

Abstract
Background and Purpose
Previous findings suggest that the growth and differentiation factor midkine (Mdk) is a negative regulator of osteoblast activity and bone formation, thereby raising the possibility that a specific Mdk antagonist might improve bone formation during fracture healing.
Experimental Approach
In the present study, we investigated the effects of a monoclonal anti‐Mdk antibody (Mdk‐Ab) on bone healing using a standardized femur osteotomy model in mice. Additional in vitro experiments using chondroprogenitor and preosteoblastic cells were conducted to analyse the effects of recombinant Mdk and Mdk‐Ab on differentiation markers and potential binding partners in these cells.
Key Results
We demonstrated that treatment with Mdk‐Ab accelerated bone healing in mice based on increased bone formation in the fracture callus. In vitro experiments using preosteoblastic cells showed that Mdk‐Ab treatment abolished the Mdk‐induced negative effects on the expression of osteogenic markers and Wnt/β‐catenin target proteins, whereas the differentiation of chondroprogenitor cells was unaffected. Phosphorylation analyses revealed an important role for the low‐density lipoproteinLDL receptor‐related protein 6 in Mdk signalling in osteoblasts.
Conclusions and Implications
We conclude that Mdk‐Ab treatment may be a potential novel therapeutic strategy to enhance fracture healing in patients with orthopaedic complications such as delayed healing or non‐union formation.
Abbreviations
- Acan
aggrecan
- Alpl
alkaline phosphatase
- BMD
bone mineral density
- BV/TV
bone volume to tissue volume ratio
- LRP‐1
low‐density lipoprotein receptor‐related protein 1
- LRP‐6
low‐density lipoprotein receptor‐related protein 6
- Mdk
midkine
- Mdk‐Ab
Mdk antibody
- PTPRz
protein tyrosine phosphatase ζ (RTP type Z)
- TV
tissue volume
Tables of Links
| TARGETS | |
|---|---|
| Other protein targets a | Catalytic receptors c |
| α‐Tubulin | PTPRz (RTP type Z1) |
| GPCRs b | Enzymes d |
| F4/80 (ADGRE1) | GSK3β |
| LIGANDS | |
|---|---|
| β‐Catenin | Pleiotrophin |
| Insulin | TGFβ1 |
These Tables list key protein targets and ligands in this article which are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Southan et al., 2016) and are permanently archived in the Concise Guide to PHARMACOLOGY 2015/16 (a,b,c, dAlexander et al., 2015a, 2015b, 2015c, 2015d).
Introduction
Although the treatment of long‐bone fractures has clearly improved over recent decades, the rate of delayed bone healing or even non‐union formation remains at up to 10% (King et al., 2007; Cadet et al., 2013). Currently, the therapeutic options available to treat fracture‐associated complications include the application of growth factors, for example, bone morphogenetic protein 2, bone grafts and non‐invasive mechanical interventions, including low‐intensity pulsed ultrasound (Busse et al., 2002; Einhorn, 2003; Giannoudis and Dinopoulos, 2010). However, the success of these therapies appears to be variable, with inconsistent results reported in the literature (Poynton and Lane, 2002). Therefore, there is still a need for an effective, robust, well‐characterized and non‐invasive systemic therapy to improve fracture healing.
One growth factor that may be a promising molecular drug target during bone healing is midkine (Mdk). Mdk belongs, together with pleiotrophin, to the unique family of heparin‐binding growth and differentiation factors (Muramatsu, 1993). Mdk is expressed in the tooth germ of murine embryos and during embryonic limb development (Mitsiadis et al., 1995; Ohta et al., 1999). Interestingly, Mdk deficiency positively impacts bone remodelling in the adult organism, whereby Mdk‐deficient mice display an increased trabecular bone formation rate in the vertebral bodies at 12 and 18 months of age (Neunaber et al., 2010). Additionally, Mdk‐deficient mice were protected from ovariectomy‐induced bone loss. In vitro stimulation of osteoblasts using recombinant Mdk induced several genes that encode proteins related to extracellular matrix mineralization. Moreover, Mdk was shown to inhibit Wnt/β‐catenin signalling in mechanically‐stimulated osteoblasts through the putative Mdk receptor, protein tyrosine phosphatase ζ (PTPRz), while Mdk‐deficient mice exhibited increased cortical bone formation in response to mechanical load (Liedert et al., 2011).
We previously demonstrated that Mdk is expressed in chondrocytes during femoral fracture healing and that Mdk‐deficient mice display delayed early fracture healing due to delayed cartilage formation in the fracture callus (Haffner‐Luntzer et al., 2014). However, because the Mdk‐deficient mice used already displayed an altered bone phenotype, the possibility that the fracture healing process was non‐physiological in these animals due to an adaption to a complete absence of Mdk during bone development and remodelling could not be ruled out. Additionally, less is known about the role of locally expressed versus circulating Mdk during fracture healing. For this reason, the aim of the present study was firstly to investigate the effects of an Mdk antibody (Mdk‐Ab), which inhibits circulating Mdk during fracture healing in WT mice, and secondly to elucidate the molecular mechanisms and signalling pathways of Mdk in chondrocytes and preosteoblastic cells, which are involved in bone regeneration.
The results of the present study demonstrated that the Mdk‐Ab significantly accelerated fracture healing in mice, presumably based on increased β‐catenin signalling in osteoblasts caused by decreased levels of circulating Mdk. Therefore, Mdk‐Ab treatment may be a potential novel therapeutic strategy to enhance fracture healing in patients with orthopaedic complications.
Methods
Study approval
All animal experiments were in compliance with international regulations for the care and use of laboratory animals (Directive 2010/63/EU) with the approval of the Local Ethical Committee (No. 1079, Regierungspräsidium Tübingen, Germany). Animal studies are reported in compliance with the ARRIVE guidelines (Kilkenny et al., 2010; McGrath & Lilley, 2015).
Animal experiments
Female, 9‐month‐old C57BL/6J mice were provided by the University of Ulm and were maintained in groups of two to four animals per cage (370 cm2) on a 14 h light and 10 h dark circadian rhythm with water and food available ad libitum.
Surgery and Mdk antibody treatment
The surgical procedure was approved by the Local Animal Care Committee and has been published previously (Rontgen et al., 2010; Haffner‐Luntzer et al., 2014). Briefly, all mice received a standardized osteotomy at the midshaft of the right femur using a 0.4 mm Gigli saw (RISystem, Davos, Switzerland) stabilized using an external fixator (axial stiffness of 3.0 N·mm−1; RISystem). The mice were randomly divided into three groups. The Mdk‐Ab (mouse anti‐human Mdk IgG1 monoclonal antibody, cross reactive to murine Mdk; elisa EC50 to human Mdk: 31.7 ng·mL−1; and elisa EC50 to mouse Mdk: 37.8 ng·mL−1, provided by Cellmid Ltd., Sydney, Australia) was administered s.c. at 25 mg·kg−1 twice weekly for 3 weeks; this treatment was initiated immediately post‐operatively to the animals of group 1. The animals of group 2 were treated in parallel using the vehicle PBS. Another group of mice were injected with equal amounts of mouse IgG1 isotype control (Ultra‐LEAF purified mouse IgG1, #401408; BioLegend, San Diego, CA, USA) to validate the PBS control. The mice were killed 4, 10, 21 or 28 days after surgery using carbon dioxide (n = 6–8 per group at each time point). Blood samples were collected from mice killed at day 0, 4, 10 or 21. The fractured and intact femurs were removed from all mice for further analysis.
Serum analysis
The Mdk protein level in the serum was determined using a human Mdk elisa kit (provided by Cellmid Ltd.) as described in the manufacturer's protocol, which was cross reactive with murine Mdk.
Biomechanical testing
Biomechanical testing of the intact and fractured femurs of the mice killed on days 21 and 28 was performed using a non‐destructive three‐point bending test as described previously (Rontgen et al., 2010). Briefly, after removal of the fixator, a bending load (maximum 4 N) was applied to the top of the cranio‐lateral callus side. The flexural rigidity of the bones was calculated using the slope of the load–deflection curve. The relative flexural rigidity of the fractured femur was calculated as the ratio between the fractured and intact femur from the same mouse.
Micro‐computed tomography (μCT)
The femurs were analysed using a micro‐CT (μCT) scanning device (Bruker, Skyscan 1172; Kontich, Belgium) operating at a voxel resolution of 8 μm (50 kV, 200 mA). The volume of interest covered the periosteal callus between the fractured cortices. Bone mineral density (BMD) was assessed using two phantoms with a defined hydroxyapatite density (250 and 750 mg·cm−3) within each scan. The apparent BMD of the fracture callus was evaluated without a threshold, whereas the bone volume to tissue volume ratio (BV/TV) was determined using a global threshold of 642 mg hydroxyapatite cm−3 as described previously and in accordance with the American Society for Bone and Mineral Research guidelines for μCT analysis (Bouxsein et al., 2010; Wehrle et al., 2015).
Histomorphometry of undecalcified femora
The amounts of bone, cartilage and fibrous tissue in the whole callus between the two inner pin holes were determined using undecalcified histological sections of fractured femurs explanted on days 21 and 28, as described previously (Haffner‐Luntzer et al., 2014). Femurs were fixed in 4% formalin, dehydrated in an ascending ethanol series and embedded in methyl methacrylate. Cross sections of 7 μm were prepared and stained using Giemsa for histomorphometric analysis. The amounts of bone, cartilage and fibrous tissue were determined using image analysis software (Leica mmaf 1.4.0 Imaging System; Leica, Heerbrugg, Switzerland). To identify osteoblasts and osteoblast surface, sections were stained using toluidine blue and analysed under 400‐fold magnification. Tartrate‐resistant acid phosphatase staining was used to identify osteoclast numbers.
Histomorphometry of decalcified femora, immunohistochemistry and immunofluorescence
Femurs of mice killed 4, 10 or 21 days post‐surgery were fixed in 4% formalin, decalcified using 20% EDTA (pH 7.2–7.4) for 10–12 days and embedded in paraffin after dehydration in an ascending ethanol series. Longitudinal cross sections of 7 μm thickness were prepared and stained using Safranin O for tissue quantification. Immunohistochemical staining of Mdk and β‐catenin was performed using the following antibodies: polyclonal goat anti‐mouse Mdk‐Ab (sc‐1398; Santa Cruz Biotechnology, Dallas, TX, USA; cross reactive to human, mouse and rat Mdk), polyclonal rabbit anti‐mouse β‐catenin antibody (AB19022; EMD Millipore Corporation, Merck, Darmstadt, Germany), HRP‐conjugated streptavidin (Zytomed Systems, Berlin, Germany), donkey anti‐goat IgG F(ab′)2 biotin‐conjugated (sc‐3854; Santa Cruz Biotechnology) and goat anti‐rabbit IgG biotin‐conjugated (B2770; Invitrogen, Thermo Fisher Scientific, Waltham, MA, USA). Species‐specific non‐targeting immunoglobulins were used as isotype controls. 3‐Amino‐9‐ethylcarbazol (Zytomed Systems) was used as the chromogen, and the sections were counterstained using haematoxylin (Waldeck, Münster, Germany) as described previously (Haffner‐Luntzer et al., 2014). Quantification of the positively stained regions for β‐catenin was performed using the image analysis software Adobe Photoshop CS4 (Adobe, Dublin, Ireland). The colour gamut of positive staining was determined with the colour picker tool and a tolerance of 40. The positively stained pixels were counted in the histogram and calculated against all pixels of the image to determine the percentage of positively stained area. Immunofluorescence double staining for Mdk and F4/80 (also known as ADGRE1) was performed using rat anti‐mouse F4/80 (ab6640; AbD Serotec, Kidlington, UK), rabbit anti‐rat IgG Alexa Fluor 594‐conjugated (A‐21211; Molecular Probes, Thermo Fisher Scientific, Waltham, MA, USA), mouse anti‐human Mdk (Cellmid; cross reactive to murine Mdk) and rabbit‐anti mouse IgG FITC‐conjugated (ab6724, Abcam, Cambridge, UK). Species‐specific non‐targeting immunoglobulins were used as isotype controls. The M.O.M basic kit (mouse antibody on mouse tissue detection kit; Vector Laboratories, Burlingame, CA, USA) was used for blocking the sections.
Cell culture
Preosteoblastic MC3T3‐E1 cells were provided by Sigma‐Aldrich (Taufkirchen, Germany) and were cultured in α‐minimum essential medium (Gibco, Thermo Fisher Scientific, Waltham, MA, USA) containing 10% fetal calf serum (FCS) (PAA Laboratories, Cölbe, Germany), 1% penicillin/streptomycin (Gibco) and 1% l‐glutamine (Biochrom, Merck, Berlin, Germany). Osteogenic differentiation was induced by adding 10 mM β‐glycerophosphate and 0.2 mM ascorbate‐2‐phosphate (both Sigma‐Aldrich) to the culture medium. Cells were seeded in 6‐ or 24‐well plates at 20 000 cells cm−2 for differentiation experiments.
ATDC5 chondroprogenitor cells were provided by Sigma‐Aldrich and cultured as described previously (Shukunami et al., 1996). Cells were seeded in 6‐ or 24‐well plates at 10 000 cells cm−2. Chondrogenic differentiation was induced by supplementing a normal culture medium with 10 μg·mL−1 human insulin (Sigma‐Aldrich) and 5 ng·mL−1 human transforming growth factor β1 (TGFβ1; R&D Systems, Minneapolis, MN, USA). Recombinant Mdk was added to the culture medium for 6 h on day 5 of differentiation at 100 ng·mL−1 (Dianova, Hamburg, Germany) and Mdk‐Ab at 2 μg·mL−1. All experiments were performed three times in duplicate or triplicate.
The murine mesenchymal cell line C3H10T1/2 (ATCC, Manassas, VA, USA) was used as a control for the analysis of Mdk expression in different cell lines. The cells were cultured in α‐minimum essential medium (Gibco) supplemented with heat‐inactivated 10% FCS (PAA Laboratories), 1% l‐glutamine (Biochrom) and 1% penicillin–streptomycin (Gibco). Murine macrophage‐like RAW 264.7 cells (from LGC Standards GmbH/American Type Culture Collection, Wesel, Germany) were grown at 1500 cells cm−2 in DMEM (Gibco) containing 10% FCS (PAA), 1% l‐glutamine (Biochrom) and 1% penicillin–streptomycin (Gibco). The culture medium was replaced twice a week. Mdk expression was analysed by real‐time RT‐PCR in undifferentiated C3H10T1/2, MC3T3‐E1 and RAW 264.7 cells.
Real time – RT‐PCR
Cells were lysed in RLT buffer (Qiagen, Hilden, Germany) containing 10 μL·mL−1 β‐mercaptoethanol (Sigma‐Aldrich). Lysates were homogenized using QIAshredder columns, and total RNA was isolated using the RNeasy Mini Kit (both Qiagen), whereas DNA was digested using the RNase‐free DNase Kit (Qiagen). We transcribed 1 μg of total RNA into cDNA using the Omniscript RT Kit (Qiagen) in a total volume of 20 μL according to the manufacturer's instructions. Quantitative PCR was performed using Brilliant Sybr Green QPCR Master Mix Kit (Stratagene, Amsterdam, Netherlands) according to the manufacturer's protocols. β2‐microglobulin was used as housekeeping gene (F: 5′‐ATA CGC CTG CAG AGT TAA GCA‐3′ and R: 5′‐TCA CAT GTC TCG ATC CCA GT‐3′). Osteogenic and chondrogenic cell differentiation was analysed using specific primers for alkaline phosphatase (Alpl; F: 5′‐GCT GAT CAT TCC CAC GTT TT‐3′ and R: 5′‐GAG CCA GAC CAA AGA TGG AG‐3′), Lef1 (F: 5′‐TCA TCA CCT ACA GCG ACG AG‐3′ and R: 5′‐TGA CAT CTG ACG GGA TGT GT‐3′) and aggrecan (Acan; F: 5′‐AAC TTC TTT GCC ACC GGA GA‐3′ and R: 5′‐GGT GCC CTT TTT ACA CGT GAA‐3′). Mdk expression in MC3T3‐E1 cells and mouse macrophage‐like RAW 264.7 cells was analysed using (Mdk; F: 5′‐AGA CCA TCC GCG TGA CTA‐AG‐3′ and R: 5′‐GGC TTT GGT CTT TGA CTT GG‐3′). Relative gene expression was calculated using the δ–δCT method with PCR efficiency correction using LinRegPCR (Academic Medical Centre, Amsterdam, Netherlands) software as previously described (Ramakers et al., 2003).
Western blot analysis
We resolved 10 μg of cellular lysate protein using SDS‐PAGE and transferred this to a nitrocellulose membrane (BioRad, Hercules, CA, USA). The membranes were incubated with antibody to α‐tubulin, cFOS, β‐catenin, phospho‐β‐catenin (Ser33/37/Thr41) (all Cell Signaling, Merck Millipore, Darmstadt, Germany), Alpl (R&D Systems), low‐density lipoprotein receptor‐related protein 1 (LRP‐1) (Abcam), low‐density lipoprotein receptor‐related protein 6 (LRP‐6), phospho‐LRP‐6 (both Cell Signaling) or Mdk (Santa Cruz Biotechnologies) overnight at 4°C respectively. Protein bands were visualized as described previously (Liedert et al., 2011).
Co‐immunoprecipitation
Cells were differentiated for 5 days in six‐well plates and incubated with Mdk for 1 h in ice‐cold PBS. Cross‐linker solution (10 mM DSP dithiobis (succinimidyl propionate)) was added for 30 min at room temperature. Cells were incubated with stop solution (1 M Tris) for 15 min, washed twice and lysed using IP‐lysis buffer (Pierce, Thermo Fisher Scientific, Rockford, IL, USA). Cell debris was removed by centrifugation at 12 000× g for 10 min at 4°C. Protein A‐sepharose beads coupled with either goat IgG or goat Mdk‐Ab (Santa Cruz Biotechnology) were added to the solution and incubated overnight at 4°C. Complexes were centrifuged at 12 000× g for 1 min and washed with lysis buffer. Protein complexes were lysed from the beads by incubating in SDS sample buffer (125 mM Tris/HCl + 8.5% glycerine + 1% SDS + 0.1% DTT) for 5 min at 96°C and for 30 min at 37°C. Co‐immunoprecipitated proteins were visualized by western blotting.
Data and statistical analysis
Sample size was calculated based on a previous fracture healing study for the main outcome parameter flexural rigidity in the fractured femur (power: 80%, α = 0.05) (Wehrle et al., 2015). Statistical analysis for the in vivo experiments was performed using the non‐parametric Mann–Whitney U‐test with SPSS software (SPSS Inc., Chicago, IL, USA). The results of the in vitro experiments were analysed for significance using either the Kruskal–Wallis test with Dunn's post hoc test or the Mann–Whitney U‐test. All results are presented as box plots with median, first and third quartiles and maximum and minimum values. Values of P < 0.05 were considered to be statistically significant. The data and statistical analysis comply with the recommendations on experimental design and analysis in pharmacology (Curtis et al., 2015).
Results
Mdk‐Ab treatment increased the mechanical competence and bone formation in the fracture callus
Biomechanical testing and μCT analysis revealed that the injection of IgG isotype control antibody did not affect fracture healing compared with PBS injection (Supporting Information Fig. S1a and b). These results validated the use of PBS as an appropriate vehicle control for further experiments.
Biomechanical testing demonstrated that Mdk‐Ab treatment significantly increased the relative and absolute flexural rigidity of the fractured femurs after both 21 and 28 days of healing compared with the control group (Figure 1A and Supporting Information Fig. S1c). Flexural rigidity of the intact femurs was unaffected by the treatment (Supporting Information Fig. S1d). μCT analysis of the fracture callus revealed an enhanced BV/TV at these time points, whereas the data reached statistical significance only on day 21, possibly due to enhanced callus remodelling in the treatment group on day 28 (Figure 1B). Callus size and geometry were unaffected at an intermediate time point of fracture healing (day 21), whereas the tissue volume (TV) was significantly lower in Mdk‐Ab‐treated animals after 28 days of healing (Figure 1C, D). These data also support the conclusion that there was an enhanced callus remodelling at that time point. Histomorphometric analysis of the fracture callus tissue composition showed that Mdk‐Ab‐treated mice displayed a significantly increased amount of newly formed bone after both 10 and 21 days (Figure 2). No significant differences were observed in the fracture callus tissue composition at the late stage of fracture healing (day 28, data not shown).
Figure 1.
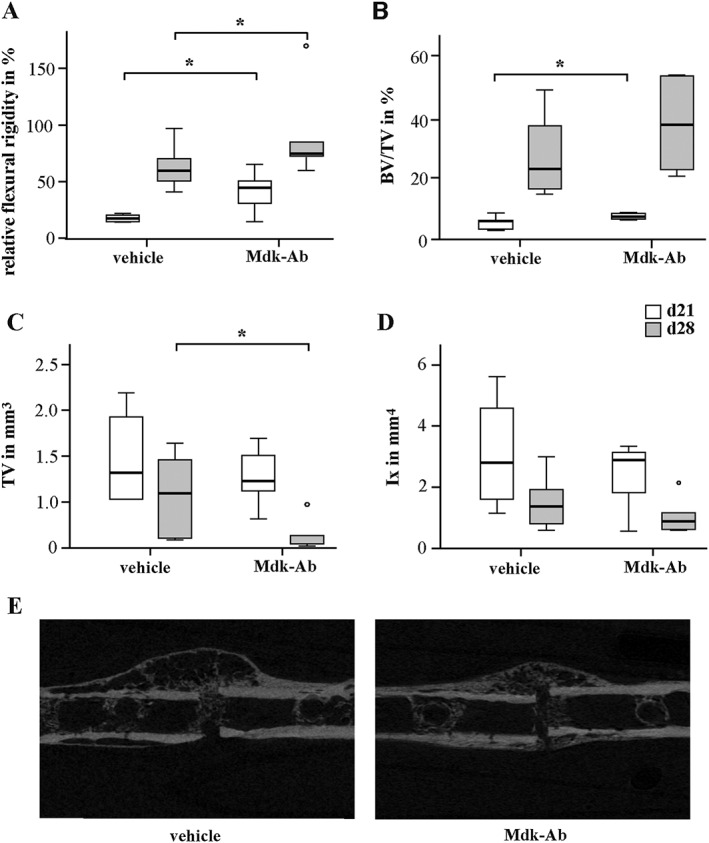
Mdk‐Ab treatment accelerated fracture healing in mice. Biomechanical and μCT analysis of the fractured femurs on days 21 (d21) and 28 (d28). (A) Relative flexural rigidity of the fractured femur in comparison with intact femur. (B) BV/TV of the fracture callus. (C) TV of the fracture callus at the osteotomy gap. (D) Moment of inertia (Ix) of the fracture callus in bending axis x. (E) Representative μCT images from the fracture calli on day 21. *Significantly different from vehicle (P < 0.05) by Mann–Whitney U‐test. (n = 6–8 per group).
Figure 2.
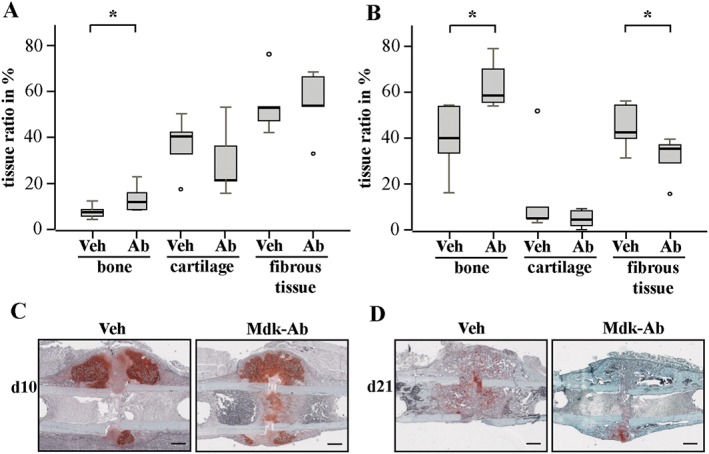
Antagonizing Mdk‐Ab increased bone formation in the fracture callus. Histological slices of fractured femurs were analysed for different tissue‐type contents using Safranin O staining at days 10 and 21. Histomorphometric analysis of the whole callus between the two inner pin holes after (A) 10 and (B) 21 days of healing. Representative images of sections from decalcified femurs stained with Safranin O are shown for (C) days 10 and (D) 21. Scale bar: 500 μm. *Significantly different from vehicle (Veh) (P < 0.05) by Mann–Whitney U‐test (n = 6–8 per group).
Mdk protein expression was differentially affected by Mdk‐Ab treatment in several areas of the fracture callus
We next analysed the impact of Mdk‐Ab treatment on Mdk expression in the fracture callus (Figure 3). On day 4 after surgery, Mdk was expressed weakly in the periosteal region of the fracture callus, which was attenuated by Mdk‐Ab treatment (Figure 3A). Some Mdk‐positive cells were found in the marrow cavities at this early time point. Mdk expression peaked 10 days after surgery in vehicle‐treated mice. The protein was located intracellularly in proliferating and hypertrophic chondrocytes and extracellularly in areas of new bone formation. Mdk protein expression was high around the vessels in the periosteal fracture callus; this was attenuated by the Mdk‐Ab. The Mdk‐Ab did not show effects on the intracellular Mdk protein in chondrocytes (Figure 3B). Additionally, we observed a high staining intensity of Mdk in the marrow cavities on day 10. Immunofluorescence double staining for Mdk and F4/80 detected Mdk‐positive macrophages in the marrow cavities proximal to the osteotomy gap (Figure 3C), and expression analysis of Mdk in mouse macrophage‐like cells revealed high Mdk expression as compared with the low Mdk expression in preosteoblastic MC3T3‐E1 cells (Figure 3D). At day 21, Mdk expression was only observed in scattered chondrocytes and was almost absent in areas of new bone formation. There were no obvious differences between vehicle‐ and antibody‐treated mice at this time point (Supporting Information Fig. S2a).
Figure 3.
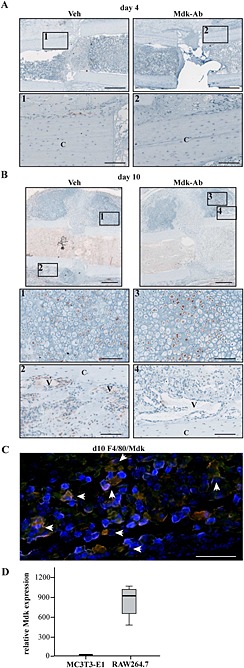
Mdk expression was decreased in areas of neovascularization and new bone formation by Mdk‐Ab treatment. (A) Mdk staining of fracture calli on day 4. Upper lane, scale bar: 500 μm. Lower lane, scale bar: 100 μm. Images 1 and 2 showing the periosteal region of the fracture callus. (B) Mdk staining of fracture calli on day 10. Upper lane scale bar: 500 μm. Two lower lanes, scale bar: 100 μm. Images 1 and 2 showing the chondrogenic part of the fracture callus and images 3 and 4 showing areas of neovascularization and new bone formation. C, cortex; V, vessels. (C) Representative image of immunofluorescence double staining for Mdk (green) and F4/80 (macrophages; red) of a fractured femur from a vehicle (Veh)‐treated mouse on day 10 after fracture. The area displayed is marked with an asterisk in (B). Cells positive for both F4/80 and Mdk appeared in yellow (marked with white arrows). DAPI (blue) was used for nucleus staining. Scale bar: 100 μm. (D) Expression levels of Mdk in preosteoblastic MC3T3‐E1 cells and macrophage‐like RAW 264.7 cells determined by RT‐PCR. B2M was used as the housekeeping gene.
Osteotomy increased Mdk serum levels
Because we suggested that the Mdk protein in areas of neovascularization in the fracture callus might reflect circulating Mdk, we next investigated Mdk serum levels before and after the osteotomy. Untreated mice on day 0 displayed only low Mdk serum levels. In control animals, Mdk serum levels were more than doubled on day 4, peaked on day 10 and returning to pre‐operation levels by day 21 (Table 1). Antibody treatment decreased the Mdk serum levels on days 4 and 10 compared with vehicle‐treated mice.
Table 1.
Mdk serum levels (pg·mL−1) during fracture healing in 9‐month‐old mice
| Days after operation | Vehicle | Mdk‐Ab |
|---|---|---|
| 0 | 7.2 ± 12.3 | |
| 4 | 63.8 ± 4.9* | 8.9 ± 15.4 |
| 10 | 86.4 ± 7.3* | 29.7 ± 31.7# |
| 21 | 9.6 ± 16.7 | n.d. |
n.d., not detectable; day 0, pre‐operation value. (n = 4–7 per group; results presented as mean ± SD).
P < 0.05 for effect of fracture.
P < 0.05 for effect of Mdk‐Ab; Mann–Whitney U‐test.
Mdk‐Ab treatment increased β‐catenin expression and osteoblast activity
We next evaluated the effects of the Mdk‐Ab on β‐catenin signalling during fracture healing. Proliferating chondrocytes and osteoblasts were positively stained for β‐catenin on day 10 after fracture (Figure 4A), whereas hypertrophic chondrocytes were β‐catenin negative. On day 10, the percentage of β‐catenin‐positive area was significantly increased in the fracture calli of the Mdk‐Ab‐treated mice (Figure 4B). In particular, the β‐catenin expression in the osteoblasts around the new trabecular bone was increased (Figure 4A). This observation was also found on day 21 after fracture (Supporting Information Fig. S2b).
Figure 4.
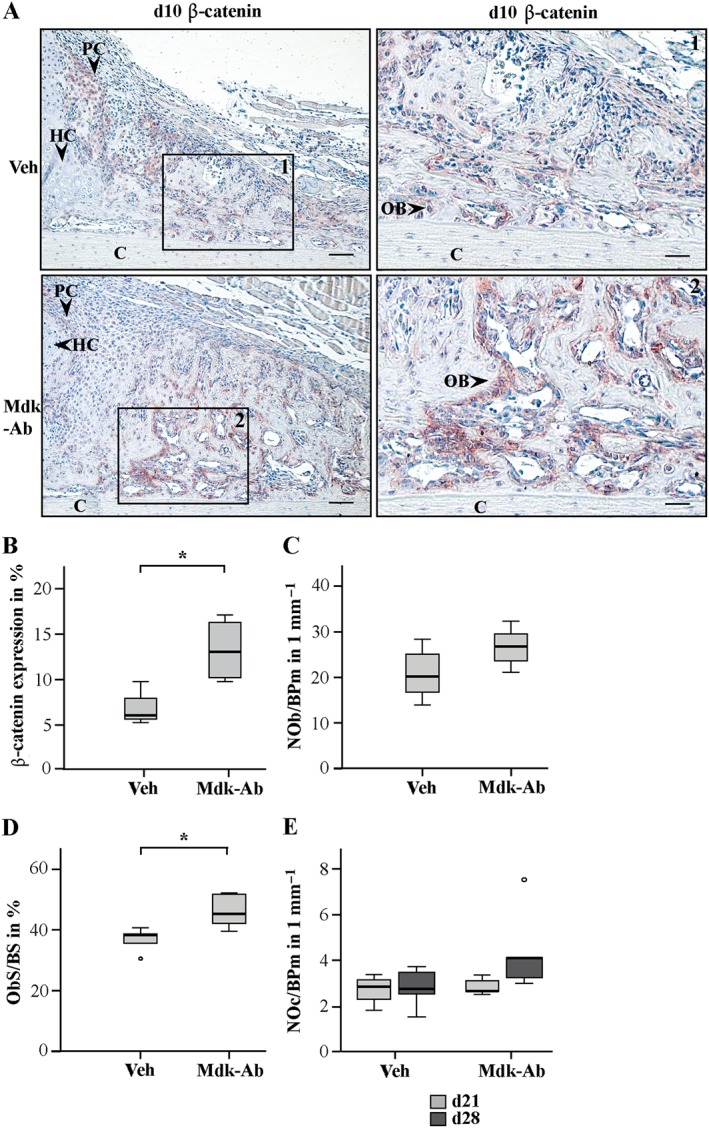
β‐catenin‐positive area and osteoblast (OB) surface were increased in Mdk‐Ab‐treated animals. (A) Sections of fractured femurs from four mice for each time point and group were stained for β‐catenin and counterstained using haematoxylin. Representative images are shown: Veh, vehicle; C, cortex; HC; hypertrophic chondrocytes; PC, proliferating chondrocytes. Images showing the periosteal callus at day 10 after fracture. Scale bar, left column: 100 μm. Scale bar, right column: 50 μm. (B) Quantification of the β‐catenin‐positive area in % (n = 4 per group). (C) Toluidine blue‐stained sections were analysed for the number of osteoblasts per bone perimeter (NOb/BPm) on day 21 and (D) osteoblast surface per bone surface at day 21. (E) Number of osteoclasts in the periosteal callus on days 21 (d21) and 28 (d28). *Significantly different from Veh (P > 0.05). NOb/BPm, osteoblast number/bone perimeter; ObS/BS, osteoblast surface/bone surface; NOc/BPm, osteoclast number/bone perimeter.
Because Mdk‐deficient mice were previously shown to display no differences in the number of osteoclasts or osteoblasts in the fracture callus (Haffner‐Luntzer et al., 2014), we also analysed these parameters in Mdk‐Ab‐treated animals. On day 21, the number of osteoclasts was similar in both groups (Figure 4E), while the number of osteoblasts was only slightly increased after antibody treatment (P = 0.151; Figure 4C). In contrast, the osteoblast surface was significantly increased after Mdk‐Ab treatment (Figure 4D). On day 28, the number of osteoclasts was slightly increased in the antibody‐treated mice (P = 0.082; Figure 4E).
Mdk‐Ab treatment diminished the negative influence of Mdk on β‐catenin signalling in osteogenic cells
Because osteoblast activity appeared to be enhanced after Mdk‐Ab treatment during fracture healing in mice, the effects of Mdk‐Ab treatment on preosteoblastic MC3T3‐E1 cells were evaluated in vitro. Because preosteoblastic cells express only low levels of endogenous Mdk (Figure 3D), the cells were stimulated with recombinant Mdk to test the effects of the Mdk‐Ab. On day 5 of differentiation, stimulation with recombinant Mdk significantly down‐regulated Alpl gene and protein expression (Figure 5A, B). As expected, additional treatment with the Mdk‐Ab abolished the Mdk‐induced effects. Because we found no differences in cartilaginous callus formation after Mdk‐Ab treatment, we wanted to verify whether Mdk and Mdk‐Ab treatment has no influence on differentiation of chondroprogenic ATDC5 cells. In fact, neither Mdk nor the Mdk‐Ab influenced the expression of Acan during chondrogenic differentiation (Figure 5C).
Figure 5.
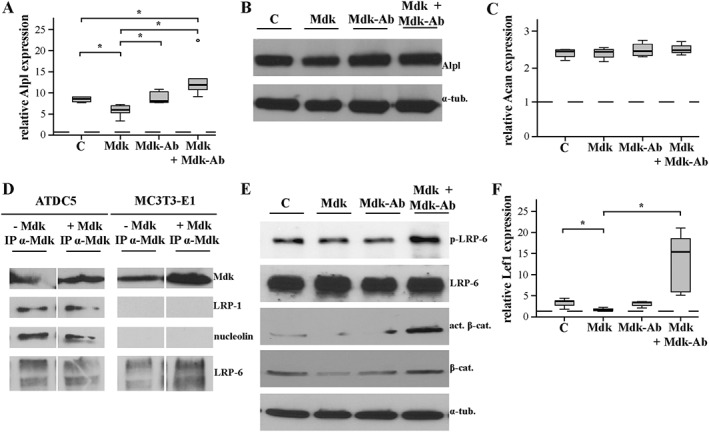
Mdk‐Ab treatment diminished the negative influence of Mdk on β‐catenin signalling in preosteoblastic cells. (A) Alpl gene expression in MC3T3‐E1 cells on day 5 of differentiation after 6 h of treatment with Mdk and the Mdk‐Ab. B2M was used as the housekeeping gene, and gene expression values were normalized to the pre‐differentiation values (dotted line). n = 6. (B) Alpl protein expression in MC3T3‐E1 cells on day 5 after 6 h of stimulation. α‐Tubulin was used as control. n = 3. (C) Acan gene expression in ATDC5 cells on day 5 of differentiation after 6 h of stimulation with Mdk and the Mdk‐Ab. B2M was used as the housekeeping gene, and gene expression values were normalized to the unstimulated control (dotted line). (D) ATDC5 and MC3T3‐E1 cells were incubated without or with recombinant Mdk for 1 h, and immunoprecipitation was performed with Mdk‐Ab. n = 6. (E) Phospho‐LRP‐6, LRP‐6, β‐catenin and active β‐catenin protein expression in MC3T3‐E1 cells after 6 h of incubation. n = 3. (F) Lef1 gene expression in MC3T3‐E1 cells on day 5 of differentiation after 6 h of treatment with Mdk and the Mdk‐Ab. B2M was used as the housekeeping gene, and gene expression values were normalized to the pre‐differentiation values (dotted line). n = 6.
To investigate putative intracellular and extracellular Mdk receptors during fracture healing, immunoprecipitation was performed using both ATDC5 cells and MC3T3‐E1 cells without and with incubation with recombinant Mdk for 1 h. Interestingly, the previously described intracellular Mdk‐interacting proteins LRP‐1 and nucleolin were immunoprecipitated with Mdk only in ATDC5 cells, whereas the canonical Wnt signalling receptor LRP‐6 was immunoprecipitated with Mdk in both cell types (Figure 5E). The amount of Mdk‐bound LRP‐6 protein was increased after incubation with exogenous Mdk in MC3T3‐E1 cells only (Figure 5D). Therefore, we further investigated the role of LRP‐6 in Mdk‐induced effects on these cells. We demonstrated that LRP‐6 phosphorylation was decreased after Mdk stimulation (Figure 5E). Additionally, total β‐catenin and active β‐catenin protein expression were decreased following Mdk stimulation as well as Lef1 gene expression, these effects being attenuated by Mdk‐Ab (Figure 5F).
Discussion
Fractures are the most common injuries of the musculoskeletal system, resulting in a high number of affected patients worldwide (Claes et al., 2012). About 10% of all fractures are still reported to show delayed healing (King et al., 2007; Cadet et al., 2013); therefore, there is a high clinical need for a new treatment strategy to enhance the bone regeneration process. One promising molecular drug target might be the growth and differentiation factor Mdk, because it was shown to negatively influence osteoblast activity (Liedert et al., 2011). However, previous studies reported that Mdk is important during chondrogenesis and that complete Mdk deficiency resulted in impaired fracture healing based on delayed cartilaginous callus formation (Ohta et al., 1999; Haffner‐Luntzer et al., 2014). Distinct expression patterns of Mdk in the cartilaginous and bony region of the fracture callus suggested different effects of locally expressed or circulating Mdk. However, less is known about the endocrine distribution of Mdk and the distinct roles of Mdk during bone regeneration. In contrast to a complete Mdk deficiency, Mdk‐Ab treatment is able to target specifically circulating Mdk. Therefore, the aims of the present study were to investigate the influence of systemic Mdk‐Ab treatment on fracture healing in mice and to further identify the involved Mdk signalling pathways. Biomechanical testing demonstrated that the fractured femurs of antibody‐treated animals displayed significantly greater flexural rigidity than those of vehicle‐treated mice. μCT analysis showed that the fracture calli of Mdk‐Ab‐treated animals contained significantly more bone than those of the control group at day 21. The total callus volume was reduced at day 28 in the Ab‐treated mice, indicating more rapid callus remodelling in these animals. This was further supported by a tendency for increased osteoclast number in the fracture calli at day 28. This enhanced callus remodelling might be the cause that the bone volume fraction was not significantly but only by trend increased at day 28. Histomorphometric analysis revealed a significant increase in the amount of bone even during the early stage of fracture healing (day 10), but no changes in the cartilage tissue ratio. Thus, fracture healing was accelerated in Mdk‐Ab‐treated mice. Because we showed previously that Mdk‐deficient mice displayed delayed early fracture healing through retarded chondrogenesis during endochondral ossification (Haffner‐Luntzer et al., 2014), we assumed that antagonizing Mdk using an antibody may have a different impact than a complete absence of Mdk by genetic modification through to different molecular mechanisms. Thus, we compared Mdk protein expression in the callus of antibody‐ and vehicle‐treated mice. As described previously (Haffner‐Luntzer et al., 2014), Mdk protein was localized intracellularly in chondrocytes and extracellularly in the periosteum and in areas of new bone formation. Mdk‐Ab treatment did not affect Mdk expression in the intracellular compartment of chondrocytes, whereas Mdk expression was decreased in the periosteal region at day 4 and in areas of neovascularization and new bone formation at day 10, assuming an endocrine signalling by circulating Mdk. The Mdk‐Ab bound circulating Mdk and the antibody–antigen complexes were cleared possibly via hepatic endothelial cells, with salvage of the antibody by the FcRn receptor located on endothelial cells (Mould and Sweeney, 2007), thereby interfering with Mdk's half‐life and interaction with receptors (Chames et al., 2009). However, because Mdk‐Ab could not bind to intracellularly located antigen, intracellular Mdk protein was not degraded, which can act directly on the producer cells through intracrine signalling pathways, as shown previously for several other cell types (Take et al., 1994; Re and Cook, 2008). Therefore, we actually found fundamental differences between Mdk‐deficient mice (complete absence of Mdk protein) and Mdk‐Ab‐treated mice (no circulating Mdk, whereas locally expressed Mdk in chondrocytes was still present). We further confirmed that the Mdk‐Ab decreased circulating Mdk protein levels by evaluation of Mdk serum levels pre‐ and post‐osteotomy. A fracture‐induced increase in the Mdk serum level in vehicle‐treated mice until day 10 was significantly attenuated by the antibody treatment. Indeed, no previous data for mouse serum or blood Mdk concentrations are available, while clinical studies reported elevated Mdk serum levels in patients suffering from rheumatoid arthritis (Maruyama et al., 2004). These data and our in vivo data indicate that circulating Mdk may play a role during tissue damages, including fractures, in mice and humans. Additionally, serum Mdk levels were shown to be increased in patients suffering from systemic inflammation and sepsis (Krzystek‐Korpacka et al., 2011), indicating that Mdk may be one factor among many responsible for delayed fracture healing after systemic inflammation (Pape et al., 2010; Claes et al., 2012). One source for circulating Mdk after tissue injury was reported as inflammatory cells like macrophages and neutrophils (Narita et al., 2008; Weckbach et al., 2012), which are also recruited after fracture (Claes et al., 2012). We observed Mdk‐positive macrophages in the marrow cavities proximal to the osteotomy gap at day 10, indicating that the increase in Mdk serum level after osteotomy is, at least in part, due to the enhanced recruitment of inflammatory cells to the blood stream. This finding was supported by an additional in vitro analysis of Mdk expression in undifferentiated mouse macrophage‐like cells, which showed high levels of Mdk expression. Maruyama et al., (2004) also showed Mdk expression in both human and murine macrophages.
Because we hypothesized that Mdk‐Ab treatment increased osteoblast activity by antagonizing circulating Mdk, we investigated its effect on osteoclasts and osteoblasts as well as on β‐catenin expression during callus maturation in fracture healing. Although Mdk‐Ab did not influence osteoclast or osteoblast number, it did significantly increase the osteoblast surface at day 21, indeed indicating greater osteoblast activity. The β‐catenin‐positive area was increased in the bony fracture callus of Ab‐treated animals at days 10 and 21. Wnt/β‐catenin signalling appears to be critically involved in fracture healing, being activated during the early and intermediate phases in mice (Hadjiargyrou et al., 2002) and humans (Chen et al., 2007). Conditional knockout of β‐catenin in osteoblasts led to significantly reduced calcified callus formation in mice (Chen et al., 2007). In contrast, Wnt/β‐catenin signalling activation significantly accelerated fracture repair in mice, rats and non‐human primates (Chen et al., 2007; Li et al., 2011; Ominsky et al., 2011). These results further corroborate our hypothesis that circulating Mdk acts as an inhibitor of Wnt/β‐catenin signalling in osteoblasts during fracture healing and that antagonizing Mdk accelerates fracture healing based on enhanced Wnt/β‐catenin signalling.
We next aimed to clarify the distinct role of Mdk during the differentiation of osteoblasts and chondrocytes, because the in vivo data from this study and the previously published study using Mdk‐deficient mice (Haffner‐Luntzer et al., 2014), led us to the hypothesis that Mdk may act differentially on both cell types. Therefore, we first investigated the direct effects of exogenously added Mdk and the Mdk‐Ab on preosteoblastic cells as well as on chondroprogenitor cells in vitro. We stimulated the cells with recombinant Mdk to mimic the effects of exogenous Mdk, because preosteoblastic cells displayed only low levels of endogenous Mdk. Recombinant Mdk reduced the expression of β‐catenin target genes associated with osteogenic differentiation, as shown previously by Liedert et al. (2011), while additional treatment with the Mdk‐Ab abolished these negative effects in osteoblasts. In contrast, we found no influence of either Mdk or the Mdk‐Ab on Acan expression in chondrogenic cells, indicating that exogenous Mdk does not have an influence on chondrogenic differentiation. Indeed, we showed in a previous study that endogenous Mdk knockdown decreased differentiation and β‐catenin signalling in chondrocytes, indicating that endogenous Mdk expression is crucial for chondrogenic differentiation (Haffner‐Luntzer et al., 2014). Therefore, we conclude that endogenous Mdk may have an effect on chondrogenic differentiation, whereas exogenous Mdk may not. This conclusion was also drawn by Ohta and colleagues; they found that recombinant Mdk did not affect chondrogenesis, whereas transfection of Mdk cDNA into the chondrocytes did (Ohta et al., 1999). However, other groups reported about the proliferative effects of recombinant Mdk on articular chondrocytes in vivo and an attenuated dedifferentiation of primary chondrocytes in vitro. (Zhang et al., 2010; Xu et al., 2011) Therefore, Mdk seems to play a complex role during cartilage formation and maturation.
Because the different effects of recombinant Mdk on either osteogenic or chondrogenic cells were very interesting and indicated distinct signalling pathways, we further investigated the participating receptors in the Mdk signalling in both cell types. Indeed, we demonstrated that the endocytosis‐related receptor LRP‐1 and the nuclear shuttle protein nucleolin were Mdk‐interacting proteins in ATDC5 chondroprogenitor cells but not in MC3T3‐E1 preosteoblastic cells. It has been shown previously that Mdk can bind to LRP‐1 and nucleolin in both an autocrine fashion and an intracrine fashion (Take et al., 1994; Shibata et al., 2002; Lee et al., 2012) and exerts anti‐apoptotic effects through binding to these proteins (Shibata et al., 2002; Lee et al., 2012). In addition, LRP‐1 has been shown to be involved in mediating Wnt/β‐catenin signalling in several cell types, although the results were variable and require further evaluation (Terrand et al., 2009; Kawata et al., 2010). Therefore, intracrine binding of locally expressed Mdk to LRP‐1 and nucleolin may be important for β‐catenin signalling in chondrocytes, but not in osteoblasts (Figure 6). We further demonstrated that the canonical Wnt/β‐catenin signalling‐related receptor LRP‐6 was co‐immunoprecipitated with Mdk in both cell types. Stimulation of MC3T3‐E1 cells with recombinant Mdk diminished LRP‐6 activation, indicating a regulatory role of this receptor during Mdk‐induced effects in osteoblasts. It has been shown previously that the receptor PTPRz is also involved in Mdk‐induced decreased Wnt/β‐catenin signalling in osteogenic cells (Liedert et al., 2011) and that LRP‐6 and PTPRz form a receptor complex for Mdk in other cell types (Muramatsu et al., 2004). Therefore, we assume that endocrine Mdk exerts its negative effects on Wnt/β‐catenin signalling in osteoblasts through binding to the PTPRz/LRP‐6 receptor complex and that Mdk‐Ab abolishes this interaction (Figure 6).
Figure 6.
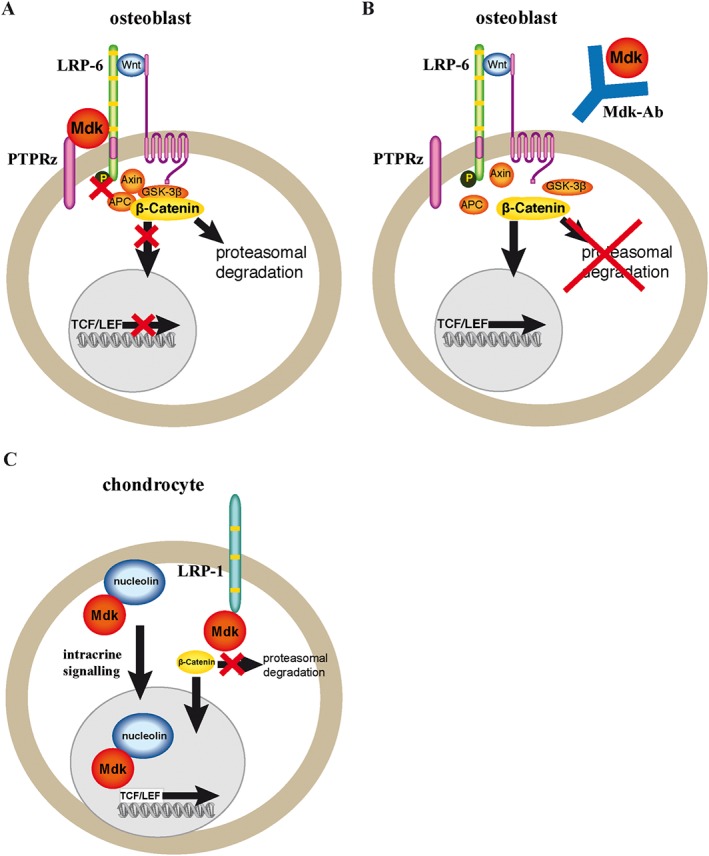
Model of the signalling pathways modulated by Mdk and the effect of Mdk‐Ab treatment on osteoblasts and chondrocytes. (A) The circulating Mdk protein may bind to a receptor complex of LRP‐6 and PTPRz, decreasing LRP‐6 phosphorylation and leading to stabilization of the β‐catenin degradation complex. Wnt/β‐catenin signalling is decreased in the presence of Mdk. (B) Mdk‐Ab treatment abolishes the interaction of Mdk with its receptor complex, leading to increased translocation of β‐catenin to the cell nucleus and increased Wnt/β‐catenin signalling in osteoblasts. (C) Locally expressed Mdk may act positively on Wnt/β‐catenin signalling in chondrocytes via intracrine signalling after binding to LRP‐1 and nucleolin. In this model, Mdk‐Ab treatment would not have an effect on chondrocytes. APC, adenomatous polyposis coli; GSK‐3b, glycogen synthase kinase‐3b; TCF/LEF, T cell factor/lymphoid enhancer factor.
In conclusion, the findings of the present study indicate that there is a strong therapeutic potential for the Mdk‐Ab to enhance fracture healing in patients with bone healing complications such as delayed healing or non‐union formation. Further studies are needed to clarify the role of circulating Mdk in human fracture patients, as well as a potential involvement of Mdk in systemic inflammation‐induced delayed fracture healing, which occurs, for example, in patients suffering from polytrauma (Karladani et al., 2001).
Author contributions
M.H.L., A.L., A.I., T.S. and M.A. designed the study; M.H.L., A.H. and A.E.R. conducted the study; M.H.L., A.H., A.E.R. and R.R. collected the data; M.H.L. and R.R. analysed the data; M.H.L., A.L., A.H., A.E.R., R.R. and A.I. interpreted the data; M.H.L., A.L. and A.I. drafted the manuscript; M.H.L., A.H., A.E.R., A.L., A.I., T.S., M.A., R.R. revised the content of the manuscript; M.H.L., A.L., A.I., A.H., A.E.R., R.R., T.S. and M.A. approved the final version of the manuscript.
Conflict of interest
The authors declare no conflicts of interest.
Declaration of transparency and scientific rigour
This Declaration acknowledges that this paper adheres to the principles for transparent reporting and scientific rigour of preclinical research recommended by funding agencies, publishers and other organisations engaged with supporting research.
Supporting information
Figure S1 Mdk‐Ab treatment accelerated fracture healing, whereas control IgG did not affect fracture healing. Biomechanical analysis of the fractured femurs at days 21 (white bars) and 28 (grey bars). (A) Relative flexural rigidity of the fractured femurs of PBS‐treated and control IgG‐treated mice. (B) Bone volume fraction of the fracture callus of PBS‐treated and control IgG‐treated mice. (C) Flexural rigidity of the fractured femurs of vehicle‐treated (PBS) or Mdk‐Ab‐treated mice. (D) Flexural rigidity of the intact femurs of vehicle‐treated (PBS) or Mdk‐Ab‐treated mice. *Significantly different from vehicle (PBS) (P < 0.05) by Mann–Whitney U‐test.
Figure S2 Mdk is rarely expressed in the fracture callus at day 21 after fracture. (A) Representative images from Mdk immunostaining of fracture calli at day 21. Upper lane, scale bar: 500 μm. Lower lane, scale bar: 100 μm. Images 1 and 2 showing the bony callus near the osteotomy gap. (B) Representative images from β‐catenin immunostaining of fracture calli at day 21. Scale bar: 100 μm. Images showing the periosteal fracture callus. C, cortex; Veh, vehicle.
Supporting info item
Supporting info item
Acknowledgements
Cellmid Ltd. is gratefully acknowledged for providing the anti‐Mdk antibody and the Mdk elisa. We thank Iris Baum, Helga Bach, Marion Tomo, Sevil Essig and Uschi Maile for excellent technical assistance. This work was supported by grants from the German Research Foundation (IG 18–3/3 and AM103/10–3) and from the Elsbeth Bonhoff Foundation.
Haffner‐Luntzer, M. , Heilmann, A. , Rapp, A. E. , Roessler, R. , Schinke, T. , Amling, M. , Ignatius, A. , and Liedert, A. (2016) Antagonizing midkine accelerates fracture healing in mice by enhanced bone formation in the fracture callus. British Journal of Pharmacology, 173: 2237–2249. doi: 10.1111/bph.13503.
References
- Alexander SPH, Kelly E, Marrion N, Peters JA, Benson HE, Faccenda E et al. (2015a). The Concise Guide to PHARMACOLOGY 2015/16: Overview. Br J Pharmacol 172: 5729–5143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Davenport AP, Kelly E, Marrion N, Peters JA, Benson HE et al. (2015b). The Concise Guide to PHARMACOLOGY 2015/16: G protein‐coupled receptors. Br J Pharmacol 172: 5744–5869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Fabbro D, Kelly E, Marrion N, Peters JA, Benson HE et al. (2015c). The Concise Guide to PHARMACOLOGY 2015/16: Catalytic receptors. Br J Pharmacol 172: 5979–6023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Fabbro D, Kelly E, Marrion N, Peters JA, Benson HE et al. (2015d). The Concise Guide to PHARMACOLOGY 2015/16: Enzymes. Br J Pharmacol 172: 6024–6109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouxsein ML, Boyd SK, Christiansen BA, Guldberg RE, Jepsen KJ, Muller R (2010). Guidelines for assessment of bone microstructure in rodents using micro‐computed tomography. J Bone Miner Res 25: 1468–1486. [DOI] [PubMed] [Google Scholar]
- Busse JW, Bhandari M, Kulkarni AV, Tunks E (2002). The effect of low‐intensity pulsed ultrasound therapy on time to fracture healing: a meta‐analysis. CMAJ 166: 437–441. [PMC free article] [PubMed] [Google Scholar]
- Cadet ER, Yin B, Schulz B, Ahmad CS, Rosenwasser MP (2013). Proximal humerus and humeral shaft nonunions. J Am Acad Orthop Surg 21: 538–547. [DOI] [PubMed] [Google Scholar]
- Chames P, Van Regenmortel M, Weiss E, Baty D (2009). Therapeutic antibodies: successes, limitations and hopes for the future. Br J Pharmacol 157: 220–233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y, Whetstone HC, Lin AC, Nadesan P, Wei Q, Poon R et al. (2007). Beta‐catenin signalling plays a disparate role in different phases of fracture repair: implications for therapy to improve bone healing. PLoS Med 4: e249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Claes L, Recknagel S, Ignatius A (2012). Fracture healing under healthy and inflammatory conditions. Nat Rev Rheumatol 8: 133–143. [DOI] [PubMed] [Google Scholar]
- Curtis MJ, Bond RA, Spina D, Ahluwalia A, Alexander SP, Giembycz MA et al. (2015). Experimental design and analysis and their reporting: new guidance for publication in BJP. Br J Pharmacol 172: 3461–3471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Einhorn TA (2003). Clinical applications of recombinant human BMPs: early experience and future development. J Bone Joint Surg Am 85‐A (Suppl 3): 82–88. [DOI] [PubMed] [Google Scholar]
- Giannoudis PV, Dinopoulos HT (2010). BMPs: options, indications, and effectiveness. J Orthop Trauma 24 (Suppl 1): S9–16. [DOI] [PubMed] [Google Scholar]
- Hadjiargyrou M, Lombardo F, Zhao S, Ahrens W, Joo J, Ahn H et al. (2002). Transcriptional profiling of bone regeneration. Insight into the molecular complexity of wound repair. J Biol Chem 277: 30177–30182. [DOI] [PubMed] [Google Scholar]
- Haffner‐Luntzer M, Heilmann A, Rapp AE, Beie S, Schinke T, Amling M et al. (2014). Midkine‐deficiency delays chondrogenesis during the early phase of fracture healing in mice. PLoS One 9: e116282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karladani AH, Granhed H, Karrholm J, Styf J (2001). The influence of fracture etiology and type on fracture healing: a review of 104 consecutive tibial shaft fractures. Arch Orthop Trauma Surg 121: 325–328. [DOI] [PubMed] [Google Scholar]
- Kawata K, Kubota S, Eguchi T, Moritani NH, Shimo T, Kondo S et al. (2010). Role of the low‐density lipoprotein receptor‐related protein‐1 in regulation of chondrocyte differentiation. J Cell Physiol 222: 138–148. [DOI] [PubMed] [Google Scholar]
- Kilkenny C, Browne W, Cuthill IC, Emerson M, Altman DG (2010). Animal research: reporting in vivo experiments: the ARRIVE guidelines. Br J Pharmacol 160: 1577–1579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- King AR, Moran SL, Steinmann SP (2007). Humeral nonunion. Hand Clin 23: 449–456, vi. [DOI] [PubMed] [Google Scholar]
- Krzystek‐Korpacka M, Mierzchala M, Neubauer K, Durek G, Gamian A (2011). Midkine, a multifunctional cytokine, in patients with severe sepsis and septic shock: a pilot study. Shock (Augusta, Ga) 35: 471–477. [DOI] [PubMed] [Google Scholar]
- Lee SH, Suh HN, Lee YJ, Seo BN, Ha JW, Han HJ (2012). Midkine prevented hypoxic injury of mouse embryonic stem cells through activation of Akt and HIF‐1alpha via low‐density lipoprotein receptor‐related protein‐1. J Cell Physiol 227: 1731–1739. [DOI] [PubMed] [Google Scholar]
- Li C, Ominsky MS, Tan HL, Barrero M, Niu QT, Asuncion FJ et al. (2011). Increased callus mass and enhanced strength during fracture healing in mice lacking the sclerostin gene. Bone 49: 1178–1185. [DOI] [PubMed] [Google Scholar]
- Liedert A, Mattausch L, Rontgen V, Blakytny R, Vogele D, Pahl M et al. (2011). Midkine‐deficiency increases the anabolic response of cortical bone to mechanical loading. Bone 48: 945–951. [DOI] [PubMed] [Google Scholar]
- Maruyama K, Muramatsu H, Ishiguro N, Muramatsu T (2004). Midkine, a heparin‐binding growth factor, is fundamentally involved in the pathogenesis of rheumatoid arthritis. Arthritis Rheum 50: 1420–1429. [DOI] [PubMed] [Google Scholar]
- McGrath JC, Lilley E (2015). Implementing guidelines on reporting research using animals (ARRIVE etc.): new requirements for publication in BJP. Br J Pharmacol 172: 3189–3193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitsiadis TA, Muramatsu T, Muramatsu H, Thesleff I (1995). Midkine (MK), a heparin‐binding growth/differentiation factor, is regulated by retinoic acid and epithelial–mesenchymal interactions in the developing mouse tooth, and affects cell proliferation and morphogenesis. J Cell Biol 129: 267–281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mould DR, Sweeney KR (2007). The pharmacokinetics and pharmacodynamics of monoclonal antibodies–mechanistic modeling applied to drug development. Curr Opin Drug Discov Devel 10: 84–96. [PubMed] [Google Scholar]
- Muramatsu H, Zou P, Suzuki H, Oda Y, Chen GY, Sakaguchi N et al. (2004). alpha4beta1‐ and alpha6beta1‐integrins are functional receptors for midkine, a heparin‐binding growth factor. J Cell Sci 117: 5405–5415. [DOI] [PubMed] [Google Scholar]
- Muramatsu T (1993). Midkine (MK), the product of a retinoic acid responsive gene, and pleiotrophin constitute a new protein family regulating growth and differentiation. Int J Dev Biol 37: 183–188. [PubMed] [Google Scholar]
- Narita H, Chen S, Komori K, Kadomatsu K (2008). Midkine is expressed by infiltrating macrophages in in‐stent restenosis in hypercholesterolemic rabbits. J Vasc Surg 47: 1322–1329. [DOI] [PubMed] [Google Scholar]
- Neunaber C, Catala‐Lehnen P, Beil FT, Marshall RP, Kanbach V, Baranowsky A et al. (2010). Increased trabecular bone formation in mice lacking the growth factor midkine. J Bone Miner Res 25: 1724–1735. [DOI] [PubMed] [Google Scholar]
- Ohta S, Muramatsu H, Senda T, Zou K, Iwata H, Muramatsu T (1999). Midkine is expressed during repair of bone fracture and promotes chondrogenesis. J Bone Miner Res 14: 1132–1144. [DOI] [PubMed] [Google Scholar]
- Ominsky MS, Li C, Li X, Tan HL, Lee E, Barrero M et al. (2011). Inhibition of sclerostin by monoclonal antibody enhances bone healing and improves bone density and strength of nonfractured bones. J Bone Miner Res 26: 1012–1021. [DOI] [PubMed] [Google Scholar]
- Pape HC, Marcucio R, Humphrey C, Colnot C, Knobe M, Harvey EJ (2010). Trauma‐induced inflammation and fracture healing. J Orthop Trauma 24: 522–525. [DOI] [PubMed] [Google Scholar]
- Poynton AR, Lane JM (2002). Safety profile for the clinical use of bone morphogenetic proteins in the spine. Spine 27: S40–S48. [DOI] [PubMed] [Google Scholar]
- Ramakers C, Ruijter JM, Deprez RH, Moorman AF (2003). Assumption‐free analysis of quantitative real‐time polymerase chain reaction (PCR) data. Neurosci Lett 339: 62–66. [DOI] [PubMed] [Google Scholar]
- Re RN, Cook JL (2008). The basis of an intracrine pharmacology. J Clin Pharmacol 48: 344–350. [DOI] [PubMed] [Google Scholar]
- Rontgen V, Blakytny R, Matthys R, Landauer M, Wehner T, Gockelmann M et al. (2010). Fracture healing in mice under controlled rigid and flexible conditions using an adjustable external fixator. J Orthop Res 28: 1456–1462. [DOI] [PubMed] [Google Scholar]
- Shibata Y, Muramatsu T, Hirai M, Inui T, Kimura T, Saito H et al. (2002). Nuclear targeting by the growth factor midkine. Mol Cell Biol 22: 6788–6796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shukunami C, Shigeno C, Atsumi T, Ishizeki K, Suzuki F, Hiraki Y (1996). Chondrogenic differentiation of clonal mouse embryonic cell line ATDC5 in vitro: differentiation‐dependent gene expression of parathyroid hormone (PTH)/PTH‐related peptide receptor. J Cell Biol 133: 457–468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Southan C, Sharman JL, Benson HE, Faccenda E, Pawson AJ, Alexander SP et al. (2016). The IUPHAR/BPS Guide to PHARMACOLOGY in 2016: towards curated quantitative interactions between 1300 protein targets and 6000 ligands. Nucl. Acids Res. 44: D1054–D1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Take M, Tsutsui J, Obama H, Ozawa M, Nakayama T, Maruyama I et al. (1994). Identification of nucleolin as a binding protein for midkine (MK) and heparin‐binding growth associated molecule (HB‐GAM). J Biochem 116: 1063–1068. [DOI] [PubMed] [Google Scholar]
- Terrand J, Bruban V, Zhou L, Gong W, El Asmar Z, May P et al. (2009). LRP1 controls intracellular cholesterol storage and fatty acid synthesis through modulation of Wnt signaling. J Biol Chem 284: 381–388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weckbach LT, Groesser L, Borgolte J, Pagel JI, Pogoda F, Schymeinsky J et al. (2012). Midkine acts as proangiogenic cytokine in hypoxia‐induced angiogenesis. Am J Physiol Heart Circ Physiol 303: H429–H438. [DOI] [PubMed] [Google Scholar]
- Wehrle E, Liedert A, Heilmann A, Wehner T, Bindl R, Fischer L et al. (2015). The impact of low‐magnitude high‐frequency vibration on fracture healing is profoundly influenced by the oestrogen status in mice. Dis Model Mech 8: 93–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu C, Zhang Z, Wu M, Zhu S, Gao J, Zhang J et al. (2011). Recombinant human midkine stimulates proliferation and decreases dedifferentiation of auricular chondrocytes in vitro. Exp Biol Med (Maywood) 236: 1254–1262. [DOI] [PubMed] [Google Scholar]
- Zhang ZH, Li HX, Qi YP, Du LJ, Zhu SY, Wu MY et al. (2010). Recombinant human midkine stimulates proliferation of articular chondrocytes. Cell Prolif 43: 184–194. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1 Mdk‐Ab treatment accelerated fracture healing, whereas control IgG did not affect fracture healing. Biomechanical analysis of the fractured femurs at days 21 (white bars) and 28 (grey bars). (A) Relative flexural rigidity of the fractured femurs of PBS‐treated and control IgG‐treated mice. (B) Bone volume fraction of the fracture callus of PBS‐treated and control IgG‐treated mice. (C) Flexural rigidity of the fractured femurs of vehicle‐treated (PBS) or Mdk‐Ab‐treated mice. (D) Flexural rigidity of the intact femurs of vehicle‐treated (PBS) or Mdk‐Ab‐treated mice. *Significantly different from vehicle (PBS) (P < 0.05) by Mann–Whitney U‐test.
Figure S2 Mdk is rarely expressed in the fracture callus at day 21 after fracture. (A) Representative images from Mdk immunostaining of fracture calli at day 21. Upper lane, scale bar: 500 μm. Lower lane, scale bar: 100 μm. Images 1 and 2 showing the bony callus near the osteotomy gap. (B) Representative images from β‐catenin immunostaining of fracture calli at day 21. Scale bar: 100 μm. Images showing the periosteal fracture callus. C, cortex; Veh, vehicle.
Supporting info item
Supporting info item