

Abstract
UV-sensitive syndrome is an autosomal recessive disorder characterized by hypersensitivity to UV light and deficiency in transcription-coupled nucleotide excision repair (TC-NER), a subpathway of nucleotide excision repair that rapidly removes transcription-blocking DNA damage. UV-sensitive syndrome consists of three genetic complementation groups caused by mutations in the CSA, CSB, and UVSSA genes. UV-stimulated scaffold protein A (UVSSA), the product of UVSSA, which is required for stabilization of Cockayne syndrome group B (CSB) protein and reappearance of the hypophosphorylated form of RNA polymerase II after UV irradiation, forms a complex with ubiquitin-specific peptidase 7 (USP7). In this study, we demonstrated that the deubiquitination activity of USP7 is suppressed by its interaction with UVSSA. The interaction required the tumor necrosis factor receptor-associated factor domain of USP7 and the central region of UVSSA and was disrupted by an amino acid substitution in the tumor necrosis factor receptor-associated factor-binding motif of UVSSA. Cells expressing mutant UVSSA were highly sensitive to UV irradiation and defective in recovery of RNA synthesis after UV irradiation. These results indicate that the interaction between UVSSA and USP7 is important for TC-NER. Furthermore, the mutant UVSSA was rapidly degraded by the proteasome, and CSB was also degraded after UV irradiation as observed in UVSSA-deficient cells. Thus, stabilization of UVSSA by interaction with USP7 is essential for TC-NER.
Keywords: deubiquitylation (deubiquitination), genetic disease, nucleotide excision repair, protein degradation, protein stability, protein-protein interaction
Introduction
Nucleotide excision repair (NER)3 is a versatile DNA repair system that removes a wide range of structurally unrelated bulky, helix-distorting DNA lesions, including UV-induced cyclobutane pyrimidine dimers (CPDs) and pyrimidine-pyrimidone (6-4) photoproducts (6-4PPs) (1). NER occurs via two subpathways, global genome NER (GG-NER) and transcription-coupled NER (TC-NER); the choice of subpathway is determined by the location of the lesion. As its name implies, GG-NER operates throughout the entire genome, whereas TC-NER specifically removes lesions from the transcribed strands of actively transcribed genes (2–4). The primary difference between the two is the mechanism by which the DNA lesion is recognized. In GG-NER, the XPC-RAD23B-CETN2 complex recognizes lesions with the help of the UV-damaged DNA-binding protein complex. By contrast, in TC-NER, RNA polymerase II (Pol II) stalled at a lesion on the transcribed strand serves as the damage recognition signal. Subsequently, TC-NER-specific factors, including CSA, CSB, UV-stimulated scaffold protein A (UVSSA), and XAB2, are recruited to the lesion site (2, 4). Following the specific recognition events of GG-NER and TC-NER, the subpathways merge into a common pathway involving several steps: unwinding of damaged DNA, introduction of dual incisions in the damaged strand, removal of the damage-containing oligonucleotide, repair synthesis in the resulting gap, and ligation of the repair patch to the contiguous parental DNA strand (2, 3).
Several autosomal recessive NER deficiency disorders exist in humans, including xeroderma pigmentosum, Cockayne syndrome (CS), trichothiodystrophy, and UV-sensitive syndrome (UVSS) (1). CS and UVSS patients are specifically deficient in TC-NER. CS is characterized by photosensitivity, growth failure, progressive neurodevelopmental disorder, and premature aging but no predisposition to skin cancer (5). CS is classified into two genetic complementation groups, CS-A and CS-B (6, 7); the causative genes are CSA (8) and CSB (9), respectively. Conversely, UVSS is characterized by photosensitivity and mild freckles but no neurological abnormalities or skin tumors (10–13). Three complementation groups have been identified among UVSS patients defined by mutations in CSA (14), CSB (15), and UVSSA (16–18). Cells from CS and UVSS patients exhibit hypersensitivity to UV light and reduced recovery of RNA synthesis after UV irradiation (6, 11–13, 19, 21).
UVSSA, the product of UVSSA, consists of 709 amino acid residues (Fig. 1A). UVSSA contains a Vps27/Hrs/STAM (VHS) domain and a conserved domain of unknown function (DUF) 2043 in the N-terminal and C-terminal regions, respectively. UVSSA interacts with ubiquitin-specific peptidase 7 (USP7), CSA, and TFIIH (16–18, 22). After UV irradiation, UVSSA stabilizes CSB and restores the hypophosphorylated form of Pol II (Pol IIa) in collaboration with USP7 (16, 17). USP7 has deubiquitination activity, and its catalytic domain is formed by residues 208–564. In addition, USP7 has a tumor necrosis factor receptor-associated factor (TRAF) domain in its N-terminal region and five ubiquitin-like domains in its C-terminal region (Fig. 1A).
FIGURE 1.
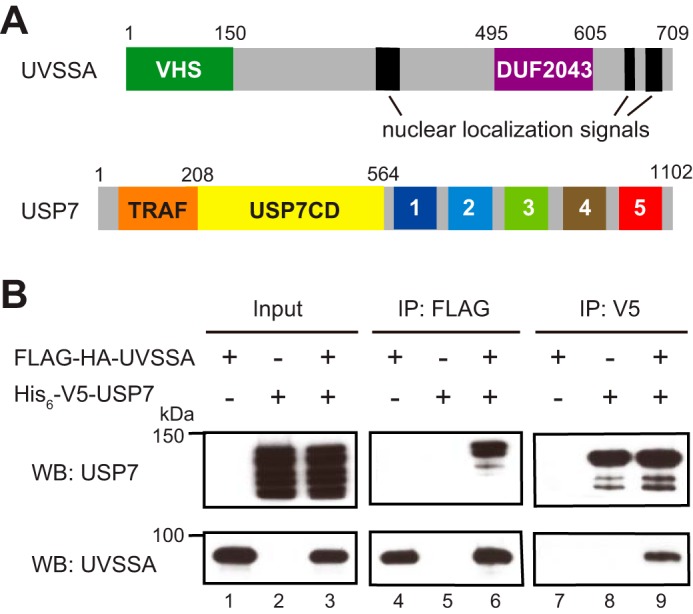
Interaction between UVSSA and USP7. A, schematic representation of human UVSSA and USP7. UVSSA contains a VHS domain (green box), a DUF2043 domain (purple box), and nuclear localization signals (black box). USP7 contains a TRAF domain (orange box), a catalytic domain (yellow box), and five ubiquitin-like domains (numbered boxes 1–5). B, Sf9 cells were infected with recombinant baculovirus for FLAG-HA-UVSSA or His6-V5-USP7 or co-infected with both viruses. Lysates were prepared from the infected cells (lanes 1–3) followed by immunoprecipitation with anti-FLAG beads (lanes 4–6) or anti-V5 beads (lanes 7–9). Western blots (WB) using anti-USP7 and anti-UVSSA antibodies are shown.
In this study, we examined the effect of interaction with UVSSA on the deubiquitination activity of USP7 and identified the regions in both proteins required for binding. Moreover, we revealed that the interaction between UVSSA and USP7 is critical for TC-NER.
Experimental Procedures
Generation of Plasmids and Baculoviruses for Protein Expression
For expression of WT and truncated UVSSA, DNA sequence encoding FLAG and HA tags was attached to the 5′-end of each UVSSA cDNA, and the resultant fragments were inserted between the NotI and XbaI sites of vector pFastBac-1 (Invitrogen). For expression of WT and truncated USP7, DNA sequence encoding the V5 tag was attached to the 5′-end of each USP7 cDNA, and the resultant fragments were inserted between the BamHI and XhoI sites of vector pFastBac HTb, which contains DNA sequence encoding the His6 tag. Recombinant baculoviruses were isolated according to the protocol for the Bac-to-Bac Baculovirus Expression System (Invitrogen). Briefly, the plasmids were transformed into DH10Bac for transposition into the bacmid. Sf9 cells were transfected with the bacmid DNA, and recombinant baculoviruses were obtained and amplified.
To generate mammalian expression constructs for UVSSA mutants, point mutations were introduced into pcDNA3-FLAG-HA-UVSSA (16) using the QuikChange site-directed mutagenesis kit (Agilent). The resultant plasmid was sequenced to rule out non-targeted mutations elsewhere in the cDNA.
Protein Expression in Sf9 Cells
Sf9 cells were cultured at 27 °C in Sf-900 II serum-free medium (Invitrogen) supplemented with 10% FCS and antibiotics. Sf9 cells were infected with the appropriate baculovirus(es) and incubated for 72 h. Infected cells were harvested by centrifugation and stored at −80 °C.
Purification of UVSSA and UVSSA-USP7 Complex
All purification steps were carried out at 4 °C. Cells infected with recombinant baculovirus for FLAG-HA-UVSSA or co-infected with viruses for FLAG-HA-UVSSA and His6-V5-USP7 were lysed in NETN buffer (50 mm Tris-HCl, pH 7.6, 150 mm NaCl, 1 mm EDTA, 1% Nonidet P-40, 1 mm DTT, and Complete protease inhibitor mixture (Roche Applied Science)) at 4 °C for 30 min. The cell lysates were clarified by centrifugation at 17,600 × g for 10 min and incubated with protein G-Sepharose (GE Healthcare) at 4 °C for 1 h. The lysates were centrifuged at 3,800 × g for 1 min, and the resultant supernatant was incubated with anti-FLAG M2 antibody-conjugated agarose (Sigma) at 4 °C for 2 h. The resin was washed five times with NETN buffer, and bound proteins were eluted with NETN buffer containing 0.2 mg/ml FLAG peptide. The eluted samples were further purified using Superose 12 PC 3.2/30 (GE Healthcare) equilibrated with a buffer containing 20 mm Tris-HCl, pH 8.0, 300 mm NaCl, 10% glycerol, 0.1% Tween 20, and 10 mm 2-mercaptoethanol. The concentration of purified UVSSA-USP7 complex was 231 ng/μl.
Purification of USP7
All purification steps were carried out at 4 °C. Cells infected with recombinant baculovirus for His6-V5-USP7 were lysed in TALON buffer (20 mm Tris-HCl, pH 8.0, 300 mm NaCl, 0.5% Triton-X-100, 10% glycerol, and Complete protease inhibitor mixture) at 4 °C for 30 min. The cell lysates were clarified by centrifugation at 17,600 × g for 10 min, and the resultant supernatant was incubated with protein G-Sepharose at 4 °C for 1 h. The samples were centrifuged at 3,800 × g for 1 min, and the resultant supernatant was incubated with nickel-Sepharose (GE Healthcare) at 4 °C for 1 h. The resin was washed five times with TALON buffer, and bound proteins were eluted with TALON buffer containing 200 mm imidazole. The eluted samples were further purified using Superose 12 PC 3.2/30 (GE Healthcare) equilibrated with a buffer containing 20 mm Tris-HCl, pH 8.0, 300 mm NaCl, 10% glycerol, 0.1% Tween 20, and 10 mm 2-mercaptoethanol. The concentration of purified USP7 was 757 ng/μl.
Deubiquitination Assay
Ubiquitin-rhodamine 110 (Life Sensors) was incubated with USP7 or UVSSA-USP7 complex in 100 μl of reaction buffer (200 mm Tris-HCl, pH 7.6, 20 mm MgCl2, 2 mm DTT, and 100 μm BSA) at 25 °C. The increase in fluorescence (excitation, 485 nm; emission, 535 nm) due to release of rhodamine 110 was followed on a multimode reader (Mithras LB 940). Reactions were performed in duplicate.
Expression of UVSSA in Kps3 Cells
The Kps3 cell line was derived from a UVSS-A patient and immortalized with SV40 large T antigen and human telomerase reverse transcriptase (16). Kps3 cells were cultured in DMEM containing 10% FCS and antibiotics at 37 °C under 5% CO2. The cells were transfected with FLAG-HA-UVSSA expression constructs using the Effectene transfection reagent (Qiagen). For transient expression, cells were harvested 24 h after transfection. To isolate stable transfectants, transfected cells were incubated in the presence of G418 (500 μg/ml) for 2 weeks. The G418-resistant clones were assayed for expression of FLAG-HA-UVSSA by Western blotting with anti-UVSSA or anti-HA antibody.
Immunoprecipitation
Cell lysates were prepared using NETN buffer as described above. After preclearance with protein G-Sepharose (GE Healthcare), the lysates were incubated with anti-FLAG M2 antibody-conjugated agarose (Sigma) or anti-V5 antibody-conjugated agarose (Sigma) at 4 °C for 2 h. The resin was washed five times with NETN buffer, and bound proteins were eluted with NETN buffer containing the corresponding peptide.
UV Survival
Exponentially growing cells were inoculated in 100-mm dishes at a density of 500 cells/dish. After 12 h, cells were washed with PBS; irradiated with UV light at 0, 4, 8, or 12 J/m2; and then incubated for 1–2 weeks. The resultant colonies were fixed with 3.7% formaldehyde, stained with 0.1% crystal violet, and counted using a stereomicroscope.
Measurement of UV-induced Photoproducts in Genomic DNA by Slot-blot Analysis
Cells were irradiated with 10 J/m2 of UV light and incubated for various times. Genomic DNA was isolated using the DNeasy Blood and Tissue kit (Qiagen). For the control, DNA was prepared from unirradiated cells. DNA was denatured by heating at 95 °C for 5 min followed by rapid chilling in an ice bath. To quantify the DNA lesions, denatured DNA (100 ng for CPD and 200 ng for 6-4PP) was spotted onto Hybond-N+ (GE Healthcare) with 5× SSC using a slot-blot apparatus (Schleicher & Schüll). The filter was baked at 80 °C for 2 h. Quantification of CPDs and 6-4PPs was carried out using anti-CPD antibody (Kamiya Biomedical; KTM53) and anti-6-4PP antibody (Cosmo Bio; 64M-2). Antibodies bound to CPDs and 6-4PPs were detected using Pierce Western Blotting Substrate Plus (Thermo Scientific) and analyzed by ImageQuant LAS-4000 (GE Healthcare).
Recovery of RNA Synthesis after UV Irradiation
To measure RNA synthesis after UV irradiation, two sets of cells were seeded in 35-mm dishes at a density of 2 × 105 cells/dish. After 12 h of incubation, cells were washed with PBS and irradiated with UV light at 10 J/m2. After 24 h of incubation, the cells in one set were counted. The other set of cells was labeled with 370 kBq/ml [3H]uridine for 30 min to quantitate RNA synthesis. Labeling was terminated by the addition of NaN3 to a final concentration of 200 μg/ml. Cells were washed twice with PBS containing 200 μg/ml NaN3 and lysed in 0.8% SDS for 30 min at room temperature. Next, an equal volume of 10% trichloroacetic acid containing 0.1 m sodium pyrophosphate was added to the lysates and incubated on ice for 1 h. Acid-insoluble materials were collected on GF-C glass microfiber filters (Whatman), and radioactivity was measured with a liquid scintillation counter. Radioactivity was normalized to cell number. The ratio of radioactivity of UV-irradiated cells to that of non-irradiated cells was taken to reflect the recovery of RNA synthesis after UV irradiation.
RNA Interference
CSB silencing was performed by transfection with siRNA (Dharmacon; M-004888) using Lipofectamine RNAiMAX Transfection Reagent (Invitrogen) according to the manufacturer's instructions. Reverse transfection was performed first, and forward transfection was performed 24 h later. Cells were irradiated with UV light 24 h after forward transfection and then incubated for the indicated amount of time.
Antibodies
Anti-UVSSA (N1N2; GTX106751) was from GeneTex. Anti-USP7 (4833) was from Cell Signaling Technology. Anti-CSB (E-18; sc-10459), anti-CSA (W-16; sc-10997), anti-Pol II (A-10; sc-17798), and anti-lamin B (C20; sc-6216) were from Santa Cruz Biotechnology. Anti-HA (3F10; 1867423) was from Roche Applied Science. Anti-V5 (46-0705) was from Invitrogen.
Results
Interaction of UVSSA with USP7 Using Baculovirus Expression System
UVSSA forms a complex with USP7 (16, 17). We analyzed the UVSSA-USP7 complex using a baculovirus expression system. Recombinant baculoviruses for N-terminally FLAG- and HA-tagged UVSSA (FLAG-HA-UVSSA) and N-terminally His6- and V5-tagged USP7 (His6-V5-USP7) were generated. Sf9 cells were infected with one or both viruses, and cell lysates were prepared. Both proteins were detected in the lysates (Fig. 1B, lanes 1–3). Smaller bands detected with anti-USP7 antibody were considered to represent degradation products (lanes 2 and 3). Immunoprecipitation of UVSSA and USP7 from the lysates was performed with anti-FLAG and anti-V5 antibody, respectively. UVSSA was precipitated by anti-FLAG antibody (lanes 4 and 6), and USP7 was co-precipitated with UVSSA from the lysates of co-infected Sf9 cells (lane 6). Similarly, USP7 was precipitated by anti-V5 antibody (lanes 8 and 9), and UVSSA was co-precipitated with USP7 from lysates of co-infected Sf9 cells (lane 9). These observations confirm that the UVSSA-USP7 interaction could be detected in the baculovirus expression system.
Purification of USP7 and UVSSA-USP7 Complex
To examine the effects of UVSSA on the deubiquitination activity of USP7, we purified USP7 and UVSSA-USP7 complex. Sf9 cells were infected with recombinant baculovirus for His6-V5-USP7 or co-infected with viruses for His6-V5-USP7 and FLAG-HA-UVSSA, and cell lysates were prepared from the infected cells. USP7 and UVSSA-USP7 complex were purified from the lysates using nickel-Sepharose and anti-FLAG-agarose, respectively. UVSSA was also purified by the same method used for the UVSSA-USP7 complex. Samples were further purified by gel filtration chromatography. The purified samples were subjected to SDS-PAGE followed by Western blotting with the appropriate antibodies (Fig. 2A). USP7 and UVSSA bands were detected at the predicted molecular sizes. To determine the purity of the samples, USP7 and UVSSA-USP7 complex were subjected to SDS-PAGE followed by silver staining (Fig. 2B). Bands corresponding to USP7 (lane 2) and to USP7 and UVSSA (lane 3) were detected as predicted. Under these conditions, the intensities of the USP7 bands in both samples were almost identical. No other bands were detected. Hence, these purified samples were used for subsequent experiments.
FIGURE 2.

Deubiquitination activity of USP7 is suppressed by the interaction with UVSSA. A, UVSSA, USP7, and UVSSA-USP7 complex were affinity-purified from Sf9 cells infected with the corresponding baculoviruses. Western blots (WB) of the purified samples are shown. B, purified USP7 and UVSSA-USP7 complex were analyzed by silver staining. C, deubiquitination activity was measured using ubiquitin-rhodamine 110 as a substrate. USP7 or UVSSA-USP7 complex (3 nm) was incubated with 1 μm substrate, and deubiquitination was monitored by measuring the change in fluorescence intensity. Vertical bars indicate S.D. D, deubiquitination activity was measured using various concentrations (0.1, 0.3, 0.5, 0.7, 1, or 4 μm) of substrate, and velocity of deubiquitination was plotted in the linear range. Vertical bars indicate S.D. Asterisks indicate a statistically significant difference between USP7 and UVSSA-USP7 values (*, p < 0.05; Student's t test). E, a double reciprocal Lineweaver-Burk plot is shown. Km and Vmax were calculated from the plot. Vertical bars indicate S.D. FI, fluorescence intensity.
Deubiquitination Activity of USP7 Is Suppressed by the Interaction with UVSSA
The deubiquitination activity of UVSSA-USP7 complex was compared with that of USP7 using ubiquitin-rhodamine 110 as a substrate. Cleavage between the C-terminal glycine of ubiquitin and rhodamine by USP7 results in an increase in rhodamine fluorescence (23). In these experiments, USP7 or UVSSA-USP7 complex (3 nm) was incubated with 1 μm substrate, and deubiquitination was monitored by measuring the increase in fluorescence intensity (Fig. 2C). For the first 20 min of the reaction, the fluorescence intensity was lower in the UVSSA-USP7 than in the sample with USP7 alone; at 10 min, the fluorescence intensity of UVSSA-USP7 complex was 48% of that of USP7. Ultimately, the fluorescence intensities reached almost the same level. Next, we ran the same reaction using substrate concentrations of 0.1, 0.3, 0.5, 0.7, 1, and 4 μm. Based on the fluorescence data, we plotted the velocity of deubiquitination in the linear range of the reaction (Fig. 2D) and calculated the kinetic parameters from the Lineweaver-Burk plot (Fig. 2E). Km did not differ significantly between USP7 and the UVSSA-USP7 complex (1.0 μm), but the Vmax of USP7 (11.1 × 102 fluorescence intensity/min) was 1.3-fold higher than that of UVSSA-USP7 (8.3 × 102 fluorescence intensity/min). Therefore, UVSSA does not influence the affinity of USP7 for the substrate but instead decreases the catalytic turnover. These data indicate that the deubiquitination activity of USP7 is inhibited by UVSSA in a non-competitive manner.
UVSSA Interacts with the N-terminal Region of USP7
To identify the UVSSA-binding region in USP7, we prepared recombinant baculoviruses for His6-V5-USP7 and truncated variants. USP7(1–564), USP7(208–1102), and USP7(1–208) contain the TRAF and catalytic domains, the catalytic and ubiquitin-like domains, and the TRAF domain alone, respectively (Fig. 3A). Sf9 cells were co-infected with viruses for full-length or truncated USP7 and FLAG-HA-UVSSA, and immunoprecipitation of UVSSA was performed as described above. USP7(1–564) and USP7(1–208) as well as full-length USP7 (USP7(1–1102)) co-precipitated with UVSSA, whereas USP7(208–1102) did not (Fig. 3B). These results indicate that UVSSA interacts with the N-terminal region of USP7 containing the TRAF domain.
FIGURE 3.

Regions responsible for the interaction between UVSSA and USP7. A, schematic representation of truncated USP7. Boxes 1–5, five ubiquitin-like domains. B, His6-V5-tagged full-length (1–1102) or each truncated (1–564, 208–1102, or 1–208) USP7 was expressed with FLAG-HA-UVSSA in Sf9 cells, and cell lysates were prepared. Immunoprecipitation (IP) was performed with anti-FLAG beads followed by Western blotting (WB) with anti-V5 and anti-UVSSA antibodies. Red dots indicate the corresponding bands. C, schematic representation of truncated UVSSA. D, FLAG-HA-tagged full-length (1–709) or truncated (151–709, 1–495, 151–495, or 496–709) UVSSA was expressed with His6-V5-USP7 in Sf9 cells, and cell lysates were prepared. Immunoprecipitation was performed with anti-V5 beads followed by Western blotting with anti-HA and anti-USP7 antibodies. Red dots indicate the corresponding bands.
USP7 Interacts with the Central Region of UVSSA
To identify the USP7-binding region in UVSSA, we performed reciprocal immunoprecipitation experiments. For this purpose, we generated recombinant baculoviruses for FLAG-HA-UVSSA and truncated variants. UVSSA(151–709) and UVSSA(1–495) lack the N-terminal VHS domain and the C-terminal region containing the DUF2043, respectively (Fig. 3C); UVSSA(151–495) and UVSSA(496–709) contain the central region and the C-terminal region containing the DUF2043 domain, respectively. UVSSA(151–709), UVSSA(1–495), and UVSSA(151–495) as well as full-length UVSSA (UVSSA(1–709)) were co-precipitated with USP7, whereas UVSSA(496–709) was not (Fig. 3D). These results indicate that USP7 interacts with the central region (residues 151–495) of UVSSA.
The Interaction between UVSSA and USP7 Is Disrupted by S254A Mutation in UVSSA
The experiments described above demonstrate that the N-terminal region (residues 1–208) containing the TRAF domain of USP7 interacts with the central region (residues 151–495) of UVSSA. USP7 interacts with p53 and MDM2 via its TRAF domain (24, 25), which recognizes a Pro/Ala-X-X-Ser motif present in its interaction partners (26–28). There are eight TRAF-binding motifs in the central region of UVSSA (Fig. 4A). Alignment of UVSSA homologs using ClustalW revealed that Pro251-Cys-Cys-Ser254 in human is highly conserved in animals. In immunoprecipitation experiments using mutant UVSSA transfectants, UVSSA(1–300) interacted with USP7, but UVSSA(1–160) did not (Fig. 4B). Residues 161–300 of UVSSA contain four TRAF-binding motifs (226–229, 237–240, 251–254, and 270–273). Therefore, we replaced Ser254 with Ala and examined the effect of this mutation on the interaction with USP7. For this purpose, we generated expression constructs for FLAG-HA-tagged UVSSA(WT), UVSSA(P251A), and UVSSA(S254A). UVSSA-deficient Kps3 cells were transfected with the constructs, and cell lysates were prepared from the transfected cells. All exogenous UVSSA proteins were expressed at comparable levels (Fig. 4C, lanes 2–4). The lysates were subjected to immunoprecipitation with anti-FLAG antibody. USP7 co-precipitated with UVSSA(P251A) (lane 7) as well as UVSSA(WT) (lane 6) but not with UVSSA(S254A) (lane 8). By contrast, CSA co-precipitated with UVSSA(WT), UVSSA(P251A), and UVSSA(S254A). These results indicate that the S254A mutation in UVSSA disrupts the interaction with USP7 but not with CSA.
FIGURE 4.

The interaction between UVSSA and USP7 is weakened by the S254A mutation in UVSSA. A, amino acid sequences of UVSSA homologs were analyzed using ClustalW. Top panel, location of the TRAF-binding motifs in the central region (residues 151–495) of UVSSA is indicated by red bars (I–VI; VI contains three successive motifs). Bottom panel, the TRAF-binding motif is shown in red. Homo sapiens, NP_065945.2; Mus musculus, NP_001074570.1; Equus caballus, XP_014695740.1; Bos taurus, XP_587703.7; Xenopus tropicalis, NP_001107306.1; Gallus gallus, XP_420845.4; Caenorhabditis elegans, NP_505012.1. B, UVSSA-deficient Kps3 cells were transfected with pCAGGS-FLAG-HA-tagged UVSSA(WT) or UVSSA deletion mutants. Cell lysates were prepared from the same number of cells. Immunoprecipitation (IP) was performed with anti-FLAG beads followed by Western blotting (WB) with anti-HA and anti-USP7 antibodies. C, UVSSA-deficient Kps3 cells were transfected with pcDNA3.1-FLAG-HA-tagged UVSSA(WT), UVSSA(P251A), or UVSSA(S254A). Immunoprecipitation was performed as described in B followed by Western blotting with anti-USP7, anti-UVSSA, and anti-CSA antibodies.
The S254A Mutation Causes Degradation of UVSSA, Degradation of CSB after UV Irradiation, and Deficiency in TC-NER
To investigate the functional importance of the interaction between UVSSA and USP7, we established Kps3 cell lines stably expressing either FLAG-HA-UVSSA(WT) (WT cells) or FLAG-HA-UVSSA(S254A) (S254A cells). In all isolated clones, the level of UVSSA(S254A) was considerably lower than that of UVSSA(WT) (Fig. 5A, lanes 3 and 5). Because UVSSA is degraded by the proteasome in cells transfected with siRNA against USP7 (16), we treated the cells with the proteasome inhibitor MG132. The level of UVSSA(WT) increased following MG132 treatment (lane 4), confirming that UVSSA is degraded by the proteasome at steady state. Moreover, the amount of UVSSA(S254A) increased substantially following MG132 treatment (lane 6). In MG132-treated cells, the level of UVSSA(S254A) was much higher than that of UVSSA(WT). This indicates that UVSSA(S254A) degradation must be higher in untreated cells despite the high level of UVSSA(S254A) in these cells. These results indicate that UVSSA is protected from proteasomal degradation by interaction with USP7.
FIGURE 5.

Stability of UVSSA is essential for TC-NER. A, Kps3 parental cells and Kps3 cells stably expressing FLAG-HA-UVSSA(WT) (WT) or FLAG-HA-UVSSA(S254A) (S254A) were either treated with 20 μm MG132 for 8 h or not treated. Cell lysates were analyzed by Western blotting (WB) using anti-USP7 and anti-UVSSA antibodies. Samples from the same number of cells were loaded in each lane of the gel. Lamin B was used as a loading control. B, Kps3, WT, and S254A cells were either irradiated with 10 J/m2 of UV light or not irradiated and then incubated for 12 h. Cell lysates were analyzed by Western blotting using the indicated antibodies. Samples from the same number of cells were loaded in each lane of the gel. Lamin B was used as a loading control. The arrow and asterisk denote CSB and CPFP protein (a fusion protein consisting of N-terminal CSB(1–465) and the piggyback transposon (20)), respectively. The intensities of CSB bands were quantitated using the ImageQuant TL software (GE Healthcare). Band intensities in UV-irradiated cells were compared with those in non-irradiated cells; relative intensities, expressed as percentages, are shown below the CSB blot. IIo and IIa denote hyperphosphorylated and hypophosphorylated forms of Pol II, respectively. C, colony-forming ability of Kps3 (circles), WT (diamonds), and S254A cells (squares) after UV irradiation. Points represent the average of three independent experiments, and vertical bars indicate S.D. D, removal of 6-4PPs and CPDs from the UV-irradiated cells was measured. Kps3 (circles), WT (diamonds), and S254A cells (squares) were irradiated with 10 J/m2 of UV light and incubated for the indicated times. Genomic DNAs were purified and used for the measurement of 6-4PP (top panel) and CPD (bottom panel) content by slot-blot analysis. Points represent the average of three independent experiments, and vertical bars indicate S.D. E, top panel, Kps3, WT, and S254A cells were transfected with siRNA against CSB (siCSB). Cont, control. Cell lysates were analyzed by Western blotting using anti-CSB and anti-UVSSA antibodies. Samples from the same number of cells were loaded in each lane of the gel. Lamin B was used as a loading control. 6-4PPs (bottom left panel) and CPDs (bottom right panel) in CSB knockdown Kps3 (circles), WT (diamonds), and S254A cells (squares) were measured as described in D. F, Kps3, WT, and S254A cells were irradiated with 10 J/m2 of UV light, and RNA synthesis was measured 24 h after UV irradiation. The relative incorporation of [3H]uridine in UV-irradiated cells was compared with that in non-irradiated cells. Points represent the average of three independent experiments, and vertical bars indicate S.D.
In UVSSA-deficient cells, CSB is degraded after UV irradiation (16–18, 22). Therefore, we investigated whether CSB is degraded in S254A cells. Following UV irradiation, the level of CSB decreased dramatically in S254A cells and Kps3 cells but barely decreased in WT cells (Fig. 5B). The level of USB7 did not change significantly after irradiation. In WT cells, the level of the hypophosphorylated form of Pol II (Pol IIa) decreased immediately after UV irradiation but returned to the pre-UV irradiation level after several hours of incubation. The return to the pre-UV irradiation level does not occur in UVSSA-deficient cells (16–18) or in S254A cells or Kps3 cells (Fig. 5B, lanes 2 and 6).
To investigate the effects of loss of the interaction between UVSSA and USP7, we measured the viability of S254A cells after UV irradiation by colony-forming assay. Like the parental Kps3 cells, S254A cells were more sensitive to UV light than WT cells (Fig. 5C). Next, we investigated the efficiency of lesion removal by determining the levels of UV-induced 6-4PPs and CPDs in WT cells, S254A cells, and Kps3 cells (Fig. 5D) as well as in the respective CSB knockdown transfectants (Fig. 5E). No significant difference in the removal of 6-4PPs and CPDs was detected between the cells. Because GG-NER is considered to contribute to lesion removal in this assay, these results indicate that S254A cells are proficient for GG-NER. Then we measured recovery of RNA synthesis after UV irradiation, which is an index of TC-NER activity. In WT cells 24 h after UV irradiation, RNA synthesis recovered almost completely, i.e. to ∼100% of the level in non-irradiated cells (Fig. 5F). By contrast, S254A and Kps3 cells did not exhibit a recovery of RNA synthesis at this time point (∼8 and ∼6% of the level in non-irradiated cells, respectively). Thus, S254A cells are deficient in TC-NER.
Discussion
To investigate the interaction between UVSSA and USP7, we used a baculovirus system to express UVSSA and USP7 and showed that the UVSSA-USP7 complex was detectable in the infected cells. In preliminary experiments using an Escherichia coli expression system, almost all UVSSA was insoluble; consequently, we were unable to purify the protein as a soluble fraction. However, the UVSSA-USP7 complex and both constituent proteins could be purified using the baculovirus system, and purified USP7 and UVSSA-USP7 had deubiquitination activity.
As shown in Fig. 2, the deubiquitination activity of USP7 was suppressed by interaction with UVSSA. Kinetic analysis revealed that this inhibition was non-competitive. These results agree with the observation that the UVSSA-binding region of USP7 is the TRAF domain rather than the catalytic domain (Fig. 3). The TRAF domain functions as a substrate-binding domain (24, 25), and we used ubiquitin-rhodamine 110 as a substrate in these experiments; however, the mode of suppression may change when physiological substrates are used. In TC-NER, it remains unclear which ubiquitinated proteins are physiological substrates for USP7. Because Pol II and CSB are ubiquitinated (29, 30), these proteins are candidate substrates. Regulation of ubiquitination by USP7 may play important roles in TC-NER, and UVSSA may influence the activity of USP7 in this pathway.
We then identified the binding regions of UVSSA and USP7. The TRAF domain of USP7 bound to the central region (residues 151–495) of UVSSA (Fig. 3), and a TRAF-binding motif of UVSSA (residues 251–254) was required for the interaction. Given that ubiquitinated p53 and MDM2 are substrates for USP7 (31–33), it is possible that UVSSA is also a USB7 substrate. Consistent with this, UVSSA is monoubiquitinated (17), and we showed here that, in the absence of the USP7 interaction, UVSSA was degraded by the ubiquitin-proteasome system. Although it remains unclear how USP7 regulates the stability of UVSSA, it is likely that USP7 protects UVSSA from degradation by directly cleaving the polyubiquitin chain of UVSSA. Alternatively, USP7 may negatively regulate the ubiquitin ligase that ubiquitinates UVSSA that remains to be identified.
In a previous study, we showed that in cells transfected with siRNA targeting USP7 (USP7 siRNA cells) the levels of both USP7 and UVSSA were reduced (16). The amount of UVSSA mRNA was not altered in USP7 siRNA cells, and UVSSA was restored to wild-type levels by treatment with MG132. Conversely, knockdown of UVSSA did not decrease the level of USP7. These results indicated that USP7 protects UVSSA from degradation by the proteasome. In this study, we showed that an amino acid substitution (S254A) in the USP7-binding region of UVSSA abolished the interaction with USP7. In UVSSA(S254A)-expressing cells, the mutant UVSSA was rapidly degraded, and the level of CSB was also greatly reduced after UV irradiation. Furthermore, these cells were hypersensitive to UV light and deficient in recovery of RNA synthesis after UV irradiation. These phenotypes resemble those of UVSSA-deficient cells (Fig. 6). Thus, these findings demonstrate that the interaction between UVSSA and USP7 is important for the stability of UVSSA and thus for efficient TC-NER.
FIGURE 6.
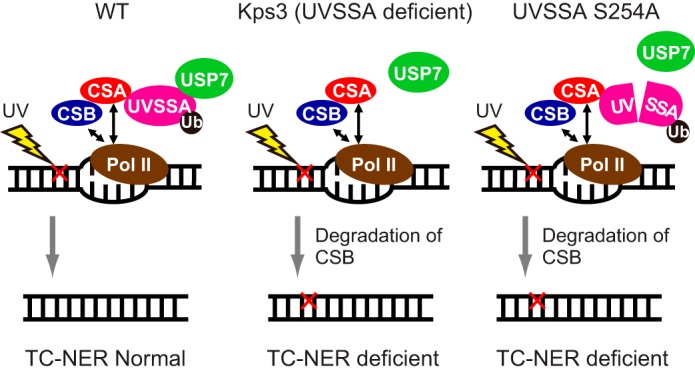
Schematic of the UVSSA function in TC-NER. The red cross indicates DNA damage by UV irradiation.
Our studies using truncated proteins revealed that the central region of UVSSA is required for the interaction with USP7. The N-terminal VHS domain of UVSSA is essential for its interaction with TFIIH and CSB following UV irradiation (18) and is also required for the interaction with CSA irrespective of UV irradiation. Moreover, the region of UVSSA containing residues 400–500 was required for the interaction with TFIIH (22). Interestingly, the interaction of UVSSA with CSA(W361C), which was derived from a UVSS patient (14), is greatly reduced, and UVSSA does not accumulate in the chromatin fraction after UV irradiation in CSA(W361C)-expressing cells (22). Taken together, our findings demonstrate that UVSSA plays a key role in connecting USP7 with TC-NER factors such as CSA, CSB, and TFIIH.
Author Contributions
M. H., X. Z., K. T., and M. S. designed the study. M. H., K. T., and M. S. wrote the manuscript. M. H, X. Z., and M. S. performed the experiments. All authors analyzed the results and approved the final version of the manuscript.
Acknowledgments
We thank all members of the Tanaka laboratory for discussion and encouragement.
This work was supported by Grant-in-aid for Scientific Research on Innovative Areas 22131009 (to K. T.) and Grant-in-aid for Scientific Research (B) 15H02820 (to M. S.) from the Japan Society for the Promotion of Science. The authors declare that they have no conflicts of interest related to the contents of this article.
- NER
- nucleotide excision repair
- TC-NER
- transcription-coupled NER
- CPD
- cyclobutane pyrimidine dimer
- 6-4PP
- pyrimidine-pyrimidone (6-4) photoproduct (6-(1,2)-dihydro-2-oxo-4-pyrimidinyl-5-methyl-2,4-(1H,3H)-pyrimidinedione)
- GG-NER
- global genome NER
- UVSS
- UV-sensitive syndrome
- CS
- Cockayne syndrome
- CSA
- Cockayne syndrome group A
- CSB
- Cockayne syndrome group B
- UVSSA
- UV-stimulated scaffold protein A
- USP7
- ubiquitin-specific peptidase 7
- Pol II
- RNA polymerase II
- VHS
- Vps27/Hrs/STAM
- DUF
- domain of unknown function
- TRAF
- tumor necrosis factor receptor-associated factor
- TFIIH
- transcription factor IIH
- XPC
- xeroderma pigmentosum group C protein.
References
- 1. Friedberg E. C., Walker G. C., Siede W., Wood R. D., Schultz R. A., and Ellenberger T. (2005) DNA Repair and Mutagenesis, 2nd Ed., pp. 267–350, ASM Press, Washington. D. C. [Google Scholar]
- 2. Hanawalt P. C., and Spivak G. (2008) Transcription-coupled DNA repair: two decades of progress and surprises. Nat. Rev. Mol. Cell Biol. 9, 958–970 [DOI] [PubMed] [Google Scholar]
- 3. Fousteri M., and Mullenders L. H. (2008) Transcription-coupled nucleotide excision repair in mammalian cells: molecular mechanisms and biological effects. Cell Res. 18, 73–84 [DOI] [PubMed] [Google Scholar]
- 4. Marteijn J. A., Lans H., Vermeulen W., and Hoeijmakers J. H. (2014) Understanding nucleotide excision repair and its roles in cancer and ageing. Nat. Rev. Mol. Cell Biol. 15, 465–481 [DOI] [PubMed] [Google Scholar]
- 5. Nance M. A., and Berry S. A. (1992) Cockayne syndrome: review of 140 cases. Am. J. Med. Genet. 42, 68–84 [DOI] [PubMed] [Google Scholar]
- 6. Lehmann A. R., Francis A. J, and Giannelli F. (1985) Prenatal diagnosis of Cockayne's syndrome. Lancet 1, 486–488 [DOI] [PubMed] [Google Scholar]
- 7. Tanaka K., Kawai K., Kumahara Y., Ikenaga M., and Okada Y. (1981) Genetic complementation groups in Cockayne syndrome. Somatic Cell Genet. 7, 445–455 [DOI] [PubMed] [Google Scholar]
- 8. Henning K. A., Li L., Iyer N., McDaniel L. D., Reagan M. S., Legerski R., Schultz R. A., Stefanini M., Lehmann A. R., Mayne L. V., and Friedberg E. C. (1995) The Cockayne syndrome group A gene encodes a WD repeat protein that interacts with CSB protein and a subunit of RNA polymerase II TFIIH. Cell 82, 555–564 [DOI] [PubMed] [Google Scholar]
- 9. Troelstra C., van Gool A., de Wit J., Vermeulen W., Bootsma D., and Hoeijmakers J. H. (1992) ERCC6, a member of a subfamily of putative helicases, is involved in Cockayne's syndrome and preferential repair of active genes. Cell 71, 939–953 [DOI] [PubMed] [Google Scholar]
- 10. Spivak G. (2005) UV-sensitive syndrome. Mutat. Res. 577, 162–169 [DOI] [PubMed] [Google Scholar]
- 11. Itoh T., Ono T., and Yamaizumi M. (1994) A new UV-sensitive syndrome not belonging to any complementation group of xeroderma pigmentosum or Cockayne syndrome. Mutat. Res. 314, 233–248 [DOI] [PubMed] [Google Scholar]
- 12. Fujiwara Y., Ichihashi M., Kano Y., Goto K., and Shimizu K. (1981) A new human photosensitive subject with a defect in the recovery of DNA synthesis after ultraviolet-light irradiation. J. Invest. Dermatol. 77, 256–263 [DOI] [PubMed] [Google Scholar]
- 13. Itoh T., Fujiwara Y., Ono T., and Yamaizumi M. (1995) UVs syndrome, a new general category of photosensitive disorder with defective DNA repair, is distinct from xeroderma pigmentosum variant and rodent complementation group I. Am. J. Hum. Genet. 56, 1267–1276 [PMC free article] [PubMed] [Google Scholar]
- 14. Nardo T., Oneda R., Spivak G., Vaz B., Mortier L., Thomas P., Orioli D., Laugel V., Stary A., Hanawalt P. C., Sarasin A., and Stefanini M. (2009) A UV-sensitive syndrome patient with a specific CSA mutation reveals separable roles for CSA in response to UV and oxidative DNA damage. Proc. Natl. Acad. Sci. U.S.A. 106, 6209–6214 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Horibata K., Iwamoto Y., Kuraoka I., Jaspers N. G., Kurimasa A., Oshimura M., Ichihashi M., and Tanaka K. (2004) Complete absence of Cockayne syndrome group B gene product gives rise to UV-sensitive syndrome but not Cockayne syndrome. Proc. Natl. Acad. Sci. U.S.A. 101, 15410–15415 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Zhang X., Horibata K., Saijo M., Ishigami C., Ukai A., Kanno S., Tahara H., Neilan E. G., Honma M., Nohmi T., Yasui A., and Tanaka K. (2012) Mutations in UVSSA cause UV-sensitive syndrome and destabilize ERCC6 in transcription-coupled DNA repair. Nat. Genet. 44, 593–597 [DOI] [PubMed] [Google Scholar]
- 17. Schwertman P., Lagarou A., Dekkers D. H., Raams A., van der Hoek A. C., Laffeber C., Hoeijmakers J. H., Demmers J. A., Fousteri M., Vermeulen W., and Marteijn J. A. (2012) UV-sensitive syndrome protein UVSSA recruits USP7 to regulate transcription-coupled repair. Nat. Genet. 44, 598–602 [DOI] [PubMed] [Google Scholar]
- 18. Nakazawa Y., Sasaki K., Mitsutake N., Matsuse M., Shimada M., Nardo T., Takahashi Y., Ohyama K., Ito K., Mishima H., Nomura M., Kinoshita A., Ono S., Takenaka K., Masuyama R., Kudo T., Slor H., Utani A., Tateishi S., Yamashita S., Stefanini M., Lehmann A. R., Yoshiura K., and Ogi T. (2012) Mutations in UVSSA cause UV-sensitive syndrome and impair RNA polymerase IIo processing in transcription-coupled nucleotide-excision repair. Nat. Genet. 44, 586–592 [DOI] [PubMed] [Google Scholar]
- 19. Mayne L. V., and Lehmann A. R. (1982) Failure of RNA synthesis to recover after UV irradiation: an early defect in cells from individuals with Cockayne's syndrome and xeroderma pigmentosum. Cancer Res. 42, 1473–1478 [PubMed] [Google Scholar]
- 20. Horibata K., Saijo M., Bay M. N., Lan L., Kuraoka I., Brooks P. J., Honma M., Nohmi T., Yasui A., and Tanaka K. (2011) Mutant Cockayne syndrome group B protein inhibits repair of DNA topoisomerase I-DNA covalent complex. Genes Cells 16, 101–114 [DOI] [PubMed] [Google Scholar]
- 21. Venema J., Mullenders L. H., Natarajan A. T., van Zeeland A. A., and Mayne L. V. (1990) The genetic defect in Cockayne syndrome is associated with a defect in repair of UV-induced DNA damage in transcriptionally active DNA. Proc. Natl. Acad. Sci. U.S.A. 87, 4707–4711 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Fei J., and Chen J. (2012) KIAA1530 protein is recruited by Cockayne syndrome complementation group protein A (CSA) to participate in transcription-coupled repair (TCR). J. Biol. Chem. 287, 35118–35126 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Hassiepen U., Eidhoff U., Meder G., Bulber J. F., Hein A., Bodendorf U., Lorthiois E., and Martoglio B. (2007) A sensitive fluorescence intensity assay for deubiquitinating proteases using ubiquitin-rhodamine110-glycine as substrate. Anal. Biochem. 371, 201–207 [DOI] [PubMed] [Google Scholar]
- 24. Hu M., Li P., Li M., Li W., Yao T., Wu J. W., Gu W., Cohen R. E., and Shi Y. (2002) Crystal structure of a UBP-family deubiquitinating enzyme in isolation and in complex with ubiquitin aldehyde. Cell 111, 1041–1054 [DOI] [PubMed] [Google Scholar]
- 25. Holowaty M. N., Sheng Y., Nguyen T., Arrowsmith C., and Frappier L. (2003) Protein interaction domains of the ubiquitin-specific protease, USP7/HAUSP. J. Biol. Chem. 278, 47753–47761 [DOI] [PubMed] [Google Scholar]
- 26. Hu M., Gu L., Li M., Jeffrey P. D., Gu W., and Shi Y. (2006) Structural basis of competitive recognition of p53 and MDM2 by HAUSP/USP7: implications for the regulation of the p53-MDM2 pathway. PLos Biol. 4, e27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Sheng Y., Saridakis V., Sarkari F., Duan S., Wu T., Arrowsmith C. H., and Frappier L. (2006) Molecular recognition of p53 and MDM2 by USP7/HAUSP. Nat. Struct. Mol. Biol. 13, 285–291 [DOI] [PubMed] [Google Scholar]
- 28. Sarkari F., La Delfa A., Arrowsmith C. H., Frappier L., Sheng Y., and Saridakis V. (2010) Further insight into substrate recognition by USP7: Structural and biochemical analysis of the HdmX and Hdm2 interactions with USP7. J. Mol. Biol. 402, 825–837 [DOI] [PubMed] [Google Scholar]
- 29. Bregman D. B., Halaban R., van Gool A. J., Henning K. A., Friedberg E. C., and Warren S. L. (1996) UV-induced ubiquitination of RNA polymerase II: a novel modification deficient in Cockayne syndrome cells. Proc. Natl. Acad. Sci. U.S.A. 93, 11586–11590 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Groisman R., Kuraoka I., Chevallier O., Gaye N., Magnaldo T., Tanaka K., Kisselev A. F., Harel-Bellan A., and Nakatani Y. (2006) CSA-dependent degradation of CSB by the ubiquitin-proteasome pathway establishes a link between complementation factors of the Cockayne syndrome. Genes Dev. 20, 1429–1434 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Li M., Chen D., Shiloh A., Luo J., Nikolaev A. Y., Qin J., and Gu W. (2002) Deubiquitination of p53 by HAUSP is an important pathway for p53 stabilization. Nature 416, 648–653 [DOI] [PubMed] [Google Scholar]
- 32. Cummins J. M., Rago C., Kohli M., Kinzler K. W., Lengauer C., and Vogelstein B. (2004) Tumor suppression: disruption of HAUSP gene stabilizes p53. Nature 428, 486–487 [DOI] [PubMed] [Google Scholar]
- 33. Meulmeester E., Maurice M. M., Boutell C., Teunisse A. F., Ovaa H., Abraham T. E., Dirks R. W., and Jochemsen A. G. (2005) Loss of HAUSP-mediated deubiquitination contributes to DNA damage-induced destabilization of Hdmx and Hdm2. Mol. Cell 18, 565–576 [DOI] [PubMed] [Google Scholar]