

Abstract
Background and Purpose
Phosphate imbalance is often present in chronic kidney disease (CKD), and it contributes to a higher cardiovascular mortality rate. A phosphate binder is typically part of a treatment strategy for controlling phosphate imbalance. However, safety concerns and low compliance are two well‐recognized disadvantages of on‐market phosphate binders. This report describes the preclinical studies of VS‐505, a non‐absorbable, calcium‐ and aluminum‐free, plant‐derived polymer currently being evaluated in haemodialysis patients in Australia.
Experimental Approach
Normal Sprague Dawley (SD) rats or uraemic SD rats induced by 5/6 nephrectomy fed a high‐phosphate diet were treated with VS‐505 or sevelamer (0.05–10% in food) for 5 and 28 days respectively.
Key Results
Urinary and serum phosphate levels were significantly elevated in untreated rats, and were decreased by VS‐505 and sevelamer. VS‐505 increased faecal phosphate levels in a dose‐dependent manner. High‐phosphate diet also caused an increase in serum FGF‐23 and parathyroid hormone in nephrectomized (NX) rats, effects prevented by VS‐505 or sevelamer. Significant aortic calcification was observed in NX rats treated with 5% sevelamer, whereas VS‐505 at all doses tested did not show effects. VS‐505 had no effects on small intestine histomorphology and intestinal sodium‐dependent phosphate cotransporter gene expression. In vitro characterizations showed that VS‐505 has a relatively high density and low expansion volume when exposed to simulated gastric fluid.
Conclusions and Implications
VS‐505 is a safe and effective phosphate binder and may offer the advantage of having a reduced pill burden and minimal GI side effects for CKD patients.
Abbreviations
- CKD
chronic kidney disease
- GI
gastrointestinal
- H–E
haematoxylin–eosin
- NX
nephrectomized
- PTH
parathyroid hormone
- Pi
phosphate
- SD
Sprague Dawley
Tables of Links
| TARGETS |
|---|
| Voltage‐gated ion channels a |
| TRPV6 (ECaC2; CaT1) |
| Transporters b |
| NPT2b (sodium phosphate 2) |
| LIGANDS |
|---|
| Paricalcitol |
| PTH |
These Tables list key protein targets and ligands in this article which are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Southan et al., 2016) and are permanently archived in the Concise Guide to PHARMACOLOGY 2015/16 (a,bAlexander et al., 2015a, 2015b).
Introduction
Many body functions such as energy metabolism, nucleic acid synthesis, bone mineralization and cell signalling require inorganic phosphate (Berndt and Kumar, 2007). Because of the critical role of the kidney in maintaining mineral homeostasis, mineral and bone disorder begins early in the course of chronic kidney disease (CKD) (Moe et al., 2009). CKD patients are known to experience a high mortality rate from cardiovascular complications. Clinical studies have linked hyperphosphataemia to a higher cardiovascular mortality in CKD (Foley et al., 2005; Bellasi et al., 2011; Eddington et al., 2010). Furthermore, Eddington et al. (2010) and a report from the Framingham offspring study (Dhingra et al., 2007) showed that higher serum Pi levels, even when they are within the normal laboratory range recommended in the Kidney Disease Outcomes Quality Initiative guidelines (Eknoyan et al., 2003), are potentially linked to an increase in mortality in stage 3/4 CKD (Eddington et al., 2010) and/or increased cardiovascular risk in the general population (Dhingra et al., 2007). In dialysis‐dependent patients with stage 5 CKD, several studies have demonstrated a robust association between elevated serum phosphorous levels and increased all‐cause mortality (Block et al., 2004; Kalantar‐Zadeh et al., 2006).
Phosphate imbalance in CKD is clinically managed by two approaches: dietary restriction of phosphate intake and the use of oral phosphate binders (Kalantar‐Zadeh et al., 2010). Dietary restriction of phosphorus intake is difficult because phosphate is abundant in many foods (Ritz et al., 2012). Thus, most CKD patients with hyperphosphataemia (based on serum phosphate levels) take phosphate binders. Oral phosphate binders, generally classified as either calcium‐containing or calcium‐free, work by binding phopsphate in the gastrointestinal (GI) tract, which in turn leads to less phosphate being available for absorption and uptake into the body. A significant clinical management issue with on‐market phosphate binders is poor patient compliance due in large part to GI intolerability and large pill burden (size and number). In addition, safety concerns such as hypercalcaemia, aluminum toxicity, negative influence on other medication and accumulation in organs have been reported for some on‐market phosphate binders (Chiu et al., 2009; Ito et al., 2014; Wang et al., 2014).
Because controlling the phosphate imbalance in CKD patients is critical, there is a need to continue searching for novel phosphate binders that better match an ‘ideal’ product profile: (1) highly efficient at binding phosphate; (2) minimal patient compliance issues; and (3) reduced side effects and safety concerns. To this end, we have discovered a series of Ca‐free, aluminum‐free and non‐absorbable novel phosphate binders that are derived from natural plant‐based polymers commonly used in the food industry. Previously, we have published preclinical study data on VS‐501, a predecessor of VS‐505 (Wu‐Wong et al., 2014). VS‐505, with significantly improved potency and physical properties over VS‐501, is currently being evaluated in a clinical study in Australia involving haemodialysis patients (ClinicalTrials.gov Identifier #: NCT02469467). In this study, we present results from preclinical studies showing that VS‐505 is effective at binding phosphate and may have advantages over current on‐market phosphate binders with regard to its potentially low pill burden and minimal side effects on the GI tract.
Methods
Normal rat studies
Normal Sprague Dawley (SD) rats were used to screen compounds so that the compounds of interest could be further evaluated in the 5/6 nephrectomized (NX) rats (as described below). Male SD rats (body weight of ~400 g) were fed a standard rodent diet in powder form (Teklad LM‐485, 7912; Harlan Laboratories, Madison, WI, USA) supplemented with 0.66% phosphate (a 2:1 mixture of dibasic and monobasic potassium phosphate by dry weight) as previously described by Sutliff et al. (2011). Rats were treated with vehicle (gum Arabic, the unprocessed plant polymer), VS‐505 or sevelamer (HCl or carbonate, concentrations as indicated) at 0.04 to 10% (by dry weight) in their food for 5 days. The doses of the phosphate binders were chosen based on previous studies (Cozzolino et al., 2003; Rosenbaum et al., 1997; Wu‐Wong et al., 2014) and adjusted upward when needed. Rats were placed in metabolic cages with one rat per cage on the first (before dosing) and last days of treatment. Faeces and/or urine samples were collected for 24 h. Due to the limited availability of metabolic cages, the studies were staged such that at each time no more than 30 rats were handled. At the end of the studies, the data were compiled. The studies were carefully conducted so that each independent study included untreated, vehicle‐treated groups and also groups treated with the test agent of interest. The animals were randomly assigned to the study groups in each independent study. The final number of rats per group was as follows: high phosphate food alone, n = 11; 1% vehicle, n = 5; sevelamer HCl at 0.2, 1, 5 and 10%, n = 8, 8, 4, 6 respectively; VS‐505 at 0.04, 0.2, 1 and 5%, n = 6, 6, 9, 6, respectively; and sevelamer carbonate at 0.04, 0.2 and 1%, n = 5, 4, 4 respectively. Blood samples from each rat were collected via the tail vein (pre‐dosing) or cardiac puncture (before death) for serum preparation. Physiological parameters were determined as shown below.
5/6 Nephrectomized uraemic rat studies
The primary focus of these studies was to test the feasibility of using VS‐505 to treat hyperphosphataemia in CKD. The male SD 5/6 NX uraemic rat model has been shown to be a highly predictive animal model for late stage 4 CKD and was used previously to test VS‐501, a predecessor of VS‐505 (Wu‐Wong et al., 2014). Briefly, nephrectomy was performed on male, SD rats weighing ~200 g with a standard two‐step surgical ablation procedure as previously described (Wu‐Wong et al., 2011; Wu‐Wong et al., 2013a). At 6 weeks after the second surgery when uraemia had been established [as indicated by elevated serum creatinine and blood urea nitrogen (BUN) levels], rats on a high‐phosphate diet (as described above) were treated with vehicle, VS‐505 or sevelamer carbonate (concentrations as indicated) at 0.2–5% (by dry weight) in their food for 4 weeks. On days 0 (pre‐dosing), 14 (week 2) and 28 (week 4), rats were placed in metabolic cages with one rat per cage, and urine and faeces samples were collected during a period of 24 h. Due to the limited availability of metabolic cages, the studies were staged such that at each time point no more than 30 rats were handled. In addition, on average, 30% of the uraemic animals died during the course of the study, and any variations from animal to animal were noted. The studies were carefully conducted so that each independent study included sham, vehicle‐treated rats and groups treated with the test agent of interest. The animals were randomly assigned to the study groups in each independent study. At the end of the studies, the data were compiled. The final number of rats per group was as follows: sham, n = 9; 1% vehicle, n = 6; 5% vehicle, n = 9; sevelamer carbonate at 0.2, 1 and 5%, n = 7, 7, 8 respectively; VS‐505 at 0.2, 1 and 5%, n = 8, 7, 13 respectively. Blood samples from each rat were collected via tail vein (before and during dosing) or cardiac puncture (before death) for serum preparation. Physiological parameters were determined as described below.
All animal studies were conducted under the auspices of the Office of Animal Care and Institutional Biosafety, University of Illinois at Chicago. The studies conformed to the Guide for the Care and Use of Laboratory Animals published by the US National Institutes of Health (NIH Publication No. 85‐23, Eighth Edition, 2011). Humane end points for animals at the end of the studies (termination of experiments) were provided according to the approved animal protocols. Animal studies are reported in compliance with the ARRIVE guidelines (Kilkenny et al., 2010; McGrath & Lilley, 2015).
Measurements of physiological parameters
Serum parathyroid hormone (PTH) was measured using a rat intact PTH elisa kit obtained from Immutopics (San Clemente, CA, USA). Serum and urine Ca were measured using a Stanbio LiquiColor Ca assay kit (Boerne, TX, USA). Serum and urine phosphorus/phosphate levels were determined using a phosphate colorimetric assay (Catalog #K410–500; BioVision, Milpitas, CA, USA). Serum FGF‐23 was determined using a rat/mouse FGF‐23 (C‐terminal) elisa kit obtained from Immutopics. The serum creatinine and BUN concentrations were measured using a chemistry analyser.
To determine faecal phosphate, each faeces sample (2 g per sample) was ashed at 800°C for 30 min, followed by extraction in 5 mL of 12 N HCl with vortexing and shaking at room temperature for ~60 min. The supernatant, collected by centrifugation, was neutralized by adding an equal volume of 12 N NaOH. The mixture was centrifuged again, and the supernatant was collected for phosphate determination as described above. Total urinary and faecal Pi levels during a 24 h period were calculated.
Urinary protein determination
Urine samples were collected as described above. Urinary protein concentrations were determined using the Thermo Scientific Pierce 660 nm Protein Assay kit (Life Technologies, Grand Island, NY, USA). Total urinary protein levels during a 24 h period were calculated.
Tissue preparation and staining
Tissue samples, fixed in formalin for 1–3 days and then transferred to 70% alcohol, were embedded in wax. They were cut into 5 μm sections and stained with haematoxylin–eosin (H–E). For calcification studies, sections of aorta were stained by the von Kossa method and counterstained with nuclear fast red (Wu‐Wong et al., 2007). Positive (brown) staining in each aorta section was scored by two investigators in a blinded manner; the staining assessments (i.e. the % of area in the section showing positive, brown staining) by the two investigators were similar.
Real‐time RT‐PCR
Real‐time RT‐PCR was performed with an ABI 7500 Fast Real‐Time PCR System (Applied Biosystems, Foster City, CA, USA). Each sample consisted of a final volume of 25 μL containing 200 ng of mRNA, 100 nM (final concentration) each of the forward and reverse PCR primers and 250 nM (final concentration) of the TaqMan™ probe (Applied Biosystems). Temperature conditions consisted of a step of 30 min at 48°C and a step of 10 min at 95°C, followed by 45 cycles of 60°C for 1 min and 95°C for 15 s. Data were collected during each extension phase of the PCR reaction and analysed by a software package (Applied Biosystems). Threshold cycles were determined for each gene. The expression levels of three genes, intestinal Calb3 (the gene encoding calbindin D9K) and TRPV6 (the gene encoding CaT1 and ECaC2) that are involved in intestinal Ca transport and intestinal type II sodium‐dependent phosphate cotransporter (NPT2b), were determined.
In vitro polymer characterization
For expansion volume determination, VS‐505 or sevelamer carbonate at 0.1 g (dry powder) was incubated with 5 mL of simulated gastric fluid [0.2% (w v‐1) NaCl, 0.7% (v v‐1) HCl, without pepsin] at 37°C. The expanded volume was recorded at different time points (20 min, 1 h, 2 h and 5 h after the addition of simulated gastric fluid) following the incubation. To determine in vitro phosphate binding capacity, VS‐505 or sevelamer carbonate at 0.1 g was incubated with 10 mL of a Pi solution containing different Pi concentrations (different amounts of 85% phosphoric acid, 3.18 g of sodium carbonate and 4.68 g of NaCl in 1 L of water) at neutral pH for 24 h at 37°C. In separate studies, VS‐505 at 0.1 g was incubated with 10 mL of a 100 mM phosphate solution (6.85 mL of 85% phosphoric acid, 3.18 g of sodium carbonate and 4.68 g of NaCl in 1 L of water) at 37°C at different pH (as indicated) for 24 h. The samples were centrifuged, and the supernatant was collected for phosphate determination as described above. The density of the dry compressed powder was determined using a helium pycnometer.
Data analysis
Statistical comparisons between two treatment groups were performed using unpaired t‐tests with 95% confidence intervals of difference. Differences among different groups were assessed using a one‐way ANOVA followed by a Dunnett's post hoc test. The D'Agostino test was carried out to confirm the normal distribution for all the groups with eight or more values. There was no significant variance in homogeneity. The data and statistical analysis comply with the recommendations on experimental design and analysis in pharmacology (Curtis et al., 2015).
Materials
VS‐505 was made by Vidasym (Chicago, Illinois, USA). The synthesis, which involves chemical modification of gum Arabic (from the acacia tree) with a molecular weight at ~250, 000 Da [devoid of a higher molecular mass (~1, 450, 000 Da) protein component and also devoid of the glycoprotein component], and detailed analytical characterizations have been published previously (Wu‐Wong, 2014). Sevelamer {as Renagel® [hydrochloride (HCl) form] or Renvela® [carbonate form]} was obtained from the University of Illinois at Chicago pharmacy (IL, USA). Other reagents were of analytical grade.
Results
Effect of VS‐505 on serum, urinary and faecal phosphate and/or calcium in normal rats on a high‐phosphate diet
We first compared the efficacy of VS‐505 with that of sevelamer (HCl or carbonate) in normal rats on a high‐phosphate diet. After 5 days on a high‐phosphate diet, serum and urinary phosphate levels were significantly elevated in untreated rats (Figure 1A, B) but were significantly lowered in the groups treated with VS‐505 and sevelamer (vs. the untreated group). Sevelamer HCl (Renagel) and carbonate (Renvela) did not show significant differences in the parameters measured in this study, although it is known that the two drugs behave differently with regard to metabolic acidosis (Grinfeld et al., 2010). No significant changes in serum and urinary phosphate levels were observed in the group treated with vehicle alone. There was no change in serum Ca in any treatment group compared with pre‐dosing values (Figure 1C). VS‐505 at 5% had a modest effect on increasing urinary Ca (a 65% increase vs. pre‐dosing), whereas sevelamer hydrochloride at 5% increased urinary Ca by 553% (Figure 1D). Figure 2 shows that VS‐505 increased faecal phosphate levels in a dose‐dependent manner, suggesting that the polymer carried phosphate into faeces.
Figure 1.
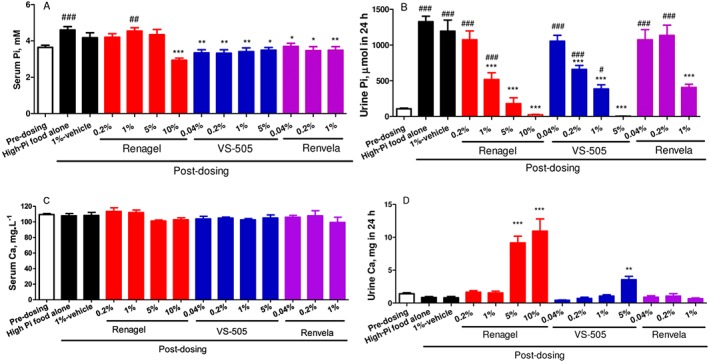
Serum and urinary phosphate (Pi) and Ca in normal rats on a high‐Pi diet. Male SD rats on a high‐Pi diet were treated with vehicle, VS‐505 or sevelamer (Renagel: sevelamer hydrochloride, and Renvela: sevelamer carbonate) in food (concentrations as indicated) for 5 days as described in the ‘Methods’ section. At two different time points (pre‐dosing and on day 5 after dosing), blood and urine samples were collected for Pi and Ca determination as described in the ‘Methods’ section. Mean ± SEM was calculated for each group (n per group as described in the ‘Methods’ section). The final number of rats per group was as follows: high‐Pi food alone, n = 11; 1% vehicle, n = 5; sevelamer HCl at 0.2, 1, 5 and 10%, n = 8, 8, 4, 6 respectively; VS‐505 at 0.04, 0.2, 1 and 5%, n = 6, 6, 9, 6 respectively; and sevelamer carbonate at 0.04, 0.2 and 1%, n = 5, 4, 4 respectively. (A) Serum Pi. (B) Urinary Pi in 24 h. (C) Serum Ca. (D) Urinary Ca in 24 h. One‐way ANOVA followed by Dunnett's test with 95% confidence intervals of difference was performed for statistical comparisons. # P < 0.05, ## P < 0.01, ### P < 0.001 versus pre‐dosing. *P < 0.05, **P < 0.01, ***P < 0.001 versus high‐Pi food alone.
Figure 2.
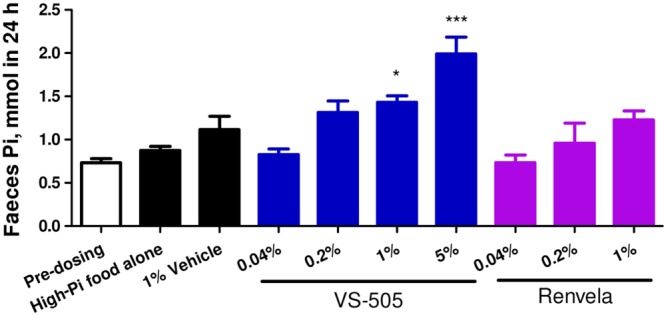
Faecal phosphate (Pi) in normal rats on a high‐Pi diet. Male SD rats were dosed with vehicle, VS‐505 or sevelamer carbonate as described in the ‘Methods’ section and in Figure 1. At two different time points (pre‐dosing and on day 5 after dosing), faeces samples were collected for Pi determination as described in the ‘Methods’ section. Mean ± SEM was calculated for each group. The final number of rats per group was as described in Figure 1. One‐way ANOVA followed by Dunnett's test with 95% confidence intervals of difference was performed for statistical comparisons. *P < 0.05, ***P < 0.001 versus high‐Pi food alone.
Effect of VS‐505 on serum, urinary and faecal phosphate and/or calcium in 5/6 NX rats on a high‐phosphate diet
VS‐505 was then evaluated in a CKD animal model. Serum BUN and creatinine levels were significantly increased in 5/6 NX rats at week 6 after surgery, indicating established uraemia (Table 1); the results are similar to those seen in our previous studies in 5/6 NX rats (Wu‐Wong et al., 2011, 2013a, 2013b, 2010a). VS‐505 or sevelamer carbonate had no significant effects on serum BUN and/or creatinine levels.
Table 1.
Serum BUN and creatinine in 5/6 nephrectomized uremic rats
| Serum BUN (mg·L−1) | Serum creatinine (mg·L−1) | |||||
|---|---|---|---|---|---|---|
| Parameters | Pre‐dosing | Week 2 | Week 4 | Pre‐dosing | Week 2 | Week 4 |
| Sham | 198.4 ± 9.6 | 178.2 ± 7.1 | 195.8 ± 7.9 | 3.7 ± 0.2 | 4.1 ± 0.6 | 3.5 ± 0.1 |
| NX‐1% vehicle | 374.3 ± 26.0# | 386.1 ± 30.3# | 416.8 ± 50.3* | 8.5 ± 1.1* | 10.6 ± 1.0* | 9.3 ± 1.5* |
| NX‐5% vehicle | 333.2 ± 28.9# | 299.3 ± 21.3 | 298.1 ± 43.7 | 10.4 ± 0.9* | 7.4 ± 0.7# | 9.9 ± 0.5* |
| 0.2% Renvela | 415.7 ± 30.2* | 438.9 ± 47.9* | 428.4 ± 66.7* | 7.7 ± 1.1# | 7.8 ± 0.5# | 8.5 ± 1.8* |
| 1% Renvela | 485.0 ± 19.9* | 424.3 ± 14.9* | 498.9 ± 28.8* | 7.7 ± 0.4* | 8.5 ± 0.2* | 9.1 ± 1.0* |
| 5% Renvela | 413.8 ± 16.8* | 388.7 ± 22.7* | 407.5 ± 32.4* | 9.4 ± 0.3* | 10.3 ± 0.4* | 10.2 ± 1.3* |
| 0.2% VS‐505 | 336.9 ± 32.0 † | 317.0 ± 30.9 | 336.0 ± 22.4 | 8.4 ± 0.5* | 9.1 ± 0.6* | 10.3 ± 1.0* |
| 1% VS‐505 | 356.9 ± 16.7 † | 371.8 ± 20.9# | 365.1 ± 23.4 † | 8.8 ± 0.6* | 9.9 ± 0.6* | 10.5 ± 0.8* |
| 5% VS‐505 | 340.0 ± 22.0# | 358.6 ± 28.2# | 364.2 ± 44.0# | 10.6 ± 0.5* | 8.6 ± 0.7* | 9.7 ± 0.3* |
Note: Pre‐dosing: week 6 after surgery. Week 2: week 8 after surgery with 2 weeks of dosing. Week 4: week 10 after surgery with 4 weeks of dosing. Sham rats were on a high‐Pi diet with no addition of vehicle or binders. Data are shown as mean ± SEM. The final number of rats per group was as follows: sham, n = 9; 1% vehicle, n = 6; 5% vehicle, n = 9; sevelamer carbonate at 0.2, 1 and 5%, n = 7, 7, 8 respectively; and VS‐505 at 0.2, 1 and 5%, n = 8, 7, 13 respectively. One‐way ANOVA followed by Dunnett's test with 95% confidence intervals of difference was performed for statistical comparisons.
P < 0.001.
P < 0.01.
P < 0.05 versus sham, pre‐dosing.
As shown in Figure 3A, B, a high‐phosphate diet led to a significant increase in urinary phosphate levels and a modest increase in serum phosphate in sham and in 5/6 NX rats treated with vehicle; the observation made on sham rats is consistent with that made in normal rats as shown above. Elevated phosphate levels were observed at both time points (at weeks 2 and 4 post‐dosing). VS‐505 or sevelamer reduced serum phosphate; both binders also reduced urinary phosphate in a dose‐dependent manner at weeks 2 and 4 post‐dosing.
Figure 3.
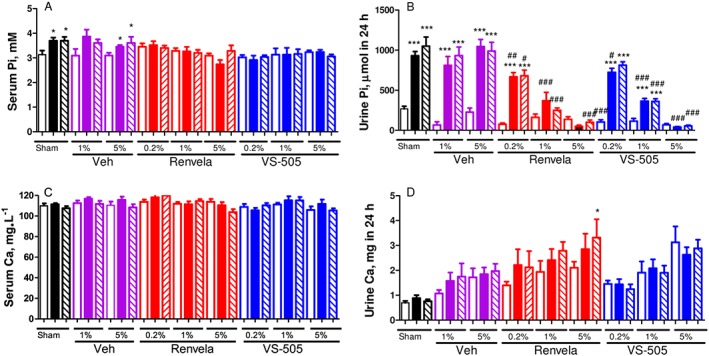
Serum and urinary phosphate (Pi) and Ca in 5/6 NX rats on a high‐Pi diet. NX rats were treated with vehicle (Veh, at 1 or 5%), VS‐505 or sevelamer carbonate (concentrations at 0.2, 1 or 5%) in food for 4 weeks. Sham rats on high‐Pi diet with no addition of Veh or binders served as control. At three different time points (pre‐dosing, at week 2 post‐dosing and at week 4 post‐dosing), blood and urine samples were collected for Pi or Ca determination as described in the ‘Methods’ section. Mean ± SEM was calculated for each group (n per group as described in the ‘Methods’ section). The final number of rats per group is shown in Table 1. (A) Serum Pi. (B) Urinary Pi in 24 h. (C) Serum Ca. (D) Urinary Ca in 24 h. One‐way ANOVA followed by Dunnett's test with 95% confidence intervals of difference was performed for statistical comparisons. *P < 0.05, ***P < 0.001 versus pre‐dosing, the same group. # P < 0.05, ## P < 0.01, ### P < 0.001 versus sham, the same time point. Open bar: pre‐dosing. Solid bar: week 2 post‐dosing. Pattern bar: week 4 post‐dosing.
Figure 3C, D shows that there was no significant difference in serum Ca across all groups. Interestingly, there was a significant increase in urinary Ca in the group treated with 5% sevelamer for 4 weeks. No significant difference was observed in faecal Ca across all groups (data not shown).
Figure 4 shows that a high‐phosphate diet led to an increase in faecal phosphate levels in sham rats and in NX rats treated with vehicle, which was observed at weeks 2 and 4 post‐dosing. VS‐505 or sevelamer further increased faecal phosphate in a dose‐dependent manner. These results suggest that both polymers carried phosphate into faeces.
Figure 4.
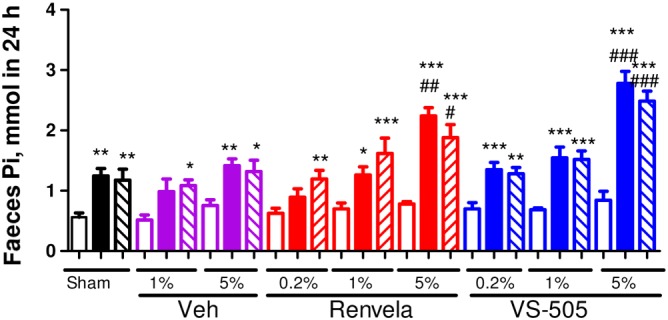
Faecal Pi in 5/6 NX rats on a high‐Pi diet. NX rats were treated with vehicle (Veh), VS‐505 or sevelamer carbonate in food for 4 weeks as described in the ‘Methods’ section and in Figure 3. Sham rats on high‐Pi diet with no addition of Veh or binders served as control. At three different time points (pre‐dosing, at week 2 post‐dosing and at week 4 post‐dosing), feces samples were collected for Pi determination as described in the ‘Methods’ section. Mean ± SEM was calculated for each group. The final number of rats per group was as presented in Table 1. One‐way ANOVA followed by Dunnett's test with 95% confidence intervals of difference was performed for statistical comparisons. *P < 0.05, **P < 0.01, ***P < 0.001 versus pre‐dosing, the same group. # P < 0.05, ## P < 0.01, ### P < 0.001 versus sham, the same time point. Open bar: pre‐dosing, the same group. Solid bar: week 2 post‐dosing. Pattern bar: week 4 post‐dosing.
Judging from the serum BUN and creatinine values (Table 1), VS‐505 and sevelamer carbonate did not significantly affect renal function, which was confirmed by the evaluation of urinary protein (Figure 5).
Figure 5.
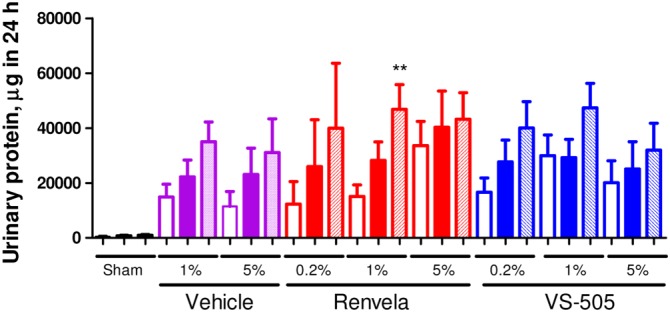
Urinary protein in 5/6 NX rats. NX rats were treated as described in Figure 3. Urine samples were collected for protein determination as described in the ‘Methods’ section. Mean ± SEM was calculated for each group. The final number of rats per group was as presented in Table 1. Differences among pre‐dosing, week 2 and week 4 groups were assessed using a one‐way ANOVA followed by a Dunnett's post hoc test. **P < 0.01 versus pre‐dosing. Open bar: pre‐dosing. Solid bar: week 2 post‐dosing. Pattern bar: week 4 post‐dosing.
Effect of VS‐505 on serum FGF‐23 and PTH in 5/6 NX rats on a high‐phosphate diet
As shown in Figure 6, serum FGF‐23 and PTH levels were significantly elevated in 5/6 NX rats, which were further elevated by the high‐phosphate diet. VS‐505 or sevelamer at 5% effectively prevented the further elevation in serum FGF‐23 and PTH levels induced by the high‐phosphate diet.
Figure 6.
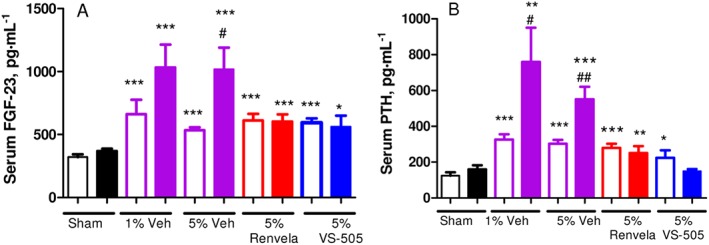
Serum PTH and FGF‐23 in 5/6 NX rats on a high‐Pi diet. NX rats were treated with vehicle (Veh), VS‐505 or sevelamer carbonate (5% in food) for 4 weeks as described in the ‘Methods’ section and in Figure 3. Sham rats on a high‐Pi diet with no addition of Veh or binders served as control. Serum samples were collected for PTH and FGF‐23 determination as described in the ‘Methods’ section. Mean ± SEM was calculated for each group. The final number of rats per group was as described in Table 1. (A) Serum FGF‐23. (B) Serum PTH. Statistical comparisons between two groups were performed by unpaired t‐test with 95% confidence intervals of difference. # P < 0.05, ## P < 0.01 versus pre‐dosing, the same group. *P < 0.05, **P < 0.01, ***P < 0.001 versus sham, pre‐dosing. Open bar: pre‐dosing. Solid bar: week 4 post‐dosing.
GI parameters in 5/6 NX rats
In an effort to investigate the effect of phosphate binders on intestinal physiology, H–E staining of intestinal samples was conducted to evaluate intestinal integrity. No significant difference across the different treatment groups was observed (data not shown). Furthermore, no significant changes in food consumption (measured every other day) were observed across different groups (data not shown).
To study further the effects of VS‐505 and sevelamer on intestinal physiology, we examined the expression of three genes involved in intestinal Ca and phosphate absorption. No significant differences were observed across the different groups in the gene expression of intestinal Calb3 (the gene encoding calbindin D9K) and TRPV6 (the gene encoding CaT1 and ECaC2) that are involved in intestinal Ca transport (data not shown). Previously, we have reported that vitamin D analogues such as calcitriol and paricalcitol induce the expression of Calb3 and TRPV6 (Nakane et al., 2007; Wu‐Wong et al., 2013b). There was also no significant difference across different groups in the intestinal type II sodium‐dependent phosphate cotransporter (NPT2b) gene expression (data not shown).
Aorta calcification in 5/6 NX rats
To investigate the effect of VS‐505 on vascular calcification in the CKD rat model, aorta samples were randomly taken from each 5/6 NX group and stained for calcification. Two representative samples of the aortic cross‐sectional area are shown in Figure 7A: the top one (0%) exhibited no positive staining (i.e. no brown staining in the whole section), and the bottom one (~50%) exhibited ~50% positive staining (i.e. ~50% of the section showing brown staining). The percentage of positive staining assessed for each treatment group was averaged and summarized in Figure 7B. There was no significant difference between the sham, NX treated with 5% vehicle, or NX treated with 5% VS‐505 groups. A significant increase in positive staining was observed in the 5% sevelamer group.
Figure 7.
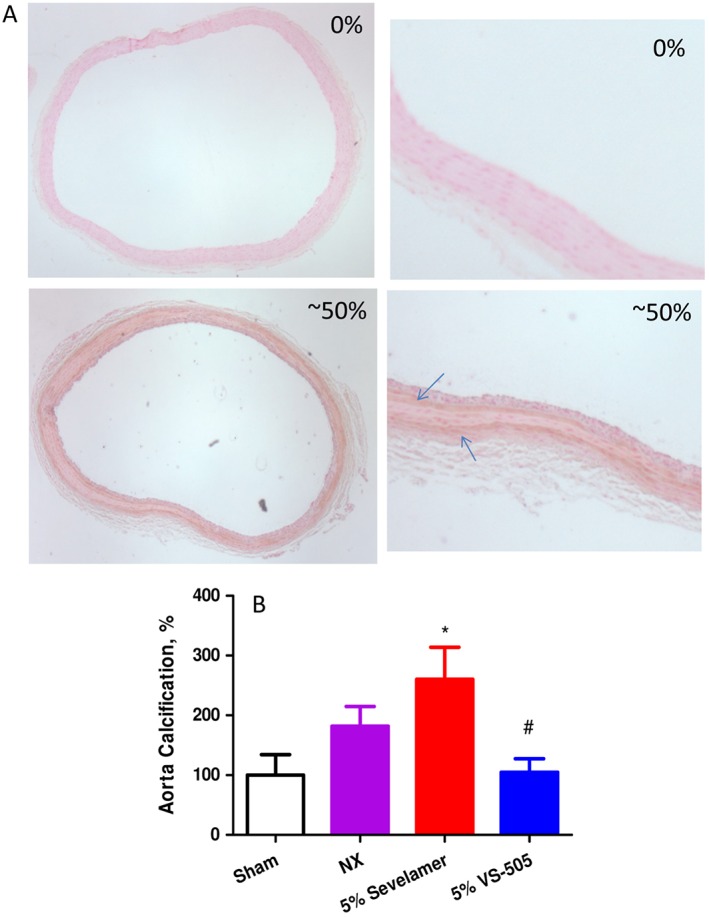
Aorta calcification determination in 5/6 NX rats on a high‐Pi diet. NX rats were treated with 5% vehicle, 5% sevelamer carbonate or 5% VS‐505 in food for 4 weeks as described above. Sham rats on a high‐Pi diet with no addition of vehicle or binders served as control. Aorta was collected and processed as described in the ‘Methods’ section (1–4 aorta rings per rat). The final number of rats per group was as described in Table 1. (A) Two representative samples of stained aortic section: the top sample (0%) with no positive staining (i.e. no brown staining in the whole section) and the bottom one (~50%) with ~50% positive staining (i.e. ~50% of the section showing brown staining). The right panel displays the whole aorta section; the left panel shows an enlarged area of part of the aorta section. Arrow: indicating the positive (brown) staining. (B) Positive (brown) staining in each aorta section was scored as described in the ‘Methods’ section. The average percentage of positive staining for each treatment group was first calculated and then normalized by sham (with an average scored value at 10.3 ± 3.5 mainly due to background) and expressed as % of sham. Mean ± SEM was calculated for each group. Statistical comparisons between two groups were performed by unpaired t‐test with 95% confidence intervals of difference. *P < 0.05 versus sham. # P < 0.05 versus sevelamer.
In vitro characterization
The phosphate binding capacity of VS‐505 was then determined in vitro. Figure 8A shows that VS‐505 binds phosphate within a wide physiologically relevant range of pH. Sevelamer (hydrochloride or bicarbonate) was tested using the same experimental conditions, and all determinations at different initial values of pH ended up within a range of pH of 6–8 (data not shown), indicating that formulated sevelamer is heavily buffered. Although we were unable to obtain sevelamer that is not formulated/buffered for direct comparison studies, a previous publication on sevelamer hydrochloride (Rosenbaum et al., 1997) reported that the optimal binding condition for sevelamer hydrochloride is at pH 7.0. Figure 8B shows that VS‐505 binds phosphate with an estimated maximal binding capacity (Bmax) of 2.5 mmol·g−1 and a K D of 56 mM (vs. sevelamer carbonate with a Bmax of 2.7 mmol·g−1 and a K D of 74 mM).
Figure 8.
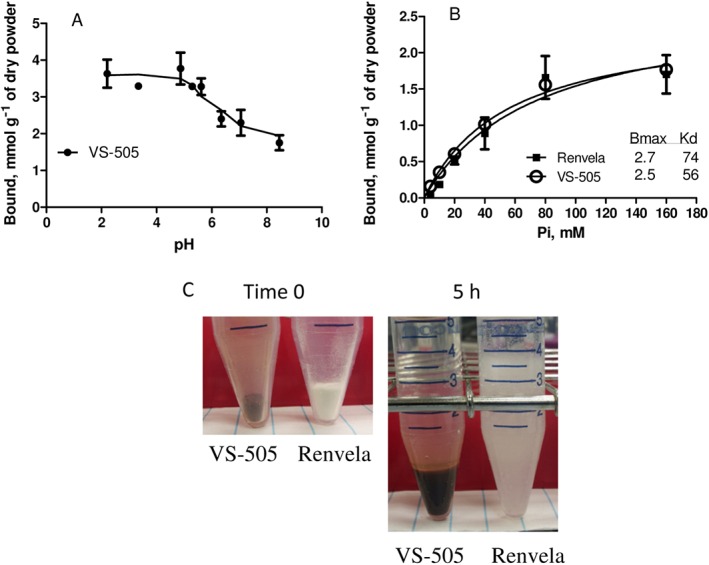
In vitro studies. (A) VS‐505 at 0.1 g was incubated with 10 mL of a 100 mM Pi solution (6.85 mL of 85% phosphoric acid, 3.18 g of sodium carbonate and 4.68 g of NaCl in 1 L of water) at 37°C at different values of pH (as indicated) for 24 h. The samples were processed for Pi determination as described in the ‘Methods’ section. Each point was triplicated, and the graph is representative of three independent experiments. (B) VS‐505 or sevelamer carbonate at 0.1 g was incubated with 10 mL of a Pi solution containing different concentrations of Pi (different amounts of 85% phosphoric acid, 3.18 g of sodium carbonate and 4.68 g of NaCl in 1 L of water) at neutral pH for 24 h at 37°C. Each point was triplicated, and the graph is representative of two independent experiments. (C) VS‐505 or sevelamer carbonate at 0.1 g (dry powder, time 0) was incubated with 5 mL of simulated gastric fluid [0.2% (w v‐1) NaCls, 0.7% (v v‐1) HCl, without pepsin] at 37°C for 5 h. The picture is representative of three independent experiments.
As mentioned above, the large pill burden associated with some phosphate binders is one factor contributing to relatively low compliance in CKD patients (Chiu et al., 2009; Wang et al., 2014). In general, a substance with a higher density will probably have a smaller size. When comparing VS‐505 with sevelamer, the density of the dry compressed powder was determined by a helium pycnometer to be 1.91 g·cm−3 for VS‐505 and 1.27 g·cm−3 for sevelamer carbonate, suggesting that, with the same mass (dose), VS‐505 is likely to have a significantly smaller pill size.
GI discomfort for some phosphate binders is another factor contributing to patient compliance issues. Unfortunately, data on food consumption and intestinal physiology from the current 5/6 NX rat studies did not provide information on this particular issue. Because the expansion volume of a non‐absorbable phosphate binder after exposure to gastric fluid may contribute to significant GI side effects (Chiu et al., 2009; Ito et al., 2014; Wang et al., 2014), the expansion volume of VS‐505 versus sevelamer was compared in vitro. When VS‐505 or sevelamer carbonate was incubated with 5 mL of simulated gastric fluid at 37°C, the expanded volume at 20 min was ~0.2 cm3 for 0.1 g of VS‐505 versus 4 cm3 for 0.1 g of sevelamer, and the expanded volume remained the same after 5 h of incubation. A representative picture of VS‐505 versus sevelamer following exposure to simulated gastric fluid for 5 h is shown in Figure 8C. These data clearly show that the expansion volume of VS‐505 is ~20‐fold less than that of sevelamer.
Discussion
The results of the present study show that VS‐505, a novel, non‐absorbable phosphate binder derived from a natural plant‐based polymer, is highly potent at binding phosphate in the GI tract and in carrying phosphate into faeces. In vitro characterization shows that the phosphate binding capacity of VS‐505 is similar to that of sevelamer (HCl or carbonate). However, whereas sevelamer (HCl) functions optimally at neutral pH (Rosenbaum et al., 1997), VS‐505 binds phosphate within a wide range of physiologically relevant pH, which may explain why VS‐505 is slightly more efficacious in carrying phosphate into the faeces in 5/6 NX rats (as shown in Figure 4).
A serious concern associated with some phosphate binders to provide effective control of phosphate imbalance in CKD patients is related to their low patient compliance, which is probably caused by large pill size/number as well as GI discomfort (Chiu et al., 2009; Waheed et al., 2013; Ito et al., 2014; Wang et al., 2014). Compared with sevelamer (HCl or carbonate), VS‐505 has a significantly higher density, suggesting its potential for being of reduced pill size/number. Furthermore, VS‐505 has a significantly lower volume of expansion when it is exposed to simulated gastric fluid. In the GI system, foods undergo major particle size reduction starting from chewing in the mouth to transit through the stomach for further processing (Kong and Singh, 2008). The secretion of gastric juice and stomach contraction are essential for mixing and homogenizing food in order to transform it into small particles of 1–2 mm in size (Kong and Singh, 2008). For phosphate binders that are not digestible, volume expansion in the GI tract is thought to be associated with GI intolerability (Ito et al., 2014). Therefore, the unique attributes of VS‐505, such as high density and low expansion volume, may serve as the basis to potentially improve patient compliance and thus provide for more effective management of phosphate imbalance.
Hyperphosphataemia in CKD is known to contribute to progressive vascular calcification and increase cardiovascular complications (Latif et al., 2013). Non‐Ca‐containing phosphate binders have been shown to be associated with a beneficial effect on vascular calcification progression in both preclinical and clinical studies (Frazao and Adragao, 2008; Raggi et al., 2010; Iida et al., 2013; Phan et al., 2013). However, Block et al. (2012) reported that the phosphate binders currently used in clinical settings, sevelamer included, can potentially cause an increase in vascular calcification. The same report (Block et al., 2012) also hinted that worsened vascular calcification may actually be a ‘class effect’ for phosphate binders. This subject is controversial, and more studies will be needed to provide some clarifications. For this reason, we investigated whether VS‐505 would affect vascular calcification. The results suggest that a propensity to increase vascular calcification may not necessarily be a ‘class effect’ associated with all phosphate binders because VS‐505 did not show any significant effect on aorta calcification. Although it was not the focus of the current study to investigate the mechanism of action for vascular calcification associated with sevelamer, a possible explanation may be found in the observations on urinary Ca in normal and 5/6 NX rats; sevelamer seems to interfere with Ca homeostasis, which may ultimately contribute to increased vascular calcification. The observations are consistent with those previously reported by Behets et al. (2014) that sevelamer increases serum and urinary Ca more than lanthanum, especially in normal rats.
Data from this study show that serum FGF‐23, a phosphorus regulating factor (Wolf, 2010), was higher in the 5/6 NX rats than in sham rats and was further elevated by the high‐phosphate diet; both sevelamer carbonate and VS‐505 prevented the effect of the high‐phosphate diet on FGF‐23. These results are consistent with clinical observations reported for patients with CKD that phosphate binders prevent the increase in FGF‐23 (Gupta et al., 2004; Martin and Gonzalez, 2011; Karczmarewicz et al., 2012). Although both sevelamer carbonate and VS‐505 prevented any further increase in serum FGF‐23, only sevelamer appeared to exacerbate aortic calcification. The existence of a direct link between FGF‐23 and vascular calcification is an issue that is still being debated (Masai et al., 2013; Moldovan et al., 2013; Ozkok et al., 2013). Our results suggest that, under the conditions in this study, vascular calcification may occur independently of serum FGF‐23 levels.
Serum PTH was significantly higher in the 5/6 NX rats, and it also became progressively more elevated when rats were fed the high‐phosphate diet, an effect prevented by both sevelamer carbonate and VS‐505. VS‐505 seems to exhibit a larger (but not statistically significant) effect on serum PTH than sevelamer. Sevelamer HCl has been shown to reduce serum PTH in an earlier study (Chertow et al., 1999), but a recent meta‐analysis study by Liu et al. (2014) reported that patients treated with non‐Ca‐based phosphate binders (NCPB) including sevelamer exhibit significantly lower serum Ca levels and higher iPTH levels than those treated with Ca‐based Pi binders. Furthermore, the osteoid volume and osteoblast numbers were significantly higher in the NCPB group (Liu et al., 2014). Because serum phosphorus, Ca and PTH play important roles in bone remodelling, additional studies are needed to further investigate the effects of VS‐505 on the bone.
NPT2b is known to play a key role in phosphate absorption and homeostasis (Sabbagh et al., 2009). It was previously shown that an adaptive increase in NaPi‐2b expression occurs in the duodenum as a response to an acute switch from a low‐to‐high phosphate diet (Giral et al., 2009). It seems that a change in the phosphate concentrations in the GI tract may potentially impact the expression of NaPi‐2b. The implication is that long‐term usage of phosphate binders in CKD patients can potentially alter GI physiology and/or NPT2b gene expression. Block et al. (2012) also discussed this issue in their paper, stating that ‘if phosphate binders were to upregulate expression of intestinal NPT2b and enhance paracellular transport, the efficacy of binders could be limited, particularly after missed doses’. Results from the current study suggest that administration of VS‐505 or sevelamer carbonate in food for 1 month in 5/6 NX rats had no significant effects on small intestinal NPT2b gene expression and intestinal histomorphology in general, which seems to support the current practice of long‐term usage of phosphate binders (Raggi et al., 2010).
In summary, the current preclinical report shows that VS‐505 is effective in binding phosphate in the GI tract and in carrying phosphate into faeces. Moreover, its effect is accomplished with a potentially reduced pill burden and minimal volume expansion in the GI tract. Clinical studies in haemodialysis patients are ongoing in Australia to confirm the potential of VS‐505 to treat phosphate imbalance and to improve long‐term patient outcomes.
Author contributions
Y.‐w.C. participated in research design, conducted experiments, contributed new reagents or analytic tools and performed data analysis. J.L.W. participated in research design, conducted experiments, contributed new reagents or analytic tools, performed data analysis and wrote or contributed to the writing of the manuscript. J.R.W.‐W. participated in research design, conducted experiments, contributed new reagents or analytic tools, performed data analysis and wrote or contributed to the writing of the manuscript. J.T.W. conducted experiments and contributed new reagents or analytic tools.
Conflict of interest
Y.‐w.C., J.L.W. and J.R.W.‐W. work for Vidasym. J.T.W. was a summer intern at Vidasym in 2013 and 2014.
Declaration of transparency and scientific rigour
This Declaration acknowledges that this paper adheres to the principles for transparent reporting and scientific rigour of preclinical research recommended by funding agencies, publishers and other organisations engaged with supporting research.
Acknowledgements
The project described was supported by grant number SBIR 1R43DK096698‐01 from the NIH. Its contents are solely the responsibility of the authors and do not necessarily represent the official views of the NIH awarding component.
This manuscript is original work not previously published in any substantial part and is not under consideration of publication elsewhere. Some of the data in this manuscript have been presented in a poster at 2014 Kidney Week (SA‐PO049). The manuscript has been read and approved for submission by all authors. The signature of the corresponding author is on behalf of all the authors.
Wu‐Wong, J. R. , Chen, Y. , Wong, J. T. , and Wessale, J. L. (2016) Preclinical studies of VS‐505: a non‐absorbable highly effective phosphate binder. British Journal of Pharmacology, 173: 2278–2289. doi: 10.1111/bph.13510.
References
- Alexander SPH, Catterall WA, Kelly E, Marrion N, Peters JA, Benson HE et al. (2015a). The Concise Guide to PHARMACOLOGY 2015/16: Voltage‐gated ion channels. Br J Pharmacol 172: 5904–5941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Kelly E, Marrion N, Peters JA, Benson HE, Faccenda E et al. (2015b). The Concise Guide to PHARMACOLOGY 2015/16: Transporters. Br J Pharmacol 172: 6110–6202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Behets GJ, Dams G, Damment SJ, Martin P, De Broe ME, D'Haese PC (2014). Differences in gastrointestinal calcium absorption after the ingestion of calcium‐free phosphate binders. Am J Physiol Renal Physiol 306: F61–F67. [DOI] [PubMed] [Google Scholar]
- Bellasi A, Mandreoli M, Baldrati L, Corradini M, Di Nicolo P, Malmusi G et al. (2011). Chronic kidney disease progression and outcome according to serum phosphorus in mild‐to‐moderate kidney dysfunction. Clin J Am Soc Nephrol 6: 883–891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berndt T, Kumar R (2007). Phosphatonins and the regulation of phosphate homeostasis. Annu Rev Physiol 69: 341–359. [DOI] [PubMed] [Google Scholar]
- Block GA, Klassen PS, Lazarus JM, Ofsthun N, Lowrie EG, Chertow GM (2004). Mineral metabolism, mortality, and morbidity in maintenance hemodialysis. J Am Soc Nephrol 15: 2208–2218. [DOI] [PubMed] [Google Scholar]
- Block GA, Wheeler DC, Persky MS, Kestenbaum B, Ketteler M, Spiegel DM et al. (2012). Effects of phosphate binders in moderate CKD. J Am Soc Nephrol 23: 1407–1415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chertow GM, Dillon M, Burke SK, Steg M, Bleyer AJ, Garrett BN et al. (1999). A randomized trial of sevelamer hydrochloride (RenaGel) with and without supplemental calcium. Strategies for the control of hyperphosphatemia and hyperparathyroidism in hemodialysis patients. Clin Nephrol 51: 18–26. [PubMed] [Google Scholar]
- Chiu YW, Teitelbaum I, Misra M, de Leon EM, Adzize T, Mehrotra R (2009). Pill burden, adherence, hyperphosphatemia, and quality of life in maintenance dialysis patients. Clin J Am Soc Nephrol 4: 1089–1096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cozzolino M, Staniforth ME, Liapis H, Finch J, Burke SK, Dusso AS et al. (2003). Sevelamer hydrochloride attenuates kidney and cardiovascular calcifications in long‐term experimental uremia. Kidney Int 64: 1653–1661. [DOI] [PubMed] [Google Scholar]
- Curtis MJ, Bond RA, Spina D, Ahluwalia A, Alexander SP, Giembycz MA et al. (2015). Experimental design and analysis and their reporting: new guidance for publication in BJP. Br J Pharmacol 172: 3461–3471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dhingra R, Sullivan LM, Fox CS, Wang TJ, D'Agostino RB, Gaziano JM et al. (2007). Relations of serum phosphorus and calcium levels to the incidence of cardiovascular disease in the community. Arch Intern Med 167: 879–885. [DOI] [PubMed] [Google Scholar]
- Eddington H, Hoefield R, Sinha S, Chrysochou C, Lane B, Foley RN et al. (2010). Serum phosphate and mortality in patients with chronic kidney disease. Clin J Am Soc Nephrol 5: 2251–2257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eknoyan G, Levin A, Levin NW (2003). K/DOQI clinical practice guidelines for bone metabolism and disease in chronic kidney disease. Am J Kidney Dis 42 (4 Suppl 3): S1–201. [PubMed] [Google Scholar]
- Foley RN, Wang C, Collins AJ (2005). Cardiovascular risk factor profiles and kidney function stage in the US general population: the NHANES III study. Mayo Clin Proc 80: 1270–1277. [DOI] [PubMed] [Google Scholar]
- Frazao JM, Adragao T (2008). Treatment of hyperphosphatemia with sevelamer hydrochloride in dialysis patients: effects on vascular calcification, bone and a close look into the survival data. Kidney Int Suppl 74: S38–S43. [DOI] [PubMed] [Google Scholar]
- Giral H, Caldas Y, Sutherland E, Wilson P, Breusegem S, Barry N et al. (2009). Regulation of rat intestinal Na‐dependent phosphate transporters by dietary phosphate. Am J Physiol Renal Physiol 297: F1466–F1475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grinfeld J, Inaba A, Hutchison AJ (2010). Update and critical appraisal of sevelamer in the management of chronic renal failure. Open Access J Urol 2: 161–170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta A, Winer K, Econs MJ, Marx SJ, Collins MT (2004). FGF‐23 is elevated by chronic hyperphosphatemia. J Clin Endocrinol Metab 89: 4489–4492. [DOI] [PubMed] [Google Scholar]
- Iida A, Kemmochi Y, Kakimoto K, Tanimoto M, Mimura T, Shinozaki Y et al. (2013). Ferric citrate hydrate, a new phosphate binder, prevents the complications of secondary hyperparathyroidism and vascular calcification. Am J Nephrol 37: 346–358. [DOI] [PubMed] [Google Scholar]
- Ito K, Takeshima A, Shishido K, Wakasa M, Kumata C, Matsuzaka K et al. (2014). Treatment of hyperphosphatemia with bixalomer in Japanese patients on long‐term hemodialysis with gastrointestinal symptoms. Ther Apher Dial 18 (Suppl 2): 19–23. [DOI] [PubMed] [Google Scholar]
- Kalantar‐Zadeh K, Gutekunst L, Mehrotra R, Kovesdy CP, Bross R, Shinaberger CS et al. (2010). Understanding sources of dietary phosphorus in the treatment of patients with chronic kidney disease. Clin J Am Soc Nephrol 5: 519–530. [DOI] [PubMed] [Google Scholar]
- Kalantar‐Zadeh K, Kuwae N, Regidor DL, Kovesdy CP, Kilpatrick RD, Shinaberger CS et al. (2006). Survival predictability of time‐varying indicators of bone disease in maintenance hemodialysis patients. Kidney Int 70: 771–780. [DOI] [PubMed] [Google Scholar]
- Karczmarewicz E, Czekuc‐Kryskiewicz E, Lorenc RS (2012). Pathologies of calcium–phosphate homeostasis. Postepy Biochem 58: 474–477. [PubMed] [Google Scholar]
- Kilkenny C, Browne W, Cuthill IC, Emerson M, Altman DG (2010). Animal research: Reporting in vivo experiments: the ARRIVE guidelines. Br J Pharmacol 160: 1577–1579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kong F, Singh RP (2008). Disintegration of solid foods in human stomach. J Food Sci 73: R67–R80. [DOI] [PubMed] [Google Scholar]
- Latif F, Khalid MM, Khan F, Omar Z, Ali FA (2013). Role of hyperphosphatemia‐mediated vascular calcification in cardiovascular outcomes and its management: a review. J Cardiovasc Med (Hagerstown) 14: 410–415. [DOI] [PubMed] [Google Scholar]
- Liu L, Wang Y, Chen H, Zhu X, Zhou L, Yang Y (2014). The effects of non‐calcium‐based phosphate binders versus calcium‐based phosphate binders on cardiovascular calcification and bone remodeling among dialysis patients: a meta‐analysis of randomized trials. Ren Fail 36: 1244–1252. [DOI] [PubMed] [Google Scholar]
- Martin KJ, Gonzalez EA (2011). Prevention and control of phosphate retention/hyperphosphatemia in CKD‐MBD: what is normal, when to start, and how to treat? Clin J Am Soc Nephrol 6: 440–446. [DOI] [PubMed] [Google Scholar]
- Masai H, Joki N, Sugi K, Moroi M (2013). A preliminary study of the potential role of FGF‐23 in coronary calcification in patients with suspected coronary artery disease. Atherosclerosis 226: 228–233. [DOI] [PubMed] [Google Scholar]
- McGrath JC, Lilley E (2015). Implementing guidelines on reporting research using animals (ARRIVE etc.): new requirements for publication in BJP. Br J Pharmacol 172: 3189–3193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moe SM, Drueke TB, Block GA (2009). KDIGO clinical practice guideline for the diagnosis, evaluation, prevention, and treatment of chronic kidney disease‐mineral and bone disorder (CKD‐MBD). Kidney Int Suppl 76: S1–130. [DOI] [PubMed] [Google Scholar]
- Moldovan D, Moldovan I, Rusu C, Kacso I, Patiu IM, Gherman‐Caprioara M (2013). FGF‐23, vascular calcification, and cardiovascular diseases in chronic hemodialysis patients. Int Urol Nephrol 46: 121–128. [DOI] [PubMed] [Google Scholar]
- Nakane M, Ma J, Rose AE, Osinski MA, Wu‐Wong JR (2007). Differential effects of vitamin D analogs on calcium transport. J Steroid Biochem Mol Biol 103: 84–89. [DOI] [PubMed] [Google Scholar]
- Ozkok A, Kekik C, Karahan GE, Sakaci T, Ozel A, Unsal A et al. (2013). FGF‐23 associated with the progression of coronary artery calcification in hemodialysis patients. BMC Nephrol 14: 241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phan O, Maillard M, Peregaux C, Mordasini D, Stehle JC, Funk F et al. (2013). PA21, a new iron‐based noncalcium phosphate binder, prevents vascular calcification in chronic renal failure rats. J Pharmacol Exp Ther 346: 281–289. [DOI] [PubMed] [Google Scholar]
- Raggi P, Vukicevic S, Moyses RM, Wesseling K, Spiegel DM (2010). Ten‐year experience with sevelamer and calcium salts as phosphate binders. Clin J Am Soc Nephrol 5 (Suppl 1): S31–S40. [DOI] [PubMed] [Google Scholar]
- Ritz E, Hahn K, Ketteler M, Kuhlmann MK, Mann J (2012). Phosphate additives in food–a health risk. Dtsch Arztebl Int 109: 49–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenbaum DP, Holmes‐Farley SR, Mandeville WH, Pitruzzello M, Goldberg DI (1997). Effect of RenaGel, a non‐absorbable, cross‐linked, polymeric phosphate binder, on urinary phosphorus excretion in rats. Nephrol Dial Transplant 12: 961–964. [DOI] [PubMed] [Google Scholar]
- Sabbagh Y, O'Brien SP, Song W, Boulanger JH, Stockmann A, Arbeeny C et al. (2009). Intestinal npt2b plays a major role in phosphate absorption and homeostasis. J Am Soc Nephrol 20: 2348–2358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sutliff RL, Walp ER, El‐Ali AM, Elkhatib S, Lomashvili KA, O'Neill WC (2011). Effect of medial calcification on vascular function in uremia. Am J Physiol Renal Physiol 301: F78–F83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Southan C, Sharman JL, Benson HE, Faccenda E, Pawson AJ, Alexander SPH et al. (2016). The IUPHAR/BPS Guide to PHARMACOLOGY in 2016: towards curated quantitative interactions between 1300 protein targets and 6000 ligands. Nucleic Acids Res 44: D1054–D1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waheed AA, Pedraza F, Lenz O, Isakova T (2013). Phosphate control in end‐stage renal disease: barriers and opportunities. Nephrol Dial Transplant 28: 2961–2968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang S, Alfieri T, Ramakrishnan K, Braunhofer P, Newsome BA (2014). Serum phosphorus levels and pill burden are inversely associated with adherence in patients on hemodialysis. Nephrol Dial Transplant 29: 2092–2099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolf M (2010). Forging forward with 10 burning questions on FGF23 in kidney disease. J Am Soc Nephrol 21: 1427–1435. [DOI] [PubMed] [Google Scholar]
- Wu‐Wong JR (2014). metal ion‐functional fiber component complex compositions, preparation and uses thereof. WO 2014/138016 A1 (published September 12, 2014).
- Wu‐Wong JR, Chen YW, Gaffin R, Hall A, Wong JT, Xiong J et al. (2014). VS‐501: a novel, non‐absorbed, calcium‐ and aluminum‐free, highly effective phosphate binder derived from natural plant polymer. Pharmacol Res Perspect 2: e00042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu‐Wong JR, Kawai M, Chen YW, Nakane M (2011). VS‐105: a novel vitamin D receptor modulator with cardiovascular protective effects. Br J Pharmacol 164: 551–560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu‐Wong JR, Kawai M, Chen YW, Wessale JL, Huang CJ, Wu MT et al. (2013a). Two novel vitamin D receptor modulators with similar structures exhibit different hypercalcemic effects in 5/6 nephrectomized uremic rats. Am J Nephrol 37: 310–319. [DOI] [PubMed] [Google Scholar]
- Wu‐Wong JR, Nakane M, Chen YW (2013b). Mapping the time‐dependent effects of paricalcitol on serum calcium, phosphorus and parathyroid hormone levels in 5/6 nephrectomized uremic rats. Life Sci 92: 161–166. [DOI] [PubMed] [Google Scholar]
- Wu‐Wong JR, Nakane M, Gagne GD, Brooks KA, Noonan WT (2010a). Comparison of the pharmacological effects of paricalcitol and doxercalciferol on the factors involved in mineral homeostasis. Int J Endocrinol 2010 (Article ID 621687): 1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu‐Wong JR, Nakane M, Ma J, Ruan X, Kroeger PE (2007). Elevated phosphorus modulates vitamin D receptor‐mediated gene expression in human vascular smooth muscle cells. Am J Physiol Renal Physiol 293: F1592–F1604. [DOI] [PubMed] [Google Scholar]