

Abstract
Calcitonin is a peptide hormone consisting of 32 amino acid residues and the calcitonin receptor is a Class B G protein-coupled receptor (GPCR). The crystal structure of the human calcitonin receptor ectodomain (CTR ECD) in complex with a truncated analogue of salmon calcitonin ([BrPhe22]sCT(8–32)) has been determined to 2.1-Å resolution. Parallel analysis of a series of peptide ligands showed that the rank order of binding of the CTR ECD is identical to the rank order of binding of the full-length CTR, confirming the structural integrity and relevance of the isolated CTR ECD. The structure of the CTR ECD is similar to other Class B GPCRs and the ligand binding site is similar to the binding site of the homologous receptors for the calcitonin gene-related peptide (CGRP) and adrenomedulin (AM) recently published (Booe, J. M., Walker, C. S., Barwell, J., Kuteyi, G., Simms, J., Jamaluddin, M. A., Warner, M. L., Bill, R. M., Harris, P. W., Brimble, M. A., Poyner, D. R., Hay, D. L., and Pioszak, A. A. (2015) Mol. Cell 58, 1040–1052). Interestingly the receptor-bound structure of the ligand [BrPhe22]sCT(8–32) differs from the receptor-bound structure of the homologous ligands CGRP and AM. They all adopt an extended conformation followed by a C-terminal β turn, however, [BrPhe22]sCT(8–32) adopts a type II turn (Gly28-Thr31), whereas CGRP and AM adopt type I turns. Our results suggest that a type II turn is the preferred conformation of calcitonin, whereas a type I turn is the preferred conformation of peptides that require RAMPs; CGRP, AM, and amylin. In addition the structure provides a detailed molecular explanation and hypothesis regarding ligand binding properties of CTR and the amylin receptors.
Keywords: crystal structure, G protein-coupled receptor (GPCR), peptide conformation, peptide hormone, peptide interaction, amylin, calcitonin
Introduction
Calcitonin (CT)3 is a peptide hormone that possesses the ability to lower the calcium plasma concentration (1). CT is predominantly produced in C cells of the thyroid gland and it is secreted upon increases in the serum calcium concentration preventing hypercalcemia. Salmon CT (sCT) has been utilized for 40 years as a therapeutic agent for treatment of metabolic bone diseases such as osteoporosis and Paget's disease, due to its high potency; 40–50 times higher compared with human CT. The calcitonin family of peptides also comprises amylin, adrenomedullin (AM), and two calcitonin gene-related peptides (α-CGRP and β-CGRP) (2). These peptides all have an amino-terminal disulfide bond connecting residues 1 and 7 (residues 2 and 7 for amylin), a stretch of amphipathic α-helix, and an amide at the carboxyl-terminal residue.
The CT receptor (CTR) is a Class B G protein-coupled receptor (GPCR) and it is expressed in many different cell types and tissues (3). Class B GPCRs are characterized by having a large ectodomain (ECD) of 100–160 amino acid residues with a conserved tertiary structure stabilized by three conserved disulfide bridges. The ECD binds the carboxyl-terminal region of the peptide ligand and thereby positions the amino-terminal part of the ligand for binding to the transmembrane domain to initiate signaling, a mechanism referred to as the two-domain binding model (reviewed in Ref. 4). The structures of several Class B GPCR ECDs in complex with and without ligands have been determined including the ECDs of corticotropin-releasing factor receptor (5), pituitary adenylate cyclase-activating polypeptide receptor (6), glucose-dependent insulinotropic polypeptide receptor (7), parathyroid hormone-1 receptor (8), glucagon-like peptide-1 receptor (9), calcitonin receptor-like receptor (CLR) (10), and the glucagon receptor (11). The crystal structures of the transmembrane domains of the glucagon receptor (12) and the type 1 corticotropin releasing factor receptor (13) were determined recently but the structure of a full-length Class B receptor remains to be determined.
The pharmacology of the CT family of peptides is complex due to heterodimerization of CTR or CLR with a membrane-bound receptor activity-modifying protein (RAMP). There are three different variants of RAMPs: RAMP-1, RAMP-2, and RAMP-3, which have ∼30% amino acid sequence identity and the ligand specificity of CTR and CLR is determined by the interaction (or not) with the RAMPs. CTR is on its own the receptor for CT, whereas in combination with RAMP-1 or RAMP-3 it changes ligand specificity and becomes the amylin receptor (14, 15). The heterodimer of CLR and RAMP-1 is the receptor for CGRP, whereas the heterodimer of CLR and RAMP-2 or RAMP-3 are the receptors for AM. Previously, crystal structures of the CLR ECD in complex with the RAMP-1 ECD have been determined, both as a heterodimer and as ternary complexes with two different small molecule receptor antagonists (10). More recently, the crystal structures of the peptide ligand-bound CGRP receptor ECD (CLR/RAMP-1ECD) and AM receptor ECD (CLR/RAMP-2ECD) were published (16). These structures provided the first molecular insights into ligand binding and specificity of this subclass of Class B GPCRs.
The molecular details that govern ligand binding and specificity of CTR have not previously been clarified by high resolution structure determination. Likewise the change of ligand specificity of CTR enforced by the interaction with RAMP is not fully understood. In this study we determined the crystal structure of the CTR ECD in complex with the peptide ligand [BrPhe22]sCT8–32 ([BrPhe22] = 4-bromo-l-phenylalanine at position 22). The molecular details of the interaction between sCT and the CTR ECD is revealed by the structure and provides new information regarding the structure of sCT in the receptor-bound state and the molecular determinants of ligand specificity of this Class B receptor subclass, in particular the receptors for calcitonin and amylin.
Experimental Procedures
Peptide Synthesis
Human calcitonin was acquired from Sigma (H-2250). All other peptides were synthesized on a Liberty (CEM) or a Prelude (PTI) synthesizer by standard Fmoc chemistry on Rink-AM or Rink-ChemMatrix resins using Fmoc-amino acids with standard protecting groups, diisopropylcarbodiimide/HOAt for coupling, and 20% piperidine for deprotection. For the peptides with a disulfide bond, Cys was incorporated as Fmoc-Cys(Trt)-OH, and after completion of the synthesis, the disulfide was formed on-resin by treatment with an excess of 1% iodine in N-methylpyrrolidone for 15 min. Cleavage was performed with trifluoroacetic acid/water/triisopropylsilane (95:2.5:2.5) for 1 h followed by precipitation with diethyl ether. The peptides were purified by RP-HPLC on a C18 column in TFA-acetonitrile and characterized by LC-MS and UPLC.
Cloning, Expression, and Refolding of CTR ECD
An oligonucleotide sequence encoding the 120-aa ECD part of the human CTR (Isoform II) was designed, codon optimized for expression in Escherichia coli, and ordered from GeneArt (GmbH Germany). A His6 purification tag was introduced in the amino-terminal of the CTR ECD sequence using PCR with forward and reverse oligonucleotide primers, to introduce NdeI/BamHI restriction enzyme sites. The PCR product was ligated into a TOPOBlunt-End vector (Invitrogen) according to the manufacturer's instructions and the sequence was verified by DNA sequencing (DNA Technology). The NdeI/BamHI fragment comprising His6-CTR ECD was ligated into a pET11a E. coli expression vector (pACSH438)(Novagen) resulting in fusion protein with the following sequence: MGSSHHHHHHSSGLVPRGSAFSNQTYPTIEPKPFLYVVGRKKMMDAQYKCYDRMQQLPAYQGEGPYCNRTWDGWLCWDDTPAGVLSYQFCPDYFPDFDPSEKVTKYCDEKGVWFKHPENNRTWSNYTMCNAFTPEKLKN. Due to the location of the thrombin cleavage site (underlined), two residues, Gly-Ser, were introduced in the amino terminus of the CTR ECD. E. coli BL21(DE3) were transfected with pACSH438 and plated on AMP containing LB Agar plates to obtain the expression strain (eMD712). Expression in a 1-liter shaker flask was performed using LB medium with 100 mg/ml of AMP for 3 h at 37 °C and using 0.5 mm isopropyl 1-thio-β-d-galactopyranoside and expression of a 14-kDa insoluble protein corresponding to His6-CTR ECD was verified using Coomassie-stained SDS-PAGE gels.
Strain emD712 was propagated on a solid agar surface (LB-agar + 100 mg/liter of AMP) at 30 °C overnight. A 10-liter fed-batch fermentation was subsequently performed in a 20-liter stainless steel bioreactor using a complex medium with glycerol feeding according to a predefined feeding profile ensuring glycerol-limited growth. Throughout the fermentation, pH was kept at 6.9 by titration of 10% H3PO4 and 10% NH4OH, and the temperature was controlled at 37 °C. The culture was aerated at a flow of 2 vessel/volume/min of air and a backpressure of 0.3–0.45 bar. After 16 h of elapsed fermentation time, at A600 of around 50, isopropyl 1-thio-β-d-galactopyranoside (0.5 mm) was added to induce expression of His6-CTR ECD. Approximately 6 h after induction, the fermentation was terminated. The final biomass concentration was A600 = 80.5 or 30.65 g/liter cell dry weight. The culture broth was directly homogenized at 1500 bar at a single pass through the APV2000 high-pressure homogenizer. A total of 253 g of inclusion bodies were sedimented by centrifugation of the homogenate for 40 min at 4500 rpm (6056 × g) and 4 °C.
The CTR ECD inclusion bodies were isolated, denatured, and refolded as previously described for the glucagon-like peptide 1 receptor ECD (17). The refolded protein was purified using Ni2+ affinity chromatography using the following procedure: 1) buffer exchange using a Sephadex G25 XK 50/100 column running in binding buffer (20 mm Tris, pH 7.5, 0.1 m Na2SO4, 0.1 m l-Arg, 20 mm imidazole); 2) Ni2+ affinity chromatography using a 5-ml HisTrap column eluted in 20 mm Tris, pH 7.5, 0.1 m Na2SO4, 0.1 m l-Arg, 500 mm imidazole; and 3) buffer exchange using a Sephadex G25 XK 50/100 column running in binding buffer. For binding studies the protein was further purified by size exclusion chromatography (SEC) using a Superdex 75 26/60 column running in 10 mm Tris, pH 7.5, 0.1 m Na2SO4, 2% glycerol. For structural studies the amino-terminal His tag was cleaved off by thrombin and the protein was further purified by ligand affinity chromatography as follows: 1) the ligand-affinity column was prepared by loading a biotinylated analogue of sCT onto a streptavidin column (HiTrap Streptavidin HP) and equilibrated in 10 mm Tris, pH 7.5, 0.1 m Na2SO4, 2% glycerol; 2) 10 mg of CTR ECD was loaded on the ligand-affinity column and equilibrated in 20 mm Tris, pH 7.5, 50 mm NaCl; and 3) the nCTR ECD was eluted in a ligand-bound form by loading of the ligand [BrPhe22]sCT(8–32), at a high concentration (500 μm) resulting in an excess of free ligand in the eluted sample. All columns were from GE Healthcare. The eluted sample was prone to aggregate at temperatures higher than 4 °C and was hard to concentrate without massive precipitation. The addition of freshly prepared urea to a final concentration of 2 m solved the solubility problem and this protein solution was concentrated on an ultrafiltration centrifugation device with 3000-kDa cutoff to 10 mg/ml.
Binding to the Human CTR (Whole Cell Binding)
The binding assay was performed using a BHK cell line stably transfected with the human CTR, and a CREB-binding protein-responsive luciferase reporter gene. The day before the assay the cells were seeded into poly-d-lysine-coated 384W Opaque White, BD BioCoat plates (10000 cells/well) and incubated overnight at 37 °C, 5% CO2, 95% humidity. Then, the cells were washed in Hanks' balanced salt solution (4 °C), and incubated overnight at 4 °C in a binding buffer containing test compounds, 25 pm 125I-sCT (PerkinElmer Life Sciences, catalog number NEX423), a calcitonin, Dulbecco medium without phenol red (500 ml) (Gibco, 11880-028), 0.1% ovalbumin (5 ml of 10% ovalbumin) (Sigma number A5503), 10 mm Hepes (5 ml, 1 m) (Gibco number 15630-056) 1× glutamine (5 ml, ×100) (Gibco number 35050087) 1% penicillin-streptomycin (5 ml, 100%) (Gibco number 15140-122), Complete (1 tablet/50 ml) (Roche number 5056489001) 0.1% Pluronic F68® (Gibco number 24040-032). The morning after, the plates were washed three times in Hanks' balanced salt solution (4 °C), and lysed in Lysis buffer (0.1 m NaOH (VWR number 1.09136.1000), 1% SDS (Cayman Chemical Company number 600216). Then MicroScint40 was added and the plates were shaken at 500 rpm for a short period. The plate was then incubated at room temperature in the dark for 1 h and read on a TopCounter. The IC50 was calculated using (one site binding competition analysis) GraphPad Prism5.
Binding to CTR ECD
Binding to the CTR ECD was assessed in a bead-based proximity assay (AlphaScreen®) with His6-CTR ECD binding to Ni Chelate Acceptor beads and biotinylated sCT binding to Streptavidin Donor Beads (PerkinElmer, catalog number 6760619C). When biotinylated sCT binds to His6-CTR ECD, acceptor and donor beads are brought close together and a signal is created. The binding of compounds to CTR ECD was assessed indirectly by the ability to displace binding between CTR ECD and biotinylated sCT. The CTR ECD, biotinylated sCT, and compounds were diluted in buffer containing 25 mm Hepes, 0.1 m NaCl, 0.1% BSA (Sigma A-7888), pH 7.4. 5 μl of compound, 5 μl of CTR ECD (final concentration 10 nm), and 5 μl of biotinylated sCT (final concentration 10 nm) was added per well in a 384-well plate and the plate incubated for 1 h at room temperature. 5 μl of acceptor beads (final concentration 20 μg/ml) and 5 μl of donor beads (final concentration 20 μg/ml) were added to the wells and the plate was incubated at 4 °C overnight. The plate was read on Envision and results drawn using GraphPad Prism.
Crystallization and Diffraction Data Collection
Crystals of the CTR ECD·[BrPhe22]sCT(8–32) complex were grown at 18 °C using sitting drop vapor diffusion, mixing equal volumes of protein (10 mg/ml of protein complex in 20 mm Tris, pH 7.5, 50 mm sodium chloride, 2 m urea) and precipitant (0.1 m Hepes, pH 7.5, 4.3 m sodium chloride). The crystal was cooled in liquid nitrogen using a cryoprotectant consisting of a 1:4 (v/v) mixture of ethylene glycol and precipitant. Diffraction data were collected at 100 K on a CCD detector from MAR Research using a MicroMaxTM-007 HF x-ray generator. Autoindexing, integration, and scaling were performed using the HKL suite (18).
Structure Determination and Refinement
Molecular replacement with PHASER (19), implemented in the program suite PHENIX (20), was performed using the ECD of the CLR from PDB entry 3N7S, chain A (10), as search model. A solution with three CTR ECD chains in the asymmetric unit was found, the side chains were altered to the CTR ECD amino acid sequence and the structure was further refined and model built using PHENIX and COOT (21), respectively. For each CTR ECD molecule, there was a clear difference electron density for amino acid residues from the [BrPhe22]sCT(8–32) peptide that could be model built (residues 21–32 in two of the chains and residues 24–32 in the third). All graphical illustrations of structures were made using PyMol (Schrödinger). The coordinates and the structure factors have been deposited in the PDB with accession number 5II0.
Modeling of CTR ECD Bound Antagonistic Peptide sCT(8–32)
A hybrid model of the CTR ECD-bound antagonistic peptide sCT(8–32) was made by combining residues 8–23 from a sCT NMR structure (PDB entry 2GLH (31)) and residues 24–32 from [BrPhe22]sCT(8–32) (chain D in this work). The angle between the two peptide parts was chosen so that Val8 from sCT approaches Met49 of CTR.
Calculation of Interaction Areas
The area covered on the GPCR Class B ECDs due to binding of their respective peptide ligands were calculated for the CTR ECD·[BrPhe22]sCT(8–32) (chain A and D), corticotropin-releasing factor receptor 2α ECD·urocortin 1 (PDB entry 3N96, chain A and G), CLR·AM (PDB entry 4RWF, chains A and B), and glucagon-like peptide-1 receptor ECD·glucagon-like peptide-1 (PDB entry 3IOL, chain A and B) complexes. The program ArealMol from the CCP4 suite of programs (22) with a probe radius of 1.4 Å was used to calculate the total surface area of the receptor, AR, the total surface area of the ligand, AL, and the total surface area of the receptor·ligand complex, ARL. The interaction area was calculated as (AR + AL − ARL)/2.
Results
Crystal Structure of the CTR ECD·[BrPhe22]sCT(8–32) Complex
The crystal structure of the CTR ECD in complex with [BrPhe22]sCT(8–32) was determined and refined to 2.1-Å resolution (Fig. 1a, statistics of the model are found in Table 1). The [BrPhe22]sCT(8–32) analogue was originally chosen for phasing of the crystal structure. This was not applicable, probably due to disorder of the 4-bromophenylalanine residue of the analogue, and molecular replacement was instead applied for phasing. The CLR ECD was used as the model for molecular replacement (CLR ECD PDB entry 3N7S, chain A only), and it shares ∼60% amino acid residue sequence identity with the crystallized CTR ECD of this study. The overall structures of the CTR and CLR ECDs are very similar with a root mean square deviation of 1.03 Å for 92 aligned Cα atoms as determined using default parameters in the program COOT (PDB code 3N7S chain A aligned with CTR ECD, chain A). The CTR ECD shares the fold with the other Class B GPCR ECD structures as illustrated in Fig. 2. The structure of the ECD-bound [BrPhe22]sCT(8–32) is most similar to the structure of a CGRP analogue bound to the CLR·RAMP-1 ECD complex, which on the other hand is quite different from the structure of other receptor-bound peptides of Class B GPCRs, such as glucagon-like peptide-1, glucose-dependent insulinotropic polypeptide, and parathyroid hormone. The only secondary structure element of ECD-bound [BrPhe22]sCT(8–32) is a type II β turn, which consists of residues Gly28-Thr31. Specifically, the backbone carbonyl oxygen of Gly28 forms a hydrogen bond across the turn structure with the hydroxyl group of Thr31 and the glycine residue at position 30 is compatible with the type II turn conformation (Fig. 3).
FIGURE 1.

Schematic of CTR ECD in complex with [BrPhe22]sCT(8–32) and peptide ligand sequences. a, CTR ECD in complex with [BrPhe22]sCT(8–32), chains A (gray schematic and surface representation) and D (yellow stick representation) displayed with PHENIX 2Fo − Fc composite omit map (light blue) at σ = 1 level for chain D. b, the three complexes in the asymmetric unit superimposed (chains A and D in yellow, chains B and E in red, and chains C and F in dark blue) illustrating the overall similarity and the amino-terminal differences. c, sequence alignment using the MUSCLE algoritm of the CT peptides used in this work and other members of the calcitonin family of peptides. All peptides contain a carboxyl-terminal amide (marked by ●) and [BrPhe22]sCT(8–32) furthermore, contains a 4-bromo-l-phenylalanine residue, which is marked as Z. In full-length calcitonin the first and the 7th residues are forming a disulfide and in the amylins, CGRPs, and AM, the 2nd and 7th residues are forming a disulfide.
TABLE 1.
Diffraction data and refinement statistics
| Space group | C2 |
|---|---|
| Unit cell | |
| a, b, c (Å) | 96.6, 113.2, 55.4 |
| α, β, γ (°) | 90, 114.8, 90 |
| Resolution (Å) | 24.94–2.097 (2.172–2.097)a |
| Total reflections | 238,084 (23,266) |
| Unique reflections | 31,627 (2,878) |
| Multiplicity | 7.5 (7.4) |
| Completeness (%) | 97 (100) |
| 〈I〉/σ(I) | 15.99 (2.86) |
| Wilson B-factor | 29.59 |
| Rmerge (%) | 8.1 (54.4) |
| Rmeas (%) | 8.7 (58.5) |
| Reflections used in refinement | 30,547 (2878) |
| Reflections used for Rfree | 1,935 (185) |
| Rwork (%) | 16.98 (21.24) |
| Rfree (%) | 20.55 (24.33) |
| Number of non-hydrogen atoms | 3088 |
| Room mean square (bonds) | 0.008 |
| Room mean square (angles) | 1.11 |
| Ramachandran favored (%) | 98 |
| Ramachandran allowed (%) | 1.5 |
| Ramachandran outliers (%) | 0 |
| Average B-factor (Å2) | 39.55 |
| Average B-factor protein atoms (Å2) | 39.26 |
| Average B-factor solvent (Å2) | 42.48 |
a Values within parentheses refer to the highest resolution shell.
FIGURE 2.
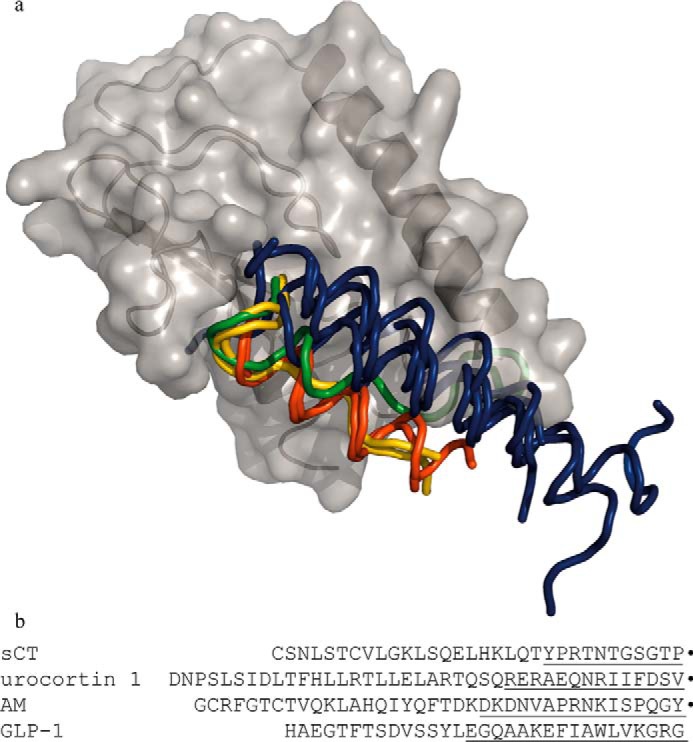
Structural alignment of GPCR Class B ECDs illustrating the variety of peptide ligand fold and binding. a, schematic displaying CTR ECD (gray) and a ribbon representation of the [BrPhe22]sCT(8–32) ligand (yellow). The ECDs of the following structures have been superposed on the CTR ECD and the peptide ligands are displayed as ribbons and peptides binding in a similar mode are colored in the same color. The other Class B GPCR ECDs used for structural alignment are: CLR in complex with AM (green) (PDB 4RWF) and a variant of CGRP (yellow) (PDB 4RWG, RAMP1 and -2 have been left out) (16), glucose-dependent insulinotropic polypeptide receptor in complex with glucose-dependent insulinotropic polypeptide (dark blue) (PDB 2QKH) (7), parathyroid hormone receptor in complex with parathyroid hormone-related protein (dark blue) (PDB 3C4M) (8), parathyroid hormone receptor in complex with parathyroid hormone (dark blue) (PDB 3H3G) (35), glucagon-like peptide-1 receptor in complex with glucagon-like peptide-1 (dark blue) (PDB 3IOL) (36), corticotropin-releasing factor receptor 2α in complex with urocortin 1 (red) (PDB 3N96) (32), and corticotropin releasing factor receptor 1 in complex with corticotropin-releasing factor (red) (PDB 3EHU) (5). The codes within parentheses are the Protein Data Bank accession numbers of the displayed coordinates. b, full-length sequences of the peptide ligands of one representative from each binding mode where the part within 4 Å from their respective ECD are underlined. Carboxyl-terminal amide residues are marked by ●.
FIGURE 3.

Interactions between CTR ECD and [BrPhe22]sCT(8–32). a, schematic of CTR ECD (gray) with the side chains of amino acid residues within 4 Å from [BrPhe22]sCT(8–32) (yellow) in stick representation. Hydrogen bonds are marked with dotted lines. b, closeup on the β turn and the carboxyl-terminal prolineamide of [BrPhe22]sCT(8–32). The hydrogen bonds between the carboxyl-terminal amide and peptide backbone of residue Ser129 from CTR ECD are marked with black dotted lines, the hydrogen bond between the Thr31 hydroxyl group and the carboxyl-terminal amide is marked with a red dotted line and the internal β turn hydrogen bond is marked with a cyan dotted line. Residues are labeled with their respective residue number, the ones belonging to [BrPhe22]sCT(8–32) are in italics.
The asymmetric unit contains three copies of the CTR ECD·[BrPhe22]sCT(8–32) complex with different numbers of disordered amino acid residues at the amino and carboxyl termini. The crystallized CTR ECD corresponds to residues 25–144 in the CTR isoform II sequence with a 2-amino acid residue (Gly-Ser) amino-terminal extension derived from the engineered thrombin cleavage site used as part of the protein production and the following residues were model built: CTR ECD residues 40–138 (chain A), CTR ECD residues 34–136 (chain B), CTR ECD residues 39–138 (chain C), [BrPhe22]sCT(8–32) residues 21–32 (chain D), [BrPhe22]sCT(8–32) residues 24–32 (chain E), and [BrPhe22]sCT(8–32) residues 21–32 (chain F). These complexes (chains A+D, chains B+E, and chains C+F) are all very similar except from the amino-terminal residues of the CTR ECD (residues 34–43), which considerably differ and so do [BrPhe22]sCT(8–32) residues 21–23 (Fig. 1b). Residues 41–43 from the C chain is forming a β strand as part of a crystal packing interaction with the CTR ECD β sheet of chain B. This crystal contact interaction probably explains the differences to chain A, where the same residues are part of a longer amino-terminal α helix and in chain C these residues are disordered. The different conformations of these residues also propagates into the [BrPhe22]sCT(8–32) residues 21–23 conformation. All amino acid residues lies within the allowed regions of the Ramachandran plot and additional to these residues there are 293 water molecules and one urea molecule modeled in the structure. The urea is added at 2 m concentration to the crystallized protein for increased solubility and the well ordered molecule is found bound to the backbone of Val42 in CTR ECD chain C and [BrPhe22]sCT(8–32) chain F, residues Pro23, carbonyl oxygen, and Thr25 side chain.
The interactions between [BrPhe22]sCT(8–32) and CTR ECD are illustrated in Fig. 3. Residues 24–27 from [BrPhe22]sCT(8–32) match the shape of the CTR ECD and hydrogen bonds are formed between Arg24 ([BrPhe22]sCT(8–32)) and Asn135 (CTR ECD), Thr25 ([BrPhe22]sCT(8–32)), and Asp101 (CTR ECD) and also between Thr27 ([BrPhe22]sCT(8–32)) and Trp128 (CTR ECD). The type II β turn formed by residues 28–31 gives a complementary shape to the CTR ECD loop consisting of residues 121–128. Finally, the carboxyl-terminal prolineamide of the peptide ligand is forming a hydrogen bond to the carbonyl oxygen of residue Ser129 of the CTR ECD, whereas the carbonyl oxygen at the carboxyl-terminal end of the ligand is involved in a hydrogen bond with the amide from the same Ser129 in CTR ECD. This complementarity explains the necessity of the amino-terminal amide for ligand binding to the receptor. The carboxyl-terminal prolineamide (Pro32) of [BrPhe22]sCT(8–32) stacks with Trp79 of the CTR ECD. For comparison, the interaction surface of the peptide ligand and receptor ECD has been calculated for the complexes shown in Fig. 2, b–e. The surfaces are of comparable size ranging from 586 Å2 for glucagon-like peptide 1 in complex with its receptor ECD to 938 Å2 for the AM·CLR ECD·RAMP-2 ECD complex with the [BrPhe22]sCT(8–32)·CTR ECD interaction surface being 603 Å2 and 802 Å2 for urocortin 1·corticotropin-releasing factor receptor 2α ECD.
Peptide Binding
To confirm that the binding property of the isolated CTR ECD was representative of the binding property of the full-length CTR, we analyzed a series of peptide ligands in parallel. Binding to the full-length receptor in a cellular environment was measured in a binding competition experiment on intact BHK cells stably transfected with the human CTR using 25 pm 125I-sCT as the tracer ligand. In this assay all analogues were able to fully compete the binding of 125I-sCT, with the following rank order sCT > sCT(8–32) > [BrPhe22]sCT(8–32) ≫ hCT > [Tyr32]sCT(8–32) ≫ pramlintide (Table 2, Fig. 4a). To analyze the ability of the selected analogues to bind the CTR ECD, a bead-based proximity binding assay was established. As seen in Fig. 4b, pramlintide, human CT, and [Tyr32]sCT(8–32) were not able to displace the biotinylated sCT, whereas sCT, sCT(8–32), and [BrPhe22]sCT(8–32) resulted in full displacement. For pramlintide, hCT, and [Tyr32]sCT(8–32) there was an unspecific increase of the binding signal. We do not know what causes this increase of signal, but one explanation may be that unspecific binding of the coated assay beads is inhibited by the high peptide concentrations of pramlintide, hCT, and [Tyr32]sCT(8–32) making more beads available for interaction with each other and thereby increasing the signal. However, the rank order is sCT > sCT(8–32) > [BrPhe22]sCT(8–32) ≫ hCT = [Tyr32]sCT(8–32) > pramlintide and resembles the rank order found with full-length receptor (Table 2, Fig. 4b). These results confirmed that the binding property of the isolated CTR ECD used for the structural studies is representative of the binding property of the full-length CTR.
TABLE 2.
Ligand binding, whole cell analysis, and CTR ECD
pIC50 values, mean ± S.E. from whole cell competition binding assay using 25 pm 125I-sCT as the tracer ligand and increasing amounts of unlabeled calcitonin analogue. Experiments were conducted on intact BHK cells stably transfected with the human calcitonin receptor. Experimental details can be found under “Experimental Procedures.” Representative data are depicted in Fig. 4. Number of experiments = 3 for all measurements.
| Peptide | Ligand binding, whole cell analysis |
Ligand binding, CTR ECD |
||||
|---|---|---|---|---|---|---|
| pIC50 | S.E. | IC50 | pIC50 | S.E. | IC50 | |
| nm | nm | |||||
| Pramlintide | 6.99 | ±0.08 | 100 | >10,000 | ||
| sCT | 9.67 | ±0.06 | 0.21 | 7.4 | ±0.06 | 40 |
| sCT(8–32) | 9.55 | ±0.14 | 0.28 | 7.3 | ±0.11 | 50 |
| [Tyr32]sCT(8–32) | 7.94 | ±0.11 | 12 | >10,000 | ||
| [BrPhe22]sCT(8–32) | 9.38 | ±0.13 | 0.42 | 7.1 | ±0.13 | 80 |
| hCT | 8.28 | ±0.26 | 5.2 | >10,000 | ||
FIGURE 4.

125I-sCT whole cell competition binding and CTR ECD binding. a, normalized competition binding curves from all three experiments using 25 pm 125I-sCT as the tracer ligand and increasing amounts of unlabeled CT analogue. Experiments were conducted on intact BHK cells stably transfected with the human CTR. For each experiment each analogue curve was normalized in GraphPad Prism by defining the fitted bottom value as 0% and top value as 100%. b, binding to the CTR ECD was assessed in a bead-based proximity assay (AlphaScreen®) with His6-CTR ECD binding to nickel chelate acceptor beads and biotinylated sCT binding to the streptavidin donor beads. The binding of compounds to CTR ECD is assessed indirectly by the ability to displace the binding between CTR ECD and biotinylated sCT. The experiments were repeated 3 times and the experimental details can be found under ”Experimental Procedures.“ Data are depicted as mean ± S.E. The summarized data of all experiments are reported in Table 2.
Discussion
The crystal structure presented here shows that sCT in the receptor-bound state forms a type II turn at the carboxyl terminus, which consists of the following residues: Gly28 (i), Ser29 (i+1), Gly30 (i+2), and Thr31 (i+3). Both hCT and sCT have a Gly in position i+2, which is typical of type II turns and therefore hCT probably also assumes a type II turn conformation. The different binding strength of sCT and hCT observed in this and previous studies is at least partially explained by the structure of [BrPhe22]sCT(8–32) bound to the CTR ECD. Several divergent residues in the carboxyl-terminal part of sCT and hCT are sensitive to Ala substitutions and these effects can be rationalized in the context of the new structural information, assuming a similar structure and binding mode of the two ligands (23) (Figs. 3 and 5a). These are Thr27 and Arg24, which are substituted by an Ile and Gln residue, respectively, in hCT (Fig. 1c). The Ile27 residue in hCT does not have any side chain hydrogen binding potential and may even be repulsive and the Arg to Gln replacement at position 24 would possibly lead to a less favorable hydrogen bonding partner. Asn26 is quite solvent exposed and may form an intramolecular hydrogen bond with the backbone amide of Gly28. A hydrophobic residue in this position, like Ala26 of hCT, seems less favorable considering the lack of hydrogen bond potential and full solvent exposure of a hydrophobic residue. In the turn of sCT, the hydroxyl group of Ser29 is facing away from the receptor and a hydrophobic residue in this position, like Val29 of hCT, seems less favorable considering the hydrophilic surroundings of the receptor (His121, Glu123, and Asn124). Finally, Thr31 of sCT may form hydrogen bonds to both Gly28 across the turn and the amide of the carboxyl-terminal prolineamide. The corresponding alanine residue in position 31 of hCT is unable to stabilize this particular conformation by hydrogen bonds. There are more amino acid sequence differences between sCT and hCT that may contribute to the difference in binding capability, but these are not observed in this crystal structure of the CTR ECD.
FIGURE 5.

Hypothetical models of CTR-bound carboxyl-terminal part of hCT and CTR ECD-bound antagonistic peptide sCT(8–32). a, the sCT residues Arg24, Asn26, Thr27, Ser29, and Thr31 of the CTR ECD bound [BrPhe22]sCT(8–32) (chain D in this work) have been replaced by the corresponding hCT residues (Gln24, Ala26, Ile27, Val29, and Ala31) and these side chains are shown in green stick representation together with the CTR ECD·[BrPhe22]sCT(8–32) structure in gray schematic and surface for the ECD and yellow for the stick representation of the peptide. Hydrogen bonds likely to be conserved compared with Fig. 3b are shown as dotted lines. b, hybrid model of CTR ECD-bound antagonistic peptide sCT(8–32) (yellow schematic) based on residues 8–23 from a sCT NMR structure (PDB entry 2GLH) and residues 24–32 from [BrPhe22]sCT(8–32) (chain D in this work). Residues implied in receptor interaction by cross-linking studies (Val8, Leu16, Leu19, and Asn26) are shown with side chains represented in red sticks. CTR ECD (Chain A) is shown as a gray schematic with a transparent surface and residues Met49 and Thr138 are shown as blue sticks marking sites of interactions on the CTR as determined by cross-linking studies.
The solution structure of sCT has been studied by NMR in different solvents: 90% methanol (24), 90% trifluoroethanol (25), and in sodium dodecyl sulfate micelles (26). These structures all show an α-helical segment for residues 8–22, whereas residues 23–32 are in an extended conformation reaching back to the α-helical part. In the structure of the ligand-bound CTR ECD presented here the corresponding α-helical fragment is not visible in the difference electron density map due to flexibility. It is, however, likely that an α-helix is formed by residues 8–22 upon binding of sCT to the full-length CTR. The α-helix formation thereby defines the distances to correctly position the amino-terminal residues, including the disulfide bridge, which is necessary for receptor activation. Flexibility was also observed at the amino-terminal part of the receptor as indicated by different conformations in the three complexes of the asymmetric unit illustrated in Fig. 1b. It is, however, quite interesting to observe these possible conformations and to speculate on the impact of these different possibilities for the full-length receptor although it is not possible to judge the actual conformation(s) in the full-length CTR. Furthermore, the 11 amino-terminal residues of the crystallized CTR ECD construct do not have any difference electron density. A potential functional role of the flexible N-terminal region of the CTR ECD is not easy to predict based on the structures alone.
Several studies using cross-linking by incorporation of photolabile p-benzoyl-l-phenylalanine (BPA) residues in hCT and sCT have been published. This was done for hCT (agonist), at positions 8, 16, and 26 (27, 28), as well as sCT (agonist) at position 19, and sCT(8–32) (antagonist) at position 8 (29, 30). The results show at which residues of the CTR the BPA substituted amino acid residues from these peptides cross-link upon binding. The BPA in position 26 of hCT was shown to cross-link with Thr30 of CTR, which is positioned in the flexible amino-terminal region of the ECD not resolved in the present crystal structure. This particular proximity would require that the flexible N-terminal part of the CTR ECD folds back toward the C-terminal part of sCT in a somewhat surprising orientation. Another interesting cross-link resulted from BPA in position 8 of the antagonist sCT(8–32), which suggested proximity with Met49 on the α-helix of the CTR ECD. We made a hypothetical hybrid model of sCT(8–32) based on the crystal structure of [BrPhe22]sCT(8–32) presented here and the solution NMR structure of sCT previously published (PDB entry 2GLH (31)). This crude hybrid model of sCT(8–32) nicely accounts for the distances and cross-link and represents an inactive conformation given that sCT(8–32) is an antagonist (Fig. 5b). In contrast, BPA in position 8 of the agonist hCT forms a cross-link to the third extracellular loop (ECL3) of CTR indicating a conformational difference between inactive and active states as previously suggested (30). BPA in positions 16 (hCT) and 19 (sCT) cross-link with residues at the carboxyl-terminal part of the CTR ECD (Phe137 and the fragment Cys134-Lys141, respectively), which is not easy to reconcile with the cross-link of the antagonist BPA8-sCT(8–32) and therefore also support a conformational difference between inactive and active states.
The recent structures of the ligand-bound CGRP and AM receptor ECDs together with the structure of the ligand-bound CTR ECD presented here give a more advanced understanding of how the structure and sequence of the carboxyl-terminal part of the ligands are involved in receptor binding and specificity. In the calcitonin peptide family, the carboxyl-terminal amide is critical for receptor binding and the crystal structures revealed similar molecular details of this particular interaction among the different ligands and receptors. The requirement for RAMPs is linked to the nature of the residue in the most carboxyl-terminal position of the ligands. In the ligands that require RAMPs this position is occupied by either Tyr (amylin and AM) or Phe (CGRP), whereas in hCT and sCT, which do not require RAMPs this position is occupied by Pro. Assuming that the amylin receptor ECD is similar to the CGRP and AM receptor ECDs, it seems that the RAMPs do not directly prevent sCT from binding to the extracellullar domain of the amylin receptor. The binding and functional properties of the RAMP-CTR heterodimers support this observation by showing that sCT maintains high affinity and potency in the presence of RAMPs and therefore a primary effect of the RAMPs is to increase affinity and potency for amylin as suggested previously (23, 33, 37). Fig. 6 shows a comparison of the carboxyl-terminal parts of [BrPhe22]sCT(8–32), CGRP analogue (PDB entry 4RWG), and AM (PDB entry 4RWF) and their receptor ECDs superimposed, including the RAMP-1 and RAMP-2 ECDs. The carboxyl-terminal residues of the CGRP and AM analogues, Phe and Tyr, respectively, make perpendicular π stacking interactions with the CLR ECD Trp72 side chain (corresponding to Trp79 in CTR ECD). The stacking interaction with Trp72 is complemented by residues from the respective RAMP-1 and RAMP-2 ECDs and thereby the RAMPs may serve the role of protecting the hydrophobic elements of Phe or Tyr side chains from solvent exposure. The RAMPs of the amylin receptor may accommodate the carboxyl-terminal tyrosine amide residue by a mechanism similar to the RAMPs of the CGRP and AM receptors. The carboxyl-terminal proline amide residue of [BrPhe22]sCT(8–32) forms a stacking interaction with Trp79 of the CTR ECD, which is potentially a strong interaction. The interaction energy of this kind of proline/tryptophan side chain stacking has been elucidated in a computational study and found to be ∼7 kcal mol−1, despite the fact that only one of the components has an aromatic character (34). It is also illustrated by the decreased binding affinity of [Tyr32]sCT(8–32) compared with sCT(8–32) caused by substituting the proline amide residue at position 32 by a tyrosine amide residue. Decrease in binding is seen for full-length CTR binding in whole cell competition binding for [Tyr32]sCT(8–32) (Fig. 4) and for the CTR ECD bead-based assay binding could not be determined. The carboxyl-terminal tyrosine amide residue of amylin appears critical for both the low affinity of amylin for binding to CTR and the high affinity of amylin for binding to the amylin receptor.
FIGURE 6.

Comparison of the carboxyl-terminal parts of [BrPhe22]sCT(8–32) (yellow), the CGRP analogue (brown; PDB entry 4RWF), and AM (green; PDB entry 4RWG) binding to their respective receptor ECDs. a, peptide ligands are displayed as colored schematics with the carboxyl-terminal amide residues in stick representation. Selected side chains from the receptor ECD are displayed in stick representation in the color belonging to the respective peptide and the ECD schematics are displayed in gray with darker gray for the RAMP-1 and RAMP-2 ECDs in PDB 4RWF and 4RWG complexes, respectively. In b–d one complex at the time is shown and here the colors of the peptide sticks follow their respective receptor ECD.
The structures of the ligand-bound CTR, CGRP, and AM receptor ECDs enable a prediction of potential structural rearrangements induced by the RAMPs on the CTR ECD of the amylin receptor (RAMP-1/CTR and RAMP-3/CTR) and how that might affect the ligand binding properties of the CTR and amylin receptor ECD. Structural alignment of the CTR ECD and the CGRP receptor ECD suggests that RAMP-1 has a direct impact on the structure of the CTR ECD, specifically related to Arg126 (Arg119 in the CGRP and AM receptors) in the receptor loop just above the ligand (residues 121–126 of CTR). In the CTR complex, Arg126 adopts a position remote from the ligand, whereas in the CGRP receptor complex, Phe83 of RAMP-1 induced another conformation of Arg119, which thereby moves into close proximity of the ligand (Fig. 6, b and c). In the AM receptor complex, where RAMP-2 is the binding partner, Arg119 assumes a third conformation because RAMP-2 has a glycine in position 110, which due to the lack of side chain does not interfere with Arg119 unlike Phe83 of RAMP-1. Comparing the structures of the CGRP and AM receptor ECDs shows that the conformation of Arg119 has a direct impact on the ligands causing a ∼1.5-Å displacement of CGRP relative to AM in the binding site. In addition to the direct effect on the side chain conformation of Arg119, Phe83 of RAMP-1 also causes a subtle shift of the entire receptor loop (CLR(114–119) and CTR(121–126)). Assuming the overall structure of the amylin receptor ECD is similar to the structures of the CGRP and AM receptor ECD, Phe83 of RAMP-1 or the corresponding Tyr83 of RAMP-3 probably drive Arg126 of the CTR ECD into a conformation similar to that of Arg119 of the CGRP receptor complex, which may assist binding of amylin to its receptor. The type II turn of [BrPhe22]sCT(8–32) bound to the CTR ECD has a Gly in position i+2 (Gly30), which is typical of type II turns. CGRP, AM, and amylin have different residues in the corresponding position of the turn sequence and specifically amylin has an Asn in this position, which is a favored residue of type I turns (see sequence alignment in Fig. 1c). These structural observations suggest that the carboxyl-terminal part of amylin assumes a type I turn conformation upon binding to the amylin receptor similar to the receptor-bound structure of the CGRP analogue.
In summary, the available crystal structures of this Class B GPCR subfamily together with the peptide sequences and the in vitro receptor pharmacology, suggest that those peptides that require RAMPs: amylin, CGRP and AM, form a type I turn upon binding to their cognate receptor, whereas sCT and probably also hCT, which do not require RAMPs, form a type II turn upon binding to the calcitonin receptor. The requirement for RAMPs for high affinity binding of amylin may result from both a direct structural effect of the RAMP on the CTR ECD enabling a favorable interaction with the type I turn conformation of amylin and also a solvent protective effect of the RAMPs on the carboxyl-terminal tyrosine amide of amylin.
Author Contributions
E. J. determined the crystal structure, J. L. H. performed cell binding assays, A. M. K. H. performed CTR ECD binding assays, A. C. S. cloned the CTR ECD, P. B. produced the CTR ECD inclusion bodies, L. S. synthesized peptides, S. R. R. developed the refolding and purification procedure and wrote the manuscript together with E. J.
Acknowledgments
We thank Thomas Seigert Harkes, Carsten Stokkebye Stenvang, and Rasmus Nørgaard Christiansen for excellent technical assistance.
The authors declare that they have no conflicts of interest with the contents of this article.
The atomic coordinates and structure factors (code 5II0) have been deposited in the Protein Data Bank (http://wwpdb.org/).
- CT
- calcitonin
- AM
- adrenomedullin
- CTR
- CT receptor
- GPCR
- G protein-coupled receptor
- ECD
- ectodomain
- RAMP
- receptor activity-modifying protein
- Fmoc
- N-(9-fluorenyl)methoxycarbonyl
- BPA
- p-benzoyl-l-phenylalanine
- PDB
- Protein Data Bank
- CLR
- calcitonin receptor-like receptor.
References
- 1. Copp D. H., Cameron E. C., Cheney B. A., Davidson A. G. F., and Henze K. G. (1962) Evidence for calcitonin: a new hormone from the parathyroid that lowers blood calcium. Endocrinology 70, 638–649 [DOI] [PubMed] [Google Scholar]
- 2. Poyner D. R., Sexton P. M., Marshall I., Smith D. M., Quirion R., Born W., Muff R., Fischer J. A., and Foord S. M. (2002) International Union of Pharmacology: XXXII, the mammalian calcitonin gene-related peptides, adrenomedullin, amylin, and calcitonin receptors. Pharmacol. Rev. 54, 233–246 [DOI] [PubMed] [Google Scholar]
- 3. Findlay D. M., and Sexton P. M. (2004) Calcitonin. Growth Factors 22, 217–224 [DOI] [PubMed] [Google Scholar]
- 4. Hoare S. R. (2005) Mechanisms of peptide and nonpeptide ligand binding to Class B G-protein-coupled receptors. Drug Discov. Today 10, 417–427 [DOI] [PubMed] [Google Scholar]
- 5. Pioszak A. A., Parker N. R., Suino-Powell K., and Xu H. E. (2008) Molecular recognition of corticotropin-releasing factor by its G-protein-coupled receptor CRFR1. J. Biol. Chem. 283, 32900–32912 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Sun C., Song D., Davis-Taber R. A., Barrett L. W., Scott V. E., Richardson P. L., Pereda-Lopez A., Uchic M. E., Solomon L. R., Lake M. R., Walter K. A., Hajduk P. J., and Olejniczak E. T. (2007) Solution structure and mutational analysis of pituitary adenylate cyclase-activating polypeptide binding to the extracellular domain of PAC1-RS. Proc. Natl. Acad. Sci. U.S.A. 104, 7875–7880 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Parthier C., Kleinschmidt M., Neumann P., Rudolph R., Manhart S., Schlenzig D., Fanghänel J., Rahfeld J. U., Demuth H. U., and Stubbs M. T. (2007) Crystal structure of the incretin-bound extracellular domain of a G protein-coupled receptor. Proc. Natl. Acad. Sci. U.S.A. 104, 13942–13947 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Pioszak A. A., and Xu H. E. (2008) Molecular recognition of parathyroid hormone by its G protein-coupled receptor. Proc. Natl. Acad. Sci. U.S.A. 105, 5034–5039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Runge S., Thøgersen H., Madsen K., Lau J., and Rudolph R. (2008) Crystal structure of the ligand-bound glucagon-like peptide-1 receptor extracellular domain. J. Biol. Chem. 283, 11340–11347 [DOI] [PubMed] [Google Scholar]
- 10. ter Haar E., Koth C. M., Abdul-Manan N., Swenson L., Coll J. T., Lippke J. A., Lepre C. A., Garcia-Guzman M., and Moore J. M. (2010) Crystal structure of the ectodomain complex of the CGRP receptor, a class-B GPCR, reveals the site of drug antagonism. Structure 18, 1083–1093 [DOI] [PubMed] [Google Scholar]
- 11. Koth C. M., Murray J. M., Mukund S., Madjidi A., Minn A., Clarke H. J., Wong T., Chiang V., Luis E., Estevez A., Rondon J., Zhang Y., Hötzel I., and Allan B. B. (2012) Molecular basis for negative regulation of the glucagon receptor. Proc. Natl. Acad. Sci. U.S.A. 109, 14393–14398 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Siu F. Y., He M., de Graaf C., Han G. W., Yang D., Zhang Z., Zhou C., Xu Q., Wacker D., Joseph J. S., Liu W., Lau J., Cherezov V., Katritch V., Wang M. W., and Stevens R. C. (2013) Structure of the human glucagon class B G-protein-coupled receptor. Nature 499, 444–449 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Hollenstein K., Kean J., Bortolato A., Cheng R. K., Doré A. S., Jazayeri A., Cooke R. M., Weir M., and Marshall F. H. (2013) Structure of class B GPCR corticotropin-releasing factor receptor 1. Nature 499, 438–443 [DOI] [PubMed] [Google Scholar]
- 14. Christopoulos G., Perry K. J., Morfis M., Tilakaratne N., Gao Y., Fraser N. J., Main M. J., Foord S. M., and Sexton P. M. (1999) Multiple amylin receptors arise from receptor activity-modifying protein interaction with the calcitonin receptor gene product. Mol. Pharmacol. 56, 235–242 [DOI] [PubMed] [Google Scholar]
- 15. Muff R., Bühlmann N., Fischer J. A., and Born W. (1999) Amylin receptor is revealed following co-transfection of a calcitonin receptor with receptor activity modifying proteins-1 or -3. Endocrinology 140, 2924–2927 [DOI] [PubMed] [Google Scholar]
- 16. Booe J. M., Walker C. S., Barwell J., Kuteyi G., Simms J., Jamaluddin M. A., Warner M. L., Bill R. M., Harris P. W., Brimble M. A., Poyner D. R., Hay D. L., and Pioszak A. A. (2015) Structural basis for receptor activity-modifying protein-dependent selective peptide recognition by a G protein-coupled receptor. Mol. Cell 58, 1040–1052 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Runge S., Schimmer S., Oschmann J., Schiødt C. B., Knudsen S. M., Jeppesen C. B., Madsen K., Lau J., Thøgersen H., and Rudolph R. (2007) Differential structural properties of GLP-1 and exendin-4 determine their relative affinity for the GLP-1 receptor N-terminal extracellular domain. Biochemistry 46, 5830–5840 [DOI] [PubMed] [Google Scholar]
- 18. Otwinowski Z., and Minor W. (1997) Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 276, 307–326 [DOI] [PubMed] [Google Scholar]
- 19. McCoy A. J., Grosse-Kunstleve R. W., Adams P. D., Winn M. D., Storoni L. C., and Read R. J. (2007) Phaser crystallographic software. J. Appl. Crystallogr. 40, 658–674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Adams P. D., Afonine P. V., Bunkóczi G., Chen V. B., Davis I. W., Echols N., Headd J. J., Hung L. W., Kapral G. J., Grosse-Kunstleve R. W., McCoy A. J., Moriarty N. W., Oeffner R., Read R. J., Richardson D. C., et al. (2010) PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr. 66, 213–221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Emsley P., Lohkamp B., Scott W. G., and Cowtan K. (2010) Features and development of Coot. Acta Crystallogr. D Biol. Crystallogr. 66, 486–501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Winn M. D., Ballard C. C., Cowtan K. D., Dodson E. J., Emsley P., Evans P. R., Keegan R. M., Krissinel E. B., Leslie A. G., McCoy A., McNicholas S. J., Murshudov G. N., Pannu N. S., Potterton E. A., Powell H. R., et al. (2011) Overview of the CCP 4 suite and current developments. Acta Crystallogr. D Biol. Crystallogr. 67, 235–242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Lee S. M., Hay D. L., and Pioszak A. A. (2016) Calcitonin and amylin receptor peptide interaction mechanisms: insights into peptide-binding modes and allosteric modulation of the calcitonin receptor by receptor activity-modifying proteins. J. Biol. Chem. 291, 8686–8700 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Meadows R. P., Nikonowicz E. P., Jones C. R., Bastian J. W., and Gorenstein D. G. (1991) Two-dimensional NMR and structure determination of salmon calcitonin in methanol. Biochemistry 30, 1247–1254 [DOI] [PubMed] [Google Scholar]
- 25. Meyer J. P., Pelton J. T., Hoflack J., and Saudek V. (1991) Solution structure of salmon calcitonin. Biopolymers 31, 233–241 [DOI] [PubMed] [Google Scholar]
- 26. Motta A., Pastore A., Goud N. A., and Castiglione Morelli M. A. (1991) Solution conformation of salmon calcitonin in sodium dodecyl sulfate micelles as determined by two-dimensional NMR and distance geometry calculations. Biochemistry 30, 10444–10450 [DOI] [PubMed] [Google Scholar]
- 27. Dong M., Pinon D. I., Cox R. F., and Miller L. J. (2004) Molecular approximation between a residue in the amino-terminal region of calcitonin and the third extracellular loop of the class B G protein-coupled calcitonin receptor. J. Biol. Chem. 279, 31177–31182 [DOI] [PubMed] [Google Scholar]
- 28. Dong M., Pinon D. I., Cox R. F., and Miller L. J. (2004) Importance of the amino terminus in secretin family G protein-coupled receptors: intrinsic photoaffinity labeling establishes initial docking constraints for the calcitonin receptor. J. Biol. Chem. 279, 1167–1175 [DOI] [PubMed] [Google Scholar]
- 29. Pham V., Wade J. D., Purdue B. W., and Sexton P. M. (2004) Spatial proximity between a photolabile residue in position 19 of salmon calcitonin and the amino terminus of the human calcitonin receptor. J. Biol. Chem. 279, 6720–6729 [DOI] [PubMed] [Google Scholar]
- 30. Pham V., Dong M., Wade J. D., Miller L. J., Morton C. J., Ng H. L., Parker M. W., and Sexton P. M. (2005) Insights into interactions between the α-helical region of the salmon calcitonin antagonists and the human calcitonin receptor using photoaffinity labeling. J. Biol. Chem. 280, 28610–28622 [DOI] [PubMed] [Google Scholar]
- 31. Andreotti G., Méndez B. L., Amodeo P., Morelli M. A., Nakamuta H., and Motta A. (2006) Structural determinants of salmon calcitonin bioactivity: the role of the Leu-based amphipathic α-helix. J. Biol. Chem. 281, 24193–24203 [DOI] [PubMed] [Google Scholar]
- 32. Pal K., Swaminathan K., Xu H. E., and Pioszak A. A. (2010) Structural basis for hormone recognition by the Human CRFR2α G protein-coupled receptor. J. Biol. Chem. 285, 40351–40361 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Tilakaratne N., Christopoulos G., Zumpe E. T., Foord S. M., and Sexton P. M. (2000) Amylin receptor phenotypes derived from human calcitonin receptor/RAMP coexpression exhibit pharmacological differences dependent on receptor isoform and host cell environment. J. Pharmacol. Exp. Ther. 294, 61–72 [PubMed] [Google Scholar]
- 34. Biedermannova L., E Riley K., Berka K., Hobza P., and Vondrasek J. (2008) Another role of proline: stabilization interactions in proteins and protein complexes concerning proline and tryptophane. Phys. Chem. Chem. Phys. 10, 6350–6359 [DOI] [PubMed] [Google Scholar]
- 35. Pioszak A. A., Parker N. R., Gardella T. J., and Xu H. E. (2009) Structural basis for parathyroid hormone-related protein binding to the parathyroid hormone receptor and design of conformation-selective peptides. J. Biol. Chem. 284, 28382–28391 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Underwood C. R., Garibay P., Knudsen L. B., Hastrup S., Peters G. H., Rudolph R., and Reedtz-Runge S. (2010) Crystal structure of glucagon-like peptide-1 in complex with the extracellular domain of the glucagon-like peptide-1 receptor. J. Biol. Chem. 285, 723–730 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Sexton P. M., Morfis M., Tilakaratne N., Hay D. L., Udawela M., Christopoulos G., and Christopoulos A. (2006) Complexing receptor pharmacology. Ann. N. Y. Acad. Sci. 1070, 90–104 [DOI] [PubMed] [Google Scholar]