic success rate of 96%; combining this estimate with a similar 5% rate for symptom failure yields a 91% composite (anatomic and symptom) success rate. All analyses also assumed that statistical tests would be conducted with a Type I error rate of 0.05 with no adjustment for multiple comparisons. Sample size estimates assumed that the 2-year success rate in the mesh-augmented arm is in the range of 90% to 93% at 24 months (Note: this represents a hazard ratio in the range of 0.33 to 0.47 under the assumed exponential survival model), that the enrollment time for the study is 2 years, the loss-to-follow-up on both arms is no more than 5% per year, and the total study duration from last participant enrolled to stopping the study for final analysis is 36 months with one interim analyses for efficacy when the last participant reaches the 24-month follow-up. Based on these calculations, a total of 180 participants will be randomized at a 1:1 ratio to the two treatment arms. This sample size will provide a power of 0.89 to detect an additive difference of 10% in the 2-year success rate (i.e. a hazard ration 0f 0.472) and a power of 0.95 to detect additive differences of 11% or greater in the 2-year success rate (hazard ratios of 0.423 or less).
Data analysis
Surgical failure rates will be compared using survival analysis approaches appropriate for interval censored data (classic log-rank tests and survival models using a generalized linear model approach with a complementary log-log link) and secondary outcomes will be reported as rates in each group or as group means and evaluated with the appropriate parametric or nonparametric statistical tests. For the primary analysis, we first generate a standard log-rank test to provide an overall test of difference of the two treatment regimens. We will then conduct model based analyses using a generalized linear model with a complementary log-log link that examines failure risk as a function of treatment controlling for appropriate design variables (site and age cohort). The model-based analyses will be used to generate an overall test of treatment difference using a two-sided hypothesis test with an overall Type I error rate of 0.05, and point and interval estimates of 12-, 24- and 36-month surgical success rates on each treatment arm as well as well as differences in these rates on the two arms. The study has been designed for a single interim analysis when the last participant enrolled reaches the 24-month follow-up time using a Lan-DeMets alpha spending function with O’Brien-Fleming type boundaries.
A number of secondary outcome measures that include both continuous and binary measures will be collected periodically across the study. To account for the correlation among the multiple measures on each study and to account for missing data associated with differential follow-up time associated with the primary design, appropriate model-based approaches (linear mixed models for continuous outcomes and generalized linear models for binary measures) will be used to compare the effects of treatment.
J. Study Design Concerns
Physician preference against using transvaginal mesh
Physicians may be reluctant to perform a mesh-augmented procedure instead of the traditional vaginal hysterectomy and sutured apical suspension. Some physicians and patients have strong beliefs against the use of mesh, especially in light of the July 2011 FDA safety communication and current litigation.9 However, the superiority of either native tissue or mesh-augmented apical suspension in terms of efficacy or safety remains unclear as discussed in the Background section. Furthermore, a survey of American Urogynecologic Society (AUGS) members suggests that many pelvic surgeons are still performing mesh-augmented procedures,21 highlighting that this study addresses a timely and challenging clinical question. To balance potential ethical concerns about the use of transvaginal mesh, the study team made several key decisions. First, we chose the Uphold® LITE device which minimizes mesh load and utilizes a wide pore light weight polypropylene mesh which is knitted. Meshes with these characteristics have been shown to have less of a negative impact on the vagina in a primate model than their heavier weight, less porous counterparts. 22–24 Second, all additional prolapse repairs performed in this trial will be native tissue further minimizing possible mesh-load and possible mesh-related complications
Participant preference
Women may have strong preferences regarding removing or conserving their uterus, which could result in recruitment difficulty and/or a skewed study population limiting the external validity of the study. We believed that evidence-based counseling on the potential known and unknown pros and cons of both options with emphasis on the importance of studying the issue would help minimize this potential problem. Indeed, previous randomized trials have similarly encountered and overcome this issue, specifically trials evaluating supracervical vs. total hysterectomy.8, 15 Similarly, a previous successful trial examined hysterectomy compared with endometrial ablation of dysfunctional uterine bleeding. (ClinicalTrials.gov Identifier NCT00114088). Though we were concerned about the prospect of not meeting recruitment goals and frequently discussed contingency plans, the protocol committee rejected a patient preference arm or trial design due to serious concerns about participant bias regarding pelvic symptoms, body image and sexual function after surgery if they were unmasked to their surgery.
Perception of commercial bias
NIH-funded networks have studied specific products in randomized trials including, Botox®25, Interstim®26 in the PFDN; Gynecare TVT®, TVT-O® and AMS Monarc® in the Urinary Incontinence Treatment Network TOMUS study27). The team considered the use of a mesh bridge configured from a surgeon-tailored piece of commercially available mesh but felt that if this proved to be inferior, there could be potential concern that the findings were due to surgical variability of a non-standardized device and that our study would lack generalizability as this is not typically done in clinical practice. For this reason we favored a hysteropexy kit that lends itself to standardization across centers. After the protocol team decided on this product, the Boston Scientific Company who produces Uphold LITE® was approached for a possible public-private partnership. The company did review the protocol through its usual research program procedures and provide comments that were discussed by the protocol team, but the study protocol was finalized independently by the protocol team and approved by the PFDN Steering Committee, Advisory Board, and Data and Safety Monitoring Board. The company partially supported the study through their independent research program both with funding and agreements for device deliveries to participants so that this cost would not appear on the procedure billing paperwork. The company also worked separately with the DCC to use the study to address FDA requirements for studies to address a change in device classification. Reports from the clinical sites and the reports to the FDA were reviewed on a regular basis by the DSMB. Boston Scientific did not participate in study design, choice of outcome measures or data collection, and will not participate in data analysis, or manuscript preparation.
K. Data and Safety Monitoring Board
The PFDN has an established Data Safety Monitoring Board (DSMB) to oversee all studies in the network. Members of the DSMB are independent of the study and clinical site investigators and include representatives with urology, urogynecology, and biostatistics expertise and a lay member. The DSMB meets every three months or more frequently if requested by the Chair, either in person or by teleconference. The DSMB monitors study progress, reviews reports sent to the FDA by Boston Scientific, and has the ability to recommend to NICHD that the trial be stopped for safety, futility, or efficacy.
Discussion
Uterovaginal prolapse is the most common indication for hysterectomy in postmenopausal women.28 Most uterovaginal prolapse is managed with vaginal hysterectomy in combination with colporrhaphy and apical suspension. Historically, justification for concomitant hysterectomy has included: improved visualization of apical support ligaments, concern for cervical elongation, future development of uterine or cervical pathology, creating a sufficient wound to facilitate fibrosis and scarring of the apex-ligament approximation, and almost all prolapse surgery outcome data came from studies where hysterectomy was routinely performed. The majority of uteri extirpated for prolapse are devoid of uterine pathology. 29, 30 It is not clear that removal of the uterus enhances durability of prolapse repairs and it may be deleterious as it disrupts the natural uterosacral attachments. It has also been associated with a 2-fold risk of accelerated ovarian failure. 31
As with other benign gynecologic conditions historically addressed by hysterectomy, alternative uterine sparing approaches have been described for management of uterovaginal prolapse. Native tissue, suture suspension of the uterus to the sacrospinous ligament has limited outcome data and has not been broadly adopted. More recently, transvaginal mesh systems for uterovaginal prolapse repair have been FDA approved since 2004. Systematic reviews of vaginal mesh kits for apical repair concluded that they may be more effective than native tissue repairs in restoring apical support in the short-term; however, reoperations for complications in the initial mesh kits were higher when compared with vaginal native tissue and abdominal repairs. 32, 33 Several transvaginal mesh systems included in the systematic reviews are no longer commercially available in the United States while those still utilized are under-represented in these previous analyses limiting their use in contemporary surgical planning.
In a survey of preferences regarding hysterectomy for the treatment of uterovaginal prolapse, 60% of women indicated they would decline hysterectomy if presented with an equally efficacious alternative to a hysterectomy-based prolapse repair.6 Their decision would be informed by their doctor’s opinion and knowledge of the risks of surgical complications and malignancy. To date, no comparative effectiveness trials have been conducted comparing the traditional approach of vaginal hysterectomy and native tissue apical suspension with transvaginal mesh hysteropexy.
Our goal in designing this study was to contribute meaningfully to the literature on transvaginal mesh and hysteropexy by generating high level evidence through a randomized trial. Review of existing literature leads us to conclude that either treatment group could plausibly demonstrate superiority. For this reason, we designed a superiority trial rather than a study to test for non-inferiority or equivalence.
This study was designed and implemented in the aftermath of the July, 2011 FDA notification1 on risks associated with transvaginal mesh for prolapse. Several important patient-reported secondary aims are subject to potential patient reporting bias if the participant is aware of her group assignment. This was considered particularly important as the study was being conducted during a period of aggressive media recruitment of patients for multi-district litigation against mesh manufacturers and bias against hysterectomy by certain patient advocacy groups. We took extraordinary measures in masking our study population to reduce the likelihood that they would become aware of the presence or absence of their uterus or mesh for the duration of the study.
There is concern that short-term gain in anatomic support as described by cohort studies of transvaginal mesh repairs are offset by the need for surgical management of subsequent surgery for complications. The comprehensive acquisition of office, emergency room, hospital admission visits and procedures for our planned cost-effectiveness analysis will shed light on the long-term outcomes of each surgical strategy and will specifically quantify the impact of mesh complications on costs.
There are limitations to our study. This study design will not allow us to tease out whether the results are due to the hysteropexy procedure itself or the mesh component of the hysteropexy. We considered a four arm trial with native tissue hysteropexy and hysterectomy with vaginal mesh apical suspension but felt the former option did not provide equipoise due to very limited and generally unfavorable outcome data, as well as lack of experience amongst the investigator surgeons. Additionally, a growing body of evidence suggests that concomitant hysterectomy with transvaginal mesh predisposes to mesh exposure at the vaginal cuff. 34 Another limitation is that our findings will only be applicable to this restricted population of non-menstruating women most of whom will be post-menopausal limiting the applicability of this trial to a younger population of women who may be more interested in uterine preservation.
The strengths of this study include that participants and primary outcomes assessors will remain masked. This will help to minimize potential bias and is critical during this time period when there can be strong opinions about both transvaginal mesh use and uterine preservation. Also, because the surgical procedures are standardized and described here in detail, they can be more easily adopted in the future by practicing experienced pelvic surgeons, depending on our findings.
In conclusion, we present the design of a randomized controlled trial comparing two very different approaches to surgical repair of uterovaginal prolapse. Our study design is intended to limit bias and provide robust data to inform decision making by women and their surgeons regarding uterovaginal prolapse repair.
Acknowledgments
Source of Funding: Supported by the Eunice Kennedy Shriver National Institute of Child Health and Human Development grants HD054214, HD069031, HD054215, HD069025, HD041261, HD041267, HD069006, HD069013, HD069010, and the National Institutes of Health Office of Research on Women’s Health. Partial support for the PFDN SUPeR trial is being supplied by Boston Scientific Corporation through an investigator initiated grant to Dr. Wallace through the Pelvic Floor Disorders Network Data Coordinating Center.
Footnotes
Conflicts of Interest Drs. Nager, Zyczynski, Rogers, Barber, Richter, Visco, Rardin, Harvie, Wallace, and Meikle have no relevant conflicts of interest related to this work or manuscript.
References
- 1.FDA. Urogynecologic Surgical Mesh: Update on the Safety and Effectiveness of Vaginal Placement for Pelvic Organ Prolapse. 2011 http://www.fda.gov/downloads/medicaldevices/safety/alertsandnotices/UCM262760.pdf.
- 2.Brown JS, Waetjen LE, Subak LL, Thom DH, Van den Eeden S, Vittinghoff E. Pelvic organ prolapse surgery in the United States, 1997. Am J Obstet Gynecol. 2002;186:712–6. doi: 10.1067/mob.2002.121897. [DOI] [PubMed] [Google Scholar]
- 3.Boyles G, Moore AD, Edwards QT. Health practices of male Department of Defense health care beneficiaries: a follow-up on prostate cancer screening in the national capital area. Mil Med. 2003;168:992–6. [PubMed] [Google Scholar]
- 4.Gutman R, Maher C. Uterine-preserving POP surgery. Int Urogynecol J. 2013;24:1803–13. doi: 10.1007/s00192-013-2171-2. [DOI] [PubMed] [Google Scholar]
- 5.Korbly NB, Kassis NC, Good MM, et al. Patient preferences for uterine preservation and hysterectomy in women with pelvic organ prolapse. Am J Obstet Gynecol. 2013;209:470, e1–6. doi: 10.1016/j.ajog.2013.08.003. [DOI] [PubMed] [Google Scholar]
- 6.Frick AC, Barber MD, Paraiso MF, Ridgeway B, Jelovsek JE, Walters MD. Attitudes toward hysterectomy in women undergoing evaluation for uterovaginal prolapse. Female Pelvic Med Reconstr Surg. 2013;19:103–9. doi: 10.1097/SPV.0b013e31827d8667. [DOI] [PubMed] [Google Scholar]
- 7.Dietz V, van der Vaart CH, van der Graaf Y, Heintz P, Schraffordt Koops SE. One-year follow-up after sacrospinous hysteropexy and vaginal hysterectomy for uterine descent: a randomized study. Int Urogynecol J. 2010;21:209–16. doi: 10.1007/s00192-009-1014-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Vu MK, Letko J, Jirschele K, et al. Minimal mesh repair for apical and anterior prolapse: initial anatomical and subjective outcomes. Int Urogynecol J. 2012;23:1753–61. doi: 10.1007/s00192-012-1780-5. [DOI] [PubMed] [Google Scholar]
- 9.Jirschele K, Seitz M, Zhou Y, Rosenblatt P, Culligan P, Sand P. A multicenter, prospective trial to evaluate mesh-augmented sacrospinous hysteropexy for uterovaginal prolapse. Int Urogynecol J. 2015;26:743–8. doi: 10.1007/s00192-014-2564-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Begg C, Cho M, Eastwood S, et al. Improving the quality of reporting of randomized controlled trials. The CONSORT statement. JAMA. 1996;276:637–9. doi: 10.1001/jama.276.8.637. [DOI] [PubMed] [Google Scholar]
- 11.Bump RC, Mattiasson A, Bo K, et al. The standardization of terminology of female pelvic organ prolapse and pelvic floor dysfunction. American Journal of Obstetrics & Gynecology. 1996;175:10–7. doi: 10.1016/s0002-9378(96)70243-0. [DOI] [PubMed] [Google Scholar]
- 12.Shull BL, Bachofen C, Coates KW, Kuehl TJ. A transvaginal approach to repair of apical and other associated sites of pelvic organ prolapse with uterosacral ligaments. Am J Obstet Gynecol. 2000;183:1365–73. doi: 10.1067/mob.2000.110910. discussion 1373–4. [DOI] [PubMed] [Google Scholar]
- 13.Barber MD, Brubaker L, Burgio KL, et al. Comparison of 2 transvaginal surgical approaches and perioperative behavioral therapy for apical vaginal prolapse: the OPTIMAL randomized trial. JAMA : the journal of the American Medical Association. 2014;311:1023–34. doi: 10.1001/jama.2014.1719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Barber MD, Walters MD, Bump RC. Short forms of two condition-specific quality-of-life questionnaires for women with pelvic floor disorders (PFDI-20 and PFIQ-7) Am J Obstet Gynecol. 2005;193:103–13. doi: 10.1016/j.ajog.2004.12.025. [DOI] [PubMed] [Google Scholar]
- 15.Thakar R, Ayers S, Clarkson P, Stanton S, Manyonda I. Outcomes after total versus subtotal abdominal hysterectomy. N Engl J Med. 2002;347:1318–25. doi: 10.1056/NEJMoa013336. [DOI] [PubMed] [Google Scholar]
- 16.Learman LA, Summitt RL, Jr, Varner RE, et al. A randomized comparison of total or supracervical hysterectomy: surgical complications and clinical outcomes. Obstet Gynecol. 2003;102:453–62. doi: 10.1016/s0029-7844(03)00664-1. [DOI] [PubMed] [Google Scholar]
- 17.ACOG Practice Bulletin no. 109: Cervical cytology screening. Obstet Gynecol. 2009;114:1409–20. doi: 10.1097/AOG.0b013e3181c6f8a4. [DOI] [PubMed] [Google Scholar]
- 18.New Cervical Cancer Screening Recommendations from the U.S. Preventive Services Task Force and the American Cancer Society/American Society for Colposcopy and Cervical Pathology/American Society for Clinical Pathology http://www.acog.org/About_ACOG/Announcements/New_Cervical_Cancer_Screening_Recommendations.
- 19.Brubaker L, Barber MD, Nygaard I, et al. Quantification of vaginal support: are continuous summary scores better than POPQ stage? Am J Obstet Gynecol. 2010;203:512, e1–6. doi: 10.1016/j.ajog.2010.06.071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Barber MD, Neubauer NL, Klein-Olarte V. Can we screen for pelvic organ prolapse without a physical examination in epidemiologic studies? Am J Obstet Gynecol. 2006;195:942–8. doi: 10.1016/j.ajog.2006.02.050. [DOI] [PubMed] [Google Scholar]
- 21.Clemons JL, Weinstein M, Guess MK, et al. Impact of the 2011 FDA transvaginal mesh safety update on AUGS members’ use of synthetic mesh and biologic grafts in pelvic reconstructive surgery. Female Pelvic Med Reconstr Surg. 2013;19:191–8. doi: 10.1097/SPV.0b013e31829099c1. [DOI] [PubMed] [Google Scholar]
- 22.Brown BN, Mani D, Nolfi AL, Liang R, Abramowitch SD, Moalli PA. Characterization of the host inflammatory response following implantation of prolapse mesh in rhesus macaque. Am J Obstet Gynecol. 2015;213:668e1–668 e10. doi: 10.1016/j.ajog.2015.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Liang R, Zong W, Palcsey S, Abramowitch S, Moalli PA. Impact of prolapse meshes on the metabolism of vaginal extracellular matrix in rhesus macaque. Am J Obstet Gynecol. 2015;212:174e1–7. doi: 10.1016/j.ajog.2014.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Liang R, Abramowitch S, Knight K, et al. Vaginal degeneration following implantation of synthetic mesh with increased stiffness. BJOG. 2013;120:233–43. doi: 10.1111/1471-0528.12085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Brubaker L, Richter HE, Visco A, et al. Refractory idiopathic urge urinary incontinence and botulinum A injection. J Urol. 2008;180:217–22. doi: 10.1016/j.juro.2008.03.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Amundsen CL, Richter HE, Menefee S, et al. The Refractory Overactive Bladder: Sacral NEuromodulation vs. BoTulinum Toxin Assessment: ROSETTA trial. Contemp Clin Trials. 2014;37:272–83. doi: 10.1016/j.cct.2014.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Richter HE, Albo ME, Zyczynski HM, et al. Retropubic versus transobturator midurethral slings for stress incontinence. N Engl J Med. 2010;362:2066–76. doi: 10.1056/NEJMoa0912658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wilcox LS, Koonin LM, Pokras R, Strauss LT, Xia Z, Peterson HB. Hysterectomy in the United States, 1988–1990. Obstet Gynecol. 1994;83:549–55. doi: 10.1097/00006250-199404000-00011. [DOI] [PubMed] [Google Scholar]
- 29.Jacobson GF, Shaber RE, Armstrong MA, Hung YY. Hysterectomy rates for benign indications. Obstet Gynecol. 2006;107:1278–83. doi: 10.1097/01.AOG.0000210640.86628.ff. [DOI] [PubMed] [Google Scholar]
- 30.Whiteman MK, Hillis SD, Jamieson DJ, et al. Inpatient hysterectomy surveillance in the United States, 2000–2004. Am J Obstet Gynecol. 2008;198:34e1–7. doi: 10.1016/j.ajog.2007.05.039. [DOI] [PubMed] [Google Scholar]
- 31.Moorman PG, Myers ER, Schildkraut JM, Iversen ES, Wang F, Warren N. Effect of hysterectomy with ovarian preservation on ovarian function. Obstet Gynecol. 2011;118:1271–9. doi: 10.1097/AOG.0b013e318236fd12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Feiner B, Jelovsek JE, Maher C. Efficacy and safety of transvaginal mesh kits in the treatment of prolapse of the vaginal apex: a systematic review. BJOG. 2009;116:15–24. doi: 10.1111/j.1471-0528.2008.02023.x. [DOI] [PubMed] [Google Scholar]
- 33.Diwadkar GB, Barber MD, Feiner B, Maher C, Jelovsek JE. Complication and reoperation rates after apical vaginal prolapse surgical repair: a systematic review. Obstet Gynecol. 2009;113:367–73. doi: 10.1097/AOG.0b013e318195888d. [DOI] [PubMed] [Google Scholar]
- 34.Ehsani Nea. Risk Factors for Synthetic Mesh Extrusion Following Abdominal Sacral Colpopexy and Vaginal Mesh Procedures: A Fellows Pelvic Research Network Study. Female Pelvic Medicine and Reconstructive Surgery. 2012;18:s4. [Google Scholar]
- 35.Sandvik H, Hunskaar S, Seim A, Hermstad R, Vanvik A, Bratt H. Validation of a severity index in female urinary incontinence and its implementation in an epidemiological survey. J Epidemiol Community Health. 1993;47:497–9. doi: 10.1136/jech.47.6.497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.McCarthy M, Jr, Jonasson O, Chang CH, et al. Assessment of patient functional status after surgery. J Am Coll Surg. 2005;201:171–8. doi: 10.1016/j.jamcollsurg.2005.03.035. [DOI] [PubMed] [Google Scholar]
- 37.Ware JE, Jr, Sherbourne CD. The MOS 36-item short-form health survey (SF-36). I. Conceptual framework and item selection. Med Care. 1992;30:473–83. [PubMed] [Google Scholar]
- 38.Barber MD, Janz N, Kenton K, et al. Validation of the surgical pain scales in women undergoing pelvic reconstructive surgery. Female Pelvic Med Reconstr Surg. 2012;18:198–204. doi: 10.1097/SPV.0b013e31825d65aa. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Rogers RG, Rockwood TH, Constantine ML, et al. A new measure of sexual function in women with pelvic floor disorders (PFD): the Pelvic Organ Prolapse/Incontinence Sexual Questionnaire, IUGA-Revised (PISQ-IR) Int Urogynecol J. 2013;24:1091–103. doi: 10.1007/s00192-012-2020-8. [DOI] [PubMed] [Google Scholar]
- 40.Jelovsek JE, Barber MD. Women seeking treatment for advanced pelvic organ prolapse have decreased body image and quality of life. Am J Obstet Gynecol. 2006;194:1455–61. doi: 10.1016/j.ajog.2006.01.060. [DOI] [PubMed] [Google Scholar]