

Abstract
Glyceraldehyde-3-phosphate dehydrogenase (GAPDH), an important glycolytic enzyme, has a non-catalytic (thus a non-canonical) role in inducing mitochondrial elimination under oxidative stress. We recently demonstrated that phosphorylation of GAPDH by δ protein kinase C (δPKC) inhibits this GAPDH-dependent mitochondrial elimination. δPKC phosphorylation of GAPDH correlates with increased cell injury following oxidative stress, suggesting that inhibiting GAPDH phosphorylation should decrease cell injury. Using rational design, we identified pseudo-GAPDH (ψGAPDH) peptide, an inhibitor of δPKC-mediated GAPDH phosphorylation that does not inhibit the phosphorylation of other δPKC substrates. Unexpectedly, ψGAPDH decreased mitochondrial elimination and increased cardiac damage in an animal model of heart attack. Either treatment with ψGAPDH or direct phosphorylation of GAPDH by δPKC decreased GAPDH tetramerization, which corresponded to reduced GAPDH glycolytic activity in vitro and ex vivo. Taken together, our study identified the potential mechanism by which oxidative stress inhibits the protective GAPDH-mediated elimination of damaged mitochondria. Our study also identified a pharmacological tool, ψGAPDH peptide, with interesting properties. ψGAPDH peptide is an inhibitor of the interaction between δPKC and GAPDH and of the resulting phosphorylation of GAPDH by δPKC. ψGAPDH peptide is also an inhibitor of GAPDH oligomerization and thus an inhibitor of GAPDH glycolytic activity. Finally, we found that ψGAPDH peptide is an inhibitor of the elimination of damaged mitochondria. We discuss how this unique property of increasing cell damage following oxidative stress suggests a potential use for ψGAPDH peptide-based therapy.
Keywords: docking, oligomerization, oxidative stress, peptides, protein kinase, protein kinase C (PKC), protein structure, Pseudosubstrate, glyceraldehyde-3-phosphate dehydrogenase (GAPDH), mitochondria elimination
Introduction
Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) is a glycolytic enzyme that catalyzes the conversion of glyceraldehyde 3-phosphate to 1,3-diphosphoglycerate. We previously found that in addition to its role as a glycolytic enzyme, GAPDH is also involved in mitochondrial elimination by mitophagy (1). GAPDH binds to damaged mitochondria, and its phosphorylation by δ protein kinase C (δPKC) under oxidative stress inhibits the removal of the damaged mitochondria through mitophagy (1). We set out to determine whether δPKC-mediated GAPDH phosphorylation alone is sufficient to mediate cell death following oxidative stress, using Langendorff, an ex vivo model of heart attack.
We first designed a novel inhibitor that selectively inhibits GAPDH phosphorylation without affecting the phosphorylation of other δPKC substrates. To this end, we relied on the rationale that, similar to many other protein kinases, δPKC contains a highly conserved catalytic domain and a regulatory domain that keeps the enzyme in an inactive conformation. This inactive conformation is stabilized by several intramolecular auto-inhibitory interactions (2). The first intramolecular interaction identified was the one mediated by the pseudosubstrate site located at the N terminus of the kinase, which mimics the phospho-acceptor sequence in PKC substrates; the pseudosubstrate site binds to the substrate-binding cavity in the catalytic domain of the kinase, thus keeping the enzyme inactive (3, 4). In addition to the pseudosubstrate auto-inhibitory site, several protein kinases have a second regulatory site outside of the catalytic site, termed the substrate-docking site (5). The substrate-docking site is a short motif that mediates selective docking of a substrate to its kinase (5, 6). We reasoned that if there is a unique GAPDH-docking site on δPKC, an inhibitor of this docking site will serve as a specific inhibitor of GAPDH phosphorylation by δPKC, without affecting the phosphorylation of other δPKC substrates.
The following rationale was used to identify this inhibitor. It is likely that the GAPDH-specific docking site on δPKC is masked when δPKC is inactive, and a conformational change in δPKC to the active state exposes this GAPDH-docking site. Analogous to the pseudosubstrate concept (3), we hypothesized that there is a pseudo-GAPDH-docking site (ψGAPDH) on δPKC that masks the GAPDH-docking site. The ψGAPDH site on δPKC mimics the binding site for δPKC on GAPDH, and therefore, it may have a short sequence that is homologous to GAPDH.
Here, we describe how we used this rationale to develop a peptide corresponding to ψGAPDH, which we termed ψGAPDH peptide. Our work with ψGAPDH peptide identified phosphorylation as a novel switch controlling the oligomeric state of GAPDH, modifying its function as a metabolic enzyme and a mediator of mitochondrial elimination.
Experimental Procedures
Animal Care
Animal care and husbandry procedures were in accordance with the National Institutes of Health guidelines. The animal protocols were approved by the Stanford University Institutional Animal Care and Use Committee.
Sequence Alignments
Sequences from different species were aligned using FASTA server (7), using the following δPKC proteins: human (Q05655), mouse (P28867), rat (P09215), zebrafish (gi|47550719), chicken (gi|57524924) and chameleon (G1KHZ5); GAPDH proteins: human (P04406), mouse (P16858), rat (P04797), zebrafish (Q5XJ10), chicken (P00356), and chameleon (H9GBL1); 2-hydroxyacyl-CoA lyase 1 (HACL1) proteins: human (Q9UJ83), mouse (Q9QXE0), rat (Q8CHM7), and zebrafish (Q6NYI5); neuritin-like (NRN1L) proteins: human (Q496H8) and mouse (Q8C4W3); ADP/ATP translocase 1 (ADT1) proteins: human (P12235), mouse (P48962), and rat (Q05962); ADP/ATP translocase 2 (ADT2) proteins: human (P05141), mouse (P51881), and rat (Q09073); ADP/ATP translocase 3 (ADT3) proteins: human (P12236) and rat (G5BQT2); ADP/ATP translocase 4 (ADT4) proteins: human (Q9H0C2), mouse (Q3V132), and rat (G5AKY3); breast cancer metastasis-suppressor 1 (BRMS1) proteins: human (Q9HCU9), mouse (Q99N20), rat (Q5M7T3), zebrafish (Q4V8V1), and chicken (Q5ZLL9); pleckstrin homology domain-containing family H member 2 (PLEKHH2 and PKHF2) proteins: human (Q8IVE3) and mouse (Q8C115); Ras association domain-containing protein 3 (RASF5) proteins: human (Q86WH2), mouse (Q99P51), and rat (Q99P51); α-centractin (ACTZ) proteins: human (Q86WH2), mouse (Q99P51), and rat (G5B0X2); β-centractin (ACTY) proteins: human (P42025) and mouse (Q8R5C5); 2–5A-dependent ribonuclease (RN5A) proteins: human (Q05823), mouse (Q05921), and rat (G5BE40); ϵPKC protein: human (Q02156); and θPKC protein: human (Q04759).
Peptide Synthesis
In Brief
Peptides were synthesized on solid support using a fully automated microwave peptide synthesizer (Liberty, CEM Corp.) (8) using the Fmoc3/tert-butyl protocol. The final cleavage and side chain deprotection were done manually. Peptides were analyzed by analytical reverse-phase high pressure liquid chromatography (RP-HPLC) (Shimadzu) and matrix-assisted laser desorption/ionization (MALDI) mass spectrometry (MS) and purified by preparative RP-HPLC (Shimadzu).
Further Details
All commercially available solvents and reagents were used without further purification. Dichloromethane, N-methyl-2-pyrrolidone, triisopropylsilane, N,N-diisopropylethylamine, O-benzotriazole-N,N,N′,N′-tetramethyl-uronium-hexafluoro-phosphate, 1-hydroxybenzotriazole, and trifluoroacetic acid (TFA) were purchased from Sigma; dimethylformamide (DMF) was purchased from Alfa Aesar; piperidine was purchased from Anaspec; rink amide AM resin was purchased from CBL Biopharma LLC (substitution 0.49 mmol/g); Fmoc-protected amino acids were purchased from Advanced ChemTech and GL Biochem (Shanghai, China). Side chains of the amino acids used in the synthesis were protected as follows: t-butoxycarbonyl (Lys/Trp), tert-butyl (Ser/Thr/Tyr), tert-butyl ester (Asp/Glu), 2,2,4,6,7-pentamethyl-dihydrobenzofuran-5-sulfonyl (Arg), and trityl (Asn/Cys/Gln/His).
Peptides were chemically synthesized using Liberty Microwave Peptide Synthesizer (CEM Corp.) on solid support with an additional module of Discover (CEM Corp.) equipped with fiber-optic temperature probe for controlling the microwave power delivery following the Fmoc/tert-butyl method in a 30-ml Teflon reaction vessel. Each deprotection and coupling reaction was performed with microwave energy and nitrogen bubbling.
Fmoc deprotection was performed in two steps, 30 and 180 s, both at 45 watts, 75 °C using piperidine (20%) in DMF with 1-hydroxybenzotriazole (0.1 m) solution. Coupling reactions were performed by repetition of the following cycle conditions: 300 s, 25 watts, 75 °C, with O-benzotriazole-N,N,N′,N′-tetramethyl-uronium-hexafluoro-phosphate (0.11 m) in DMF, amino acids (0.12 m) in DMF, and N,N-diisopropylethylamine (0.25 m) in N-methyl-2-pyrrolidone solution. The coupling and Fmoc deprotection steps were monitored using Kaiser (ninhydrin) test (9) and small cleavage. Peptide cleavage from the resin and deprotection of the amino acid side chains were carried out with TFA/triisopropylsilane/H2O/phenol solution (90:2.5:2.5:5 v/v/v/w) for 3 h at room temperature without microwave energy. The crude products were precipitated with diethyl ether, collected by centrifugation, dissolved in H2O/CH3CN, and lyophilized.
Products were analyzed by analytical reverse-phase high pressure liquid chromatography (RP-HPLC) (Shimadzu LC-20 equipped with CBM-20A system controller, SPD-20A detector, CTO-20A column oven, 2× LC-20AD solvent delivery unit, SIL-20AC autosampler, DGU-20A5 degasser from Shimadzu) using an Ultro 120 5-μm C18Q (4.6 mm inner diameter × 150 mm) column (Peeke Scientific) at 1 ml/min. The solvent systems used were A (H2O with 0.1% TFA) and B (CH3CN with 0.1% TFA). A linear gradient of 5–50% B in 15 min was applied, and the detection was at 215 nm.
The synthesis products were purified by preparative RP-HPLC (Shimadzu LC-20 equipped with CBM-20A system controller, SPD-20A detector, CTO-20A column oven, 2× LC-6AD solvent delivery unit, and FRC-10A fraction collector from Shimadzu), using an XBridge Prep OBD C18 5 μm (19 mm × 150 mm) column (Waters) at 10 ml/min. The solvent systems used were A (H2O with 0.1% TFA) and B (CH3CN with 0.1% TFA). For separation, a linear gradient of 5–50% B in 45 min was applied, and the detection was at 215 nm.
Peptides were conjugated to TAT(47–57) (YGRKKR RQRRR) carrier peptide through an amide bond to the N terminus of the cargo peptide, as part of the solid-phase peptide synthesis and included a GG spacer between TAT and the cargo. δV1–1 was synthesized and conjugated to TAT as previously described (10). Note that TAT(47–57)-based delivery of a variety of peptide cargoes into cells has been used now for over 25 years and delivery of the cargo across biological membranes and into subcellular organelles has been confirmed (11–13). The peptides were dissolved in water or saline.
Interaction of GAPDH/δPKC, in Vitro
To determine the level of GAPDH binding to δPKC in the presence of ψGAPDH peptide, 200 ng of recombinant δPKC (Invitrogen) were incubated with or without the indicated peptides (1 μm) for 10 min at 4 °C, prior to adding 200 ng of recombinant GST-GAPDH (Abnova, Taiwan) for 20 min at 37 °C. GAPDH was immunoprecipitated using GAPDH antibody (mAb 6C5CA, Advanced ImmunoChemical Inc., CA) and δPKC binding to GAPDH was determined with δPKC antibody (C-17, Santa Cruz Biotechnology) and then with HRP-conjugated secondary antibody using Western blot analysis.
GAPDH Phosphorylation by δPKC and GAPDH Activity, in Vitro
To determine the level of GAPDH phosphorylation in the presence of ψGAPDH peptide, 200 ng of recombinant δPKC protein (Invitrogen) was incubated with or without the peptides (1 μm) for 10 min at 4 °C and then 200 ng of recombinant GST-GAPDH (Abnova, Taiwan) was added for 20 min at 37 °C in kinase buffer (40 μl) (20 mm Tris-HCl, 20 mm MgCl2, 1 μm DTT, 25 μm ATP, and 1 mm CaCl2) in the presence of the PKC activators, phosphatidylserine (1.25 μg) and 1,2-dioleoyl sn-glycerol (0.04 μg). The kinase assay was terminated by adding Laemmli buffer containing 5% SDS, and the samples were loaded on a SDS-10% polyacrylamide gel, and the levels of phosphorylated GAPDH protein were determined using anti-phosphothreonine (9381S and 2351S, Cell Signaling) and anti-phosphoserine PKC substrate (2261L, Cell Signaling) antibodies and then with HRP-conjugated secondary antibody using Western blot analysis. The nitrocellulose was also reprobed using GAPDH antibody (mAb 6C5CA, Advanced ImmunoChemical Inc.) to confirm that equal amounts of GAPDH were used.
GAPDH activity was determined using a KDalert GAPDH assay kit as instructed by the manufacturer (Applied Biosystems) using the same reagent as above. Briefly, δPKC protein (200 ng, Invitrogen) was incubated without or with ψGAPDH peptide (10 μm) for 10 min at 4 °C, and then GST-GAPDH (200 ng, Abnova, Taiwan) was added for 20 min at 37 °C in kinase buffer (40 μl) in the presence of the PKC activators. Samples were combined with the KDalert master mixture in 96-well plates and read at 690 nm using an UV-visible plate reader. GAPDH activity was calculated and expressed as units of GAPDH/mg of total protein.
Cell Culture, Viability Assay, Measurements of Mitochondrial Content, and siRNA Treatment
Cardiac H9C2 cells were maintained in modified Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% (v/v) fetal bovine serum (FBS) and 1% (v/v) penicillin/streptomycin. Cultured H9C2 cells in black 96-well plates were treated without or with ψGAPDH peptide (1 μm), 15 min prior to treatment with H2O2 (1 mm). After 1 h of treatment, cell death was quantified by measurement of the activity of lactate dehydrogenase (LDH) released from damaged cells into the supernatant using a cytotoxicity detection kit (Roche Diagnostics).
H9C2 cells (rat cardiac muscle cells) were grown in Dulbecco's modified Eagle's medium (DMEM) containing 10% fetal bovine serum (FBS), 1% l-glutamine, 1% antibiotic (penicillin and streptomycin) at 37 °C under 5% CO2 in air. After transfection with GAPDH or scrambled siRNA (75 nm for 48 h; AM4650, Thermo Fisher Scientific Inc.), cells were treated with 1 mm H2O2 for 1 h. 1 mm pyruvate was added into transfection medium in both siRNA-GAPDH groups and negative control.
Animal Studies
All treatments were performed between 9:00 a.m. and 4:00 p.m. by observers blinded to the treatment groups. Male rats were housed in a temperature- and light-controlled room for at least 3 days before use. All animals were randomized and assigned to testing groups to generate biological replicates for each group.
Ex Vivo Rat Heart Model of Myocardial Infarction-induced Ischemia and Reperfusion Injury
Wistar male rats, 4–6 weeks old, were treated with different peptides or control, and the levels of phosphorylation, infarct size, and CK release were measured, as described previously (14, 15). We excluded hearts if they met one of the following criteria: 1) time to perfusion prior to ischemia and reperfusion protocol exceeded 3 min; 2) coronary flow was outside the range of 9–15 ml/min; or 3) heart rate was below 240 beats/min, or severe arrhythmia was observed.
An ex vivo model of acute ischemic heart injury was carried out as described previously (10). Briefly, Wistar rats (250–275 g) purchased from Charles River were heparinized (1000 units/kg; intraperitoneal injection) and then anesthetized with Beuthanasia-D (100 mg/kg intraperitoneal injection). Hearts were rapidly excised and then perfused with an oxygenated Krebs-Henseleit buffer (120 mm NaCl, 5.8 mm KCl, 25 mm NaHCO3, 1.2 mm NaH2PO4, 1.2 mm MgSO4, 1 mm CaCl2, and 10 mm dextrose, pH 7.4) at 37 °C in a Langendorff coronary perfusion system. A constant coronary flow rate of 10 ml/min was used. Hearts were submerged into a heat-jacketed organ bath at 37 °C. After 10 min of equilibration, the hearts were subjected to 30 min of global ischemia and 60 min of reperfusion. The hearts were perfused with 1 μm peptides as indicated for 20 min after the ischemia. Normoxic control hearts were subjected to 90 min of perfusion in the absence of ischemia. Coronary effluent was collected to determine CK release during the first 30 min of the reperfusion period. At the end of the reperfusion period, hearts were sliced into 1-mm-thick transverse sections and incubated in triphenyltetrazolium chloride solution (1% in phosphate buffer, pH 7.4) at 37 °C for 15 min. Infarct size was expressed as a percentage of the risk zone (equivalent to total muscle mass). Infarct size and CK release were used to assess cardiac damage, as described previously (10).
Western Blot Analysis and Two-dimensional Gel Analysis
Rat hearts following the ischemia and reperfusion protocol were homogenized in buffer A (210 mm mannitol, 70 mm sucrose, 5 mm MOPS, and 1 mm EDTA) in the presence of protease inhibitor and phosphatase inhibitor mixtures (Sigma) followed by isolation of the mitochondrial fraction.
Heart mitochondria were isolated as described previously (16). Briefly, cardiac samples from a remote area were minced and homogenized in isolation buffer (300 mm sucrose, 10 mm Hepes, 2 mm EGTA, pH 7.2, 4 °C) containing 0.1 mg/ml type I protease (bovine pancreas) to release mitochondria from within the muscle fibers and later washed in the same buffer in the presence of 1 mg/ml bovine serum albumin. The suspension was homogenized in a 40-ml tissue grinder and centrifuged at 800 × g for 5 min. The resulting supernatant was centrifuged at 9,500 × g for 10 min. The mitochondrial pellet was washed, resuspended in isolation buffer, and submitted to a new centrifugation (10,000 × g for 10 min). The mitochondrial pellet was washed, and the final pellet was resuspended in a minimal volume of isolation buffer.
Protein content was measured with the Bradford protein assay. Cell lysates were separated by 6 or 10% SDS-PAGE and transferred onto PVDF membranes (Millipore). Membranes were incubated with anti-4-hydroxynonenal (4HNE) (HNE11-S,Alpha Diagnostic, Int.), anti-aconitase (ab71440, Abcam, Cambridge, UK), anti-GRP75 (138(B-14), Santa Cruz Biotechnology), anti-TOM20 (11415, Santa Cruz Biotechnology), anti-parkin (32282, Santa Cruz Biotechnology), and anti-enolase (15343, Santa Cruz Biotechnology) antibodies and then with HRP-conjugated secondary antibody.
For protein phosphorylation, cardiac fractions were resuspended in buffer A (210 mm mannitol, 70 mm sucrose, 5 mm MOPS, and 1 mm EDTA). Pyruvate dehydrogenase kinase (PDK) phosphorylation was determined using two-dimensional isoelectric focusing/SDS-PAGE; the samples were homogenized in buffer consisting of 7 m urea, 2 m thiourea, and 4% CHAPS in the presence of protease inhibitor and phosphatase inhibitor mixtures (Sigma). Supernatants were subjected to a first dimensional separation by an IPGphor isoelectric focus power supply using pre-cast Immobiline DryStrip pI 3–10 strips according to the manufacturer's instruction manual (Amersham Biosciences). 10% SDS-gel electrophoresis and Western blotting were carried out using anti-PDK2 (Abgent), anti-phosphothreonine (9381S and 2351S Cell Signaling), and anti-phosphoserine PKC substrate (2261L Cell Signaling) antibodies and then with HRP-conjugated secondary antibody. Phosphatase treatment confirmed that the leftward shift in pyruvate dehydrogenase kinase mobility is due to phosphorylation (16).
Phosphorylation of HSP27, MARCKS, STAT, GAPDH, and troponin I in the ex vivo model were determined on one-dimensional SDS-PAGE using anti-phospho-HSP27 (04-447, EMD Millipore) and anti-HSP27 (ADI-SPA-800-F, Enzo Life Sciences), anti-phospho-MARCKS (2741, Cell Signaling) and anti-MARCKS (6455, Santa Cruz Biotechnology), anti-phospho-STAT (8826S, Cell Signaling) and anti-STAT (346, Santa Cruz Biotechnology), anti-phospho-troponin I (4004, Cell Signaling) and anti-troponin I (31655, Santa Cruz Biotechnology) antibodies. The levels of phosphorylated substrates were normalized to total substrate and presented as a ratio to the phosphorylation in hearts subjected to ischemia and reperfusion in the presence of control treatment. Data are representative of four independent experiments.
Phosphorylation of GAPDH in the ex vivo heart attack model was determined after immunoprecipitation. Samples were incubated with GAPDH antibody (Mab6C5, Advanced Immunochemical) in buffer containing 10 mm Tris-base, pH 7.4, 150 mm NaCl, 0.1% Triton X-100, 5 mm EDTA, pH 8, and protease inhibitor overnight at 4 °C with gentle agitation. Protein A/G beads were then added, and the mixture was incubated for 2 h at 4 °C. The mixture was centrifuged for 1 min at 800 × g, and the immunoprecipitates were washed three times with buffer, analyzed by 10% SDS-PAGE, followed by Western blot using anti-phosphothreonine (9381S and 2351S, Cell Signaling), anti-phosphoserine PKC substrate (2261L, Cell Signaling), and anti-GAPDH (Mab6C5, Advanced Immunochemical) antibodies and then with HRP-conjugated secondary antibody. The levels of phosphorylated substrate were normalized to total substrate and presented as a ratio to the phosphorylation in hearts subjected to ischemia and reperfusion in the presence of control peptide. Data are representative of four independent experiments.
ATP Level Determination
Following the ex vivo ischemia and reperfusion experiment, 100 mg of heart tissue was homogenized in 1% TCA (500 μl). The lysate was spun to remove debris, and the pH was adjusted to pH 7.4. 10 μl of the lysate were used in a 200-μl assay for quantitative determination of ATP levels with recombinant firefly luciferase and its substrate, d-luciferin, according to the manufacturer's protocol (Invitrogen). Data are representative of six independent experiments.
Determination of GAPDH Activity in Samples from Whole Hearts Subjected to Ischemia and Reperfusion ex Vivo
GAPDH activity was determined using a KDalert GAPDH assay kit as instructed by the manufacturer (Applied Biosystems). Briefly, samples were combined with the KDalert master mixture in 96-well plates and read at 690 nm using a UV-visible plate reader. GAPDH activity was calculated and expressed as units of GAPDH/mg of total protein. Data are representative of at least four independent experiments.
Determination of GAPDH Oligomerization
GAPDH oligomerization was analyzed by native gel electrophoresis. The samples were loaded on a 6 or 10% native gel, and the levels of tetramer, trimer, dimer, and monomer GAPDH protein were determined using GAPDH antibody (mAb 6C5CA, Advanced ImmunoChemical Inc.) and then with HRP-conjugated secondary antibody.
Statistical Analysis
Data are provided as individual points and means ± S.E.; the number of independent experiments performed is provided in each data set. Data were tested for significance by using the two-tailed unpaired Student's t test. Differences were considered statistically significant when p values were <0.05. Sample sizes were estimated based on previous experience with similar assays and the effect of size observed in preliminary experiments. All samples were identical prior to allocation of treatments, and analysis was carried out by an observer blinded to the experimental conditions. The intensity of the spots on the gels were measured using ImageJ from National Institutes of Health (17).
Results
Rational Design of a Peptide Based on Homology between δPKC and GAPDH
We hypothesized that when δPKC is inactive, its GAPDH-docking site may be occupied by a pseudo-GAPDH (ψGAPDH) site, a GAPDH-like sequence that mimics the δPKC-binding site on GAPDH (Fig. 1A, left panel). If correct, upon δPKC activation, a conformational change in δPKC dissociates the ψGAPDH site from the GAPDH-docking site to expose the GAPDH-docking site for interaction with GAPDH (Fig. 1A, right panel). A peptide corresponding to the sequence of ψGAPDH is expected to selectively inhibit GAPDH docking to δPKC, and therefore it will diminish δPKC/GAPDH binding and the phosphorylation of GAPDH by δPKC (Fig. 1B). We also expect that ψGAPDH will not affect the binding and phosphorylation of other δPKC substrates (e.g. substrate xx, Fig. 1B).
FIGURE 1.

Rational design of an inhibitor of GAPDH phosphorylation by δPKC. Scheme depicting an inhibitor that selectively inhibits the docking and phosphorylation of GAPDH by the multisubstrate kinase δPKC. A, intramolecular interactions within δPKC are disrupted after δPKC activation, exposing the catalytic site as well as substrate-specific docking sites (shown are docking sites for GAPDH and substrate XX (sub XX) on δPKC). Docking of these substrates to the kinase increases the access of the catalytic site to the substrates, leading to their phosphorylation. In inactive δPKC (left), the GAPDH-docking site on δPKC interacts with another δPKC sequence, GIYQGF, which mimics the kinase-binding site on GAPDH (blue, GIVEGL). B, peptide corresponding to this GAPDH-like sequence on δPKC, ψGAPDH sequence, is a competitive inhibitor for docking to and phosphorylation of GAPDH by δPKC, without affecting docking and phosphorylation of other PKC substrates (e.g. substrate XX). C, sequence alignment of human δPKC and GAPDH identified a short sequence of homology, GIYQGF/GIVEGL. D, GIVEGL (light blue) in GAPDH (Protein Data Bank code 1U8F) is exposed and is located right near the δPKC phosphorylation site (Thr-246, yellow). E, conservation of GIYQGF sequence in δPKC, and F, conservation of GIVEGL in GAPDH in a variety of species. Symbols below the letters are * for identity and ∧ for homology. G, GIYQGF sequence is found in the V3 domain of δPKC. GIYQGF sequence is not present in other PKC isozymes, including θPKC, the most homologous PKC isozyme to δPKC, and ϵPKC, which is also a member of the novel PKC isozymes, which include δPKC (25). H, GIYQGF sequence is found in 15 human proteins (GAPDH, δPKC, and 13 other proteins, including four from the ADT family). A heat map of the GIYQGF conservation in orthologs of these proteins shows GIYQGF conservation only in δPKC and GAPDH.
Similar to the pseudosubstrate site on δPKC that mimics and has a similar sequence to the phospho-acceptor site on the substrate, the ψGAPDH-docking site on δPKC may have a similar sequence to GAPDH. Using a sequence homology search, Lalign (18), we searched for such a site and identified a six-amino acid stretch in GAPDH (GIVEGL; amino acids 169–174) that is 83% homologous to a sequence in δPKC (GIYQGF, amino acids 311–316; Fig. 1C). The GIVEGL sequence in the GAPDH structure is located in an exposed region in the monomeric enzyme, potentially available for protein-protein interaction (Fig. 1D). Interestingly, the GIVEGL sequence in GAPDH is adjacent to the main phosphorylation site by δPKC, Thr-246 (1) (Fig. 1D).
Sequences that are critical for protein-protein interactions are expected to be conserved in evolution (19–22). We therefore determined the evolutionary conservation of these short homologous sequences in GAPDH and δPKC. The GIYQGF sequence in δPKC and the GIVEGL sequence in GAPDH are 83% conserved in mammalian, avian, fish, and lizards (Fig. 1, E and F). We reasoned that if these sequences are specific for only δPKC-GAPDH interaction, then they are unlikely to be conserved in other proteins. Indeed, the GIYQGF sequence in δPKC is not present in other PKC isozymes, including θPKC, the most similar PKC isozyme to δPKC (Fig. 1G) (23). Next, we searched for GIYQGF sequence in other proteins in the human genome. Thirteen other proteins were identified with 83% homology to GIYQGF. However, the GIYQGF sequence was not conserved in evolution in any of these proteins (Fig. 1H). Altogether, these results suggest a functionally important and selective role of GIYQGF/GIVEGL for δPKC and GAPDH interaction.
ψGAPDH Peptide Specifically Inhibits GAPDH Phosphorylation by δPKC, in Vitro and Ex Vivo
We synthesized the peptide corresponding to the ψGAPDH site on δPKC, GIYQGF, and we tested its specificity as an inhibitor of the protein-protein interaction between δPKC and GAPDH. As a control, we synthesized a peptide with one amino acid substitution (Gln substitute to Ala, GIYAGF), and referred to it as control (Ctrl peptide). As expected, ψGAPDH peptide (1 μm) blocked δPKC binding to GAPDH, as determined by inhibition of co-immunoprecipitation (Fig. 2A). ψGAPDH peptide also inhibited δPKC-mediated GAPDH phosphorylation by over 70%, in vitro (Fig. 2, B and C), although the control peptide did not inhibit δPKC-GAPDH interaction or the phosphorylation of GAPDH by δPKC. Thus, the ψGAPDH peptide inhibited the interaction between GAPDH and δPKC and therefore inhibited GAPDH phosphorylation by δPKC in vitro.
FIGURE 2.

ψGAPDH peptide inhibits the interaction between δPKC and GAPDH in vitro. A, ψGAPDH peptide (1 μm) inhibited GAPDH-δPKC interaction in vitro, determined by co-immunoprecipitation (IP) and Western blotting (WB) analysis (Ctrl = control; a ψGAPDH peptide analog with a Gln substitute to an Ala, GIYAGF) (n = 3). B and C, δPKC-mediated GAPDH phosphorylation in vitro was inhibited by ψGAPDH peptide (1 μm) relative to control peptide (n = 3). Data are representative of three independent experiments. Statistical analysis was performed using two-tailed unpaired Student's t test. ***, p < 0.005 compared with control.
If ψGAPDH inhibits the docking of GAPDH to δPKC, it should be a selective inhibitor of phosphorylation of only GAPDH and should not affect the phosphorylation of other δPKC substrates. To test this prediction, we used an ex vivo Langendorff model of heart attack, an oxidative stress model that we previously showed results in δPKC activation and phosphorylation of many protein substrates (16, 24). In this model, rat hearts are subjected to 30 min of ischemia, followed by 60 min of reperfusion (ischemia and reperfusion protocol; Fig. 3A), mimicking a heart attack. ψGAPDH peptide was conjugated to TAT(47–57), to enable entry of the peptide into cells (11–13), and was added to the hearts during the reperfusion period, when δPKC activation is maximal (15).
FIGURE 3.
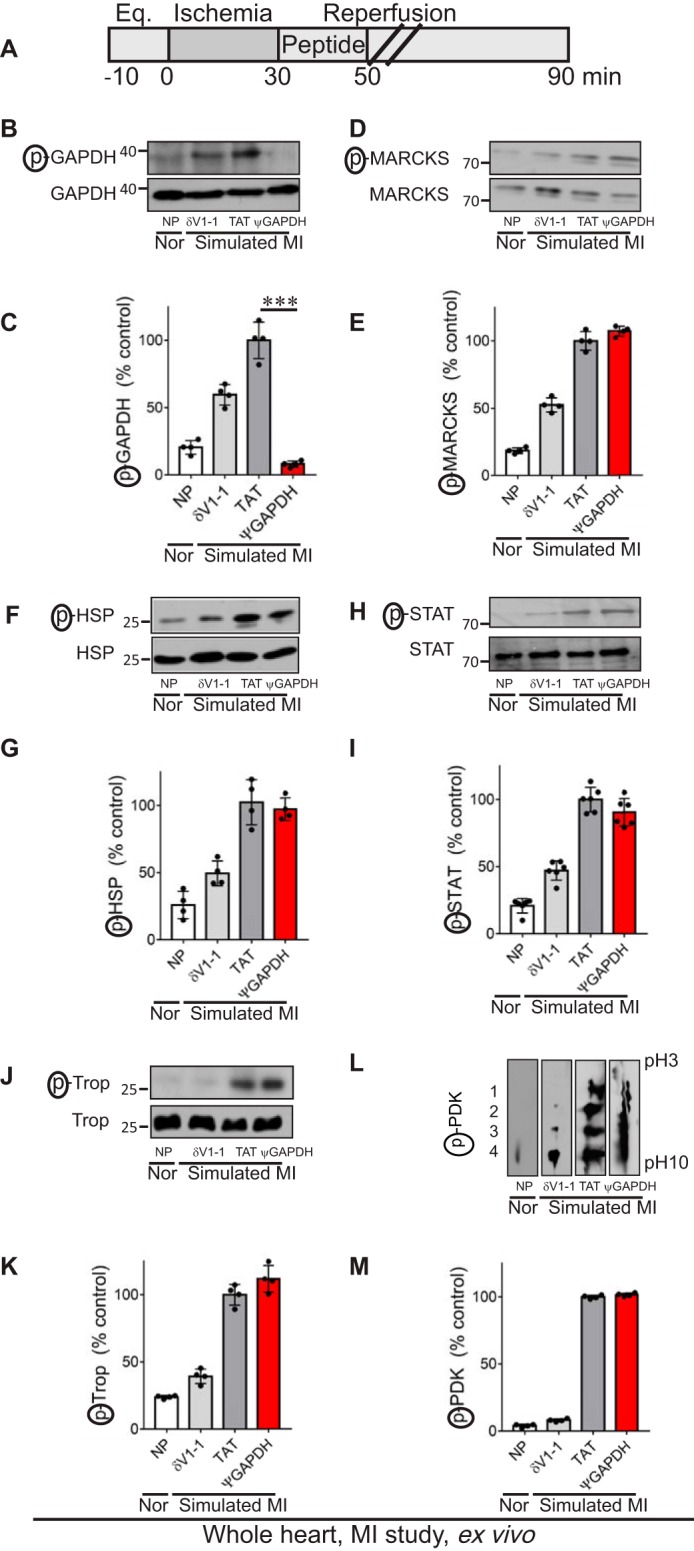
ψGAPDH peptide selectively inhibits phosphorylation of GAPDH, ex vivo. A, protocol of myocardial infarction model using isolated hearts subjected to ischemia and reperfusion (a model of simulated myocardial infarction (MI)) or normoxia (Nor). Bars indicate the length (in minutes) of each treatment (Eq. = equilibration). Rat hearts were subjected to 30 min of ischemia followed by 60 min of reperfusion without or with peptide treatment. The treatment was carried out for the first 20 min only during reperfusion. B and C, Western blot showing phosphorylation of GAPDH in heart extracts after ischemia and reperfusion without peptide (NP) and in the presence of control (TAT, 1 μm), δV1-1 (general inhibitor of δPKC phosphorylation, 1 μm), or ψGAPDH peptide (1 μm) using antibodies to phosphorylated serine and threonine (n = 4 hearts per treatment). The level of phosphorylation was measured by the level of phosphorylated protein (p) divided by the level of protein. Phosphorylation of MARCKS, D and E; HSP27, F and G; STAT, H and I; troponin I, J and K; or PDK, L and M; in heart extracts after ischemia and reperfusion without peptide (NP) and in the presence of control (TAT, 1 μm), δV1-1 (1 μm), or ψGAPDH peptide (1 μm), was determined using antibodies to the corresponding phosphorylated proteins (n ≥4 hearts per treatment). To determine the level of PDK phosphorylation, cardiac homogenates were analyzed by two-dimensional isoelectric focusing gel, and phosphorylation of PDK was evaluated by a shift toward lower pH. Phosphorylation is expressed as percent change from control (TAT)-treated hearts. Data are representative of independent experiments. Statistical analysis was performed using two-tailed unpaired Student's t test. ***, p < 0.005 compared with control.
Treatment with ψGAPDH-TAT(47–57) (referred to as ψGAPDH peptide; 1 μm), but not with TAT vehicle alone, selectively abolished GAPDH phosphorylation under these conditions; a more modest inhibition was also observed by δV1-1, a peptide inhibitor of all δPKC-mediated phosphorylation (Fig. 3, B and C) (25). Importantly, δPKC-mediated phosphorylation of δPKC substrates MARCKS (26), heat shock protein 27 (27), STAT (28), troponin I (29), and pyruvate dehydrogenase kinase (16) were all unaffected by treatment with ψGAPDH peptide but were inhibited by δV1-1 treatment (Fig. 3, D–M). These data demonstrate the high selectivity of ψGAPDH peptide as a specific phosphorylation inhibitor of only one δPKC substrate, GAPDH.
ψGAPDH Peptide Treatment Does Not Reduce Damage Following Oxidative Stress Ex Vivo
We previously showed that the phosphorylation of GAPDH by δPKC following oxidative stress correlates with inhibition of GAPDH-mediated mitochondrial elimination, as well as a 70% increase in cell damage (1). We therefore predicted that ψGAPDH peptide treatment should increase mitochondrial elimination and reduce cell damage.
We therefore determined the effect of ψGAPDH peptide treatment on the response of the heart to ischemia and reperfusion injury, using the Langendorff isolated heart model (ex vivo; Fig. 4A). Unexpectedly, treatment with 1 μm ψGAPDH peptide increased, rather than decreased, cardiac injury following ischemia and reperfusion, as measured by infarct size (Fig. 4, B and C). Furthermore, when measuring the release of CK into the perfusate, a measure of cardiac myocyte lysis, an increase in cardiac injury was observed relative to TAT (vehicle) and non-peptide controls (Fig. 4D). In addition, ψGAPDH peptide increased oxidative stress as measured by 4HNE, a marker of lipid peroxidation (Fig. 4E).
FIGURE 4.

ψGAPDH peptide treatment causes damage in an ex vivo model of myocardial infarction. A, protocol of myocardial infarction (MI) model using isolated hearts subjected to ischemia and reperfusion or normoxia (Nor). Bars indicate the length (in minutes) of each treatment (Eq. = equilibration). Rat hearts were subjected to 30 min of ischemia followed by 60 min of reperfusion without (NP) or with peptide treatment. The peptide treatment (1 μm) was carried out for the first 20 min only during reperfusion. B and C, infarct size in rat heart following simulated myocardial infarction was determined by triphenyltetrazolium chloride staining. Red indicates live tissue and white indicates dead tissue (n = 4 hearts per treatment). D, effect of ψGAPDH peptide (1 μm) treatment on total CK release following simulated myocardial infarction during the 30 min of reperfusion (n = 6 hearts per treatment). E, 4HNE levels in hearts subjected to ischemia and reperfusion in the presence of ψGAPDH peptide (1 μm) or TAT control (1 μm, vehicle) (n = 3 hearts per treatment). Statistical analysis was performed using two-tailed unpaired Student's t test. *, p < 0.05 compared with TAT control.
ψGAPDH Peptide Treatment Does Not Induce Mitochondrial Elimination in the Myocardial Infarction Model
During cardiac ischemia and reperfusion injury, excessive dysfunctional and damaged mitochondria are removed by processes of mitochondrial elimination such as autophagy and mitophagy (30–32). This selective removal is a protective process that prevents the release of cytotoxic substances and reactive oxygen species from the damaged mitochondria, thus preventing damage to the healthy/functional mitochondria and neighboring cells and enabling the recovery of ATP generation in the tissue (33). Previously, we have shown that GAPDH mediates mitochondrial elimination by mitophagy after cardiac ischemia and reperfusion injury and that inhibition of GAPDH phosphorylation by δPKC increases GAPDH-mediated elimination of damaged mitochondria (1).
First, we confirmed that ψGAPDH treatment caused an inhibition of GAPDH phosphorylation (Fig. 5, A and B). We also found that GAPDH translocation to the mitochondria was completely blocked by ψGAPDH treatment (Fig. 5, C and D). ψGAPDH peptide treatment inhibited mitochondrial elimination as assessed by measuring the levels of GRP75 and aconitase (two mitochondrial matrix proteins) and TOM20 (a mitochondrial outer membrane protein) relative to a cytosolic protein, enolase (Fig. 5, E–J). These unexpected data suggest that mitochondrial elimination by δPKC under oxidative stress is impaired by the ψGAPDH peptide, thus contributing to the increased cell damage described above.
FIGURE 5.

ψGAPDH peptide reduces mitochondrial elimination following ischemia and reperfusion injury, ex vivo. The level of GAPDH and phosphorylated GAPDH on the mitochondria is shown. A and B, Western blot showing the levels of phosphorylated GAPDH in cardiac mitochondria after normoxia (Nor) or after ischemia and reperfusion (simulated myocardial infarction (MI)) without peptide (NP) and in the presence of control (TAT, 1 μm) or ψGAPDH peptide (1 μm) using antibodies to phosphorylated serine and threonine (n = 4 hearts per treatment). The level of phosphorylation is measured by the level of phosphorylated GAPDH (p) divided by the total level of GAPDH. C and D, Western blot showing the level of GAPDH in heart extracts after ischemia and reperfusion in the presence of control (TAT, 1 μm) or ψGAPDH peptide (1 μm) (n = 4 hearts per treatment). The level of GAPDH was measured by the level of the protein divided by the level of aconitase (mitochondrial matrix protein). E–J, effect of ψGAPDH peptide (1 μm) on the elimination of mitochondria was determined by analyses of the levels of GRP75, aconitase, and TOM20 (n ≥5 hearts per treatment). The levels of mitochondrial matrix proteins, GRP75 (E and F) and aconitase (G and H), as well as outer membrane protein, TOM20 (I and J), were elevated under indicated conditions. K and L, effect of ψGAPDH peptide (1 μm) on canonical autophagy was determined by analyses of the levels of parkin (n = 3 hearts per treatment). Mitochondrial parkin levels were determined by Western blot analysis under the above conditions (K and L). M, effect of ψGAPDH peptide (1 μm) on the levels of tissue ATP, expressed as nanomoles of ATP per cardiac wet weight (n ≥5 hearts per treatment). Statistical analysis was performed using two-tailed unpaired Student's t test. **, p < 0.01, and ***, p < 0.005 compared with TAT control.
Because mitochondrial elimination can also occur by activating the canonical autophagy process, we determined the levels of mitochondrial Parkin, which marks damaged mitochondria for autophagy. We found an increase in Parkin association with the mitochondrial fraction (Fig. 5, K and L) following simulated ischemia in the presence of ψGAPDH, indicating that there was an increase in mitochondrial damage (34).
ψGAPDH Peptide Treatment Does Not Improve ATP Levels Following Ischemia and Reperfusion Injury
To confirm that the increase in mitochondrial content does not correlate with an increase in functional mitochondria, we measured ATP levels after ischemia and reperfusion in the Langendorff ex vivo model of heart attack. Even an hour after the return of oxygen and nutrients to the heart by reperfusion, ATP levels were ∼30% lower than the levels under normoxic conditions, and these were not altered by ψGAPDH peptide treatment (Fig. 5M). Together, these data suggest that ψGAPDH peptide inhibited mitochondrial elimination following oxidative stress, but these additional mitochondria are unlikely to be functional.
ψGAPDH Peptide Treatment Reduces GAPDH Activity and GAPDH Tetramerization, in Vitro and Ex Vivo
Following ischemia and reperfusion injury, GAPDH phosphorylation by δPKC increases (1), and GAPDH activity is reduced (35). We therefore expected that treatment with ψGAPDH peptide, which inhibits δPKC phosphorylation of GAPDH (Figs. 2, B and C, and 3, B and C), should increase GAPDH activity in the myocardial infarction model. Surprisingly, relative to TAT-treated hearts, ψGAPDH treatment of hearts subjected to ischemia and reperfusion, a model of myocardial infarction, decreased GAPDH activity further, by over 25% (Fig. 6A). Because ψGAPDH is homologous to the GAPDH sequence, we then determined whether the inhibition of GAPDH activity is due to a direct effect of this peptide on GAPDH. Indeed, in the presence of ψGAPDH peptide (and in the absence of δPKC), the activity of recombinant GAPDH decreased by 80% in vitro, as compared with vehicle control (Fig. 6B).
FIGURE 6.
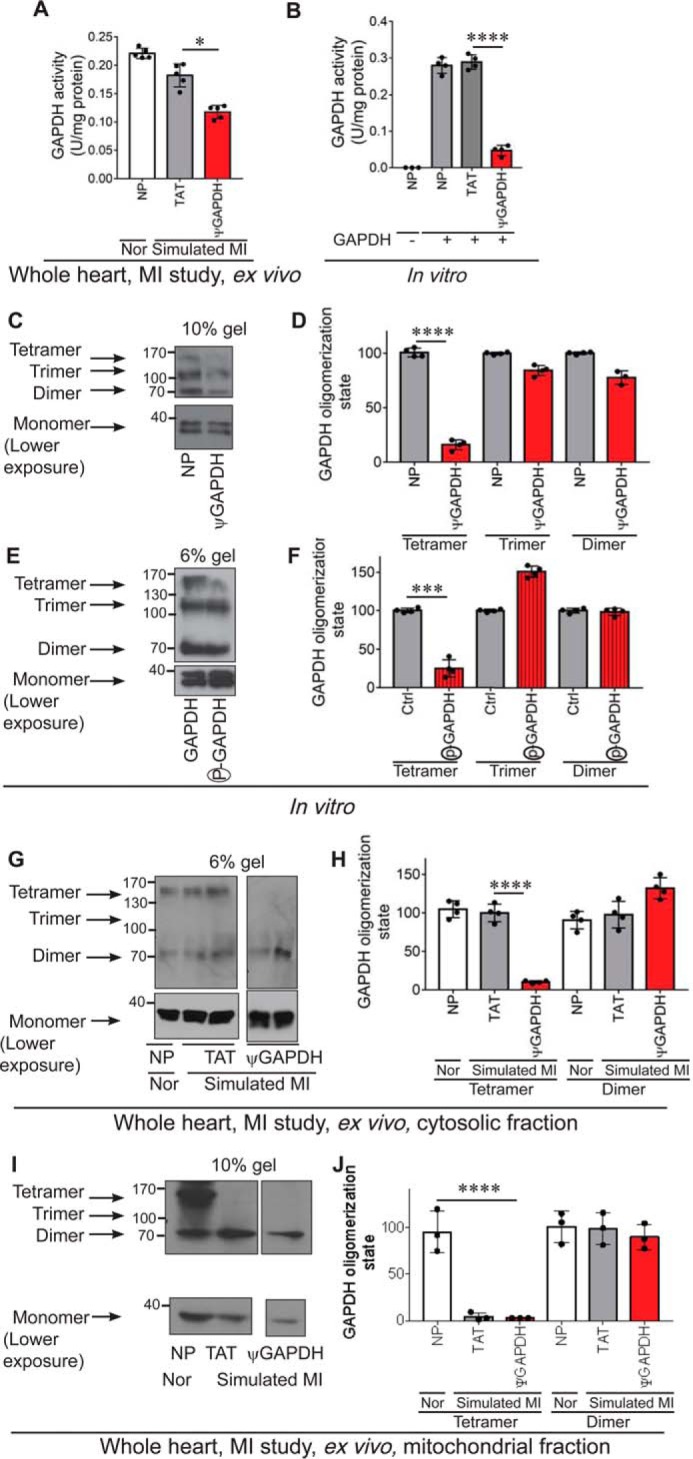
ψGAPDH peptide treatment inhibits GAPDH protein activity and tetramerization, in vitro and ex vivo. Measurement of GAPDH activity ex vivo (A) (n ≥4) in total heart extracts and in vitro (no peptide = NP) (B) (n ≥4). C and D, ψGAPDH peptide significantly reduced the tetramer formation of GAPDH enzyme, when GAPDH samples were separated on non-denaturing gel electrophoresis, in vitro (n ≥4). E and F, phosphorylation of GAPDH by δPKC significantly reduced the tetramer formation of GAPDH enzyme, when GAPDH samples were separated on non-denaturing gel electrophoresis (n ≥4). G and H, treatment of whole heart ex vivo with ψGAPDH peptide significantly reduced the tetramer formation of GAPDH enzyme (n ≥4). I and J, ψGAPDH peptide treatment of whole heart ex vivo significantly reduced the tetramer formation of GAPDH enzyme at the mitochondria (n ≥4). Statistical analysis was performed using two-tailed unpaired Student's t test. ****, p < 0.001; ***, p < 0.005; *, p < 0.05 compared with control.
The most active form of GAPDH is a tetramer (36, 37). Using a native gel, we therefore determined the oligomerization state of recombinant GAPDH with and without the treatment of ψGAPDH peptide in vitro. After a 30-min incubation with ψGAPDH peptide, the tetrameric form of GAPDH was almost eliminated as compared with control-treated GAPDH, in vitro (Fig. 6, C and D). A similar decline in the tetrameric form of GAPDH was observed after in vitro phosphorylation of GAPDH by δPKC (Fig. 6, E and F). Finally, ischemia and reperfusion injury in the presence of ψGAPDH peptide also greatly reduced the level of tetrameric GAPDH relative to tetrameric GAPDH from ischemic hearts treated with control peptide, both in the cytosol (Fig. 6, G and H) and at the mitochondria (Fig. 6, I and J).
We then examined the crystal structure of GAPDH. Tetrameric GAPDH is a dimer of dimers with three non-equivalent P, Q, and R interfaces (Fig. 7A). The R axis marks the dimer-dimer interface; the P axis is the interface with the most contacts between two monomeric subunits, and these contacts are highly conserved in evolution (38, 39). Relevant to our study, we found that the sequence from which the ψGAPDH peptide was derived as well as the phosphorylation site Thr-246 are both found on the P axis (Fig. 7B). At the dimerization interface between subunits O and P (Fig. 7A), Glu-179 (the fourth amino acid in the δPKC-like sequence, GIVEGL) in subunit O makes contact with Asn-304 in subunit P, and Leu-181 sits in a hydrophobic pocket on subunit P, stabilizing the dimer formation (Fig. 7C). These interactions may explain how the presence of ψGAPDH peptide inhibits GAPDH oligomerization, leading to a decrease in GAPDH activity.
FIGURE 7.

ψGAPDH sequence and Thr-246 lie at an oligomerization interface of GAPDH. A and B, ribbon drawing of the GAPDH tetramer. Broken lines represent the symmetry axes; Q axis is perpendicular to the plane of the paper. The organization of the four subunits O, P, Q, and R are indicated by letters and colors dark green, light green, light pink, and magenta, respectively. Monomers O and P form one dimer subunit, and Q and R form the other dimer subunit. The ψGAPDH sequence is indicated on each subunit in cyan, and Thr-246 in yellow. C, detailed view of the ψGAPDH sequence at the dimerization interface. The ψGAPDH sequence and Thr-246 are shown as sticks; Glu-179 in the GIVEGL sequence contacts Asn-304 (also shown as sticks) across the dimerization interface. The structure was modeled based on that of human GAPDH (Protein Data Bank code 4WNC) (66) using the PyMOL Molecular Graphics System software (67).
ψGAPDH Peptide Treatment Following Oxidative Stress in Cultured H9C2 Cells Recapitulates the Data Observed in Whole Hearts under Oxidative Stress
To confirm our data, we also used H9C2 cardiac cells in culture exposed to H2O2. H2O2 is a stressor that induces reactive oxygen species, thus leading to cell death. Cell death was measured by the release of LDH, a cytoplasmic enzyme, to the cell culture medium (Fig. 8A). As in the heart attack model, treatment with ψGAPDH peptide (1 μm) under these conditions increased (rather than decreased) LDH release (Fig. 8A). Mitochondrial elimination was also inhibited by ψGAPDH treatment as determined by measuring the cellular levels of aconitase and TOM20 (Fig. 8, B–E).
FIGURE 8.

ψGAPDH peptide reduces mitochondrial elimination following oxidative stress injury in cultured H9C2 cells. A, LDH release (a measure of cell death) post-oxidative stress (H2O2, 1 mm, 60 min) was measured in H9C2 cells (no peptide = NP) (n = 4). B and C, measurement of mitochondrial elimination. Western blot showing the level of aconitase in H9C2 cells after oxidative stress in the presence of control (TAT, 1 μm) or ψGAPDH peptide (1 μm) (n = 4). The level of aconitase is measured by the level of the protein divided by the level of β-actin. D and E, Western blot showing the level of TOM20 in H9C2 cells after oxidative stress in the presence of control (TAT, 1 μm) or ψGAPDH peptide (1 μm) (n = 4). The level of TOM20 is measured by the level of the protein divided by the level of β-actin. F and G, ψGAPDH peptide significantly reduced the tetramer formation of GAPDH enzyme in H9C2 cells after oxidative stress (samples were separated on non-denaturing gel electrophoresis; n = 2). H and I, ψGAPDH peptide (1 μm) treatment reduced nuclear translocation of GAPDH following oxidative stress in culture, as measured by immunofluorescence; I provides quantitation of the immunofluorescence data. J and K, siRNA knockdown reduced GAPDH levels by ∼50% (n = 4). L and M, ψGAPDH peptide had no effect on mitochondrial content in GAPDH-silenced H9C2 cells after oxidative stress (n = 4). N, ψGAPDH did not increase cell death (as measured by LDH release) in cells subjected to oxidative stress, when GAPDH was silenced (n = 6). Statistical analysis was performed using two-tailed unpaired Student's t test. **, p < 0.01; *, p < 0.05 compared with control.
We then determined the effect of ψGAPDH on GAPDH polymerization and found that cellular tetrameric GAPDH was almost eliminated (Fig. 8, F and G). GAPDH has been implicated in other functions, including change in transcription under oxidative stress, following its translocation into the nucleus (40, 41). Although the time scale for changes in gene expression is too long to affect cell death measured in this study, we determined GAPDH translocation into the nucleus (42). We found that ψGAPDH also inhibited GAPDH translocation into the nucleus (Fig. 8, H and I), indicating that tetramerization of GAPDH is also required for its non-canonical role in regulating gene expression in the nucleus.
Finally, to confirm that ψGAPDH selectively affects GAPDH-mediated functions, we knocked down GAPDH in H9C2 cells, then exposed them to H2O2, and determined the effect of ψGAPDH treatment in the presence of pyruvate as a carbon source. In cells with GAPDH siRNA, GAPDH protein levels were reduced by 50% (Fig. 8, J and K), and the ∼50% increase in mitochondrial content (which occurred even under basal conditions) was not increased by ψGAPDH treatment following simulated ischemia; if anything, there was a trend toward a declined in mitochondrial content (Fig. 8, L–M). Consistent with the lack of ψGAPDH in cells treated with GAPDH siRNA, cell viability in these cells also was unaffected by ψGAPDH as compared with TAT (Fig. 8N). Finally, treatment with scrambled siRNA did not affect any of these measurements (Fig. 8, J–N).
Discussion
Kinases have evolved at least two mechanisms to optimize substrate specificity and affinity. The first involves coordinated binding of a substrate and its kinase onto scaffolding or adaptor proteins (43). The second involves a direct interaction between the kinase and its substrates, mediated by sequences outside the catalytic site on the enzyme, at what is referred to as the substrate-docking site (5). These latter interactions have often been found to increase the affinity of kinases for their substrates (44, 45). Such docking sites are prevalent in a number of serine/threonine kinases, including JNKs, cyclin-dependent kinases, and MAPKs (46–53), and peptides derived from these docking sequences can be used as inhibitors of the phosphorylation of the corresponding substrate. For example, Lee et al. (54) identified a six-amino acid substrate-docking site on C-terminal Src kinase (Csk), and a peptide mimicking the docking site inhibited Csk phosphorylation of Src (IC50 = 21 μm) and only moderately inhibited Src kinase activity on other substrates.
These kinase-substrate interaction sites, and the inhibitors derived from them, are often identified by extensive mutagenesis analysis and co-crystallization studies. Here, we used a simple and fast rational approach to identify the interaction site between GAPDH and δPKC, and we showed that a sequence derived from this site is an inhibitor of GAPDH docking to δPKC. This inhibitor, ψGAPDH peptide, is selective for binding to and phosphorylation of GAPDH by δPKC; it does not affect the phosphorylation of five other δPKC substrates in the same preparation (Figs. 2 and 3).
We previously showed that inhibition of GAPDH phosphorylation by δPKC correlates with increased elimination of damaged mitochondria and increased cytoprotection under oxidative stress (1). Unexpectedly, although ψGAPDH peptide inhibited δPKC-mediated GAPDH phosphorylation, mitochondrial elimination did not increase; ATP levels did not increase, and cytoprotection was not achieved (Figs. 4, 5, and 8). These unexpected results are likely attributed to the fact that ψGAPDH directly decreased the activity of GAPDH by reducing the levels of the tetrameric active form of GAPDH (Fig. 6). Therefore, our identification of a novel GAPDH inhibitor elucidated the role of GAPDH tetramerization in glycolysis and in mitochondrial elimination. However, it is still to be determined whether this effect is only due to the reduced activity of GAPDH or possibly by direct action of δPKC on mitochondrial function. Currently, we are developing additional δPKC-specific inhibitors for other δPKC substrates using the same rational design approach that may enable us to address this important possibility.
Both the δPKC-mediated phosphorylation site on GAPDH (Thr-246) and the potential GAPDH-δPKC interaction site (amino acids 169–174 in GAPDH) are adjacent to each other (Fig. 1D), and both are at the interface between monomers in the tetrameric structure (Fig. 7). Therefore, our data are consistent with a dynamic oligomerization of GAPDH, in which either ψGAPDH peptide or phosphorylation of Thr-246 by δPKC interferes with monomer-monomer interaction, thus decreasing the levels of the catalytically competent GAPDH tetramer. Our data are also consistent with tetrameric GAPDH as a mediator of mitochondrial elimination, as decreased tetramerization by both ψGAPDH and Thr-246 phosphorylation resulted in decreased mitochondrial elimination (Figs. 5 and 8) (1) and increased cell injury (Figs. 4 and 8).
In addition to its role in glycolysis, GAPDH plays a critical role in nuclear events. GAPDH translocation to the nucleus has been associated with change in gene expression, leading to apoptosis in a number of cell systems; blocking this translocation was found to be sufficient to reduce cytotoxicity (55, 56). Transfection with GAPDH-specific siRNA resulted in substantial reduction of GAPDH expression to <55% of control as shown in Fig. 8, J and K. GAPDH silencing had no effect on cell viability but showed a general increase in mitochondrial content. ψGAPDH peptide had no effect in silenced H9C2 cells. Our data also suggest that although nuclear translocation of GAPDH is essential for initiating apoptosis, translocation is regulated by the GAPDH oligomerization state and that blocking with ψGAPDH significantly reduced nuclear GAPDH. These data indicate that the oligomerization state of GAPDH is critical for at least two non-canonical functions of GAPDH outside glycolysis.
GAPDH is a vital glycolytic enzyme that is involved in DNA repair (57), tRNA export (58), membrane fusion and transport (59), cytoskeletal dynamics (60), cell death (55), as well as in elimination of damaged mitochondria following oxidative stress (1). GAPDH has been implicated in various human diseases; increased nuclear localization of GAPDH correlates with increased neuronal apoptosis in Parkinson disease (61), Huntington disease (62), and Alzheimer disease (63), and increased glycolytic activity correlates with cancer cell survival (64). GAPDH is also an attractive target for drugs against protozoan parasites, whose bloodstream forms depend solely on glycolysis for energy (65). Therefore, an inhibitor of GAPDH glycolytic activity that also increases oxidative stress-induced injury by damaged mitochondria, such as ψGAPDH, may be an attractive drug lead for cancer as well as an anti-parasitic drug.
Author Contributions
N. Q. and D. M. R. conceived the experiments, analyzed the data, and wrote the article, which was read by all the authors. N. Q., A. U. J., and J. C. B. F. performed the experiments, and A. D. C. analyzed the GAPDH crystal structure.
Acknowledgments
We thank Dr. Marie-Helene Disatnik for critical advice and help with the design and interpretation of the study and Dr. Sunhee Hwang and Dr. Marie-Helene Disatnik for reviewing the manuscript.
This work was supported by National Institutes of Health Grant HL52141 (to D. M.-R.). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
- Fmoc
- fluorenylmethoxycarbonyl
- 4HNE
- 4-hydroxynonenal
- DMF
- dimethylformamide
- CK
- creatine kinase
- MARCKS
- myristoylated alanine-rich protein kinase C substrate
- STAT
- signal transducer and activator of transcription
- LDH
- lactate dehydrogenase
- PDK
- pyruvate dehydrogenase kinase.
References
- 1. Yogalingam G., Hwang S., Ferreira J. C., and Mochly-Rosen D. (2013) Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) phosphorylation by protein kinase Cδ (PKCδ) inhibits mitochondria elimination by lysosomal-like structures following ischemia and reoxygenation-induced injury. J. Biol. Chem. 288, 18947–18960 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Steinberg S. F. (2008) Structural basis of protein kinase C isoform function. Physiol. Rev. 88, 1341–1378 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. House C., and Kemp B. E. (1987) Protein kinase C contains a pseudosubstrate prototope in its regulatory domain. Science 238, 1726–1728 [DOI] [PubMed] [Google Scholar]
- 4. Soderling T. R. (1990) Protein kinases–regulation by autoinhibitory domains. J. Biol. Chem. 265, 1823–1826 [PubMed] [Google Scholar]
- 5. Ubersax J. A., and Ferrell J. E. Jr. (2007) Mechanisms of specificity in protein phosphorylation. Nat. Rev. Mol. Cell Biol. 8, 530–541 [DOI] [PubMed] [Google Scholar]
- 6. Reményi A., Good M. C., and Lim W. A. (2006) Docking interactions in protein kinase and phosphatase networks. Curr. Opin. Struct. Biol. 16, 676–685 [DOI] [PubMed] [Google Scholar]
- 7. Pearson W. R., and Lipman D. J. (1988) Improved tools for biological sequence comparison. Proc. Natl. Acad. Sci. U.S.A. 85, 2444–2448 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Merrifield R. B. (1963) Solid phase peptide synthesis I. The synthesis of a tetrapeptide. J. Am. Chem. Soc. 85, 2149–2154 [Google Scholar]
- 9. Kaiser E., Colescott R. L., Bossinger C. D., and Cook P. I. (1970) Color test for detection of free terminal amino groups in solid-phase synthesis of peptides. Anal. Biochem. 34, 595–598 [DOI] [PubMed] [Google Scholar]
- 10. Chen L., Wright L. R., Chen C. H., Oliver S. F., Wender P. A., and Mochly-Rosen D. (2001) Molecular transporters for peptides: delivery of a cardioprotective ϵPKC agonist peptide into cells and intact ischemic heart using a transport system, R(7). Chem. Biol. 8, 1123–1129 [DOI] [PubMed] [Google Scholar]
- 11. Begley R., Liron T., Baryza J., and Mochly-Rosen D. (2004) Biodistribution of intracellularly acting peptides conjugated reversibly to TAT. Biochem. Biophys. Res. Commun. 318, 949–954 [DOI] [PubMed] [Google Scholar]
- 12. Rizzuti M., Nizzardo M., Zanetta C., Ramirez A., and Corti S. (2015) Therapeutic applications of the cell-penetrating HIV-1 Tat peptide. Drug Discov. Today 20, 76–85 [DOI] [PubMed] [Google Scholar]
- 13. Lönn P., and Dowdy S. F. (2015) Cationic PTD/CPP-mediated macromolecular delivery: charging into the cell. Expert Opin. Drug Deliv. 12, 1627–1636 [DOI] [PubMed] [Google Scholar]
- 14. Inagaki K., Hahn H. S., Dorn G. W. 2nd, and Mochly-Rosen D. (2003) Additive protection of the ischemic heart ex vivo by combined treatment with δ-protein kinase C inhibitor and ϵ-protein kinase C activator. Circulation 108, 869–875 [DOI] [PubMed] [Google Scholar]
- 15. Inagaki K., Chen L., Ikeno F., Lee F. H., Imahashi K., Bouley D. M., Rezaee M., Yock P. G., Murphy E., and Mochly-Rosen D. (2003) Inhibition of δ-protein kinase C protects against reperfusion injury of the ischemic heart in vivo. Circulation 108, 2304–2307 [DOI] [PubMed] [Google Scholar]
- 16. Churchill E. N., Murriel C. L., Chen C. H., Mochly-Rosen D., and Szweda L. I. (2005) Reperfusion-induced translocation of δPKC to cardiac mitochondria prevents pyruvate dehydrogenase reactivation. Circ. Res. 97, 78–85 [DOI] [PubMed] [Google Scholar]
- 17. Abràmoff M. D., Magalhães P. J., and Ram S. J. (2004) Image processing with ImageJ. Biophotonics Int. 11, 36–43 [Google Scholar]
- 18. Huang X. Q., and Miller W. (1991) A time-efficient, linear-space local similarity algorithm. Adv. Appl. Math. 12, 337–357 [Google Scholar]
- 19. Thomas P. D., Campbell M. J., Kejariwal A., Mi H., Karlak B., Daverman R., Diemer K., Muruganujan A., and Narechania A. (2003) PANTHER: a library of protein families and subfamilies indexed by function. Genome Res. 13, 2129–2141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Caffrey D. R., Somaroo S., Hughes J. D., Mintseris J., and Huang E. S. (2004) Are protein-protein interfaces more conserved in sequence than the rest of the protein surface? Protein Sci. 13, 190–202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Guharoy M., and Chakrabarti P. (2010) Conserved residue clusters at protein-protein interfaces and their use in binding site identification. BMC Bioinformatics 11, 286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Qvit N., and Mochly-Rosen D. (2010) Highly specific modulators of protein kinase C localization: applications to heart failure. Drug Discov. Today Dis. Mech. 7, e87–e93 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Baier G., Telford D., Giampa L., Coggeshall K. M., Baier-Bitterlich G., Isakov N., and Altman A. (1993) Molecular cloning and characterization of PKCθ, a novel member of the protein kinase C (PKC) gene family expressed predominantly in hematopoietic cells. J. Biol. Chem. 268, 4997–5004 [PubMed] [Google Scholar]
- 24. Murriel C. L., Churchill E., Inagaki K., Szweda L. I., and Mochly-Rosen D. (2004) Protein kinase Cδ activation induces apoptosis in response to cardiac ischemia and reperfusion damage: a mechanism involving BAD and the mitochondria. J. Biol. Chem. 279, 47985–47991 [DOI] [PubMed] [Google Scholar]
- 25. Chen L., Hahn H., Wu G., Chen C. H., Liron T., Schechtman D., Cavallaro G., Banci L., Guo Y., Bolli R., Dorn G. W. 2nd, and Mochly-Rosen D. (2001) Opposing cardioprotective actions and parallel hypertrophic effects of δPKC and ϵPKC. Proc. Natl. Acad. Sci. U.S.A. 98, 11114–11119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Li J., O'Connor K. L., Greeley G. H. Jr., Blackshear P. J., Townsend C. M. Jr., and Evers B. M. (2005) Myristoylated alanine-rich C kinase substrate-mediated neurotensin release via protein kinase C-δ downstream of the Rho/ROK pathway. J. Biol. Chem. 280, 8351–8357 [DOI] [PubMed] [Google Scholar]
- 27. Kim E. H., Lee H. J., Lee D. H., Bae S., Soh J. W., Jeoung D., Kim J., Cho C. K., Lee Y. J., and Lee Y. S. (2007) Inhibition of heat shock protein 27-mediated resistance to DNA damaging agents by a novel PKC δ-V5 heptapeptide. Cancer Res. 67, 6333–6341 [DOI] [PubMed] [Google Scholar]
- 28. Novotny-Diermayr V., Zhang T., Gu L., and Cao X. (2002) Protein kinase C δ associates with the interleukin-6 receptor subunit glycoprotein (gp) 130 via Stat3 and enhances Stat3-gp130 interaction. J. Biol. Chem. 277, 49134–49142 [DOI] [PubMed] [Google Scholar]
- 29. Noland T. A. Jr., Raynor R. L., and Kuo J. F. (1989) Identification of sites phosphorylated in bovine cardiac troponin I and troponin T by protein kinase C and comparative substrate activity of synthetic peptides containing the phosphorylation sites. J. Biol. Chem. 264, 20778–20785 [PubMed] [Google Scholar]
- 30. Eltzschig H. K., and Eckle T. (2011) Ischemia and reperfusion-from mechanism to translation. Nat. Med. 17, 1391–1401 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Hoshino A., Matoba S., Iwai-Kanai E., Nakamura H., Kimata M., Nakaoka M., Katamura M., Okawa Y., Ariyoshi M., Mita Y., Ikeda K., Ueyama T., Okigaki M., and Matsubara H. (2012) p53-TIGAR axis attenuates mitophagy to exacerbate cardiac damage after ischemia. J. Mol. Cell. Cardiol. 52, 175–184 [DOI] [PubMed] [Google Scholar]
- 32. Quinsay M. N., Thomas R. L., Lee Y., and Gustafsson A. B. (2010) Bnip3-mediated mitochondrial autophagy is independent of the mitochondrial permeability transition pore. Autophagy 6, 855–862 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Ma S., Wang Y., Chen Y., and Cao F. (2015) The role of the autophagy in myocardial ischemia/reperfusion injury. Biochim. Biophys. Acta 1852, 271–276 [DOI] [PubMed] [Google Scholar]
- 34. van der Bliek A. M., Shen Q., and Kawajiri S. (2013) Mechanisms of mitochondrial fission and fusion. Cold Spring Harb. Perspect. Biol. 5, a011072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Eaton P., Wright N., Hearse D. J., and Shattock M. J. (2002) Glyceraldehyde phosphate dehydrogenase oxidation during cardiac ischemia and reperfusion. J. Mol. Cell. Cardiol. 34, 1549–1560 [DOI] [PubMed] [Google Scholar]
- 36. Mazzola J. L., and Sirover M. A. (2003) Subcellular localization of human glyceraldehyde-3-phosphate dehydrogenase is independent of its glycolytic function. Biochim. Biophys. Acta 1622, 50–56 [DOI] [PubMed] [Google Scholar]
- 37. Hoagland V. D. Jr., and Teller D. C. (1969) Influence of substrates on the dissociation of rabbit muscle d-glyceraldehyde 3-phosphate dehydrogenase. Biochemistry 8, 594–602 [DOI] [PubMed] [Google Scholar]
- 38. Roitel O., Vachette P., Azza S., and Branlant G. (2003) P but not R-axis interface is involved in cooperative binding of NAD on tetrameric phosphorylating glyceraldehyde-3-phosphate dehydrogenase from Bacillus stearothermophilus. J. Mol. Biol. 326, 1513–1522 [DOI] [PubMed] [Google Scholar]
- 39. Seidler N. W. (2012) GAPDH: Biological Properties and Diversity, pp. 207–242, Springer Science & Business Media, Dordrecht, Heidelberg, Germany [Google Scholar]
- 40. Dastoor Z., and Dreyer J. L. (2001) Potential role of nuclear translocation of glyceraldehyde-3-phosphate dehydrogenase in apoptosis and oxidative stress. J. Cell Sci. 114, 1643–1653 [DOI] [PubMed] [Google Scholar]
- 41. Hara M. R., Agrawal N., Kim S. F., Cascio M. B., Fujimuro M., Ozeki Y., Takahashi M., Cheah J. H., Tankou S. K., Hester L. D., Ferris C. D., Hayward S. D., Snyder S. H., and Sawa A. (2005) S-Nitrosylated GAPDH initiates apoptotic cell death by nuclear translocation following Siah1 binding. Nat. Cell Biol. 7, 665–674 [DOI] [PubMed] [Google Scholar]
- 42. Zheng L., Roeder R. G., and Luo Y. (2003) S phase activation of the histone H2B promoter by OCA-S, a coactivator complex that contains GAPDH as a key component. Cell 114, 255–266 [DOI] [PubMed] [Google Scholar]
- 43. Pawson T., and Scott J. D. (1997) Signaling through scaffold, anchoring, and adaptor proteins. Science 278, 2075–2080 [DOI] [PubMed] [Google Scholar]
- 44. Biondi R. M., and Nebreda A. R. (2003) Signalling specificity of Ser/Thr protein kinases through docking-site-mediated interactions. Biochem. J. 372, 1–13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Holland P. M., and Cooper J. A. (1999) Protein modification: docking sites for kinases. Curr. Biol. 9, R329–R331 [DOI] [PubMed] [Google Scholar]
- 46. Adams P. D., Li X., Sellers W. R., Baker K. B., Leng X., Harper J. W., Taya Y., and Kaelin W. G. Jr. (1999) Retinoblastoma protein contains a C-terminal motif that targets it for phosphorylation by cyclin-cdk complexes. Mol. Cell. Biol. 19, 1068–1080 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Adams P. D., Sellers W. R., Sharma S. K., Wu A. D., Nalin C. M., and Kaelin W. G. Jr. (1996) Identification of a cyclin-cdk2 recognition motif present in substrates and p21-like cyclin-dependent kinase inhibitors. Mol. Cell. Biol. 16, 6623–6633 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Chang C. I., Xu B. E., Akella R., Cobb M. H., and Goldsmith E. J. (2002) Crystal structures of MAP kinase p38 complexed to the docking sites on its nuclear substrate MEF2A and activator MKK3b. Mol. Cell 9, 1241–1249 [DOI] [PubMed] [Google Scholar]
- 49. Kallunki T., Deng T., Hibi M., and Karin M. (1996) c-Jun can recruit JNK to phosphorylate dimerization partners via specific docking interactions. Cell 87, 929–939 [DOI] [PubMed] [Google Scholar]
- 50. Lee T., Hoofnagle A. N., Kabuyama Y., Stroud J., Min X., Goldsmith E. J., Chen L., Resing K. A., and Ahn N. G. (2004) Docking motif interactions in MAP kinases revealed by hydrogen exchange mass spectrometry. Mol. Cell 14, 43–55 [DOI] [PubMed] [Google Scholar]
- 51. Luciani M. G., Hutchins J. R., Zheleva D., and Hupp T. R. (2000) The C-terminal regulatory domain of p53 contains a functional docking site for cyclin A. J. Mol. Biol. 300, 503–518 [DOI] [PubMed] [Google Scholar]
- 52. Schulman B. A., Lindstrom D. L., and Harlow E. (1998) Substrate recruitment to cyclin-dependent kinase 2 by a multipurpose docking site on cyclin A. Proc. Natl. Acad. Sci. U.S.A. 95, 10453–10458 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Tanoue T., Maeda R., Adachi M., and Nishida E. (2001) Identification of a docking groove on ERK and p38 MAP kinases that regulates the specificity of docking interactions. EMBO J. 20, 466–479 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Lee S., Lin X., Nam N. H., Parang K., and Sun G. (2003) Determination of the substrate-docking site of protein tyrosine kinase C-terminal Src kinase. Proc. Natl. Acad. Sci. U.S.A. 100, 14707–14712 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Sawa A., Khan A. A., Hester L. D., and Snyder S. H. (1997) Glyceraldehyde-3-phosphate dehydrogenase: nuclear translocation participates in neuronal and nonneuronal cell death. Proc. Natl. Acad. Sci. U.S.A. 94, 11669–11674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Saunders P. A., Chalecka-Franaszek E., and Chuang D. M. (1997) Subcellular distribution of glyceraldehyde-3-phosphate dehydrogenase in cerebellar granule cells undergoing cytosine arabinoside-induced apoptosis. J. Neurochem. 69, 1820–1828 [DOI] [PubMed] [Google Scholar]
- 57. Meyer-Siegler K., Mauro D. J., Seal G., Wurzer J., deRiel J. K., and Sirover M. A. (1991) A human nuclear uracil DNA glycosylase is the 37-kDa subunit of glyceraldehyde-3-phosphate dehydrogenase. Proc. Natl. Acad. Sci. U.S.A. 88, 8460–8464 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Singh R., and Green M. R. (1993) Sequence-specific binding of transfer RNA by glyceraldehyde-3-phosphate dehydrogenase. Science 259, 365–368 [DOI] [PubMed] [Google Scholar]
- 59. Tisdale E. J. (2001) Glyceraldehyde-3-phosphate dehydrogenase is required for vesicular transport in the early secretory pathway. J. Biol. Chem. 276, 2480–2486 [DOI] [PubMed] [Google Scholar]
- 60. Kumagai H., and Sakai H. (1983) A porcine brain protein (35 K protein) which bundles microtubules and its identification as glyceraldehyde 3-phosphate dehydrogenase. J. Biochem. 93, 1259–1269 [DOI] [PubMed] [Google Scholar]
- 61. Tatton N. A. (2000) Increased caspase 3 and Bax immunoreactivity accompany nuclear GAPDH translocation and neuronal apoptosis in Parkinson's disease. Exp. Neurol. 166, 29–43 [DOI] [PubMed] [Google Scholar]
- 62. Hwang S., Disatnik M. H., and Mochly-Rosen D. (2015) Impaired GAPDH-induced mitophagy contributes to the pathology of Huntington's disease. EMBO Mol. Med. 12, 1307–1326 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. El Kadmiri N., Slassi I., El Moutawakil B., Nadifi S., Tadevosyan A., Hachem A., and Soukri A. (2014) Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) and Alzheimer's disease. Pathol. Biol. 62, 333–336 [DOI] [PubMed] [Google Scholar]
- 64. Guo C., Liu S., and Sun M. Z. (2013) Novel insight into the role of GAPDH playing in tumor. Clin. Transl. Oncol. 15, 167–172 [DOI] [PubMed] [Google Scholar]
- 65. Werbovetz K. A. (2000) Target-based drug discovery for malaria, leishmaniasis, and trypanosomiasis. Curr. Med. Chem. 7, 835–860 [DOI] [PubMed] [Google Scholar]
- 66. White M. R., Khan M. M., Deredge D., Ross C. R., Quintyn R., Zucconi B. E., Wysocki V. H., Wintrode P. L., Wilson G. M., and Garcin E. D. (2015) A dimer interface mutation in glyceraldehyde-3-phosphate dehydrogenase regulates its binding to AU-rich RNA. J. Biol. Chem. 290, 1770–1785 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. DeLano W. L. (2002) The PyMOL Molecular Graphics System, DeLano Scientific, San Carlos, CA [Google Scholar]