

Abstract
Background and Purpose
Chemical conjugation of therapeutic proteins with polyethylene glycol (PEG) is an established strategy to extend their biological half‐life (t 1/2) to a clinically useful range. We developed a novel uncharged and unstructured recombinant polypeptide composed of five amino acids (P, S, T, A and G), named PsTag, as another approach to extend the t 1/2 of human FGF21, with increased hydrodynamic radius.
Experimental Approach
Human FGF21 was fused with PsTag polymers of differing lengths (200 ‐ 600 residues). Three fusion proteins and native FGF21 were produced in Escherichia coli . The biophysical characteristics, metabolic stability, immunogenicity and pharmacokinetics in were assessed in first. In lean and diet‐induced obese (DIO) mice, effects on body weight, oral glucose tolerance tests and levels of relevant hormones and metabolites were studied.
Key Results
Fusion proteins were solubly expressed in E. coli and prolonged the t 1/2 from 0.34h up to 12.9 h in mice. Fusion proteins were also biodegradable, thus avoiding vacuole formation, while lacking immunogenicity in mice. In DIO mice, administration of PsTag fused to FGF21 reduced body weight, blood glucose and lipids levels and reversed hepatic steatosis.
Conclusions and Implications
The novel recombinant polypeptide, PsTag, should be useful in the development of biological drugs with properties comparable to those achievable by PEGylation, but with potentially less side effects. In mice, fusion of FGF21 to PsTag prolonged and potentiated pharmacological effects of native FGF21, and may offer greater therapeutic effects in treatment of obesity.
Abbreviations
- AKP
alkaline phosphatase
- ALT
alanine aminotransaminase
- AST
aspartate aminotransaminase
- BAT
brown adipose tissue
- CD
circular dichroism
- DIO
diet‐induced obese
- DLS
dynamic light scattering
- Fas
fatty acid synthase
- H&E
haematoxylin and eosin
- HDL‐C
HDL cholesterol
- HFD
high‐fat diet
- IEF
isoelectric focusing
- iWAT
inguinal white adipose tissue
- Klb
β‐Klotho
- LDL‐C
LDL cholesterol
- OGTT
oral glucose tolerance test
- PEG
polyethylene glycol
- pI
isoelectric point
- PGC‐1α
PPARγ‐coactivator‐1α
- SCD‐1
stearoyl‐CoA desaturase 1
- SD
standard diet
- UCP
uncoupling protein
Tables of Links
| TARGETS |
|---|
| Transporters a |
| UCP, uncoupling protein |
| Nuclear hormone receptors b |
| PGC‐1α, PPARγ‐coactivator‐1α |
These Tables list key protein targets and ligands in this article which are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Southan et al., 2016) and are permanently archived in the Concise Guide to PHARMACOLOGY 2015/16 (a,bAlexander et al., 2015a, 2015b).
Introduction
Proteins and peptides have been successfully used as highly effective therapeutic agents for almost three decades. Rapid clearance from blood circulation through renal filtration is a typical property shared by most proteins and peptides, except antibodies and vaccines. One established strategy to increase the in vivo biological half‐life (t 1/2) of therapeutic proteins is chemical conjugation of the biological component with the synthetic polymer polyethylene glycol (PEG) (Jain and Jain, 2008; Pasut and Veronese, 2012). However, such PEGylation has several disadvantages, including the expense of commercial PEG derivatives and the additional purification steps required by the in vitro covalent coupling to a protein or peptide, thus reducing yield and increasing costs. Furthermore, PEG has some potential safety risks, such as generation of antibodies against PEG and the very limited biodegradability of PEG, which can cause side effects, such as vacuolation of kidney epithelium (Knop et al., 2010; Veronese et al., 2009).
In spite of these problems, PEGylation has proven to be bio‐effective and has already resulted in 11 FDA‐approved therapeutic agents (Zhang et al., 2014). Here we describe a novel recombinant polypeptide with the advantages of PEGylation but without its defects. Tis polypeptide is a hydrophilic, uncharged and unstructured biological polymers with biophysical properties remarkably similar to PEG (Figure 1). Genetic fusion of known therapeutic proteins and peptides with conformationally disordered polypeptide sequences currently has been emerging as a potential strategy to extend plasma t 1/2 without any need for either chemical or post‐translational modification of the fusion product (Kontermann, 2011; Kontermann, 2012).
Figure 1.
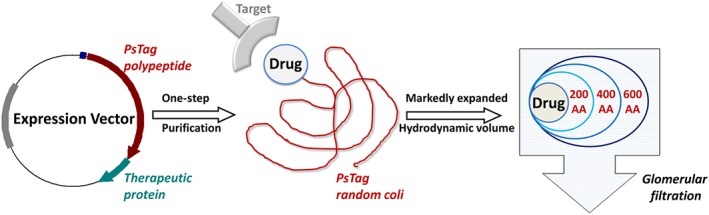
PsTag sequences can be fused to the pharmaceutically active protein using recombinant DNA technology, to produce a fusion protein in a single expression step. Like PEG, PsTag tags with different sequences exhibit stable random coil conformation in physiological solution, so they generate a large hydrodynamic volume that retards renal filtration of a PsTag fused to active protein.
Up to now, all unstructured polypeptides are designed to change the physical size and charge in order to retard renal filtration (Kontermann, 2012; Hu et al., 2015). Most of these methods yield either highly immunogenic products or only moderately extend the t 1/2 (Alvarez et al., 2004; Schlapschy et al., 2007; MacEwan and Chilkoti, 2010). The most striking example of the more advanced recombinant peptide polymer technology is XTEN which comprises six amino acids (A, E, G, P, S and T) (Schellenberger et al., 2009). XTEN contains 144 Glu residues in the XTEN sequence of 864 residues, resulting in a high overall negative charge and a very low isoelectric point (pI) value for the XTEN fusion protein. This approach takes advantage of the repulsion from negatively charged glomerular basement membrane, which leads to an increased plasma t 1/2, as well as reduced tissue penetration of fused drug, because of the negatively charged glycocalyx of endothelial cells. However, the latter is problematic because most biological drugs exert their maximum therapeutic effects only after entering the interstitial space and the underlying tissues. In addition, electrostatic repulsion tends to decrease binding of drugs, as most drug receptors are expressed at the negatively charged cell surface, which reduces efficacy of the drug. A different approach, PASylation technology, is based on three amino acids (P, A and S) and was was established by XL‐Protein (Schlapschy et al., 2013). Long PAS‐encoding gene cassettes were synthesized by ligation of only one hybridized oligodeoxynucleotide building block (encoding 20 amino acid). Hence, PAS polymers have defined sequences due to the repetition of the long building block.
In our approach, using five amino acids (P, S, T, A and G), we generated libraries of short peptide segments comprising repeatable sequences. Through a series of screening processes, we obtained novel uncharged recombinant polypeptides of different lengths, with biophysical properties highly similar to those of PEG and have called them PsTag . To assess the utility and properties of fusion with PsTag sequences, we selected human FGF21, one distinct member of the FGF family, which has been extensively investigated for its potential as a treatment for diabetes and obesity (Kharitonenkov et al., 2005; Zhang and Li, 2014). The plasma t 1/2 of native FGF21 is very short, ranging from 0.5 to 5 h depending on the route of administration and species. Therefore, frequent injection or continuous infusion of the native protein is required for effective in vivo bioactivity (Smith et al., 2013) and would pose a major challenge in the use of native FGF21 as a therapeutic protein. As variants of long‐acting FGF21 analogues had been developed with reduced dosing frequency by several groups (Gimeno and Moller, 2014), we expected that fusing PsTag to FGF21 would yield a protein with improved pharmacokinetic and pharmacodynamic properties. Long‐acting FGF21 analogues had been widely assessed for the treatment of diabetes mellitus (Huang et al., 2011; Hecht et al., 2012). We evaluated our fusion products using high‐fat diet (HFD)‐induced obese (DIO) mice and lean mice, hoping to provide a broader measurement of a range of relevant pharmacodynamic parameters.
Methods
Animals
All animal care and experimental protocols complied with the Laboratory Animal Management Regulations in China and the ethical committee at China Pharmaceutical University. They were approved by the College of Veterinary Medicine Yangzhou University and Use Committee (license No. SCXK (Su) 2012–0004). The animal studies are reported as recommended by the ARRIVE guidelines (Kilkenny et al., 2010; McGrath & Lilley, 2015).
For pharmacokinetic studies, male C57BL/6 mice (20–25 g; 8 weeks) were used. For testing immunogenicity, female BALB/c mice (20–25 g; 8 weeks) were used. In pharmacological studies, male C57BL/6 mice were used at 3 weeks of age. All animals were provided by the Animal Facility at the College of Veterinary Medicine, Yangzhou University. The mice were kept as follows: n = 5 per cage for pharmacokinetic studies in C57BL/6 mice; n = 4 per cage for a single dose pharmacodynamics study in DIO mice; n = 3 per cage for a 30 days pharmacological study in DIO mice; n = 3 per cage for a 30 days pharmacological study in C57BL/6 mice; n = 3 per cage for immunogenicity test in BALB/c mice. All animals were housed in cages in a temperature‐controlled (22–24°C) environment with 12 h light and 12 h dark cycle. Apart from the DIO mice, all animals received water and food ad libitum. Mice were maintained on their respective diets during the treatment and recovery periods. After 11 weeks of a diet period, either a standard diet (SD) (XIETONG, China) or a HFD (60% kJ as fat) (D12492, Research Diets) mice were randomly assigned to treatment or vehicle groups, and randomization was stratified by body weight and fed blood glucose levels.
General procedures
We chose the DIO model in mice because the metabolic characteristics of DIO mice more closely resemble those of human type 2 diabetes (Xu et al., 2009). Also DIO mice have become the most widely used model for experimental obesity and fatty liver (Sheng et al., 2011). Thus, we used DIO mice to provide measurement of a broader range of relevant metabolic parameters.
Randomization was conducted by an individual other than the operator. The animals were selected randomly from the pool of all cages eligible for inclusion in the study and randomly divided into groups.
Group sizes were as follows: n = 10 per each group for pharmacokinetic studies in C57BL/6 mice; n = 8 per each group for a single dose pharmacodynamics study in DIO mice; n = 12 per each group for a 30 days pharmacological study in DIO mice; n = 6 per each group for a 30 days pharmacological study in C57BL/6 mice; n = 6 per each group for immunogenicity test in BALB/c mice. We have used the minimum possible mice to achieve statistical significance.
Experimental procedures in mice
Pharmacokinetic studies
Forty C57BL/6 mice were randomly divided into four groups, which were injected i.v. with 1 mg·kg−1 of FGF21, 2.0 mg·kg−1 of PsTag200‐FGF21, 2.7 mg·kg−1 PsTag400‐FGF21 or 3.7 mg·kg−1 of PsTag600‐FGF21 (as FGF21 equivalent of 51 nmol·kg−1). For the FGF21 group, blood samples were collected at 0.25, 0.5, 1, 2, 4, 8, 12 and 24 h after injection; for PsTag200‐FGF21 group, blood was collected at 0.25, 0.5, 1, 4, 8, 12, 24 and 48 h after injection; for PsTag400‐FGF21 group, blood was collected at 0.5, 1, 4, 8, 12, 24, 48 and 72 h after injection; and for the PsTag600‐FGF21 group, blood was collected at 0.5, 1, 4, 12, 24, 48, 72 and 96 h after injection. Serum was obtained by centrifugation at 4°C and 900 x g for 15 min and then stored at −80°C until analysis. The serum concentrations of FGF21 and variant PsTag fusion protein were measured with a sandwich elisa (DF2100; R&D Systems, Minneapolis, MN, USA). A solid‐phase elisa was designed to measure full‐length human FGF21 in serum. Plasma concentration data was analyzed with PKSolver (China Pharmaceutical University, Nanjing, China) (Zhang et al., 2010).
In vivo biology
In a single dose pharmacodynamic study, DIO mice (n=40) were randomly divided into five groups, which were, respectively, injected i.p. with vehicle, 1 mg·kg−1 of FGF21, 2.0 mg·kg−1 of PsTag200‐FGF21, 2.7 mg·kg−1 PsTag400‐FGF21 or 3.7 mg·kg−1 of PsTag600‐FGF21 (as FGF21 equivalent of 51 nmol·kg−1). Body weight was measured daily until 7 days after the single injection.
In a 30 day pharmacological study, FGF21 and roglitazone were given to DIO mice at 1 or 4 mg·kg−1·day−1 for 15 days. The mice were i.p. injected with FGF21 twice daily (at one‐half of the daily dose) to achieve the indicated daily dose. Injections (i.p.) of vehicle, 3.7 mg·kg−1 PsTag600‐FGF21 (as FGF21 equivalent of 51 nmol·kg−1) were administered every 3 days for 15 days. Body weight and food and water intake were recorded every day throughout the study. Body composition was analyzed on day 12 using MesoMR23‐060H‐I imaging instrument (Shanghai Niumag Corporation). At day 14, plasma was collected from the retro‐orbital sinus of each unanaesthetized mouse to assay blood glucose, glucagon, insulin, leptin, triglyceride, cholesterol, free fatty acid, adiponectin, HDL cholesterol (HDL‐C), LDL cholesterol (LDL‐C), alanine aminotransaminase (ALT), aspartate aminotransaminase (AST) and alkaline phosphatase (AKP) levels. An oral glucose tolerance test (OGTT) was performed on day 15 with an intragastric administration of a 2 g·kg−1 glucose solution after a 12 h fast. At day 16, half the mice in each group were selected randomly and killed. Tissues were collected for lipid analysis, mRNA expression analysis and histological examinations. Only the body weight and food intake in the remainder of each group were measured after drugs were removed at day 16, until the end of this study.
In lean mice, 16 lean mice were randomly divided into two groups, which were, respectively, injected i.p. every 3 days for 15 days with vehicle, 3.7 mg·kg−1 of PsTag600‐FGF21. Body composition was analyzed on day 12 using MesoMR23‐060H‐I imaging instrument (Shanghai Niumag and Technology Corporation, Ltd., Shanghai, China). Body weight was measured every day for 30 days. Plasma was collected to assay glucose, leptin, insulin and free fatty acid levels on day 14 and the OGTT was performed on day 15.
Other methods
Establishment of libraries and gene screening for the PsTag amino acid sequences
DNA digestion and molecular cloning were performed following standard procedures (Sambrook et al., 1989). Plasmids or DNA fragments were isolated from Escherichia coli cells or agarose gels with TIANprep Mini Plasmid Kit (Tiangen Biotech, Beijing, China), or TIANgel Midi Purification Kit (Tiangen Biotech) respectively.
A library of 10 amino‐acid segments is listed in Supporting Information Table S1. Gene fragments encoding building blocks of 10 amino acids were obtained by hybridization of two phosphorylated and complementary oligodeoxynucleotides. One GGC codon was used to create complementary sticky ends of three nucleotides on both sides of the gene cassette. Thus, after hybridization of two corresponding synthetic oligodeoxynucleotides, these two 5′‐protruding but non‐palindromic ends could be used for efficient unidirectional ligation. These annealed oligonucleotides were added to the ligation mixture to form segment multimerization and allow us to obtain nucleotide fragments with different lengths, as we needed. Oligodeoxynucleotides were purchased from GenScript (Nanjing, China). Enzymatic phosphorylation was performed by mixing 100 pmol of both oligodeoxynucleotides in 50 μL 50 pmol ATP, 10 U polynucleotide kinase (Thermo Fisher Scientific Inc., Waltham, MA, USA) and incubation for 30 min at 37°C. After denaturation for 10 min at 75°C, the mixture was cooled to room temperature overnight to achieve hybridization. Then, 50 μL of this solution was ligated by adding 0.06 U T4 DNA ligase (Thermo Fisher Scientific Inc.) in a total volume of 100 μL and incubation for 5 min at 4°C. After 10 min heat inactivation at 70°C, the ligation products were separated by 2% (w/v) agarose gel electrophoresis in the presence of Tris/borate/edta buffer (90 mmol·L−1 Tris, 90 mmol·L−1 boric acid, and 2 mmol·L−1 edta) and 1 μg·mL−1 Goldview™ (ZSGB‐bio, Beijing, China). The ligation reaction produced a ladder of different lengths, and the DNA fragments of the desired length were isolated (Supporting Information Figure S1). PsTag‐encoding fragments were cloned into an expression vector (derived from pET28a(+); Invitrogen, Carlsbad, CA, USA), named DMT, that deleted BspQIin the vector backbone region and inserted the gene for GFP (NCBI accession no. AAX07425.1), a short sequence with multiple restriction enzyme sites (Figure 2B). First, 60 nucleotide fragments encoding 20 amino acids were isolated on an agarose gel (Supporting Information Figure S1A). The DMT vector was cut with BspQI(New England Biolabs, Ipswich, MA, USA), dephosphorylated with alkaline phosphatase (Thermo Fisher Scientific Inc.) and ligated with the DNA fragments from above. After transformation of E. coli BL21(DE3) (Invitrogen), the library encoding 20‐amino‐acids, PsTag20, was screened for GFP expression, and isolates with the highest fluorescence were sequenced. The fluorescence was measured using a Tecan infinite M200 PRO reader (excitation 397 nm, emission 506 nm, gain 100, i‐control 1.8 software; Tecan Group Ltd., Männedorf, Switzerland). This process yielded two PsTag20 fragments with the highest fluorescence intensity. Then, the two 60 bp oligodeoxynucleotides were synthesized from GenScript as shown in Figure 2A and ligated using the above strategy (Supporting Information Figure S1B). The synthetic gene fragments of 200 amino acids were cloned into BspQI in DMT for digestion and screened for the GFP fluorescence. This process yielded a PsTag200 fragment and a vector, named DMT‐PsTag200. Finally, based on these two PsTag20 fragments and the DMT‐PsTag200 vector, the screening process of PsTag200 was repeated to get a PsTag400 fragment of 400 amino acids. Finally, the gene encoding a PsTag600 sequence (NCBI accession no KT964028) with 600 amino acids was screened by repeating the screening process of PsTag200 three times (Figure 2B).
Figure 2.
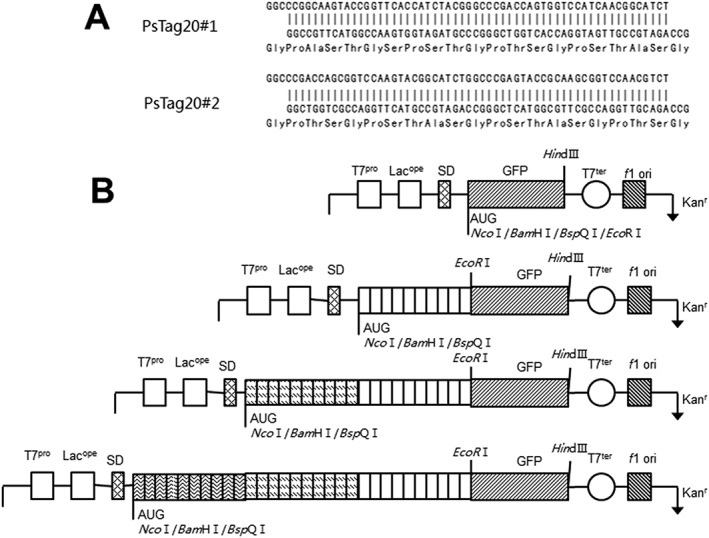
Gene design and cloning strategy for PsTag sequences. (A) Nucleotide and encoded amino acid sequences of the building blocks for PsTag20#1 and PsTag20#2 gene cassettes obtained by hybridization of two complementary oligodeoxynucleotides, with two sticky ends that can be cloned with BspQI restriction sites. (B) Schematic presentation of the features and sequence elements of the plasmids used in this study. The synthetic gene fragments of several building blocks were cloned into BspQI in DMT for digestion and screened for the fluorescence of GFP. This process could be repeated until the length of amino acid sequences had met requirements.
Construction of expression plasmids for the production of human FGF21, fused with different PsTag sequences
The coding sequence of human FGF21 (NCBI accession no AAQ89444.1) was synthesized by GenScript. The FGF21 gene was digested with enzymes EcoRI and HindIII and ligated into the pET28a(+) vector, generating pET28a‐FGF21. DMT‐PsTag200, DMT‐PsTag400 and DMT‐PsTag600 were digested with enzymes NcoI and EcoRI and inserted into the pET28a‐FGF21 vector, generating pET28a‐PsTag200‐FGF21, pET28a‐PsTag400‐FGF21 and pET28a‐PsTag600‐FGF21 respectively. His6‐tag was introduced into these three vectors at the 5′‐end of the PsTag sequences. All constructs were maintained in E. coli DH5α with kanamycin as selection pressure for subsequent expression in E. coli BL21 (DE3).
Recombinant protein production and purification
The recombinant strains were cultured in Lysogeny broth (LB) medium containing 100 μg·mL−1 kanamycin, and the cells were grown at 37°C and 220 rpm until OD600 reached to 0.8–1.0. The cultures were further incubated with isopropyl‐β‐d‐thiogalactoside (Bio Basic, Markham, ON, CA) at a final concentration of 0.8 mg·L−1, and cells were grown at 37°C for additional 5 h. The cells were pelleted, and the protein was purified by Ni2 + affinity chromatography, followed by Q Sepharose Fast Flow column, both according to the manufacturer's protocol (GE Healthcare, Pittsburgh, PA, USA). SDS‐PAGE (12%) and Western blot were used for analyzing the expression levels of each fusion proteins. The Western blot was carried out with a rabbit anti‐human FGF21 antibody (1:1000 dilutions, ab64857; Abcam, Cambridge, MA, USA) as primary antibody and HRP‐conjugated goat anti‐rabbit Ig antibody (1:5000 dilutions, Sangon Biotech, Shanghai, China) as secondary antibody for detection of FGF21. The protein concentration was determined using the Bradford Protein Assay Kit (Beyotime Institute of Biotechnology, Shanghai, China).
Circular dichroism analysis
The circular dichroism (CD) spectra of proteins (0.1 mg·mL−1 in 20 mmol·L−1 sodium phosphate pH 7.4) was measured by a Jasco‐720 spectropolarimeter (Jasco, Groß‐Umstadt, Germany) over a range of wavelength of 190–250 nm using a 0.2 cm cell.
Isoelectric focusing
Isoelectric focusing (IEF) was performed with the Multiphor II Electrophoresis System (GE Healthcare). The pH range 3–10 IEF Standard (161–0310; Bio‐Rad, Hercules, CA, USA) and the pH range 2.5–6.5 IEF Standard (17–0472‐01, GE) were applied as standard. The samples were treated by ultra‐filter for desalting. The loaded strips of 10 μg protein samples were focused at 15°C on a Multiphor II Electrophoresis System using an initial focusing step at 700 V for 20 min and then a gradient to 500 V for 20 min and a gradient to 2000 V for 90 min. After electrophoresis, the gel was transferred into fixative solution for at least 30 min. Then, it was transferred to the staining solution at least 120 min, and then destained with destaining solution. The gel was scanned after the background was clear, using imagequant tl version 7.0 (GE Healthcare) software to determine the pI value
Mass spectroscopy
Mass spectra were acquired using 5800 MALDI‐TOF/TOF (AB Sciex, Framingham, MA, USA). Load 1 μL protein sample to the MALDI plate after the desalination process by ZipTip C4 (ZTC04S096, Millipore, Billerica, MA, USA) and 0.6 μL SA matrix solution (85429; Fluka Billerica, MA, USA) to the corresponding target location after natural drying. Use the same method in adjacent position of sample for ProteoMass Peptide & Protein MALDI‐MS Calibration Kit (MSCAL1; Sigma, St. Louis, MO, USA). Samples were analyzed using 5800 MALDI‐TOF/TOF system in positive high mass linear mode and saved by 4000 Series Explorer V3.5 (AB Sciex, Framingham, MA, USA).
Size exclusion chromatography
Analytical SEC was performed on a Zenix‐C 300 column (Sepax Technologies, Inc., Newark, DE, USA) at a flow rate of 1 mL·min−1 using high‐performance liquid chromatography system (Agilent Technologies, Santa Clara, CA, USA) equipped with an autosampler. The purified proteins were applied with 150 mmol·L−1 sodium phosphate buffer (pH 7.0), and respective elution volumes were measured. The apparent molecular masses were estimated by interpolation from a calibration line (R 2 = 0.9991) (Supporting Information Figure S2) obtained with the reference proteins thyroglobulin, BSA, ovalbumin, ribonuclease A and vitamin B12 (all from Sepax Technologies, Inc.).
Dynamic light scattering
Dynamic light scattering (DLS) measurements of proteins with the concentration of 2 mg·mL−1 in PBS at 25°C were detected by Dynapro Nanostar (Wyatt Technology Corporation, Beijing, China) equipped with a 45 μL quartz cuvette. Molar mass was estimated based on the estimated hydrodynamic radius, particle density and conformation model using regularization fit in software DYNAMICS 7.1.7 (Wyatt Technology Corporation, Beijing, China).
Assay of binding specificity
Binding affinities of FGF21 and PsTag fusion proteins to human β‐klotho (58 890 KB, R&D) were examined by direct binding elisa. Human β‐klotho (25 ng per well) was coated in a 96‐well elisa plate (Costar; Sigma‐Aldrich, St. Louis, MO, USA), and various concentrations of FGF21 and PsTag fusion proteins were added into the pre‐coated plate. The bound protein complexes were detected by HRP‐labelled FGF21 antibody for each concentration; FGF21 antibody was pursued from Abcam and labelled by HRP labelling kit (Thermo Fisher Scientific Inc.). The OD450nm absorbance was plotted against protein concentrations, and EC50 values were fitted with a single‐site binding model using graphpad prism 5 (GraphPad Software Inc).
Cell culture and treatment
The pre‐adipocytes celline (3 T3‐L1 cells) were from the American Type Culture Collection maintained in DMEM high glucose (Gibco Life Technologies, Grand Island, NY, USA) (25 mmol·L−1 glucose) containing 10% FBS. 3T3‐L1 fibroblasts were seeded at 25 000 cells per well density, the differentiation was induced 2 days later in DMEM supplemented with 10% FBS, 1 μmol·L−1 dexamethasone, 0.5 mmol·L−1 3‐isobutyl‐1‐methylxanthine and 1 μg·mL−1 insulin (48 h), and the medium was changed to DMEM/10% FBS/1 μg·mL−1 insulin (48 h). For glucose uptake experiments, adipocytes were starved overnight in DMEM/0.1% FBS, and then treated with FGF21, PsTag200‐FGF21, PsTag400‐FGF21 or PsTag600‐FGF21 (1 nmol·L−1, 10 nmol·L −1, 100 nmol·L −1) for 24 h. The concentrations of glucose in cell supernatant of each group were determined with the glucose assay kit (Jiancheng, Nanjing, China) following the manufacturer's instructions. Cell proliferation was measured by 3‐(4,5)‐dimethylthiahiazo (‐z‐y1)‐3,5‐di‐ phenytetrazoliumromide (MTT) assay. A 100 μL MTT solution (5 g·L−1) was added in each well. The reaction was carried out in 37°C for 4 h, removed the supernatant, terminated by addition of DMSO (200 μL per well), measured the absorbance at 490 nm of each well with Multiskan Spectrum Microplate Spectrophotometer (Thermo Scientific, Helsinki, Finland). Medium were added as negative control (Yin et al., 2002).
Immunoblotting
3T3‐L1 adipocytes were starved for 18 h, stimulated with FGF21, PsTag200‐FGF21, PsTag400‐FGF21 or PsTag600‐FGF21 (10 nmol·L −1) for 10 min and lysed in RIPA lysis buffer (Beyotime Institute of Biotechnology) containing a protease inhibitor cocktail (88668; Thermo Fisher Scientific Inc.). Protein samples were separated by SDS‐PAGE, transferred to magna nitrocellulose supported transfer membrane and probed with antibodies against the following proteins: p44/42 ERK1/2 (137F5), Phospho‐p44/42 ERK1/2 (Thr202/Tyr204) (D13.14.4E) (Cell Signaling Technology, Inc., Danvers, MA, USA.) and β‐actin (ZSGB‐Bio, Beijing, China).
Evaluation of hormones and metabolites
Plasma insulin, leptin, adiponectin and glucagon levels were determined with elisa kits (Millipore), and elisa was conducted following a protocol provided by the manufacturer. Plasma cholesterol, triglyceride, free fatty acids, HDL‐C, LDL‐C, ALT, AST and AKP were measured with an autoanalyser (Hitachi 7020; Hitachi, Tokyo, Japan). Liver lipid extraction was conducted as described previously (Gao et al., 2014), and lipid contents were measured with a cholesterol kit (A111–1, Jiancheng) and a triglyceride kit (A110–1, Jiancheng). Blood glucose was measured at 0, 15, 30, 60, 90 and 120 min with One Touch glucose Meters (Sannuo, Changsha, China). The glucose AUC during an OGTT from 0 to 120 min was calculated by the linear trapezoidal method (Graphpad prism 5; GraphPad Software Inc).
Histology
All histological analyses was carried out without knowledge of treatments (blind assessment). Tissues were fixed in 10% zinc‐formalin. For haematoxylin and eosin (H&E) staining, tissue samples were embedded into paraffin, cut at 5 μm in thickness, and stained with H&E. For Oil‐red O staining, liver samples were frozen in liquid nitrogen and sectioned at 8 μm in thickness using a cryostat (Leica Biosystems, Buffalo Grove, IL, USA).
T1‐weighted MRI
T 1‐weighted MRI was performed using MesoMR23‐060H‐I imaging instrument (Shanghai Niumag Corporation, Shanghai, China). Mice were anaesthetized with 10% chloral hydrate (4 mL·kg−1). The instrumental parameters were set as follows: a 0.55 T magnet, K space = 192 × 256 mm, section thickness = 3.5 mm, time of echo (TE) = 13.5 ms, time of waiting (TR) = 300 ms, field of view (FOV)Read =100 mm, field of view (FOV)Phase = 100 mm and number of scans (NS) = 8. The fat highlight image was analysed by ImageSystem (Shanghai Niumag Corporation).
Real‐time PCR Analysis
Total RNA was extracted from the mouse liver and white adipose tissue by using EasyPure™ RNA Kit (TransGen, Beijing, China), and cDNA was synthesized using GoTaq® 2‐Step RT qPCR System (Promega, Madison, Wisconsin, USA). The quantitative RT‐PCR was performed using SYB green (Applied Biosystems, Foster City, CA, USA) according to manufacturer's instructions (Stepone plus; Applied Biosystems). The data were analysed by using the ΔΔCt method and normalized to internal control of β‐actin mRNA. Primers were synthesized by GenScript and listed in Supporting Information Table S2.
Data and statistical analysis
The data and statistical analysis comply with the recommendations on experimental design and analysis in pharmacology (Curtis et al., 2015). Data are expressed as the mean ± SD. All statistical analyses were performed using sigmaplot 10.0 (Systat Software Inc., San Jose, CA, USA) and graphpad prism 5 (GraphPad Software Inc., San Diego, CA, USA). Data were compared by two‐way ANOVA, followed by post hoc Newman–Keuls Student's t‐tests. A P‐value below 0.05 (P < 0.05) was considered significantly different.
Results
Design of PsTag sequences
In our design of a novel recombinant polypeptide with PEG‐like properties, such as large hydrodynamic volume, high solubility and lack of charge, we first excluded hydrophobic amino acids (F, I, L, M, V, W and Y) that could give rise to aggregation and might elicit an HLA/MHC‐II‐mediated immune response. Also excluded were cysteine residues (because of possible crosslinking), the charged amino acids (H, K, R, E and D) and the amide side chains of N and Q, which were potentially prone to hydrolysis.. Consequently, only the five amino acids (P, S, T, A and G), which all were fairly hydrophilic, remained as possible components of the building block peptides.
To create a genetically stable, highly expressed, unstructured polypeptide in E. coli, we generated a library of 10‐amino‐acid segments comprising randomized sequences (with each segment comprising 2P, 3S, 2T, 1A and 2G). Notably, homo‐polymers of each of these amino acids all had a certain secondary structure preference: for example, α‐helix for A, β‐sheet for P and T and β‐turn for G and S. However, we thought that certain combination of P, S, T, A and G in the amino acid sequences could possibly counterbalance their distinct conformational preferences, which might lead to a stably disordered polypeptide. There were several principles for designing the library of 10 amino acids: first, the same amino acid must be separated by two other amino acids, thus avoiding repetition of the same amino acids, to form stable secondary structures. Secondary, the composition proportion of the five amino acids (P, S, T, A and G) in the 10‐amino‐acids segments was affected by their hydrophilicity. Finally, the randomized sequences were then assessed for criteria such as mRNA second structure, peptide second structure and protein digestion. Based on these principles, a library of 10‐amino‐acids segments was designed for further study (Supporting Information Table S1). Then, synthetic gene libraries encoding randomized sequences comprising the 10‐amino‐acids segments, which were fused to the GFP, were screened with respect to fluorescence intensity in E. coli. In other words, long PsTag genes were synthesized by ligation of hybridized oligodeoxynucleotide building blocks for 10‐amino‐acid segments. Hence, the ligation of these segments constituted the PsTag polypeptide. Because this ligation was one kind of random process, these segments might be repeatable. The PsTag polymers of encoding up to 600 residues were selected in the library and successfully produced in E. coli.
Construction and biophysical characterization of PsTag fusion FGF21
To investigate the biophysical properties of PsTag polymers and their effects on plasma t 1/2 in vivo, three PsTag sequences of different lengths were chosen for fusion with FGF21: PsTag200‐FGF21, PsTag400‐FGF21 and PsTag600‐FGF21. Native FGF21 was expressed as inclusion bodies in E. coli. All PsTag fusion proteins were produced in E. coli cytoplasm, with no detectable production of inclusion bodies. These three PsTag fusion proteins were purified by two‐column chromatography steps, resulting in homogeneous preparations (Figure 3A). The unmodified recombinant human FGF21 showed a size of approximately 19 kDa in 12% SDS‐PAGE (Figure 3A) and Western blot (Figure 3B). In contrast, the PsTag fusion proteins exhibited drastically altered electrophoretic mobility and migrated significantly more slowly than predicted from their calculated masses (Figure 3A and B), with the following apparent sizes: ~80 kDa for PsTag200‐FGF21 (calculated mass: 36.7 kDa), ~140 kDa for PsTag400‐FGF21 (53.5 kDa) and ~200 kDa for PsTag600‐FGF21 (72 kDa). On the other hand, the purities of FGF21 and PsTag600‐FGF21 were determined by reversed phase HPLC (Supporting Information Figure S3). Both profiles showed a single homogeneous peak with obviously earlier elution of PsTag600‐FGF21 (24.6% acetonitrile) in comparison with the corresponding unfused FGF21 (40.9% acetonitrile).
Figure 3.
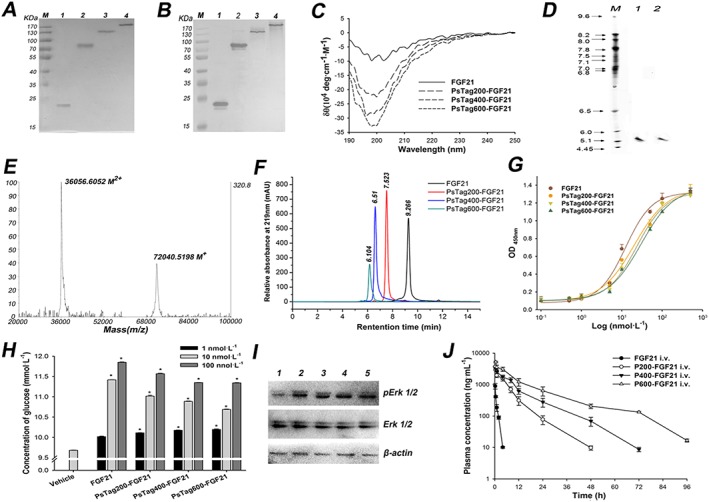
The characterization of FGF21 and fusion proteins. The unmodified FGF21 and fusion proteins were assayed by (A) 12% SDS‐PAGE and (B) Western blotting with a rabbit anti‐human FGF21 antibody (lane 1: FGF21; lane 2: PsTag200‐FGF21; lane 3: PsTag400‐FGF21; lane 4: PsTag600‐FGF21). (C) CD spectra of unmodified FGF21 and fusion proteins. (D) IEF of FGF21 and PsTag600‐FGF21 (lane 1: PsTag600‐FGF21; lane 2: FGF21). (E) MALDI‐TOF mass spectrometry of PsTag600‐FGF21 (M+ and M2 + refer to the singly and doubly charged ionic species of PsTag600‐FGF21 respectively. (F) SEC‐HPLC in the presence of 150 mmol·L−1 sodium phosphate buffer (pH 7.0) resulted in a single peak with decreasing elution time for PsTag fusion proteins with increasing number of amino acid residues. (G) Binding affinities of FGF21 and PsTag fusion proteins to human β‐klotho were examined by direct binding elisa. (H) Cellular glucose uptake stimulated by native FGF21 and PsTag fused FGF21 in 3 T3‐L1 cells. n = 3. *P < 0.05 versus vehicle control. (I) 3T3‐L1 cells were treated with vehicle (lane 1), 10 nmol·L−1 FGF21 (lane 2), 10 nmol·L−1 PsTag200‐FGF21 (lane 3), 10 nmol·L−1 PsTag400‐FGF21 (lane 4) and 10 nmol·L−1 PsTag600‐FGF21 (lane 5) for 10 min. Phospho‐specific antibody was used to determine phosphorylation of ERK. (J) Pharmacokinetic plasma profile of native FGF21 and PsTag fusion proteins intravenously injected in C57BL/6 mice (n = 10 per group).
The secondary structures of FGF21 and PsTag fusion proteins were evaluated by CD (Song et al., 2014), and the corresponding waveforms are shown in Figure 3C. Notably, the spectra of the PsTag fusion proteins showed a predominant negative minimum at 198 nm. This minimum became more marked with increasing length of the fused PsTag polypeptides. This indicated that the proportion of random coil conformation in secondary structure had increased gradually. The biophysical behavior of PsTag fusion proteins was further investigated with regard to isoelectric properties. IEF tests, carried out for FGF21 and PsTag600‐FGF21, showed that both had about the same pI, about 5.35 (Figure 3D). Because the complete absence of charged side chains in the PsTag sequence did not affect the pI of the fusion protein, it was very different from the XTEN sequence. Mass spectrometry (Figure 3E) showed a single peak of the expected mass (72 040 Da measured vs. 72 020 Da). By comparison, mass spectrometry of PEGylated proteins typically contained one considerably broader peak, because of the heterogeneity of PEG reagents. The effect of PsTag on the apparent molecular size of FGF21 in physiological solution was studied by SEC‐HPLC (Figure 3F). Both native FGF21 and PsTag fusion proteins showed a single peak, confirming the monodisperse nature of each protein preparation. Interpolation of the SEC elution time of the native FGF21 versus a set of protein standards led to an apparent molecular mass of 21 kDa, which was slightly above the theoretical mass of 19.5 kDa. However, all PsTag sequences showed much larger apparent molecular sizes of 128 kDa for PsTag200‐FGF21, 361 kDa for PsTag400‐FGF21 and 547 kDa for PsTag600‐FGF21.
The binding affinities of FGF21 and PsTag fusion proteins to human β‐klotho were determined by direct binding elisa (Figure 3G). The PsTag sequences led to an increase in EC50 values from 12.4 ± 2.0 nmol·L−1 to 30.0 ± 4.7 nmol·L−1 (Table 1). The slight loss of receptor activity was caused by the initial fusion with PsTag200 and subsequent extension of the PsTag sequences only had minor effects. We used a glucose uptake assay to evaluate the cellular function of FGF21. After treatment with different concentrations of FGF21 and PsTag fusion proteins, consumption of glucose by differentiated mouse 3T3‐L1 adipocytes increased and reached a maximum at 100 nmol·L−1 (Figure 3H). The results of the MTT assay (data not shown) showed no significant differences between each group, indicating that these proteins did not affect the viability of 3T3‐L1 cells. Meanwhile, ERK phosphorylation was increased after treatment with FGF21 and PsTag fusion proteins in 3T3‐L1 adipocytes, compared with vehicle control group (Figure 3I). Like the native FGF21, all three PsTag fusion proteins were fully capable of triggering FGF21 receptor signalling.
Table 1.
Pharmacokinetic parameters of native FGF21 and PsTag fused FGF21
| Parameter | FGF21 | PsTag200‐FGF21 | PsTag400‐FGF21 | PsTag600‐FGF21 |
|---|---|---|---|---|
| Mass (KDa) | 19.5 | 38.1 | 55 | 72 |
| Radius by DLS (nm) | 2.4 | 4.5 | 6.5 | 8.7 |
| SEC size (KDa) | 21 | 128 | 361 | 547 |
| EC50 (nmol·L−1) | 12.4 ± 2.0 | 20.3 ± 3.9 | 23.7 ± 3.8 | 30.0 ± 4.7 |
| Dose (mg·kg−1) | 1.0 | 2.0 | 2.8 | 3.7 |
| t 1/2 (h) | 0.342 ± 0.028 | 5.673 ± 0.404* | 8.802 ± 0.472* | 12.933 ± 0.446* |
| Cmax (ng·mL−1) | 895 ± 175 | 3700 ± 92* | 3289 ± 82* | 5247 ± 247* |
| AUC (ng·mL−1·h) | 667 ± 172 | 18344 ± 3376* | 30630 ± 5212* | 59725 ± 10371* |
| MRT (h) | 0.417 ± 0.048 | 6.637 ± 0.45* | 11.404 ± 0.95* | 16.965 ± 0.30* |
| Vz (mL·kg−1) | 650.4 ± 112 | 1188 ± 185* | 1308 ± 285* | 1220 ± 254* |
| Cl (mL·h−1·kg−1) | 1346.1 ± 294 | 108.6 ± 20.1* | 91.1 ± 16* | 61.6 ± 11* |
| Vss (mL·kg−1) | 549.6 ± 172 | 720.5 ± 87.1 | 1038.8 ± 88.1* | 1045.5 ± 168.4* |
Data are means ± SD from n = 10 per group. AUC, area under the plasma concentration curve; Cmax, maximum concentration; Cl, clearance; MRT, mean residence time; Vss, the apparent volume of the plasma compartment; Vz, the apparent volume of distribution during the terminal phase.
P < 0.05 versus FGF21.
Furthermore, biochemical stability of PsTag fusion proteins was studied in vitro by incubation with BALB/c mouse plasma (Supporting Information Figure S4), demonstrating resistance to serum proteases for at least 48 h at 37°C. Like PEG, PsTag sequences also improved the thermostability of the biological protein by means of a shielding mechanism (Supplementary Figure S5 and S6). In order to test the solubility and liquid stability of PsTag600‐FGF21, the protein was concentrated to 30 mg·mL−1 in PBS. It was important to note that no visible aggregates or significant viscosity was noted at this stage. In contrast, the fusion proteins were rapidly degraded during incubation with a mouse kidney homogenate (Supporting Information Figure S7).
To evaluate the immunogenic properties of the PsTag sequences, mice were subcutaneously administered with unmodified FGF21 and PsTag600‐FGF21. The proteins were injected every 2 days for 15 days, either with or without Freund's adjuvant. IgG titers specific for corresponding antigen were measured for all mice, 1 week after the last injection (Figure 4). As expected, high titers of FGF21 directed antibodies were detected in native FGF21‐immunized animals whether or not adjuvant was co‐administered. In contrast, the PsTag600‐FGF21 displayed a significantly reduced immunogenicity, even when the PsTag fusion protein was labelled with a polyhistidine tag. Antibody titers, directed against PsTag600‐FGF21, were not detectable without adjuvant co‐administration. Continuous monitoring over 5 weeks confirmed that PsTag sequences significantly reduced immunogenicity of FGF21 in mice (Figure 4D).
Figure 4.
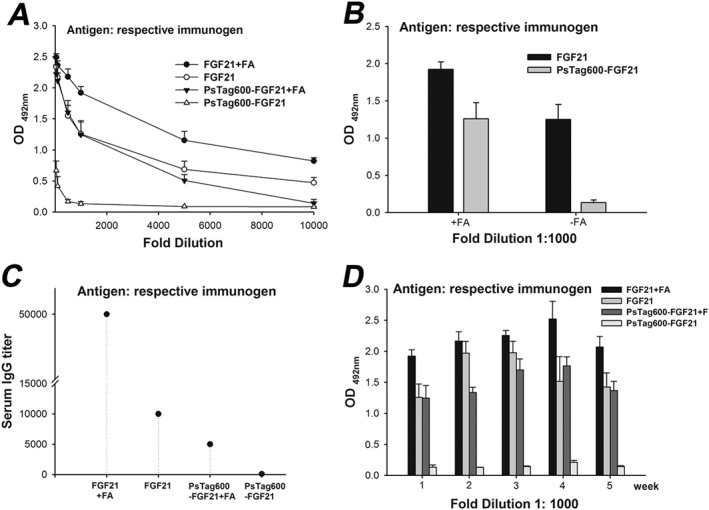
PsTag600‐FGF21 had almost no immunogenicity in mice (n = 6 per group). Plasma samples were analysed for immunogenicity according to the protocol given in the Experimental Section (Supporting Information). (A) Plasma samples were serially diluted (1:50–1:10000). (B) Data was shown for a plasma dilution at 1:1000. (C) Plasma samples were analysed for IgG titer. (D) Immunogenicity of corresponding antigen was continuously monitored for 5 weeks after the final immunization.
Pharmacokinetic analysis
The pharmacokinetics of proteins with PsTag polypeptides was determined in C57BL/6 mice. Plasma concentration data of unmodified FGF21 and PsTag fusion proteins are shown in Figure 3J, and the corresponding pharmacokinetic parameters are listed in Table 1. Curve fitting with PKSolver software revealed a rather short terminal plasma t 1/2 of 0.342 h for the unmodified FGF21, which was considerably prolonged by PsTag sequences from 5.7 h for PsTag200‐FGF21, to 8.8 h for PsTag400‐FGF21 to 12.9 h for PsTag600‐FGF21. This demonstrated a marked effect of PsTag polypeptides on in vivo pharmacokinetics, depending on the length of the polypeptide sequences. The strongly retarded renal filtration led to an almost 90‐fold increase in the AUC. Thus, by varying the length of the PsTag sequences, the terminal t 1/2 could be simply adjusted to the desired clinical requirements. The hydrodynamic radius was measured in DLS experiments (Table 1). In this case, the following hydrodynamic radius of unmodified FGF21 and PsTag fusion proteins were determined: 2.4 nm for FGF21, 4.5 nm for PsTag200‐FGF21, 6.5 nm for PsTag400‐FGF21 and 8.7 nm for PsTag600‐FGF21. Accordingly, PsTag600‐FGF21 showed a 3.6‐fold increased radius compared with the unmodified FGF21. Together with the results of SEC‐HPLC (Table 1), these findings clearly revealed PsTag polypeptides to be high flexibility, recombinant polymers with an expanded hydrodynamic radius, very similar to PEG.
PsTag fusion proteins were effective in vivo
The systemic effects of single i.p. injections of unmodified FGF21 and PsTag fusion proteins on change in body weight were assessed in the DIO mice. As shown in Figure 5A, body weight was reduced by 2.5 g at 3 days after treatment with PsTag600‐FGF21 and lasted until day 7 (P < 0.05), whereas no loss of body weight could be detected for the unmodified FGF21. Treatment with PsTag200‐FGF21 or PsTag400‐FGF21 also reduced body weight at least from days 3 to 4 (P < 0.05). The weight loss depended on the length of the PsTag sequences, consistent with the results of the pharmacokinetic study.
Figure 5.
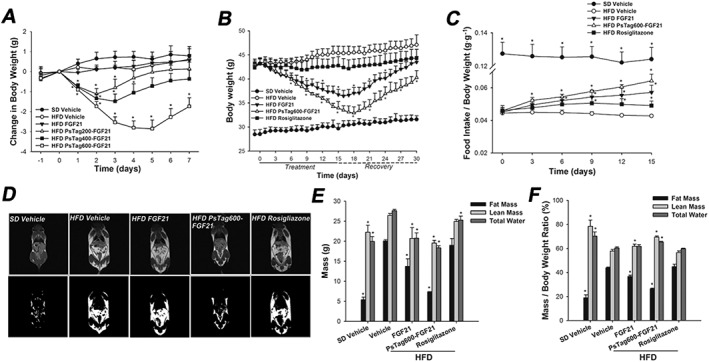
Physiological effects of FGF21 and PsTag fused FGF21 in DIO mice. (A) Body weight change was measured every day for 7 days. DIO mice were injected i.p. with a single dose on day 0. (B) In a 30 days pharmacological study, body weight was monitored throughout the treatment and recovery period. (C) Food intake was adjusted to body weight over 15 days. (D) T 1‐weighted MRI images (top) and the fat highlight images (bottom) of mice are shown. (E) Body composition analysed after 14 days of treatment. (F) Change in body composition adjusted to body weight. Data are means ± SD; n = 8 per group in A; n = 12 per group in B. * P < 0.05 versus vehicle‐treated HFD mice.
PsTag600‐FGF21 demonstrated prolonged pharmacodynamics in lean and DIO mice
To assess the repeat dose efficacy of the PsTag fusion protein in the DIO model, PsTag600‐FGF21 (3.7 mg·kg−1) was administered i.p. to DIO mice once every 3 days, in comparison with daily i.p. injections of unmodified FGF21 (1 mg·kg−1). Compared with age‐matched mice on SD, mice on HFD were clearly heavier with increased body fat mass, lean mass and total water (Figure 5E). Treatment with FGF21 and PsTag600‐FGF21 reduced body weight by about 7 and 10 g at the end of 15 days of treatment respectively (Figure 5B). Treatment with rosiglitazone for 15 days had little effect on the body weight of DIO mice compared with the vehicle‐treated HFD group (Figure 5B). When food intake was normalized to mice body weights, FGF21 and PsTag600‐FGF21 increased food intake (Figure 5C).
After 12 days of treatment, T 1‐weighted MR images were used to evaluate body fat distribution in mice. Te PsTag600‐FGF21 treated group showed significant signal reduction (darkness) in the whole body scan, compared with DIO mice (Figure 5D). In comparison, the darkened signal was only moderately reduced after treatment with unmodified FGF21. Similarly, to analyze the body composition, treatment with FGF21 and PsTag600‐FGF21 was associated with 32 and 63% reductions in body fat mass, respectively (Figure 5E). Marginal, but statistically significant, decreases in lean mass and total water were also observed (Figure 5E). Decreases in body weight mirrored reductions in body fat content (Figure 5F).
We next examined the effect of FGF21 and PsTag600‐FGF21 on glucose and lipid metabolism. Both FGF21 and PsTag600‐FGF21 significantly reduced serum glucose (Figure 6A and B), insulin (Figure 6E), cholesterol (Figure 6F), triglycerides (Figure 6G), free fatty acids (Figure 6H), leptin (Figure 6I) and glucagon (Table 2) on day 14 of treatment in comparison with the vehicle‐treated HFD group. Changes in the lipoprotein profile were also observed, including increased HDL‐C and reduced LDL‐C (Table 2). Moreover, administration of FGF21 and PsTag600‐FGF21 resulted in an about twofold increase in plasma levels of adiponectin (Table 2). An OGTT was performed on day 15 of treatment after a 12 h fast. PsTag600‐FGF21 also significantly improved glucose tolerance and was more effective than treatment with rosiglitazone (Figure 6C). The AUC for blood glucose after treatment with PsTag600‐FGF21 was reduced by 42.9% compared with vehicle control in DIO mice (Figure 6D).
Figure 6.
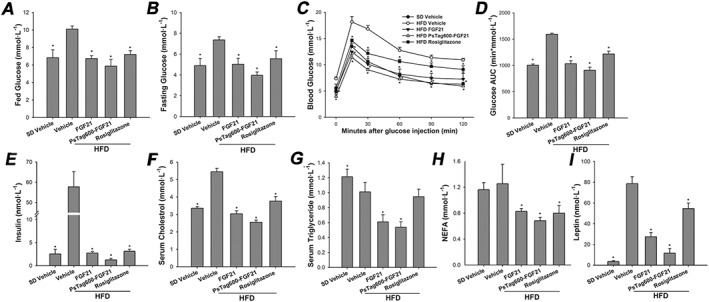
Both native FGF21 and PsTag fusion of FGF21 reduced blood glucose, insulin, non‐esterified fatty acid (NEFA), leptin, lipid levels and improves glucose tolerance in DIO mice. (A) Fed glucose. (B) Fasting glucose. (C) For OGTT, blood glucose was measured at 0, 15, 30, 60, 90 and 120 min. (D) The glucose AUC during an OGTT from 0 to 120 min. (E) Insulin. (F) Serum cholesterol. (G) Serum triglyceride. (H) NEFA. (I) Leptin. Data are means ± SD; n = 12 per group. * P < 0.05 versus vehicle‐treated HFD mice..
Table 2.
Metabolic parameters in mice fed SD or HFD and treated with vehicle, FGF21, PsTag600‐FGF21 or rosiglitazone
| SD | HFD | ||||
|---|---|---|---|---|---|
| Parameters | Vehicle | Vehicle | FGF21 | PsTag600‐FGF21 | Rosiglitazone |
| Body weight (g) | 29.7 ± 1.2* | 45.6 ± 1.4 | 36.7 ± 2.0* | 33.9 ± 1.3* | 41.9 ± 1.9 |
| Liver weight (g) | 1.1 ± 0.1* | 1.5 ± 0.2 | 1.0 ± 0.1* | 0.9 ± 0.1* | 1.3 ± 0.1 |
| iWAT weight (g) | 0.40 ± 0.23* | 2.16 ± 0.21 | 1.49 ± 0.25* | 1.21 ± 0.22* | 1.91 ± 0.21 |
| BAT weight (g) | 0.11 ± 0.03* | 0.15 ± 0.03 | 0.11 ± 0.02* | 0.1 ± 0.02* | 0.14 ± 0.02 |
| Liver triglyceride (mg·g−1) | 9.2 ± 2.2* | 122.3 ± 23.7 | 42.5 ± 5.8* | 34 ± 1.5* | 110 ± 14.2 |
| Liver cholesterol (mg·g−1) | 2.6 ± 0.6* | 6.8 ± 1.2 | 3.2 ± 0.7* | 2.2 ± 0.2* | 6.8 ± 0.7 |
| HDL‐C (mmol·L−1) | 2.0 ± 0.1* | 1.8 ± 0.1 | 2.0 ± 0.1* | 2.2 ± 0.1* | 1.6 ± 0.1 |
| LDL‐C (mmol·L−1) | 0.21 ± 0.03* | 1.24 ± 0.07 | 0.78 ± 0.06* | 0.56 ± 0.09* | 0.80 ± 0.11* |
| Plasma ALT (units·L−1) | 56.8 ± 1.4* | 162.8 ± 23.3 | 67.7 ± 15.7* | 58.7 ± 24.2* | 80.2 ± 15.8* |
| Plasma AST (units·L−1) | 58.8 ± 7.1* | 150.4 ± 14.1 | 91.1 ± 9.5* | 78.4 ± 18.5* | 134.9 ± 0.8 |
| Plasma AKP (units·L−1) | 63.7 ± 2.7* | 74.9 ± 3.2 | 27.7 ± 3.4* | 42.0 ± 2.9* | 43.8 ± 3.5* |
| Plasma glucagon (pg·mL−1) | 299 ± 7 | 293 ± 13 | 263 ± 7* | 255 ± 5* | 271 ± 5* |
| Plasma adiponectin (μg·mL−1) | 9.9 ± 2.0 | 7.7 ± 2.1 | 13.5 ± 1.2* | 14.7 ± 0.3* | 15.9 ± 3.1* |
Data are means ± SD from n = 6 per group.
P < 0.05 versus vehicle‐treated HFD mice.
Histological examination of liver sections stained with H&E (Figure 7A–E) and oil‐red‐O (Figure 7F–J). H&E staining showed more cellular vacuoles in vehicle‐treated HFD group compared with those after treatment with FGF21 and PsTag600‐FGF21. Both FGF21 and PsTag600‐FGF21 treatments resulted in a significant reversal effect of hepatocellular vacuolation and large amounts of oil‐red‐O‐stained lipid droplets (Figure 7A–J). Liver triglycerides and cholesterol contents of FGF21 and PsTag600‐FGF21 treatments were markedly reduced, accompanied by reductions of plasma ALT, AST and AKP levels (Table 2). H&E staining (Figure 7K–T) showed that treatments with FGF21 or PsTag600‐FGF21 srongly suppressed the HFD‐induced enlargement of adipocytes in inguinal white adipose tissue (iWAT) (Figure 7K–O), and a higher density of the cellular matrix in brown adipose tissue (BAT) of treated DIO mice (Figure 7P–T) compared with vehicle‐treated DIO mice. Quantification analysis of Figure 7K–T using image analytical software (Image‐Pro Plus 6.0) showed the same results (Figure 7W and X). Treatment with rosiglitazone did not change the histological parameters, compared with vehicle‐treated DIO mice.
Figure 7.
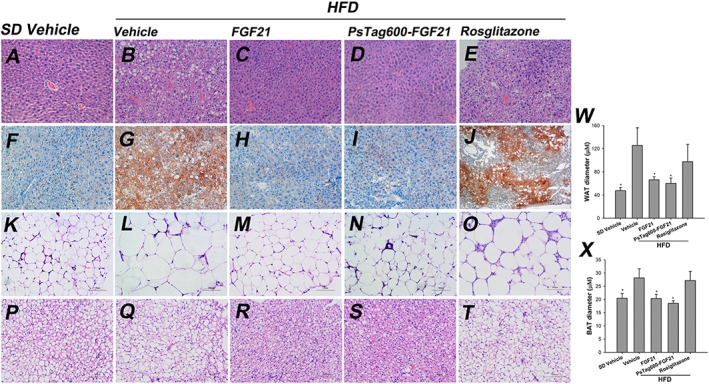
Tissue histological analysis. Tissue sections were prepared at day 16 in the study from mice fed SD and treated with vehicle (A, F, K and P) or fed HFD and treated with vehicle (B, G, L and Q), FGF21 (C, H, M and R), PsTag600‐FGF21 (D, I, N and S) or rosiglitazone (E, J, O and T). Histological examination of liver sections stained with H&E (A–E) and oil‐red‐O (F–J) staining of liver sections showed a significant reversal of hepatocellular vacuolation and large amounts of oil‐red‐O‐stained lipid droplets following treatment with FGF21 or PsTag600‐FGF21. H&E staining (K–T) exhibited that treatment with FGF21 or PsTag600‐FGF21 strongly suppressed the HFD‐induced enlargement of adipocytes in iWAT (K‐O), and a higher density of the cellular matrix in BAT of treated DIO mice (P–T) compared with vehicle‐treated DIO mice. (200× magnification). Quantification analysis of WAT (K–O) and BAT (P–T) using image analytical software (Image‐Pro Plus 6.0) show the (W) WAT diameter and (X) BAT diameter. Data are means ± SD; n = 3. * P < 0.05 versus vehicle‐treated HFD mice.
In the liver, gene expression analysis showed that treatments with FGF21 and PsTag600‐FGF21 markedly repressed transcription of key genes involved in lipogenesis, including Fas and Scd1, and significantly increased expression of Pgc‐1α (Supporting Information Figure S8A). We also examined the gene expression associated with energy expenditure in iWAT. No change in Ucp2 mRNA was detected, whereas Ucp1 mRNA level was dramatically increased (Supporting Information Figure S8B). In iWAT, a dramatic down‐regulation of mRNA from Fas and Leptin also were observed (Supporting Information Figure S8B). Additionally, FGF21 or PsTag600‐FGF21 treatment led to a significant increase in Glut1 mRNA but not Glut4 mRNA to enhance glucose uptake (Supporting Information Figure S8B). Gene expression of the FGF21 receptor signalling complex, FGF21 receptor 1 (Fgfr‐1) and β‐klotho (Klb) in liver and iWAT, was unchanged by PsTag600‐FGF21 or FGF21 administration (Supporting Information Figure S8A and B).
After 15 days of PsTag600‐FGF21 treatment in lean mice, body weight (Figure 8A) and glucose tolerance were improved (Figure 8D). Meanwhile, PsTag600‐FGF21 treatment did not change serum glucose and leptin levels but reducde insulin and non‐esterified fatty acid levels (Figure 8B, C and E). Decreases in body weight mirrored reductions in body fat content (Figure 8F). During the recovery period, body weight similarly recovered gradually in lean and DIO mice (Figures 5B and 8A).
Figure 8.
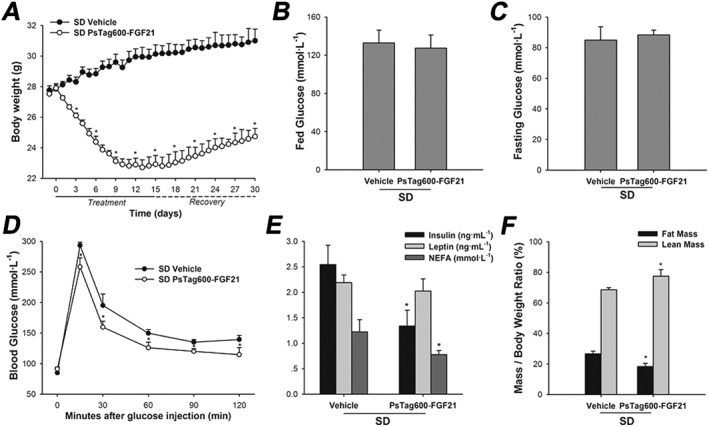
Physiological effects of PsTag600‐FGF21 in lean mice. (A) In a 30 day pharmacological study, body weight was monitored throughout the treatment and recovery period. (B) Fed glucose. (C) Fasting glucose. (D) For OGTT, blood glucose was measured at 0, 15, 30, 60, 90 and 120 min. (E) Plasma insulin, leptin and non‐esterified fatty acid (NEFA) levels. (F) Body composition analyzed after 14 days of treatment. Data are means ± SD; n = 6 per group. * P < 0.05 versus vehicle‐treated SD mice.
Discussion and conclusions
A novel approach based on a synthetic peptide with structurally disordered amino acid sequence was explored here. This approach, named PsTag, offered some advantages over chemical coupling with PEG, polysialic acid or other polymers, in particular with regard to the lower cost and by not needing downstream purification steps. PsTag fusion proteins were stable in blood plasma but were readily degradable by renal enzymes. In this way tissue accumulation, which was a common problem caused by synthetic polymers such as PEG, could be minimized. Another advantage for the development of biological drugs was the hydrophilia of the PsTag sequences. This property of the PsTag sequence tended to dominate the characteristics of any fusion protein, allowing soluble expression to solve the problem of inclusion bodies produced in E. coli. Refolding of inclusion body proteins into bioactive forms is complicated, resulting in poor recovery and accounting for the main cost in production of recombinant proteins from E. coli.
Furthermore, PsTag fusion proteins did not require any specific expression system that distinguished it from fusion with large globular proteins such as albumin and Fc fusion technology. In addition, PEG and Fc fragments can elicit undesired immunological responses. Anti‐drug antibodies were also a possible problem for albumin fusion proteins, particularly because of potential cross reactivity with endogenous albumin (Elsadek and Kratz, 2012). However, the immunogenicity of the PsTag polypeptide appeared to be negligible because of a complete absence of the hydrophobic amino acids.
A few other conformationally disordered polypeptides, in particular XTEN or PASylation (Cleland et al., 2012; Schlapschy et al., 2013), also showed potential for extending t 1/2, strategies that could replace traditional chemical polymers in biological drug development. The former included a large proportion of Glu residues, giving rise to high overall anionic charge and an extremely low pI value for the fusion protein. This property was quite different from the effects of PEGylation and had a marked effect on tissue distribution and cell surface receptor affinity. However, an XTEN fusion of human growth hormone has been in Phase‐II clinical trials and an XTEN fusion of exendin‐4 has been in Phase I clinical trials (www.amunix.com), demonstrating safety and tolerability of recombinant PEG mimetics. The latter included only three amino acids (A, S and P) that added to the difficulty of genetic manipulation, due to the highly repetitive sequences. Further, we held the opinion that the smallest amino acid, Gly, had no certain secondary structure preference. A certain amount of discontinuous Gly in the sequence would help to form unstructured or disordered polypeptides.
Here, we have established a new strategy to modulate the plasma t 1/2 of therapeutic proteins. Using this strategy, we obtained a potent and effective PsTag fused FGF21 as a potential therapeutic agent. The challenges in developing native FGF21 as a therapeutic agent for diabetes and obesity, include its short plasma t 1/2 and its tendency to rapid aggregation (Kharitonenkov and Adams, 2014). Renal filtration was probably one of the major routes for FGF21 elimination from the circulation as endocrine FGF21 levels were markedly elevated in the serum of patients with severely damaged renal functions and reduced glomerular filtration rates (Stein et al., 2009; Hecht et al., 2012). The marked increase in molecular size conferred on FGF21 by fusion with the PsTag sequences resulted in an expanded hydrodynamic radius and lowered the filtration coefficient in the kidney. The much higher AUC we observed for the PsTag fusion protein was a result of the much reduced renal filtration. An N terminally PEGylated FGF21 has a t 1/2 of about 4 h in rat (Huang et al., 2011). Similarly, the terminal t 1/2 of an Fc‐fused FGF21 analogue and a CovX‐Body conjugation of FGF21 was 11.2 h and 28 h in mice, respectively (Hecht et al., 2012; Huang et al., 2013). FGF21 has a tendency to form aggregates, mostly dimeric and oligomeric self‐associations, which are a disadvantage for a possible biotherapeutic agent because protein aggregates can induce adverse reactions and elicit immunogenicity in humans. Using recombinant polypeptide technology, the PsTag fusion with FGF21 not only greatly increased the t 1/2 to 12.9 h in mice, but also prevented aggregation of native FGF21. Another benefit of recombinant PsTag polypeptides was their easily variable length, just by regulating the coding DNA region. This allows customized metabolization characteristics of the biological medicine according to clinical requirements.
Consistent with improved pharmacokinetics, the in vivo activity of PsTag fusion FGF21 was also increased. As with previous reports (Xu et al., 2009), PsTag600‐FGF21 also reduced body weight, body fat mass, blood glucose, insulin and lipid levels and reversed hepatic steatosis in DIO mice. It was worth noting that the t 1/2 of native FGF21 in mice was extremely short and twice daily administration may not have produced full 24 h coverage, which could account for its decreased effcts on glucose and lipid metabolism, relative to those of PsTag600‐FGF21.
Taken together, our results have demonstrated that the novel recombinant polypeptide, PsTag, would be useful in the development of biological drugs with pharmacological properties comparable to those achievable by PEGylation, but with potentially less side effects. The PsTag‐FGF21 fusion protein developed in this study showed not only prolonged but also greater pharmacological effects in mice and would therefore be a better and more effective drug candidate than the native FGF21.
Author contributions
J.Y., H.T., X.G. and W.B. designed the research study. J.Y. performed most of the experiments and analysed the data. L.B. and Q.W. performed some part of the research. J.Y. and L.B. wrote the manuscript.
Conflict of interest
The authors declare no conflicts of interest.
Declaration of transparency and scientific rigour
This Declaration acknowledges that this paper adheres to the principles for transparent reporting and scientific rigour of preclinical research recommended by funding agencies, publishers and other organizations engaged with supporting research.
Supporting information
Figure S1 Analysis of the building block by 2% agarose gel electrophoresis after phosphorylation and ligation. DNAs bands were stained with Goldview™. (A) 60 nucleotide fragments encoding 20 amino acids were isolated. (B) 600 nucleotide fragments encoding 200 amino acids were isolated.
Figure S2 SEC‐HPLC of standard proteins Analytical SEC was performed on a Zenix‐C 300 column (Sepax Technologies, Inc.) at a flow rate of 1 ml·min‐1 using High Performance Liquid Chromtography System (Agilent Technologies) equipped with an autosampler. The standard proteins were applied with 150 mmol·L‐1 sodium phosphate buffer (pH 7.0) and respective elution volumes were measured. Protein standards were each run independently in successive experiments. The molecular weights (MWs) of marker proteins (thyroglobulin, 670 kDa; BSA, 66 kDa; ovalbumin, 44 kDa; ribonuclease A, 13.7 kDa; vitamin B12, 1.35 kDa) was plotted versus their elution time and fitted by a nonlinear regression model (R2 = 0.9991).
Figure S3 Purity analysis of native FGF21 and PsTag600‐FGF21 by RP‐HPLC. The purities of FGF21 and PsTag600‐FGF21 were determined on a SepaxGP‐C18 column using an elution gradient from 5% v/v acetonitrile, 0.065% v/v TFA to 60% v/v acetonitrile, 0.065% TFA. Both profiles showed a single homogeneous peak with earlier elution of PsTag600‐FGF21(24.6% acetonitrile), relative to the corresponding unfused FGF21 (40.9% acetonitrile).
Figure S4 Analysis of plasma stability for PsTag600‐FGF21 Purified PsTag600‐FGF21 was incubated in the presence of BALB/c mouse plasma at 37°C for up to 48 h. Samples taken at different time points were subject to (A) 10% SDS‐PAGE and (B) Western blotting with a rabbit anti‐human FGF21 antibody. Lane 1, serum only (as control); lane 2, PsTag600‐FGF21; lane 3‐9: samples taken at 0, 1, 3, 6, 18, 24, and 48 h, respectively. Full length PsTag600‐FGF21 protein was detectable for up to 48 h, demonstrating high stability against serum proteases. (C) Western blotting quantitative analysis of Figure S4B by Quantity One ® (Bio‐Rad).
Figure S5 Human FGF21 was stabilized by PsTag polypeptides To demonstrate the stabilization of FGF21 conferred by PsTag polypeptides, FGF21 and PsTag600‐FGF21 (5 mg·mL‐1 in PBS) were incubated at 25°C (lanes 1 & 3) and 85°C (lanes 2 & 4) for 15 min at which time any insoluble protein was rapidly removed by centrifugation at 13800 x g for 20 min. The soluble fraction was then analyzed by 12% SDS‐PAGE. PsTag600‐FGF21 remained soluble after heating while a precipitate was visible after heat treatment of FGF21 alone.
Figure S6 PsTag600‐FGF21 retained activity following heat treatment Human β‐klotho binding was measured for FGF21 and PsTag600‐FGF21 after incubation for 15 min at 85°C versus untreated controls (N = 3). Here binding activities of FGF21 and PsTag600‐FGF21 were determined by measuring the EC50 for binding to human β‐klotho (58890KB, R&D) in a direct binding ELISA. Human β‐klotho (25 ng per well) was coated in 96‐well ELISA plate (Costar, USA), and various concentrations of FGF21 and PsTag fusion proteins were added to the pre‐coated plate. The bound protein complexes was detected by HRP labeled FGF21 antibody for each concentration, FGF21 antibody was purchased from Abcam and labelled by HRP labeling kit (Thermo). The OD450nm absorbance was plotted against protein concentrations and EC50 values were fitted with a single‐site binding model using GraphPad Prism 5. All values were reported as relative to FGF21 incubated at 25°C (set to 100%). FGF21 and PsTag600‐FGF21 samples were prepared as described in Figure S5. Thermal denaturation of FGF21 resulted in a significant loss of activity consistent with the observed precipitation (Figure S5), while PsTag600‐FGF21 retained the majority of its starting activity.
Figure S7 Analysis of biodegradability for PsTag600‐FGF21 Purified PsTag600‐FGF21 was incubated with different dilutions of a mouse kidney homogenate, at 37°C for 1 h and subjected to (A) 10% SDS‐PAGE and (B) Western blotting with a rabbit anti‐human FGF21 antibody. Lane 1, PsTag600‐FGF21 with homogenate; lane 2, 1:5 dilution of homogenate (as control); lane 3, purified PsTag600‐FGF21; lane 4‐8, PsTag600‐FGF21 incubated with the kidney homogenate at 1:5, 1:10, 1:50, 1:100 and 1:1000 dilution, respectively. The band for PsTag600‐FGF21 disappears with decreasing dilution of the kidney extract, showing that PsTag600‐FGF21 was rapidly degraded. Thus, in contrast with the poorly degradable chemical polymer PEG, PsTag polypeptides should be easily metabolized.
Figure S8 Gene analysis of liver and iWAT. (A) Relative mRNA levels of key genes in liver. Fas, fatty acid synthase; Scd‐1, stearoyl‐CoA desaturase 1; Pgc‐1α, PPARγ‐coactivator‐1α; Fgfr1, fibroblast growth factor receptor 1; Klb, β‐Klotho. (B) Relative mRNA levels of key genes in WAT. Ucp, uncoupling protein; Glut, glucose transporter. (N = 6 per group. Data are means ± SD. * P < 0.05 vs vehicle‐treated HFD mice.).
Table S1 A library of 10‐amino‐acid segments.
Table S2 PCR Primer Sequences.
Supporting info item
Acknowledgements
This research was supported by the National Natural Science Foundation of China (Nos. 81273426, 81430082, 81172974 and 31200694), the Fundamental Research Funds for the Central Universities (YD2014SK0002, 2015XPT02), Ph.D. Programs Foundation of Ministry of Education of China (20120096110007), Scientific Innovation Research of College Graduate in Jiangsu Province (KYLX_0624) and A Project Funded by the Priority Academic Program Development of Jiangsu Higher Education Institutions.
Yin, J. , Bao, L. , Tian, H. , Wang, Q. , Gao, X. , and Yao, W. (2016) Genetic fusion of human FGF21 to a synthetic polypeptide improves pharmacokinetics and pharmacodynamics in a mouse model of obesity. British Journal of Pharmacology, 173: 2208–2223. doi: 10.1111/bph.13499.
References
- Alexander SPH, Kelly E, Marrion N, Peters JA, Benson HE, Faccenda E et al. (2015a). The Concise Guide to PHARMACOLOGY 2015/16: Transporters. Br J Pharmacol 172: 6110–6202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Cidlowski JA, Kelly E, Marrion N, Peters JA, Benson HE et al. (2015b). The Concise Guide to PHARMACOLOGY 2015/16: Nuclear hormone receptors. Br J Pharmacol 172: 5956–5978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alvarez P, Buscaglia CA, Campetella O (2004). Improving protein pharmacokinetics by genetic fusion to simple amino acid sequences. J Biol Chem 279: 3375–3381. [DOI] [PubMed] [Google Scholar]
- Cleland JL, Geething NC, Moore JA, Rogers BC, Spink BJ, Wang CW et al. (2012). A novel long‐acting human growth hormone fusion protein (vrs‐317): enhanced in vivo potency and half‐life. J Pharm Sci 101: 2744–2754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Curtis MJ, Bond RA, Spina D, Ahluwalia A, Alexander SP, Giembycz MA et al. (2015). Experimental design and analysis and their reporting: new guidance for publication in BJP. Br J Pharmacol 172: 3461–3471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elsadek B, Kratz F (2012). Impact of albumin on drug delivery—new applications on the horizon. J Control Release 157: 4–28. [DOI] [PubMed] [Google Scholar]
- Gao M, Ma Y, Cui R, Liu D (2014). Hydrodynamic delivery of FGF21 gene alleviates obesity and fatty liver in mice fed a high‐fat diet. J Control Release 185: 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gimeno RE, Moller DE (2014). FGF21‐based pharmacotherapy–potential utility for metabolic disorders. Trends Endocrinol Metab 25: 303–311. [DOI] [PubMed] [Google Scholar]
- Hecht R, Li Y‐S, Sun J, Belouski E, Hall M, Hager T et al. (2012). Rationale‐based engineering of a potent long‐acting FGF21 analog for the treatment of type 2 diabetes. PLoS One 7: e49345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu J, Wang G, Liu X, Gao W (2015). Enhancing pharmacokinetics, tumor accumulation, and antitumor efficacy by elastin‐like polypeptide fusion of interferon alpha. Adv Mater 27: 7320–7324. [DOI] [PubMed] [Google Scholar]
- Huang J, Ishino T, Chen G, Rolzin P, Osothprarop TF, Retting K et al. (2013). Development of a novel long‐acting antidiabetic FGF21 mimetic by targeted conjugation to a scaffold antibody. J Pharmacol Exp Ther 346: 270–280. [DOI] [PubMed] [Google Scholar]
- Huang Z, Wang H, Lu M, Sun C, Wu X, Tan Y et al. (2011). A better anti‐diabetic recombinant human fibroblast growth factor 21 (rhFGF21) modified with polyethylene glycol. PLoS One 6: e20669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jain A, Jain SK (2008). PEGylation: an approach for drug delivery. A review. Crit Rev Ther Drug Carrier Syst 25: 403–447. [DOI] [PubMed] [Google Scholar]
- Kharitonenkov A, Adams AC (2014). Inventing new medicines: the FGF21 story. Mol Metab 3: 221–229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kharitonenkov A, Shiyanova TL, Koester A, Ford AM, Micanovic R, Galbreath EJ et al. (2005). FGF‐21 as a novel metabolic regulator. J Clin Invest 115: 1627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kilkenny C, Browne W, Cuthill IC, Emerson M, Altman DG (2010). Animal research: reporting in vivo experiments: the ARRIVE guidelines. J Gene Med 12: 561–563. [DOI] [PubMed] [Google Scholar]
- Knop K, Hoogenboom R, Fischer D, Schubert US (2010). Poly (ethylene glycol) in drug delivery: pros and cons as well as potential alternatives. Angew Chem Int Ed 49: 6288–6308. [DOI] [PubMed] [Google Scholar]
- Kontermann R (2012). Therapeutic Proteins: Strategies to Modulate Their Plasma Half‐lives, Vol. 48 John Wiley & Sons: Hoboken. [Google Scholar]
- Kontermann RE (2011). Strategies for extended serum half‐life of protein therapeutics. Curr Opin Biotechnol 22: 868–876. [DOI] [PubMed] [Google Scholar]
- McGrath JC, Lilley E (2015). Implementing guidelines on reporting research using animals (ARRIVE etc.): new requirements for publication in BJP. Br J Pharmacol 172: 3189–3193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacEwan SR, Chilkoti A (2010). Elastin‐like polypeptides: biomedical applications of tunable biopolymers. Pept Sci 94: 60–77. [DOI] [PubMed] [Google Scholar]
- Pasut G, Veronese FM (2012). State of the art in PEGylation: the great versatility achieved after forty years of research. J Control Release 161: 461–472. [DOI] [PubMed] [Google Scholar]
- Sambrook J, Fritsch EF, Maniatis T (1989). Molecular Cloning, Vol. 1 Cold spring harbor laboratory press: New York. [Google Scholar]
- Schellenberger V, Wang C‐w, Geething NC, Spink BJ, Campbell A, To W et al. (2009). A recombinant polypeptide extends the in vivo half‐life of peptides and proteins in a tunable manner. Nat Biotechnol 27: 1186–1190. [DOI] [PubMed] [Google Scholar]
- Schlapschy M, Binder U, Börger C, Theobald I, Wachinger K, Kisling S et al. (2013). PASylation: a biological alternative to PEGylation for extending the plasma half‐life of pharmaceutically active proteins. Protein Eng Des Sel 26: 489–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlapschy M, Theobald I, Mack H, Schottelius M, Wester H‐J, Skerra A (2007). Fusion of a recombinant antibody fragment with a homo‐amino‐acid polymer: effects on biophysical properties and prolonged plasma half‐life. Protein Eng Des Sel 20: 273–284. [DOI] [PubMed] [Google Scholar]
- Sheng X, Wang M, Lu M, Xi B, Sheng H, Zang YQ (2011). Rhein ameliorates fatty liver disease through negative energy balance, hepatic lipogenic regulation, and immunomodulation in diet‐induced obese mice. Am J Physiol Endocrinol Metab 300: E886–E893. [DOI] [PubMed] [Google Scholar]
- Smith R, Duguay A, Weiszmann J, Stanislaus S, Belouski E, Cai L et al. (2013). A novel approach to improve the function of FGF21. BioDrugs 27: 159–166. [DOI] [PubMed] [Google Scholar]
- Song L, Zhu Y, Wang H, Belov AA, Niu J, Shi L et al. (2014). A solid‐phase PEGylation strategy for protein therapeutics using a potent FGF21 analog. Biomaterials 35: 5206–5215. [DOI] [PubMed] [Google Scholar]
- Southan C, Sharman JL, Benson HE, Faccenda E, Pawson AJ, Alexander SPH et al. (2016). The IUPHAR/BPS Guide to PHARMACOLOGY in 2016: towards curated quantitative interactions between 1300 protein targets and 6000 ligands. Nucl Acids Res 44 (Database Issue): D1054–D1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stein S, Bachmann A, Lössner U, Kratzsch J, Blüher M, Stumvoll M et al. (2009). Serum levels of the adipokine FGF21 depend on renal function. Diabetes Care 32: 126–128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veronese FM, Mero A, Pasut G (2009). Protein PEGylation, basic science and biological applications. Basic Science and Clinical Applications. Springer: Basel: In PEGylated Protein Drugs, pp. 11–31. [Google Scholar]
- Xu J, Lloyd DJ, Hale C, Stanislaus S, Chen M, Sivits G et al. (2009). Fibroblast growth factor 21 reverses hepatic steatosis, increases energy expenditure, and improves insulin sensitivity in diet‐induced obese mice. Diabetes 58: 250–259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin J, Hu R, Chen M, Tang J, Li F, Yang Y et al. (2002). Effects of berberine on glucose metabolism in vitro. Metabolism 51: 1439–1443. [DOI] [PubMed] [Google Scholar]
- Zhang F, Liu MR, Wan HT (2014). Discussion about several potential drawbacks of PEGylated therapeutic proteins. Biol Pharm Bull 37: 335–339. [DOI] [PubMed] [Google Scholar]
- Zhang J, Li Y (2014). Fibroblast growth factor 21, the endocrine FGF pathway and novel treatments for metabolic syndrome. Drug Discov Today 19: 579–589. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Huo M, Zhou J, Xie S (2010). PKSolver: An add‐in program for pharmacokinetic and pharmacodynamic data analysis in Microsoft Excel. Comput Methods Programs Biomed 99: 306–314. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1 Analysis of the building block by 2% agarose gel electrophoresis after phosphorylation and ligation. DNAs bands were stained with Goldview™. (A) 60 nucleotide fragments encoding 20 amino acids were isolated. (B) 600 nucleotide fragments encoding 200 amino acids were isolated.
Figure S2 SEC‐HPLC of standard proteins Analytical SEC was performed on a Zenix‐C 300 column (Sepax Technologies, Inc.) at a flow rate of 1 ml·min‐1 using High Performance Liquid Chromtography System (Agilent Technologies) equipped with an autosampler. The standard proteins were applied with 150 mmol·L‐1 sodium phosphate buffer (pH 7.0) and respective elution volumes were measured. Protein standards were each run independently in successive experiments. The molecular weights (MWs) of marker proteins (thyroglobulin, 670 kDa; BSA, 66 kDa; ovalbumin, 44 kDa; ribonuclease A, 13.7 kDa; vitamin B12, 1.35 kDa) was plotted versus their elution time and fitted by a nonlinear regression model (R2 = 0.9991).
Figure S3 Purity analysis of native FGF21 and PsTag600‐FGF21 by RP‐HPLC. The purities of FGF21 and PsTag600‐FGF21 were determined on a SepaxGP‐C18 column using an elution gradient from 5% v/v acetonitrile, 0.065% v/v TFA to 60% v/v acetonitrile, 0.065% TFA. Both profiles showed a single homogeneous peak with earlier elution of PsTag600‐FGF21(24.6% acetonitrile), relative to the corresponding unfused FGF21 (40.9% acetonitrile).
Figure S4 Analysis of plasma stability for PsTag600‐FGF21 Purified PsTag600‐FGF21 was incubated in the presence of BALB/c mouse plasma at 37°C for up to 48 h. Samples taken at different time points were subject to (A) 10% SDS‐PAGE and (B) Western blotting with a rabbit anti‐human FGF21 antibody. Lane 1, serum only (as control); lane 2, PsTag600‐FGF21; lane 3‐9: samples taken at 0, 1, 3, 6, 18, 24, and 48 h, respectively. Full length PsTag600‐FGF21 protein was detectable for up to 48 h, demonstrating high stability against serum proteases. (C) Western blotting quantitative analysis of Figure S4B by Quantity One ® (Bio‐Rad).
Figure S5 Human FGF21 was stabilized by PsTag polypeptides To demonstrate the stabilization of FGF21 conferred by PsTag polypeptides, FGF21 and PsTag600‐FGF21 (5 mg·mL‐1 in PBS) were incubated at 25°C (lanes 1 & 3) and 85°C (lanes 2 & 4) for 15 min at which time any insoluble protein was rapidly removed by centrifugation at 13800 x g for 20 min. The soluble fraction was then analyzed by 12% SDS‐PAGE. PsTag600‐FGF21 remained soluble after heating while a precipitate was visible after heat treatment of FGF21 alone.
Figure S6 PsTag600‐FGF21 retained activity following heat treatment Human β‐klotho binding was measured for FGF21 and PsTag600‐FGF21 after incubation for 15 min at 85°C versus untreated controls (N = 3). Here binding activities of FGF21 and PsTag600‐FGF21 were determined by measuring the EC50 for binding to human β‐klotho (58890KB, R&D) in a direct binding ELISA. Human β‐klotho (25 ng per well) was coated in 96‐well ELISA plate (Costar, USA), and various concentrations of FGF21 and PsTag fusion proteins were added to the pre‐coated plate. The bound protein complexes was detected by HRP labeled FGF21 antibody for each concentration, FGF21 antibody was purchased from Abcam and labelled by HRP labeling kit (Thermo). The OD450nm absorbance was plotted against protein concentrations and EC50 values were fitted with a single‐site binding model using GraphPad Prism 5. All values were reported as relative to FGF21 incubated at 25°C (set to 100%). FGF21 and PsTag600‐FGF21 samples were prepared as described in Figure S5. Thermal denaturation of FGF21 resulted in a significant loss of activity consistent with the observed precipitation (Figure S5), while PsTag600‐FGF21 retained the majority of its starting activity.
Figure S7 Analysis of biodegradability for PsTag600‐FGF21 Purified PsTag600‐FGF21 was incubated with different dilutions of a mouse kidney homogenate, at 37°C for 1 h and subjected to (A) 10% SDS‐PAGE and (B) Western blotting with a rabbit anti‐human FGF21 antibody. Lane 1, PsTag600‐FGF21 with homogenate; lane 2, 1:5 dilution of homogenate (as control); lane 3, purified PsTag600‐FGF21; lane 4‐8, PsTag600‐FGF21 incubated with the kidney homogenate at 1:5, 1:10, 1:50, 1:100 and 1:1000 dilution, respectively. The band for PsTag600‐FGF21 disappears with decreasing dilution of the kidney extract, showing that PsTag600‐FGF21 was rapidly degraded. Thus, in contrast with the poorly degradable chemical polymer PEG, PsTag polypeptides should be easily metabolized.
Figure S8 Gene analysis of liver and iWAT. (A) Relative mRNA levels of key genes in liver. Fas, fatty acid synthase; Scd‐1, stearoyl‐CoA desaturase 1; Pgc‐1α, PPARγ‐coactivator‐1α; Fgfr1, fibroblast growth factor receptor 1; Klb, β‐Klotho. (B) Relative mRNA levels of key genes in WAT. Ucp, uncoupling protein; Glut, glucose transporter. (N = 6 per group. Data are means ± SD. * P < 0.05 vs vehicle‐treated HFD mice.).
Table S1 A library of 10‐amino‐acid segments.
Table S2 PCR Primer Sequences.
Supporting info item