

Abstract
An elaborate network of dynamic lipid membranes, termed tubular recycling endosomes (TRE), coordinates the process of endocytic recycling in mammalian cells. The C-terminal Eps15 homology domain (EHD)-containing proteins have been implicated in the bending and fission of TRE, thus regulating endocytic recycling. EHD proteins have an EH domain that interacts with proteins containing an NPF motif. We found that NPF-containing EHD1 interaction partners such as molecules interacting with CasL-like1 (MICAL-L1) and Syndapin2 are essential for TRE biogenesis. Also crucial for TRE biogenesis is the generation of phosphatidic acid, an essential lipid component of TRE that serves as a docking point for MICAL-L1 and Syndapin2. EHD1 and EHD3 have 86% amino acid identity; they homo- and heterodimerize and partially co-localize to TRE. Despite their remarkable identity, they have distinct mechanistic functions. EHD1 induces membrane vesiculation, whereas EHD3 supports TRE biogenesis and/or stabilization by an unknown mechanism. While using phospholipase D inhibitors (which block the conversion of glycerophospholipids to phosphatidic acid) to deplete cellular TRE, we observed that, upon inhibitor washout, there was a rapid and dramatic regeneration of MICAL-L1-marked TRE. Using this “synchronized” TRE biogenesis system, we determined that EHD3 is involved in the stabilization of TRE rather than in their biogenesis. Moreover, we identify the residues Ala-519/Asp-520 of EHD1 and Asn-519/Glu-520 of EHD3 as defining the selectivity of these two paralogs for NPF-containing binding partners, and we present a model to explain the atomic mechanism and provide new insight for their differential roles in vesiculation and tubulation, respectively.
Keywords: endosome, NMR, receptor recycling, trafficking, vesicles, EHD1, EHD3, MICAL-L1, Rabankyrin-5, tubular recycling endosome
Introduction
Endocytic trafficking is key to the internalization, sorting, degradation, and recycling of macromolecules in mammalian cells (1). These processes are not only essential for the maintenance of cellular homeostasis but also vital for the regulation of diverse cellular events, including nutrient uptake, cell adhesion, cell migration, cell polarity, cytokinesis, and signal transduction. Upon receptor ligation, internalized ligand-receptor complexes reach peripheral sorting endosomes, where they dissociate. The receptors typically undergo sorting and are either sent for degradation or recycled back to the plasma membrane, enabling them to resume their roles in signal transduction (2).
Recycling of receptors to the plasma membrane can occur through distinct routes (3). A subset of the receptors are recycled directly from sorting endosomes (SE)2 in a process known as fast recycling (4). Other receptors are first trafficked to a transitory perinuclear organelle adjacent to the microtubule organizing center, known as the endocytic recycling compartment (ERC), and this process has been termed slow recycling (5). The ERC maintains cargo segregation acquired upon exit from the SE and serves as a focal point for vesicular transport to the plasma membrane (6). Key regulators of endocytic recycling are the small GTP-binding family of Rab proteins, which couple with specific effectors to promote SNARE-based membrane fusion. Although each pathway is likely regulated by multiple proteins, Rab4 and Rab11 are among the best-characterized Rabs involved in fast and slow recycling, respectively (7, 8).
The ERC is comprised of an array of dynamic, densely situated, yet largely independent tubular and vesicular recycling endosomes (6). Efficient recycling via the ERC relies on the integrity of an elaborate network of elongated, non-symmetrical endosomes known as tubular recycling endosomes (TRE) (9). Current models hold that fission of TRE-containing receptors facilitates the formation of vesicle carriers that are recycled to the plasma membrane (10–13). Because of the significance of TRE in membrane recycling, a growing number of studies have addressed a family of proteins known as the C-terminal Eps15 homology domain (EHD1–4) proteins that have been implicated in TRE generation and fission and control membrane recycling (14). EHD1–4 are hetero/homodimeric ATPases that oligomerize and influence endocytic trafficking by promoting the bending and/or fission of endosomes. Despite their high level of amino acid identity (70–86%), the EHD proteins display unique subcellular localizations and regulate distinct endocytic steps. EHD3 and EHD1 are the most closely related and share 86% identity (15). Nonetheless, we have demonstrated that they have distinct mechanistic functions. EHD1 induces the vesiculation of TRE, whereas EHD3 supports the process of membrane tubulation (11).
The hallmark of EHDs is their C-terminal Eps-15 Homology (EH) domain (16, 17). These EH domains contain a positively charged electrostatic surface that preferentially binds to proteins containing NPF motifs followed by acidic residues (18–20). Over the last decade, a variety of important EHD-interaction partners, including molecules interacting with CasL-like1 (MICAL-L1) and Syndapin2, have been identified (21, 22). Both MICAL-L1 and Syndapin2 are essential for TRE biogenesis, and impaired recruitment of either protein to membranes causes a failure of TRE biogenesis and impaired recycling (21, 22). Indeed, TRE biogenesis involves the recruitment of MICAL-L1 and Syndapin2 to membranes that have a high local concentration of phosphatidic acid, a lipid essential for TRE biogenesis (21, 22). MICAL-L1 and Syndapin2 stably interact with each other via the Syndapin2 Src homology 3 domain and proline-rich regions of MICAL-L1. The MICAL-L1-Syndapin2 interaction leads to membrane bending and tubulation. EHD3 is subsequently recruited to these membranes through the interaction of its EH domain with the NPF motifs of MICAL-L1 and/or Syndapin2. A recent model holds that EHD1 thereafter joins this complex on TRE, where it binds to both MICAL-L1 and Syndapin2, possibly replacing EHD3 within the complex to perform fission and give rise to newly formed vesicles (11).
Given the 86% amino acid identify between EHD1 and EHD3 and their disparate cellular functions, these findings frame new questions of outstanding biological significance. By what mechanism do two such remarkably similar proteins play opposing roles in the generation and vesiculation of TRE? Because EHD3 is essential for TRE biogenesis, what is its specific role in the process? Does EHD3 directly promote membrane bending as proposed for EHD2 (31)? Or does EHD3 stabilize MICAL-L1·Syndapin2 complexes on the TRE membranes and thus stabilize TRE?
Here we demonstrate that EHD3 is dispensable for TRE biogenesis but that it serves to stabilize these membrane structures when they have been generated. Moreover, we characterize the molecular and atomic bases for the differential interactions of EHD1 and EHD3 with binding partners, providing new insights into their differential roles in vesiculation and tubulation, respectively.
Experimental Procedures
Cell Lines
HeLa cells were obtained from the ATCC. HeLa cells were grown in complete DMEM containing 10% FBS, 2 mm glutamine, 100 units/ml penicillin, and 100 units/ml streptomycin.
Antibodies and Reagents
The following primary antibodies were used: mouse polyclonal anti-MICAL-L1 (Novus Biologicals, catalog no. H00085377-B01P), rabbit polyclonal anti-Syndapin2 (Abgent, catalog no. AP8088b), rabbit anti-HA epitope (Signalway, catalog no. T506-1), and goat anti-GST conjugated to HRP (GE Life Sciences, catalog no. RPN1236V). The secondary antibodies Alexa Fluor 568-conjugated goat anti-mouse, Alexa Fluor 488-conjugated goat anti-mouse, and Alexa Fluor 568-conjugated goat anti-rabbit were purchased from Invitrogen. HRP conjugated to goat anti-mouse and donkey anti-rabbit were obtained from Jackson ImmunoResearch Laboratories and GE Life Sciences, respectively. CAY 10593 and CAY 10594 were purchased from Cayman Chemical Co.
Yeast Two-hybrid Assay
The yeast two-hybrid assay was done as described previously (40). Briefly, the Saccharomyces cerevisiae strain AH109 (BD Biosciences, Clontech) was maintained on yeast extract peptone dextrose agar plates. A loop full of yeast was grown overnight at 30 °C with shaking at 250 rpm in liquid YPD medium. Transformation was done by the lithium acetate procedure as described in the instructions for the Matchmaker two-hybrid kit (BD Biosciences, Clontech). For colony growth assays, AH109 co-transformants were streaked on plates lacking leucine and tryptophan. Cells were allowed to grow at 30 °C, usually for 3 days or until the colonies were large enough for further assays. An average of three to four colonies were selected and suspended in water, equilibrated to the same optical density at 600 nm, and replated on plates lacking leucine and tryptophan (+HIS, −2 plates) as well as plates also lacking histidine (−HIS, −3 plates). The positive control used was the interaction between p53 and SV40.
GST Pulldown
50 μg of the purified GST fusion proteins EH1, EH3, or EH3(NE519AD) were incubated with GST beads in buffer containing 20 mm Tris and 300 mm NaCl containing 0.1% Triton X-100 and leupeptin and incubated for 4 h as done previously (21). GST beads were then washed four times in 20 mm Tris and 300 mm NaCl containing 0.1% Triton X-100 and leupeptin. HeLa cells were transiently transfected with HA-Rabankyrin-5 for 24 h and lysed in buffer containing 50 mm Tris-HCl (pH 7.4), 150 mm NaCl, 0.5% Triton X-100, 1.8 mg/ml iodoacetamide, and protease inhibitor mixture (23). GST beads were then incubated with the HeLa lysate overnight, washed four times in 20 mm Tris and 300 mm NaCl containing 0.1% Triton X-100 and leupeptin, eluted with 4× sample buffer, and analyzed by immunoblotting with the anti-HA antibody.
Recombinant DNA Constructs
Cloning of EHD1, EHD3, MICAL-L1, and Rabankyrin-5 into the yeast two-hybrid vectors pGBKT7 and pGADT7, GST-EH1, GST-EH3, GFP-Myc-EHD1, and GFP-Myc-EHD3 and its siRNA-resistant counterparts were described previously (22–24). The siRNA-resistant GFP-Myc-EHD3/EH1 chimera was cloned in the eGFPC3 vector. Two-hybrid control vectors (Gal4ad-SV40 large T-antigen and Gal4bd-p53) were purchased from Clontech. The following constructs were generated using the QuikChange site-directed mutagenesis kit (Stratagene, catalog no. 200518): pGADT7 EHD3 AHLL523PHLV, pGADT7 EHD3 D459N, pGADT7 EHD3 M447T, pGADT7 EHD3 KVAE523RHE, EHD3(N519A), EHD1(AD519NE), MICAL-L1(NPFDEEEEE to NPFDEEEEA), MICAL-L1(NPFDEEEEE to NPFDEEEAA), MICAL-L1(NPFDEEEEE to NPFDEEAAA), MICAL-L1(NPFDEEEEE to NPFDEAAAA), MICAL-L1(NPFDEEEEE to NPFDAAAAA), MICAL-L1(NPFDEEEEE to NPFAAAAAA), Rabankyrin-5 (NPFEDV to NPFEDE), Rabankyrin-5 (NPFEDV to NPFEDEE), Rabankyrin-5 (NPFEDV to NPFEDEEE), Rabankyrin-5(QSV to KPY), Rabankyrin-5(QSV to KSV), Rabankyrin-5(QSV to QPV), Rabankyrin-5(QSV to QSY), and GST-EHD3(NE519AD).
Transfection and siRNA Treatment
Transfection of HeLa cells for 16 h at 37 °C was performed using X-tremeGENE 9 (Roche Applied Science) according to the protocol of the manufacturer. siRNA treatment was carried out with Lipofectamine RNAiMAX (Invitrogen, catalog no. 13778-075) for 48 h according to the protocol of the manufacturer using 0.3 μm oligonucleotide (Dharmacon).
CAY Inhibitor Washout Assay
HeLa cells were grown on coverslips in 6-well plates and were subjected to siRNA against Syndapin2, EHD1, and EHD3. After 48 h, the cells were subjected to 100 μm PLD inhibitors (CAY 10593 and CAY 10594) for 30 min at 37°C. The cells were then washed three times with complete medium and allowed to recover after washout of the inhibitors for 20 min, 1 h, 4 h, and 6 h. The coverslips were stained with anti-MICAL-L1 antibodies for the assessment of TRE.
Immunofluorescence and Transferrin Uptake
Cells were either serum-starved for 30 min and incubated with 25 μg/ml Transferrin from human serum Alexa Fluor 568 conjugate (Tf-568, Thermo Fisher) for 20 min or directly fixed with 4% paraformaldehyde and immunostained as described previously (25). Images were acquired using an LSM 5 Pascal confocal microscope (Carl Zeiss Imaging) using a ×63/1.4 numerical aperture objective with the appropriate filters.
Quantification of MICAL-L1-containing Tubular Recycling Endosomes
Tubular recycling endosomes were quantified as described previously (11). Briefly, with NIH ImageJ software, the image background was reduced by adjusting the threshold. The particle size was set between 5 and 150 μm2. All MICAL-L1-containing particles in this range were counted. Ten fields of images from each treatment were analyzed.
Isothermal Titration Calorimetry (ITC)
Heat produced by the binding of MICAL-L1 peptide with the EH domains of EHD1 and EHD3 was measured by ITC using the MicroCal iTC200 isothermal titration calorimeter from Malvern (Worcestershire, UK). All proteins were equilibrated in 1× PBS (pH 7.4) by overnight dialysis. ITC binding isotherms were collected at 25 °C by injecting 20 × 2 μl of peptide (930 μm) into a solution of each of the EH domains (90 μm), representing a 1:10 molar ratio. The heat from each injection was measured by integrating the area of the injection peak, corrected for the background heat produced by the dilution of the peptide into the buffer and plotted as a function of the EH domain/MICAL-L1 peptide molar ratio. KD values were calculated by fitting the titration curves according to a single binding site mode with Origin 7 software with ITC add-ons supplied by Malvern.
Statistical Analysis
Datasets (n = 100 unless otherwise indicated) were analyzed by analysis of variance, and data were considered significantly different at p < 0.01. The p values are shown for each experiment.
Results
Our previous studies have provided a model for the biogenesis of TRE (21) and support for opposing roles of EHD3 and EHD1 in the tubulation and vesiculation of TRE, respectively (11). There is strong consensus that EHD1 vesiculates membranes (10–12, 26), recruited by MICAL-L1 and Syndapin2, with the latter proteins directly implicated in TRE biogenesis (21). However, although MICAL-L1 and Syndapin2 interact directly with EHD3, the mechanistic role of the latter EHD protein in this process is not well understood (11, 14). Because chronic EHD3 depletion leads to a loss of MICAL-L1 and Syndapin2-containing TRE, we hypothesized two potential roles for EHD3 in TRE biogenesis: EHD3 is directly required for membrane bending and TRE generation (together with Syndapin2 and MICAL-L1), or EHD3 is not required for TRE biogenesis but rather serves to stabilize TRE when they have been generated.
To determine the functional role of EHD3 in TRE biogenesis, we took advantage of our recent observation that, upon treatment of cells with 100 μm PLD inhibitors CAY 10593 and CAY 10594 for 30 min at 37°C, TRE were depleted (21). Moreover, upon washout of the inhibitors, we observed a rapid regeneration of TRE (“burst”), providing us with a system to temporally monitor the role of EHD3 in acute TRE biogenesis. It was not feasible, however, to repeat these findings using a different PLD inhibitor, 5-fluoro-2-indolyl des-chlorohalopemide because previous studies indicated that washout of this PLD inhibitor only led to 29% recovery of PLD activity 1 h after removal (27). As depicted in Fig. 1, ∼75% of mock-treated cells contained some visible MICAL-L1-decorated TRE (Mock; the pretreatment, mean tubule area is quantified in Fig. 2B). Upon PLD inhibitor treatment, TRE were almost entirely depleted from the cells, but TRE levels returned to (and exceeded) baseline levels within 20 min after inhibitor washout (Fig. 1, Mock; quantified in Fig. 2B). In comparison, upon Syndapin2 knockdown (efficiency of knockdown calculated at ∼90%, Fig. 1B), as anticipated, no TRE were observed either at pretreatment, after inhibitor treatment, or even following washout, further validating the essential role of Syndapin2 in TRE biogenesis (Fig. 1, Syndapin2-KD; quantified in Fig. 2B). On the other hand, although no TRE were observed upon chronic EHD3 knockdown (efficiency of knockdown calculated at ∼85%, Fig. 1B) or following PLD inhibitor treatment, a dramatic recovery of TRE was documented following inhibitor washout (20 min to 1 h), indicating that TRE can be generated even in the absence of EHD3 (Figs. 1 and 2, EHD3-KD; quantified in Fig. 2B). When EHD1 was knocked down in pretreated cells (efficiency of knockdown calculated at ∼90%, Fig. 1B), we observed increased levels of TRE, consistent with its role in TRE vesiculation (Fig. 1, EHD1-KD; quantified in Fig. 2B). As expected, PLD inhibitors depleted cellular TRE in EHD1 knockdown cells, but TRE levels recovered upon inhibitor washout (Fig. 1, EHD1-KD; quantified in Fig. 2B). These data suggest that Syndapin2 but not EHD3 (nor EHD1) is required for the generation of MICAL-L1-decorated TRE.
FIGURE 1.

TRE biogenesis occurs in the absence of EHD3. A, using MICAL-L1 as a marker, TRE biogenesis was assessed in mock-treated cells (first column), Syndapin2 knockdown cells (second column), EHD3 knockdown cells (third column), and EHD1 knockdown cells (fourth column). Top row, pretreatment shows TRE status under the corresponding knockdown conditions. Center row, 30-min treatment with PLD inhibitors (100 μm CAY 10593 and CAY 10594 for 30 min at 37°C). Bottom row, 30-min treatment with PLD inhibitors followed by 20-min washout of the inhibitor. A representative field of cells (in the top half of each micrograph) is also depicted by an inverted-contrast image (bottom) to better visualize TRE. Note that there is initiation of TRE formation (burst) at the 20-min washout for all treatments except Syndapin2-KD. Scale bar = 10 μm. B, immunoblot analysis depicting the efficiency of knockdown for endogenous EHD1 and Syndapin2 and transfected EHD3. Actin is shown as a loading control.
FIGURE 2.

EHD3 stabilizes TRE. A, mock-treated cells (first column), Syndapin2 knockdown cells (second column), EHD3 knockdown cells (third column), and EHD1 knockdown cells (fourth column) were subjected to PLD inhibitor treatment and washout (as described in Fig. 1) and then incubated for 1 h (top row), 4 h (center row), or 6 h (bottom row) after washout. A single red asterisk depicts the maximum biogenesis of TRE in EHD3 knockdown cells after 1-h washout. Two red asterisks (4 h after washout, center panel) highlight the disappearance of the newly formed TRE in EHD3 knockdown cells, whereas TRE in mock-treated and EHD1 knockdown cells are increasingly abundant 4–6 h after washout. A field of cells (in the top half of each micrograph) is also represented as an inverted-contrast image to better visualize TRE (bottom half of each micrograph). B, quantitative analysis of 100 cells from three independent experiments, measuring mean TRE area in PLD inhibitor washout experiments. The red and purple dashed lines denote the increase in mean TRE length for EHD1 knockdown cells over time, whereas the black dashed line depicts the increase and subsequent decrease in mean TRE length over time for EHD3 knockdown cells. Significance was assessed by analysis of variance. *, p < 0.01. Scale bar = 10 μm.
EHD3 interacts with both MICAL-L1 and Syndapin2, giving rise to the notion that this interaction may be required to stabilize the presence of these two proteins on membranes, thus stabilizing TRE. To assess whether EHD3 stabilizes TRE over time, we monitored TRE levels 1–6 h after PLD inhibitor washout in the absence of EHD3 (Fig. 2). In mock-treated cells, the mean TRE area continued to increase for up to 6 h after PLD inhibitor washout (Mock, quantified in Fig. 2B). As anticipated, no TRE generation was observed at any time point in cells lacking Syndapin2 (Syndapin2-KD, quantified in Fig. 2B). Interestingly, despite the recovery of TRE generation in EHD3-depleted cells immediately following PLD inhibitor washout, within 1–4 h after washout, the TRE levels in these cells dropped to pretreatment levels (EHD3-KD, quantified in Fig. 2B), suggesting that, although EHD3 is not needed for TRE generation, it is needed for continued stability of tubules within the cell. It is important to note, however, that these data do not provide direct information on the TRE half-life because TRE could be undergoing multiple cycles of biogenesis and vesiculation in the course of recovery. On the other hand, in the absence of EHD1, the mean TRE area continued to increase 4–6 h after washout, further promoting a role for EHD1 in TRE vesiculation (EHD1-KD, quantified in Fig. 2B). The transient recovery of TRE in EHD3 knockdown cells immediately following PLD inhibitor washout, coupled with the loss of TRE over 4–6 h post-washout, are consistent with a role for EHD3 in stabilization of TRE but indicate that this protein is not required directly for TRE biogenesis.
Despite their distinct cellular functions, it is remarkable that EHD3 and EHD1 display ∼86% amino identity. Given that their ATP hydrolysis domains are highly conserved and that the two proteins differentially interact with partners through their EH domains, we hypothesized that the EH domains may determine whether these proteins affect TRE biogenesis or vesiculation. To test the role of the EH domain in the tubulation or vesiculation of TREs, we knocked down EHD3 or EHD1 in HeLa cells and, respectively, expressed wild-type EHD3 (Fig. 3, A and B), wild-type EHD1 (Fig. 3, A and C), or a chimera of EHD3 containing the EH domain of EHD1 (EHD3-EH1, Fig. 3D) and assessed their effect on mean TRE length. Endogenous Syndapin2 served as a bona fide marker for TRE. Average TRE length is quantified in Fig. 3E, which demonstrates that, although EHD3 (wild-type) localized to long TRE that were positive for Syndapin2 (Fig. 3, B and E), EHD1 was associated with shorter and more fragmented TRE (Fig. 3C, quantified in Fig. 3E). Upon expression of the EHD3-EH1 chimera, TRE were not only shorter than those observed for wild-type EHD3 but also for those observed for wild-type EHD1 (Fig. 3D, quantified in Fig. 3E). These data support the notion that the EH domains are the prime determinants of differential EHD function.
FIGURE 3.

The EH domain is responsible for the differential function of EHD1 and EHD3 in vesiculation and TRE stabilization, respectively. A, the C-terminal EHD proteins EHD1 and EHD3 share 86% identity and have a conserved domain architecture comprised of four domains: two helical domains, a G domain, and a C-terminal EH domain. B—D, HeLa cells were treated with EHD3 siRNA (B and D) or EHD1 siRNA (C) for 48 h. Cells were then transfected with (B) siRNA-resistant GFP-myc-EHD3 (WT), (C) siRNA-resistant GFP-myc-EHD1 (WT), and (D) siRNA-resistant GFP-myc-EHD3-EH1. TRE morphology was assessed by immunostaining with endogenous Syndapin2. Note that untransfected cells in C and D lack TRE. E, quantitative analysis of mean TRE length was measured in 100 cells from three independent experiments as in B–D. Significance was assessed by analysis of variance.*, p < 0.01. Scale bar = 10 μm.
Because both EHD1 and EHD3 interact with many of the same NPF-containing proteins, such as MICAL-L1 (22, 28) and Syndapin2 (29), we hypothesized that their EH domains may have different affinities for these binding partners. For example, we rationalized that, initially, EHD3 might reside on and stabilize TRE but that the proximity of EHD1, with a potentially higher affinity for NPF-containing proteins such as MICAL-L1, might provide a mechanism for the onset of vesiculation over time. To test the affinity of EH1 and EH3 for a MICAL-L1 NPF peptide, we used ITC. As demonstrated, the KD for EH1 binding was calculated at 23.2 ± 3.2 μm (Fig. 4), very close to that observed previously for an NPF peptide from Rabankyrin-5 (23). On the other hand, the KD for EH3 binding was measured at 17.8 ± 3.7 μm (Fig. 4), which is not significantly different from that of EH1. Based on these data, although we cannot rule out different binding affinities for full-length EHD proteins or differences in affinity resulting from additional in vivo interactions and/or localizations, it appears unlikely that simple differences in binding affinity control the recruitment of EHD1 and EHD3 to TRE.
FIGURE 4.

Comparison of the binding affinity of the EHD1 EH domain and EHD3 EH domain for a MICAL-L1 NPF peptide by isothermal titration calorimetry. Solutions containing 90 μm purified EH domains of EHD1 or EHD3 were injected with 930 μm MICAL-L1 peptide, and ITC binding isotherms were collected. KD values obtained by fitting the titration curves are indicated for each EH domain.
Given the role of the EH domain in dictating EHD1 versus EHD3 function, we next asked what the specific EH domain determinants are that mediate differential function. In comparing the amino acid sequences of EH1 and EH3, we identified six potential stretches of residues with non-conserved differences between the two domains (Fig. 5A, white highlights). As a readout for differential function, we assessed the ability of EHD1, EHD3, and substitution mutants to interact with binding partners. For example, wild-type EHD1 normally interacts with both Rabankyrin-5 and MICAL-L1, whereas wild-type EHD3 binds only to MICAL-L1 (22, 23, 28) (Fig. 5B). We engineered substitution mutations in all six non-conserved regions of EH3, changing the residues to those found in EH1 (Fig. 5A), and then tested whether the EHD3 mutant protein displayed gain-of-function binding to Rabankyrin-5 (Fig. 5C). As shown in the selective two-hybrid binding assay, and as expected, wild-type EHD1 bound to both MICAL-L1 and Rabankyrin-5, whereas EHD3 bound exclusively to MICAL-L1 (Fig. 5C, dashed red rectangle). Of the six substitutions to render the EH3 domain similar to EH1, remarkably only the NE519AD change resulted in a gain-of-function binding to Rabankyrin-5 (Fig. 5C, dashed red rectangle). Moreover, each of the single mutants (EHD3 N519A and EHD3 E520D) were capable of inducing gain of binding of EHD3 to Rabankyrin-5 (Fig. 5E). Complimentary GST pulldown experiments demonstrated that the GST-EH3 NE519AD mutant also gained the ability to bind to Rabankyrin-5 in vitro, although the binding was not as robust as that seen with wild-type GST-EH1 (Fig. 5D), suggesting that additional regulation might be carried out by other residues in EH1. Indeed, the reverse substitution in EHD1 (EHD1 AD519NE, Fig. 5E) was insufficient to cause a loss of binding to Rabankyrin-5, further hinting that, in the EHD1 EH domain, there is a role for additional residues in regulating this interaction.
FIGURE 5.

Identification of EH1 and EH3 residues responsible for their differential interactions with NPF-containing partners. A, comparison of EH1 and EH3, exhibiting homologous (green) and non-homologous (white) residues. Six key regions containing non-conserved residues chosen for analysis are depicted. B, schematic showing the interaction of EHD1 with both Rabankyrin-5 and MICAL-L1, whereas EHD3 interacts only with MICAL-L1. C, the S. cerevisiae yeast strain AH109 was co-transformed with the following Gal4bd fusion constructs: Gal4bd-p53 (control), MICAL-L1 and Rabankyrin-5 along with Gal4ad fusion constructs Gal4ad-SV40 (control), Gal4ad-EHD1 (WT), Gal4ad-EHD3 (WT), Gal4ad-EHD3(NE519AD), Gal4ad-EHD3 (AHLL523PHLV), Gal4ad-EHD3(D459N), Gal4adEHD3(M447T), Gal4ad-EHD3(A437V), and Gal4ad-EHD3(KVAE523RHE). D, HeLa cells were transfected with HA-Rabankyrin-5 (R-5) and lysed after 24 h. The GST-EH domains indicated were used to pull down Rabankyrin-5 from HeLa cell lysates. The protein pulled down was detected by immunoblotting with anti-HA, and anti-GST was used as a control for equal loading of proteins. The immunoblot shown is a representative of four individual experiments. E, the S. cerevisiae yeast strain AH109 was co-transformed with the following Gal4bd fusion constructs: Gal4bd-p53 (control), MICAL-L1, and Rank-5 along with the Gal4ad fusion constructs Gal4ad-SV40 (control), Gal4ad-EHD1 (WT), Gal4ad-EHD3 (WT), Gal4ad-EHD3 (NE519AD), Gal4ad-EHD1(AD519NE), Gal4ad-EHD3(N519A), and Gal4ad-EHD3(E519D). Co-transformants from C and E were plated on non-selective (+HIS) and selective (−HIS) agar plates. Dotted white lines indicate where two different scanned agar plates have been compiled into the same image. The dashed red rectangle in C shows the gain of binding by the EHD3 NE519AD mutant. A representative from four experiments is depicted.
Given that the EHD3 NE519AD mutant is capable of binding to an EHD1-specific interaction partner (Rabankyrin-5), we next asked whether the mutant EHD3 retains EHD3 function or whether the NE-to-AD mutations render it incapable of carrying out EHD3 function. To test this, we used a previously characterized functional assay in which labeled transferrin (Tf-568) is internalized continuously for 20 min in the presence or absence of EHD3 (25). As we have demonstrated, in mock-treated cells, internalized Tf-568 displays clear accumulation at the ERC (Fig. 6, A, C, and F) regardless of whether WT EHD3 or EHD3 NE519AD was transfected (Fig. 6, B–D and E–G). Upon EHD3 knockdown, however, as we have demonstrated previously (25), Tf-568 failed to reach the ERC and instead was maintained in the periphery in somewhat enlarged endosomal structures (Fig. 6, I and L; untransfected cells without yellow borders). EHD3 function could be restored by reintroduction of WT EHD3, allowing Tf-568 to accumulate at the ERC (Fig. 6I; transfected cells with yellow borders and arrows indicating ERC accumulation). However, introduction of EHD3 NE519AD did not restore EHD3 function because transfected cells failed to show ERC accumulation and the Tf-568 remained largely in the periphery (Fig. 6L, transfected cells with yellow borders). Based on three independent experiments, these data were scored and quantified (Fig. 6, graph). In mock-treated cells, Tf-568 reached the ERC in almost 90% of cells. Upon EHD3 knockdown, less than 15% of the cells displayed Tf-568 at the ERC, whereas, upon reintroduction of WT EHD3 into the cells, ∼75% of the cells had perinuclear Tf-568. However, the EHD3 NE519AD mutant failed to rescue transferrin trafficking, and, upon transfection with this mutant, only about 20% of the cells displayed Tf-568 at the ERC (Fig. 6, graph). These data led us to argue that residues 519 and 520 of EHD3 not only regulate its binding to NPF-containing protein partners but also dictate its function in vivo.
FIGURE 6.

Wild-type EHD3, but not the EHD3 NE519AD mutant, rescues the impaired transferrin trafficking phenotype observed in EHD3 knockdown cells. HeLa cells were either mock-treated (A–G) or treated with EHD3 siRNA oligonucleotides (H–M) for 48 h. After the first 24 h, cells were transfected with either WT GFP-EHD3 (B–D and H–J) or GFP-EHD3 NE519AD (E–G and K–M) and then subjected to a 20-min uptake with Tf-568 prior to fixation and microscopic analysis. Mock-treated cells typically display accumulation of Tf-568 in the perinuclear ERC (A, C, and F). Upon EHD3 knockdown, Tf-568 failed to reach the ERC (I, non-labeled cells). I, wild-type GFP-EHD3-transfected cells are marked with a yellow border, and arrows highlight the rescued Tf-568 trafficking and arrival at the ERC. L, yellow borders indicate EHD3 KD cells transfected with GFP-EHD3 NE519AD. Arrows are not shown because Tf-568 does not reach the ERC. The graph (top right) displays quantification of the percentage of cells in which Tf reaches the ERC (within 20 min), comparing wild-type and EHD3 NE519AD-transfected cells. At least 50 cells from each treatment were scored from three individual experiments. Confirmation was done by “blind scoring.” Significance was assessed by analysis of variance. *, p < 0.01.
Having identified the EH domain residues responsible for the binding selectivity to Rabankyrin-5, we next turned to ask which residues in Rabankyrin-5 and MICAL-L1 mediate their selective binding to EHDs. Previous studies from our laboratory and others identified a requirement for an NPF motif followed by acidic residues for binding to EHDs (18–20, 30). Because the MICAL-L1 NPF motif is followed by six acidic residues (and binds to both EHD1 and EHD3), whereas the Rabankyrin-5 motif is followed by only two acidic residues (and binds only to EHD1), we postulated that EHD3 might bind to an NPF motif followed by more than two acidic residues (Fig. 7A). To test this idea, we first engineered Rabankyrin-5 proteins containing three, four, or five acidic residues after the NPF sequence. As shown by selective two-hybrid binding (Fig. 7B), wild-type Rabankyrin-5 and the mutants with additional acidic residues all bound to EHD1 as anticipated. However, no gain of binding for EHD3 was observed, even in a Rabankyrin-5 mutant with five acidic residues following its NPF motif (Fig. 7B). We also tested whether reducing the number of acidic residues following the NPF motif of MICAL-L1 would cause loss of binding to EHD3 (Fig. 7C). As demonstrated, although both EHD1 and EHD3 displayed loss of binding in the absence of any acidic residues (NPFAAAAAA), EHD3 continued to bind to MICAL-L1 even when it contained only a single acidic residue after its NPF motif, whereas EHD1 showed decreased binding to MICAL-L1 with only a single acidic residue after its NPF motif (NPFEAAAAA). Taken together, these data suggest that the number of acidic residues following the NPF motif does not determine the selectivity of binding to EHD1 and EHD3.
FIGURE 7.
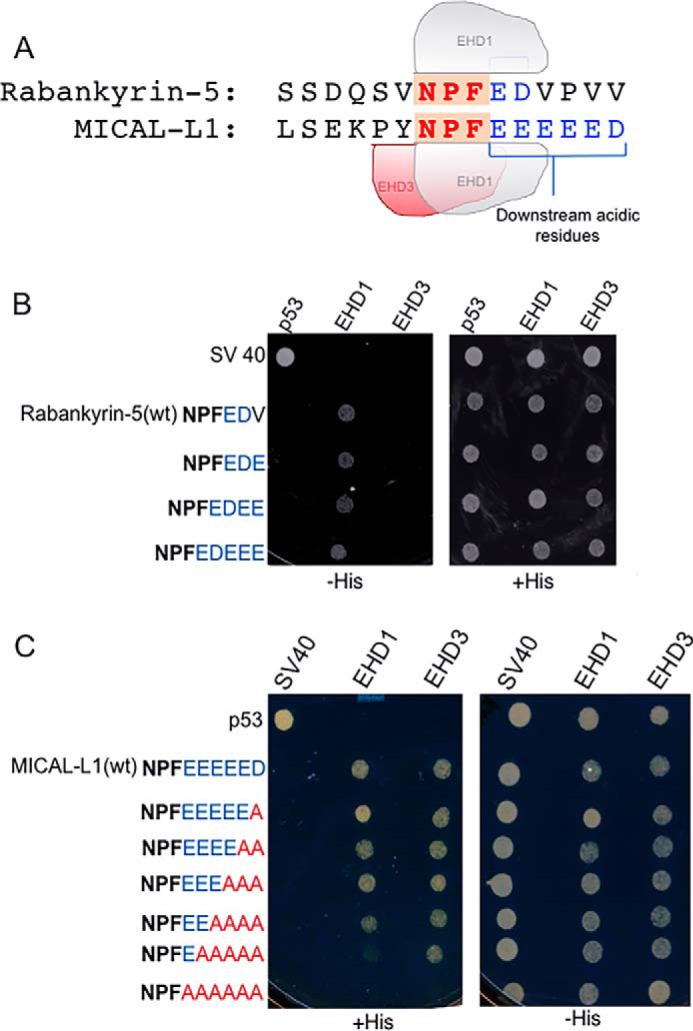
The number of acidic residues after the NPF motif does not discriminate between binding to EHD1 or EHD3. A, comparison of the number of acidic residues (blue letters) following the NPF motif (red letters) of MICAL-L1(NPFDEEEEEE) and Rabankyrin-5(NPFED). B, the S. cerevisiae yeast strain AH109 was co-transformed with the following Gal4bd fusion constructs: Gal4bd-p53 (control), Gal4bd-Rabankyrin-5 (WT) (NPFEDV), Gal4bd-Rabankyrin-5(NPFEDV to NPFEDE), Gal4bd-Rabankyrin-5 (NPFEDV to NPFEDEE), and Rabankyrin-5 (NPFEDV to NPFEDEEE) along with the Gal4ad fusion constructs Gal4ad-SV40 (control), Gal4ad-EHD1 (WT), and Gal4ad-EHD3 (WT). Co-transformants were plated on non-selective (+HIS) and selective (−HIS) agar plates. C, the S. cerevisiae yeast strain AH109 was co-transformed with the following Gal4bd fusion constructs: Gal4bd-p53 (control), Gal4bd-MICAL-L1(WT) (NPFDEEEEE), Gal4bd-MICAL-L1(NPFDEEEEE to NPFDEEEEA), Gal4bd-MICAL-L1(NPFDEEEEE to NPFDEEEAA), Gal4bd-MICAL-L1(NPFDEEEEE to NPFDEEAAA), Gal4bd-MICAL-L1(NPFDEEEEE to NPFDEAAAA), Gal4bd-MICAL-L1(NPFDEEEEE to NPFDAAAAA), and Gal4bd-MICAL-L1(NPFDEEEEE to NPFAAAAAA) along with the Gal4ad fusion constructs Gal4ad-SV40 (control), Gal4ad-EHD1 (WT) and Gal4ad-EHD3 (WT). A representative from four experiments is depicted.
Because our studies suggest that the binding selectivity of MICAL-L1 and Rabankyrin-5 for EHD proteins does not lie distal to the NPF motifs, we addressed the possibility that residues upstream of the NPF motif might dictate binding selectivity. We first hypothesized that the KPY residues immediately upstream of the MICAL-L1 NPF motif (Fig. 8A) might allow more promiscuous binding than the QSV upstream of the Rabankyrin-5 NPF motif. Accordingly, in Fig. 6B, we tested the binding of wild-type MICAL-L1, wild-type Rabankyrin-5, and a Rabankyrin-5 mutant containing KPYNPF instead of QSVNPF to EHD1 and EHD3. As expected, MICAL-L1 bound to both EHD1 and EHD3, whereas Rabankyrin-5 bound exclusively to EHD1. However, replacing the QSV with KPY upstream of the Rabankyrin-5 NPF motif caused a gain of binding to EHD3 (Fig. 8B). To further delineate the amino acid requirements upstream of the NPF motif for binding to EHD3, we then mutated individual residues in the Rabankyrin-5 QSV motif (Fig. 8C). Changing QSV to either KSV or QSY prior to the Rabankyrin-5 NPF motif was sufficient to induce gain of binding to EHD3 (in addition to EHD1). However, modifying QSV to QPV did not lead to gain of binding to EHD3. Indeed, even binding to EHD1 was lost. Collectively, these data suggest that, although NPF followed by at least one to two acidic residues is required for EHD binding, the residues upstream of the NPF motif dictate the “fine-tuning” of binding to individual EHD proteins.
FIGURE 8.
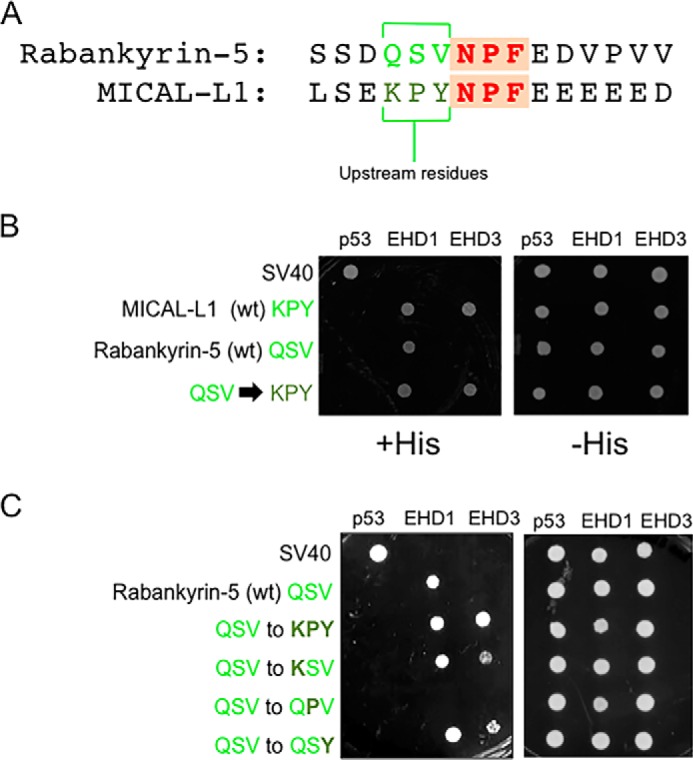
Residue differences upstream of the NPF motif govern binding difference between EHD1 and EHD3. A, comparison of the residues upstream of the NPF motif (green) between MICAL-L1 (KPY) and Rabankyrin-5 (QSV). B, the S. cerevisiae yeast strain AH109 was co-transformed with the following Gal4bd fusion constructs: Gal4bd-p53 (control) and Gal4bd-EHD1 (WT), Gal4bd-EHD3 (WT) along with the Gal4ad fusion constructs Gal4ad-SV40 (control), Gal4ad-MICAL-L1 (WT), Gal4ad-Rabankyrin-5 (WT), and Gal4ad-Rabankyrin-5 (QSV to KPY). C, the S. cerevisiae yeast strain AH109 was co-transformed with the following Gal4bd fusion constructs: Gal4bd-p53 (control), Gal4bd-EHD1 (WT), or Gal4bd-EHD3 (WT) along with the Gal4ad fusion constructs Gal4ad-SV40 (control), Gal4ad-MICAL-L1 (WT) and Gal4ad-Rabankyrin-5 (WT), Gal4ad-Rabankyrin-5 (QSV to KPY), Gal4ad-Rabankyrin-5 (QSV to KSV), Gal4ad-Rabankyrin-5 (QSV to QPV), and Gal4ad-Rabankyrin-5 (QSV to QSY). Co-transformants were plated on non-selective (+HIS) and selective (−HIS) agar plates. A representative from four experiments is depicted.
Because changing the Rabankyrin-5 QSVNPF motif to QSYNPF was sufficient to allow gain-of-function binding to EH3, we examined the three-dimensional structure of the EH1 domain and its binding to the NPF motifs of MICAL-L1 (18–20). The EH1 binding pocket is predicted to interact with the tyrosine of the NPF peptide via hydrogen bonds with glycine 494. We hypothesized that the valine residue of the Rabankyrin-5 QSVNPF motif may display a weakened interaction with glycine 494 because of the nature of its branched carbon chains. Accordingly, we predicted that changing QSVNPF to QSANPF and including the non-branched alanine in the NPF peptide might allow gain-of-function binding to EH3. As shown in Fig. 9A, yeast two-hybrid binding assays indeed demonstrated that mutating the Rabankyrin-5 NPF motif from QSVNPF to QSANPF led to binding to EHD3.
FIGURE 9.
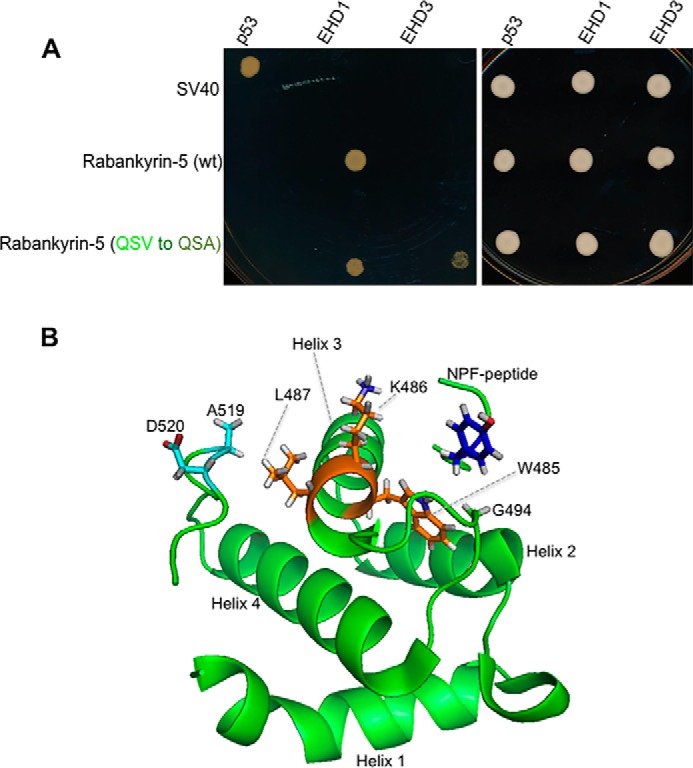
Atomic basis for the differential interaction of EHD1 and EHD3 with NPF-containing binding partners. A, the S. cerevisiae yeast AH109 strain was co-transformed with the following Gal4bd fusion constructs: Gal4bd-p53 (control), Gal4bd-EHD1 (WT), and Gal4bd-EHD3 (WT) along with the Gal4ad fusion constructs Gal4ad-SV40 (control), Gal4ad-MICAL-L1 (WT) and Rabankyrin-5 (WT), and Gal4ad-Rabankyrin-5 (QSV to QSV). Co-transformants were plated on non-selective (+HIS) and selective (−HIS) agar plates. A representative from four experiments is depicted. B, model of the EHD1 EH domain with the MICAL-L1 KPYNPFEEEEED peptide, based on the NMR solution structure (20). Red depicts oxygen atoms in the Asp-520 side chain.
Discussion
The EHDs are membrane curvature-sensing proteins that can induce membrane bending (13), and through their intrinsic ATPase activity (13, 25, 31), they have the ability to promote vesiculation (10–12, 26). Indeed, chronic knockdown of EHD3 (over 48 h) leads to lack of cellular TRE, as determined by immunostaining with antibodies for either endogenous MICAL-L1 or Syndapin2 (11), seemingly supporting a role for EHD3 in TRE biogenesis. However, because TRE are examined after chronic EHD3 loss, such studies do not allow us to discern between two distinct possibilities: failure of TRE to undergo biogenesis in the absence of EHD3 or loss of TRE stability over time upon EHD3-depletion.
Recycling tubules play an important role in endosomal trafficking (32, 33) and are thought to enhance the efficiency of membrane sorting by providing a high surface-to-volume ratio (5). TRE that are decorated by MICAL-L1, Syndapin2, and EHD proteins display greater stability than most endosomal tubules (22) and are required for efficient recycling of a variety of receptor cargos, including integrins and major histocompatibility complex class I proteins, that often take tens of minutes to recycle (9, 24, 34). Typically, we have observed that ∼75% of non-synchronized cells at steady state contain TRE decorated by endogenous MICAL-L1. Although we do not understand why all cells do not display such TRE, recent work from our laboratory leads us to suggest the possibility that the cell cycle is a factor in TRE biogenesis.3 Indeed, it was recently shown that, in the course of mitosis, cells dramatically decrease their recycling (35). Although we can only speculate that ∼25% of unsynchronized cells lacking TRE may be entering mitosis, future experiments will be needed to assess whether this is indeed the case.
In this study, we describe a unique method of inducing acute TRE biogenesis that has allowed us to address the mechanistic role of EHD3. Having observed that inhibition of phosphatidic acid generation with PLD inhibitors causes TRE depletion from cells, we also noticed that washout of the inhibitors promotes a rapid recovery (burst) of TRE within 20–30 min that goes on for several hours. Taking advantage of the acute and “synchronized” TRE biogenesis under these conditions, for the first time we were able to determine that EHD3 is not required for TRE generation (unlike MICAL-L1 and Syndapin2 (21)). Because these experiments were performed under conditions where 85–95% of EHD3 was depleted, and because we have seen that chronic EHD3 knockdown of even 50% efficiency leads to loss of TRE after 48 h, TRE recovery following washout in EHD3-depleted cells is unlikely to result from residual EHD3. On the other hand, although EHD3 depletion did not prevent TRE generation upon inhibitor washout, the TRE remained stable for only 1–2 h, suggesting that EHD3 has a role in TRE stabilization. We do not anticipate that the very same tubules are maintained for many hours but, rather, that overall TRE stability is enhanced in the presence of EHD3. We have demonstrated previously that TRE-decorating proteins such as EHD1 and MICAL-L1 can remain stably associated for 5–10 min (22), but because of dynamic fusion and fission events, which may occur at the TRE tips or along the length of the tubule, measuring the TRE life span is difficult. Although we cannot pin down the precise mechanism for TRE stabilization by EHD3, we hypothesize that its ability to interact with both MICAL-L1 and Syndapin2 via NPF-EH interactions raises the possibility that it physically stabilizes these two proteins on TRE membranes, preventing their dissociation and degradation.
An open question is what differences in NPF-containing proteins, such as MICAL-L1 and Rabankyrin-5, determine their ability to interact with different EHD proteins. Initially, we hypothesized that the number of acidic residues following the NPF motif may signify the ability to bind to certain EHDs. Indeed, the first NPF motif of MICAL-L1 is followed by six acidic residues, and it interacts with both EHD1 and EHD3. On the other hand, Rabankyrin-5 has only two acidic residues after its NPF motif, and it binds only to EHD1. However, neither reducing the number of acidic residues following the NPF motif of MICAL-L1 nor increasing the number of acidic residues following the NPF motif of Rabankyrin-5 had any impact on the binding pattern to EHD1 and EHD3. This indicates that, although NPF motifs flanked by acidic residues are necessary for EHD binding (20, 30), the precise number of acidic residues is not a factor in fine-tuning the binding to select EHDs.
The remarkable likeness of EHD1 and EHD3 raises interesting questions of how two proteins that share 86% identity carry out such different functions. Given that their ATP hydrolysis domains are nearly identical, we predicted that the distinct function of these EHD proteins may be attributed to differences in their EH domains, possibly resulting in binding to distinct subsets of interaction partners. One such difference in binding is to Rabankyrin-5. Although both EHD1 and EHD3 bind to MICAL-L1 (22) and Syndapin2 (29), Rabankyrin-5 binds exclusively to EHD1 (23). We were able to map this differential binding to a pair of residues (Ala-519/Asp-520 for EHD1 and Asn-519/Glu-520 for EHD3). Remarkably, of the entire 534 EHD residues, a simple reversal of EHD3 Asn-519/Glu-520 to AD (as found in EHD1) led to its ability to bind to Rabankyrin-5. Notably, the switching of EHD3 residues 519 and 520 from NE to AD had a direct functional effect on EHD3. Depletion of EHD3 normally causes a failure of internalized receptors, such as transferrin, to reach the perinuclear ERC from peripheral sorting endosomes (Fig. 6, I and L). Remarkably, the NE-to-AD EHD3 mutant failed to rescue this phenotype (as did wild-type EHD3), highlighting the significance of these residues functionally. Nonetheless, it remains to be determined what connection, if any, exists between Rabankyrin-5 binding and the membrane trafficking functions of these EHD proteins.
An examination of the EH1 NMR solution structure provides a possible explanation for the significance of these two residues. Both Ala-519 and Asp-520 reside within 5 Å of Leu-487, likely leading to hydrogen bond formation. Although Leu-487 does not localize to the surface of the EH1 binding pocket, it does comprise the back part (helix 3) of the binding pocket (helix 3 of the helix 2-loop-helix 3 pocket) (Fig. 9B). Indeed, such an interaction might alter the alignment of amino acids Lys-486 and the critical Trp-485 that makes direct contact with the NPF peptide. Thus, we hypothesize that hydrogen bonds between Ala-519/Asp-520 and Leu-487 impact the crucial Trp-485 and serve to stabilize the pocket in a conformation that maintains affinity for both the MICAL-L1 KPYNPF motif and the Rabankyrin-5 QSVNPF motif. Indeed, it appears as though such bonds can come from either Ala-519 or Glu-520 because reversal of Asn-519 of EHD3 to Ala or Glu-520 to Asp was sufficient to induce gain of binding to Rabankyrin-5. However, the lack of the Ala-519/Asp-520 residues in the EHD3 EH domain and the absence of the upstream tyrosine prior to the Rabankyrin-5 peptide NPF motif to interact with Gly-494 likely lead to low binding affinity between EHD3 and the peptide.
It is curious that, upon mutating EHD1 from AD to NE (at residues 519/520), we did not discern a loss of EHD1 binding to Rabankyrin-5 as we initially anticipated (Fig. 5E). Although further experimentation will be needed to fully understand the reason for this anomaly, it is clear that other residues that are distinct from EHD3 within the EHD1 EH domain must play a role in maintaining EH1 binding pocket affinity for the Rabankyrin-5 NPF peptide.
Surprisingly, we observed that the residues immediately upstream of the NPF motif are able to dictate binding to select EHDs. The QSVNPFED motif of Rabankyrin-5 bound to EHD1 but not EHD3. However, when mutated to KPYNPFED (the KPY is from the MICAL-L1 NPF motif), binding was observed with both EHD1 and EHD3. What is the atomic mechanism to explain the differential binding?
Based on our previous NMR solution structure of EH1 with NPF-containing peptides (18–20), there are hydrogen bonds between glycine 494 of the binding pocket and the tyrosine (and potentially lysine) residue of the KPYNPF motif of MICAL-L1. We anticipate that a similar mechanism explains the binding between the QSVNPF motif of Rabankyrin-5 and EH1. In the case of EH3, it is plausible that the weakened affinity of the pocket (resulting from Asn-519/Glu-520 instead of Ala-519/Asp-520) causes a failure of EH3 binding to QSVNPF. However, we also considered that the branched valine residue of the QSVNPF peptide may create a geometry that is not conducive to hydrogen bonds with Gly-494. Indeed, mutation of the Rabankyrin-5 to QSANPF without the branched valine induces a gain of binding to EH3.
Although Rabankyrin-5-binding only serves as a model to illustrate how subtle residue changes can lead to differential binding, and additional studies will need to be done to determine how such binding might affect EHD function in vesiculation and/or TRE stabilization, our findings provide a unique window to understand how two proteins that were likely generated through gene duplication evolved in higher eukaryotes to perform distinct functions.
Based on our findings in this study and previous studies by our group and others, a basic model is emerging for TRE function. TRE are essential for efficient recycling (9) and appear to regulate trafficking steps both from sorting endosomes to the ERC (6) and from the ERC to the plasma membrane (24). The biogenesis of TRE occurs when phosphatidic acid is generated on membranes by one of several pathways (10, 21, 38). The phosphatidic acid-binding proteins MICAL-L1 and Syndapin2 are then recruited to the membrane, where they interact, and the Syndapin2 F-BAR domain induces membrane curvature (21). Although the loss of TRE by chronic depletion of EHD3 initially suggested that this protein is required for TRE biogenesis, and a recent study maintains that EHD3 binds directly to phosphatidic acid and induces curvature (39), this study indicates that EHD3 is not required for TRE biogenesis and likely plays a role in stabilizing TRE, possibly by strengthening the MICAL-L1-Syndapin2 interactions.
Following cargo sorting within the TRE, the most likely scenario, based on various studies supporting a role for EHD1 in TRE fission (11, 12), is that TRE stability is decreased by a temporal replacement of EHD3 by EHD1. The mechanism and specific triggers for this EHD switch and recruitment of EHD1 remain unknown and appear to be unrelated to EHD affinity for the MICAL-L1 NPF motif because both EH1 and EH3 bind with similar affinities (Fig. 4). We speculate that in vivo modifications, such as EHD3 sumoylation (41) and, potentially, desumoylation may regulate this process. Alternatively, specific signaling triggers through receptors (i.e. epidermal growth factor) may also promote EHD1 replacement of EHD3 and TRE fission (36, 37). Ultimately, TRE are cleaved, either at their tips or potentially along the entire membrane, and the resulting vesicles are transported along microtubules to the plasma membrane.
In summary, we provide a deeper understanding of the mechanism of EHD3 function. First, we have demonstrated that EHD3 is not required for TRE biogenesis but instead is needed to maintain stable TRE over time. Next, we characterized the differential function of EHD1 and EHD3 and found important differences in EH domain residues that led to differential binding with NPF-containing proteins. These residues likely promote optimal hydrogen bonds with hydrophobic residues immediately upstream of the NPF motifs. Although future studies will be needed to determine how differential binding partners control TRE vesiculation versus stability, these results provide novel insight into the atomic mechanisms of EHD function.
Author Contributions
K. B. designed, performed, and analyzed the experiments and wrote parts of the manuscript. S. X. quantified the data in Figs. 2 and 3 and assisted with vector construction. G. S. performed and analyzed the ITC studies and prepared Fig. 4. P. S. prepared Fig. 9B, analyzed the data, and wrote parts of the discussion. N. N. helped coordinate the study, analyzed experiments, generated figures, and wrote and edited parts of the manuscript. S. C. coordinated the study overall, analyzed data, prepared figures, and wrote and edited the manuscript. All authors reviewed the results and approved the final version of the manuscript.
This work supported by National Institutes of Health Grants R01GM074876 (to S. C.) and R01GM072631 (to P. S.) and NIGMS, National Institutes of Health institutional development award P30GM106397.
S. Xie, J. B. Reinecke, K. Bahl, N. Naslavsky, and S. Caplan, unpublished data.
- SE
- sorting endosome(s)
- ERC
- endocytic recycling compartment
- TRE
- tubular recycling endosome(s)
- EHD
- Eps15 homology domain
- PLD
- phospholipase D
- ITC
- isothermal titration calorimetry
- KD
- knockdown.
References
- 1. Conner S. D., and Schmid S. L. (2003) Regulated portals of entry into the cell. Nature 422, 37–44 [DOI] [PubMed] [Google Scholar]
- 2. Jovic M., Sharma M., Rahajeng J., and Caplan S. (2010) The early endosome: a busy sorting station for proteins at the crossroads. Histol. Histopathol. 25, 99–112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Grant B. D., and Donaldson J. G. (2009) Pathways and mechanisms of endocytic recycling. Nat. Rev. Mol. Cell Biol. 10, 597–608 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Hao M., and Maxfield F. R. (2000) Characterization of rapid membrane internalization and recycling. J. Biol. Chem. 275, 15279–15286 [DOI] [PubMed] [Google Scholar]
- 5. Maxfield F. R., and McGraw T. E. (2004) Endocytic recycling. Nat. Rev. Mol. Cell Biol. 5, 121–132 [DOI] [PubMed] [Google Scholar]
- 6. Xie S., Bahl K., Reinecke J. B., Hammond G. R., Naslavsky N., and Caplan S. (2016) The endocytic recycling compartment maintains cargo segregation acquired upon exit from the sorting endosome. Mol. Biol. Cell 27, 108–126 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. van der Sluijs P., Hull M., Webster P., Mâle P., Goud B., and Mellman I. (1992) The small GTP-binding protein rab4 controls an early sorting event on the endocytic pathway. Cell 70, 729–740 [DOI] [PubMed] [Google Scholar]
- 8. Ullrich O., Reinsch S., Urbé S., Zerial M., and Parton R. G. (1996) Rab11 regulates recycling through the pericentriolar recycling endosome. J. Cell Biol. 135, 913–924 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Jović M., Kieken F., Naslavsky N., Sorgen P. L., and Caplan S. (2009) Eps15 homology domain 1-associated tubules contain phosphatidylinositol-4-phosphate and phosphatidylinositol-(4,5)-bisphosphate and are required for efficient recycling. Mol. Biol. Cell 20, 2731–2743 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Cai B., Caplan S., and Naslavsky N. (2012) cPLA2α and EHD1 interact and regulate the vesiculation of cholesterol-rich GPI-anchored protein-containing endosomes. Mol. Biol. Cell 23, 1874–1888 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Cai B., Giridharan S. S., Zhang J., Saxena S., Bahl K., Schmidt J. A., Sorgen P. L., Guo W., Naslavsky N., and Caplan S. (2013) Differential roles of C-terminal Eps15 homology domain proteins as vesiculators and tubulators of recycling endosomes. J. Biol. Chem. 288, 30172–30180 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Cai B., Xie S., Caplan S., and Naslavsky N. (2014) GRAF1 forms a complex with MICAL-L1 and EHD1 to cooperate in tubular recycling endosome vesiculation. Front. Cell Dev. Biol. 2, 22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Daumke O., Lundmark R., Vallis Y., Martens S., Butler P. J., and McMahon H. T. (2007) Architectural and mechanistic insights into an EHD ATPase involved in membrane remodelling. Nature 449, 923–927 [DOI] [PubMed] [Google Scholar]
- 14. Naslavsky N., and Caplan S. (2011) EHD proteins: key conductors of endocytic transport. Trends Cell Biol. 21, 122–131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Pohl U., Smith J. S., Tachibana I., Ueki K., Lee H. K., Ramaswamy S., Wu Q., Mohrenweiser H. W., Jenkins R. B., and Louis D. N. (2000) EHD2, EHD3, and EHD4 encode novel members of a highly conserved family of EH domain-containing proteins. Genomics 63, 255–262 [DOI] [PubMed] [Google Scholar]
- 16. Grant B. D., and Caplan S. (2008) Mechanisms of EHD/RME-1 protein function in endocytic transport. Traffic 9, 2043–2052 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Naslavsky N., and Caplan S. (2005) C-terminal EH-domain-containing proteins: consensus for a role in endocytic trafficking, EH? J. Cell Sci. 118, 4093–4101 [DOI] [PubMed] [Google Scholar]
- 18. Kieken F., Jović M., Naslavsky N., Caplan S., and Sorgen P. L. (2007) EH domain of EHD1. J. Biomol. NMR 39, 323–329 [DOI] [PubMed] [Google Scholar]
- 19. Kieken F., Jović M., Tonelli M., Naslavsky N., Caplan S., and Sorgen P. L. (2009) Structural insight into the interaction of proteins containing NPF, DPF, and GPF motifs with the C-terminal EH-domain of EHD1. Protein Sci. 18, 2471–2479 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Kieken F., Sharma M., Jovic M., Giridharan S. S., Naslavsky N., Caplan S., and Sorgen P. L. (2010) Mechanism for the selective interaction of C-terminal Eps15 homology domain proteins with specific Asn-Pro-Phe-containing partners. J. Biol. Chem. 285, 8687–8694 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Giridharan S. S., Cai B., Vitale N., Naslavsky N., and Caplan S. (2013) Cooperation of MICAL-L1, syndapin2, and phosphatidic acid in tubular recycling endosome biogenesis. Mol. Biol. Cell 24, 1776–1790, S1–15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Sharma M., Giridharan S. S., Rahajeng J., Naslavsky N., and Caplan S. (2009) MICAL-L1 links EHD1 to tubular recycling endosomes and regulates receptor recycling. Mol. Biol. Cell 20, 5181–5194 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Zhang J., Reiling C., Reinecke J. B., Prislan I., Marky L. A., Sorgen P. L., Naslavsky N., and Caplan S. (2012) Rabankyrin-5 interacts with EHD1 and Vps26 to regulate endocytic trafficking and retromer function. Traffic 13, 745–757 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Caplan S., Naslavsky N., Hartnell L. M., Lodge R., Polishchuk R. S., Donaldson J. G., and Bonifacino J. S. (2002) A tubular EHD1-containing compartment involved in the recycling of major histocompatibility complex class I molecules to the plasma membrane. EMBO J. 21, 2557–2567 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Naslavsky N., Rahajeng J., Sharma M., Jovic M., and Caplan S. (2006) Interactions between EHD proteins and Rab11-FIP2: a role for EHD3 in early endosomal transport. Mol. Biol. Cell 17, 163–177 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Jakobsson J., Ackermann F., Andersson F., Larhammar D., Löw P., and Brodin L. (2011) Regulation of synaptic vesicle budding and Dynamin function by an EHD ATPase. J. Neurosci. 31, 13972–13980 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Su W., Yeku O., Olepu S., Genna A., Park J. S., Ren H., Du G., Gelb M. H., Morris A. J., and Frohman M. A. (2009) 5-Fluoro-2-indolyl des-chlorohalopemide (FIPI), a phospholipase D pharmacological inhibitor that alters cell spreading and inhibits chemotaxis. Mol. Pharmacol. 75, 437–446 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Sharma M., Giridharan S. S., Rahajeng J., Caplan S., and Naslavsky N. (2010) MICAL-L1: an unusual Rab effector that links EHD1 to tubular recycling endosomes. Commun. Integr. Biol. 3, 181–183 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Braun A., Pinyol R., Dahlhaus R., Koch D., Fonarev P., Grant B. D., Kessels M. M., and Qualmann B. (2005) EHD proteins associate with syndapin I and II and such interactions play a crucial role in endosomal recycling. Mol. Biol. Cell 16, 3642–3658 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Henry G. D., Corrigan D. J., Dineen J. V., and Baleja J. D. (2010) Charge effects in the selection of NPF motifs by the EH domain of EHD1. Biochemistry 49, 3381–3392 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Lee D. W., Zhao X., Scarselletta S., Schweinsberg P. J., Eisenberg E., Grant B. D., and Greene L. E. (2005) ATP Binding regulates oligomerization and endosome association of RME-1 family proteins. J. Biol. Chem. 280, 17213–17220 [DOI] [PubMed] [Google Scholar]
- 32. van Weering J. R., and Cullen P. J. (2014) Membrane-associated cargo recycling by tubule-based endosomal sorting. Semin. Cell Dev. Biol. 31, 40–47 [DOI] [PubMed] [Google Scholar]
- 33. Chi R. J., Harrison M. S., and Burd C. G. (2015) Biogenesis of endosome-derived transport carriers. Cell. Mol. Life Sci. 72, 3441–3455 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Jović M., Naslavsky N., Rapaport D., Horowitz M., and Caplan S. (2007) EHD1 regulates β1 integrin endosomal transport: effects on focal adhesions, cell spreading and migration. J. Cell Sci. 120, 802–814 [DOI] [PubMed] [Google Scholar]
- 35. Boucrot E., and Kirchhausen T. (2007) Endosomal recycling controls plasma membrane area during mitosis. Proc. Natl. Acad. Sci. U.S.A. 104, 7939–7944 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Reinecke J. B., Katafiasz D., Naslavsky N., and Caplan S. (2015) Novel functions for the endocytic regulatory proteins MICAL-L1 AND EHD1 in mitosis. Traffic 16, 48–67 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Reinecke J., and Caplan S. (2014) Endocytosis and the Src family of non-receptor tyrosine kinases. Biomol. Concepts 5, 143–155 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Xie S., Naslavsky N., and Caplan S. (2014) Diacylglycerol kinase α regulates tubular recycling endosome biogenesis and major histocompatibility complex class I recycling. J. Biol. Chem. 289, 31914–31926 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Henmi Y., Oe N., Kono N., Taguchi T., Takei K., and Tanabe K. (2016) Phosphatidic acid induces EHD3-containing membrane tubulation and is required for receptor recycling. Exp. Cell Res. 342, 1–10 [DOI] [PubMed] [Google Scholar]
- 40. Naslavsky N., Boehm M., Backlund P. S. Jr., and Caplan S. (2004) Rabenosyn-5 and EHD1 interact and sequentially regulate protein recycling to the plasma membrane. Mol. Biol. Cell 15, 2410–2422 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Cabasso O., Pekar O., and Horowitz M. (2015) SUMOylation of EHD3 modulates tubulation of the endocytic recycling compartment. PLoS ONE 10, e0134053. [DOI] [PMC free article] [PubMed] [Google Scholar]