

Abstract
Background and Purpose
Prenatal exposure to dexamethasone slows down fetal linear growth and bone mineralization but the regulatory mechanism remains unknown. Here we assessed how dexamethasone regulates bone development in the fetus.
Experimental Approach
Dexamethasone (1 mg·kg−1·day−1) was injected subcutaneously every morning in pregnant rats from gestational day (GD)9 to GD20. Fetal femurs and tibias were harvested at GD20 for histological and gene expression analysis. Femurs of 12‐week‐old female offspring were harvested for microCT (μCT) measurement. Primary chondrocytes were treated with dexamethasone (10, 50, 250 and 1000 nM).
Key Results
Prenatal dexamethasone exposure resulted in accumulation of hypertrophic chondrocytes and delayed formation of the primary ossification centre in fetal long bone. The retardation was accompanied by reduced maturation of hypertrophic chondrocytes, decreased osteoclast number and down‐regulated expression of osteocalcin and bone sialoprotein in long bone. In addition, the mitogen‐inducible gene‐6 (Mig6) and osteoprotegerin (OPG) expression were stimulated, and the receptor activator of NF‐κB ligand (RANKL) expression was repressed. Moreover, dexamethasone activated OPG and repressed RANKL expression in both primary chondrocytes and primary osteoblasts, and the knockdown of Mig6 abolished the effect of dexamethasone on OPG expression. Further, μCT measurement showed loss of bone mass in femur of 12‐week‐old offspring with prenatal dexamethasone exposure.
Conclusions and Implications
Prenatal dexamethasone exposure delays endochondral ossification by suppressing chondrocyte maturation and osteoclast differentiation, which may be partly mediated by Mig6 activation in bone. Bone development retardation in the fetus may be associated with reduced bone mass in later life.
Abbreviations
- BSP
bone sialoprotein
- EGFR
EGF receptor
- GD
gestational day
- Mig6
mitogen‐inducible gene‐6
- OPG
osteoprotegerin
- RANKL
receptor activator of NF‐κB ligand
- TRAP
tartrate‐resistant acid phosphatase
- μCT
microCT
Tables of Links
| TARGETS |
|---|
| Catalytic receptors a |
| EGFR, EGF receptor |
| Osteoprotegerin (TNFRSF11B) |
| GPCRs b |
| CT calcitonin receptor |
| Enzymes c |
| Cathepsin K |
| LIGANDS |
|---|
| Dexamethasone |
| RANKL, RANK ligand |
These Tables list key protein targets and ligands in this article which are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Southan et al., 2016) and are permanently archived in the Concise Guide to PHARMACOLOGY 2015/16 (a,b,cAlexander et al., 2015a, 2015b, 2015c).
Introduction
Glucocorticoids, in particular dexamethasone, can cross the placenta readily; therefore, they have been widely used as a medication for several types of severe disorders of pregnancy, including congenital adrenal hyperplasia, premature delivery, placenta previa and multiple pregnancies (Young et al., 1980; Morales et al., 1986; Crowther et al., 2011; Maryniak et al., 2014). Also, synthetic glucocorticoids are frequently used in the prevention or treatment of pregnant women with asthma (Tan and Thomson, 2000). However, prenatal dexamethasone treatment is associated with decreased birth weight and size (Murphy et al., 2008, 2012). In addition, it is known that fetal exposure to an excess of dexamethasone slows down fetal linear growth and bone mineralization, and this negative effect of dexamethasone treatment may result in continued neonatal modification of bone tissues, thus diminishing bone quality and mechanical properties (Sliwa et al., 2010). Despite the known negative effects of prenatal dexamethasone treatment on bone development, the underlying mechanisms still remain to be elucidated. It is important to recognize that reduced fetal growth and low birth weight are associated with osteoporosis in later life (Godfrey et al., 2011). Therefore, studying the underlying mechanism of bone growth retardation induced by prenatal dexamethasone treatment will provide helpful clues to the early prevention of osteoporosis with developmental origin.
Over the past several years, a number of studies have identified that the EGF receptor (EGFR) may be one of the major modulators of bone metabolism, as it can regulate the proliferation and differentiation of osteoblasts and chondrocytes, and osteoclastogenesis (Schneider et al., 2009). EGFR belongs to the ErbB receptor family; its ligands include EGF, amphiregulin, TGF‐α, heparin‐binding EGF, betacellulin and epiregulin. Upon ligand binding, EGFR either homodimerizes or heterodimerizes with other EGFR family members. The tyrosine residues in the intracellular domain of dimerized EGFR then undergo phosphorylation, thus activating downstream signal pathways (mainly Ras/Raf/MAPK and PI3K/Akt pathways), regulating cell proliferation, differentiation and migration (Citri and Yarden, 2006). EGFR signalling in chondrocytes at the chondro‐osseous junction supports osteoclastogenesis via stimulating the expression of receptor activator of NF‐κB ligand (RANKL) and suppressing the expression of osteoprotegerin (OPG), a decoy receptor for RANKL. This signalling is essential in the remodelling of growth plate cartilage extracellular matrix into bone during endochondral ossification (Zhang et al., 2011, 2013). EGFR‐deficient mice have an expanded area of hypertrophic cartilage in the growth plate and fewer bony trabeculae because of defective osteoclast recruitment (Wang et al., 2004). Taken together, these data clearly indicate that EGFR signalling plays an important role in primary endochondral ossification and that fine tuning of EGFR signalling is required for the maintenance of bone metabolism.
The mitogen‐inducible gene‐6 (Mig6) is a known negative regulator of ErbB receptors. It can be recruited to and bind with the kinase domain of ErbB receptors, thus blocking phosphorylation of ErbB receptors and downstream signalling (Anastasi et al., 2003). Loss of Mig6 induced abnormal endochondral ossification along with enhanced EGFR signalling. Whole‐body knockout of Mig6 leads to the formation of osteochondral nodules in the knee (Zhang et al., 2005; Shepard et al., 2013). Target deletion of Mig6 in chondrocytes results in excessive proliferative activities in articular cartilage, apart from the abnormally formed osteophytes (Pest et al., 2014; Staal et al., 2014). Treatment with dexamethasone (10−6M) up‐regulated the expression of Mig6 (Xu et al., 2005; Scheving et al., 2007), which in turn suppressed basal and EGF‐associated phosphorylation of EGFR and downstream ERK and Akt, exerting a time‐dependent and redundant inhibitory effect on cell proliferation (Scheving et al., 2007). Overall, the above studies suggest that Mig6 might mediate the toxic effect of dexamethasone exposure on bone development in early stage.
In the present study, to examine the influences of prenatal dexamethasone exposure on skeletal development during morphogenesis, we treated pregnant rats with dexamethasone from gestational days (GD)9 to GD20. Using histological analyses, immunohistochemistry and quantitative measurement of gene expression, we studied whether prenatal dexamethasone up‐regulates Mig6 expression, which in turn down‐regulates EGFR signalling in chondrocytes, leading to delayed endochondral ossification and bone development. This study will be helpful to elucidate the mechanisms underlying the toxic effect of prenatal dexamethasone exposure on fetal bone development.
Methods
Animals
All animal care and experimental procedures complied with the Guidelines for the Care and Use of Laboratory Animals in Wuhan University and were approved by the Animal Welfare Committee in Wuhan University (No. 14016). This study was approved by the National Science & Technology Pillar Program of China and the National Natural Science Foundation of China. Animal studies are reported in compliance with the ARRIVE guidelines (Kilkenny et al., 2010; McGrath & Lilley, 2015). Pathogen‐free Wistar rats at 3–4 months of age weighing 180–220 g (female) or 260–300 g (male) were obtained from the Experimental Centre of Medical Scientific Academy of Hubei (No. 2006‐0005, Hubei, China). Rats were housed in temperature‐controlled room (24 to 25°C with 40–60% humidity). Pregnant rats were housed individually in cages (48 cm × 35 cm × 20 cm) with corn cob bedding. Food and water were freely available.
General procedures
We used pregnant rats to study the underlying mechanism of developmental toxicity of dexamethasone because animal models have been widely used to conduct toxicity studies as they provide strong evidence for the relationship between early‐life exposures and later‐life diseases. Rodents, including mice and rats, are useful models for investigating the biological mechanisms of the developmental origins of health and disease (McMullen and Mostyn, 2009). This study does not have any implications for replacement, refinement or reduction.
To reduce bias in animal experiments, rats were housed and treated by a technician, whereas bone sample harvesting and data analyses were all conducted by different co‐authors. Pregnant rats were randomly divided into a control group and a prenatal dexamethasone‐treated group. On GD20, 22 pregnant rats in each group were killed, and their fetuses were removed quickly from the uterus; two fetuses were selected randomly from every litter. For the experiment on adult offspring, the remaining eight pregnant rats in each group were maintained until normal delivery. After weaning, two female offspring were selected randomly from every litter and housed until 12 weeks old.
Experimental procedures
Preparation of animal models
Virgin female and male rats were used in these experiments. Two female rats were mated with one male rat overnight. The next day, the occurrence of spermatozoa in the vaginal smear was considered as GD0. As the distinct condensation of the hindlimb bud in rodents appears around GD9 (Taher et al., 2011), the pregnant rats were injected subcutaneously with dexamethasone (1 mg·kg−1 daily, n = 30) or saline as control (n = 30) every morning (between 0800h and 0900h) from GD9 to GD20. On GD20, the pregnant rats were anaesthetized with isoflurane and killed with decapitation, and then the fetuses were removed quickly from the uterus; their femurs and tibias were dissected free from soft tissue for further analysis. The 12‐week‐old female offspring rats were also killed by decapitation and their femurs were dissected and fixed in 70% ethanol for microCT (μCT) analysis.
Histology and immunohistochemistry
To study morphology and calcification of hindlimb long bones in the fetus, femurs were collected at GD20 (n = 22 per group). Femurs were fixed in 4% paraformaldehyde overnight and then processed and embedded in paraffin. Serial longitudinal sections (6 μm thick) were cut. One out of six sections was stained with haematoxylin and eosin (H&E) for quantification of the length of the primary ossification centre and hypertrophic zone. The neighbouring slices were then subjected to histochemistry and immunohistochemistry staining as described below. For von Kossa staining, sections were deparaffinized and stained with 5% AgNO3 until they become dark brown. The length of full femur and primary ossification centre was measured on von Kossa‐stained sections at ×50 magnification. For tartrate‐resistant acid phosphatase (TRAP) staining, sections were stained using a leukocyte acid phosphatase kit. TRAP+ cells at the chondro‐osseous junction and in the primary spongiosa were counted at ×100 magnification.
For immunohistochemistry analysis, sections were deparaffinized and hydrated through a graded series of ethanol (100–70%). Antigen retrieval was achieved by boiling the sections in 0.01 M sodium citrate buffer (pH 6.0) at about 95°C for 10–15 min. After antigen retrieval, the hydrated sections were then incubated in 3% H2O2 for 15 min to quench the endogenous peroxidase activity. Sections were then blocked in blocking serum at room temperature for 1 h and incubated with a primary antibody at 4°C overnight. The following primary antibodies were used: rabbit anti‐Runx2, rabbit anti‐Ki67, rabbit anti‐P57, rabbit anti‐Mig6 and rabbit anti‐phospho‐EGFR. After washing with PBS, sections were incubated with a biotinylated secondary antibody and then with an avidin‐biotinylated horseradish peroxidase complex solution according to the manufacturer's directions. Finally, peroxidase activity was revealed by immersion in diaminobenzidine (DAB). For negative controls in immunohistochemistry, immunostaining was performed on parallel sections in which the primary antibody was replaced with non‐immune rabbit IgGs. Apoptosis detection was carried out using the DeadEnd Colorimetric TUNEL System according to the manufacturer's directions. To quantify positive stained cells, the intensity of immunostaining was determined by measuring the mean optical density in six sections from different samples.
μCT measurement
The right femurs dissected from 12‐week‐old female rat offspring were fixed with 70% ethanol and then scanned and analysed with a μCT system (VivaCT 40; Scanco, Basserdorf, Switzerland). To determine trabecular bone volume per tissue volume, trabecular number, trabecular separation and trabecular thickness, 0.5–5.5 mm below the lowest point of growth plate was selected as the region of interest; cross‐sectional images were scanned at 21 μm resolution and analysed using 3D analysis.
Cell culture
Primary epiphyseal chondrocytes were isolated from the distal femoral and proximal tibial condyles of 1‐ to 3‐day‐old Wistar rats. Briefly, epiphyseal condyles were dissected, cleaned to remove connective tissue and digested in type 2 collagenase solution (2.2 mg·mL−1 in PBS) with gentle agitation at 37°C for 30 min. The condyles were collected and further digested for 4 h. The supernatant was filtered and centrifuged to collect chondrocytes. Cells were plated at a density of 4 × 105 cells per well in six‐well plates in chondrogenic medium (DMEM/F12 medium with 5% fetal bovine serum, 50 mg·mL−1 L‐ascorbic acid, 1% glutamine, 100 mg·mL−1 streptomycin and 100 U·mL−1 penicillin). Two to three days later, when cells reached 90% confluence, cultures were given fresh medium. On the next day, the cells were treated with various concentrations of dexamethasone (10, 50, 250 and 1000 nM) for 48 h based on the effect of dexamethasone on chondrocytes (Cheng et al., 2014) and then collected for further analysis.
Rat primary calvarial osteoprogenitors were obtained from neonatal calvariae by sequential digestion with collagenase and trypsin as described previously (Shalhoub et al., 1992). Briefly, calvariae were harvested from neonatal rat pups and cleaned to remove surrounding soft tissue and then digested in 2 mg·mL−1 collagenase A and 2.5 mg·mL−1 trypsin solution for 20 min; the cells liberated during the first digestion were discarded. Repeated digestion using fresh collagenase A/trypsin solution for 90 min was used. The cells liberated were then collected and plated at a density of 4 × 105 cells per well in six‐well plates in growth medium [α‐Minimum Essential Medium (MEM) with 10% fetal bovine serum, 100 mg·mL−1 streptomycin and 100 U·mL−1 penicillin]. When the cells reached 80% confluence, growth medium was switched to osteogenic differentiation medium (α‐MEM medium with 10% fetal bovine serum, 100 mg·mL−1 streptomycin, 100 U·mL−1 penicillin, 10 mM β‐glycerophosphate and 50 μg·mL−1 ascorbic acid); cells were then treated with various concentrations of dexamethasone for 48 h and harvested for analysis.
Gene expression
Tibias and femurs were dissected free of soft tissue. Under a dissection microscope, the primary ossification centre in each tibia and femur were carefully dissected out from the cartilage. To obtain total RNA, primary ossification centre tissue was homogenized in TRIzol Reagent (Invitrogen, Carlsbad, CA, USA). Total RNA was isolated according to manufacturer's instructions. The Applied Biosystems TaqMan Reverse Transcription reagents kit was used to convert mRNA into cDNA. Then, qRT‐PCR was performed using a SYBR Green qPCR Master Mix Kit and ABI StepOnePlus cycler (Applied Biosystems, Foster City, CA, USA) with 40 cycles at 95°C for 15 s and 60°C for 30 s. Relative expression of gene was calculated for each gene by the 2− ΔΔCT method with GAPDH for normalization. The rat primer sequences for the genes used in this study were as follows: GAPDH (forward, 5′‐GCAAGTTCAACGGCACAG‐3′; reverse, 5′‐GCCAGTAGAC‐TCCACGACA‐3′), osteocalcin (forward, 5′‐CAGACCTAGCAGACACCATG‐3′; reverse, 5′‐GCTTGGACATGAAGGCTTTG‐3′), bone sialoprotein (BSP) (forward, 5′‐ACGCTGGAAA‐GTTGGAGTTAG‐3′; reverse, 5′‐TCCTCTTCCTCTTCCTCTTCC‐3′), Mig6 (forward, 5′‐CCTACAATCTGAACTCCCCTG‐3′; reverse, 5′‐AGCTTGACTTTGGAGATGG‐AC‐3′), RANKL (forward, 5′‐AAACAAGCCTTTCAAGGGGC‐3′; reverse, 5′‐CACATCGAGCCACGAACCTT‐3′), OPG (forward, 5′‐TGTGTCCCTTGCCCTGACT‐ACT‐3′; reverse, 5′‐TGTTTCACGGTCTGCAGTTCC‐3′), calcitonin receptor (Calcr) (forward, 5′‐CGTGCCGTCTACTACAACGA‐3′; reverse, 5′‐AGAAGTTGACCACCAGAGCC‐3′), cathepsin K (Ctsk) (forward, 5′‐GACCCGTCTCTGTGTCCATC‐3′; reverse, 5′‐ACGGTCGCAGTTTTCGTCAT‐3′) and TRAP (forward, 5′‐GCTTCCACCCTGAGATTCGT‐3′; reverse, 5′‐ATGATGAAGTCAGCGCCCAT‐3′). For protein extraction, cells were washed twice with ice‐cold PBS and lysed in lysis buffer (10 mM HEPES, pH 7.5, 10 mM KCl, 1.5 mM MgCl2, 0.5 mM EDTA, 1 mM Na3VO4, 1 mM PMSF, 50 mM β‐glycerophosphate and 1 mM dithiothreitol) for 5 min on ice; the lysates were then collected. Equal amounts of protein lysates (30 μg per lane) were resolved by SDS‐PAGE on 10% polyacrylamide gels, transferred to polyvinylidene difluoride membranes and blotted with rabbit anti‐Mig6 or anti‐phospho‐EGFR antibody at 4°C overnight. After incubation with horseradish peroxidase‐conjugated secondary antibody, signals were developed using enhanced chemiluminescence. To normalize quantitative RT‐PCR (qRT‐PCR) data, the expression level of each target genes was normalized to the stably expressed ‘housekeeping’ genes. For each gene, relative expression of gene was calculated by the 2− ΔΔCT methods with GAPDH for normalization.
siRNA knockdown of Mig6 gene
To knockdown Mig6 expression, RNA interference technology was used. Three pairs of designed siRNA oligonucleotides against rat Mig6 were purchased from Shanghai GenePharma Co. Ltd. (Shanghai, China). The sequences of the Mig6 siRNA were GCAGAUCUUAGCUGUGCAUTT and AUGCACAGCUAAGAUCUGCTT. A pair of non‐specific oligonucleotides (nonsilencing control) was used as a negative control; the sequences of the control siRNA were UUCUCCGAACGUGUCACGUTT and ACGUGACACGUUCGGAGAATT. Prior to transfection, primary chondrocyte was seeded in six‐well plates at a density of 4 × 105 cells per well. Twenty‐four hours later, cells were then transfected with 30 nM Mig6 siRNA or control oligonucleotides using Lipofectamine 2000 (Invitrogen) in Opti‐MEM according to the manufacturer's protocol. Eight hours later, the medium was exchanged for a fresh medium, and the cells were treated with 50 or 250 nM dexamethasone. The mRNA of the cells was harvested after 48 h.
Data and statistical analysis
The data and statistical analysis comply with the recommendations on experimental design and analysis in pharmacology (Curtis et al., 2015). All data were expressed as mean ± SEM. Means between the control and prenatal dexamethasone‐treated groups were compared by independent Student's t‐test or Mann–Whitney U‐test. For cell culture experiments, all results derived from experiments were repeated independently at least six times; multiple comparisons were assessed by the Kruskal–Wallis test to identify significant differences among the groups. When the Kruskal–Wallis test indicated a significant difference, pairwise comparisons were performed using the Mann–Whitney U‐test to determine which group differed from the control. A value of P < 0.05 was considered statistically significant.
Materials
Dexamethasone (Shuanghe Pharmaceutical Company, Wuhan, China), leukocyte acid phosphatase kit (Sigma‐Aldrich, St. Louis, MO, USA), rabbit anti‐Runx2 (Santa Cruz Biotechnology, Santa Cruz, Texas, USA), rabbit anti‐Ki67, rabbit anti‐P57, rabbit anti‐Mig6 (all from Proteintech, Wuhan, China), rabbit anti‐phospho‐EGFR (Cell Signaling Technology Inc., Danvers, MA, USA), avidin‐biotinylated horseradish peroxidase complex (Vectastain ABC Kit; Vector Laboratories, Burlingame, CA, USA) and DAB (Dako, Glostrup, Denmark). Apoptosis detection was carried out using the DeadEnd Colorimetric TUNEL System (Cat. G7130; Promega, Madison, WI, USA), type 2 collagenase (Invitrogen), Tri Reagent (Sigma‐Aldrich), SYBR Green qPCR Master Mix Kit (Takara Biotechnology Co., Ltd. Dalian, China) and Lipofectamine 2000 (Invitrogen).
Results
Prenatal dexamethasone exposure retards endochondral ossification
Prenatal dexamethasone exposure at a dose of 1 mg·kg−1·day−1 markedly affected the development of epiphyseal cartilage in both femurs and tibias of fetuses, which was characterized by shortened primary ossification centre and hypertrophic chondrocyte accumulation (Figure 1A). In those fetuses treated with prenatal dexamethasone, we observed an overall 1.65‐fold increase in the length of the hypertrophic zone (Figure 1B). The expansion of the hypertrophic zone also was confirmed by Safranin O staining, which showed less staining intensity in the hypertrophic zone than in the proliferative zone (Figure 1A).
Figure 1.
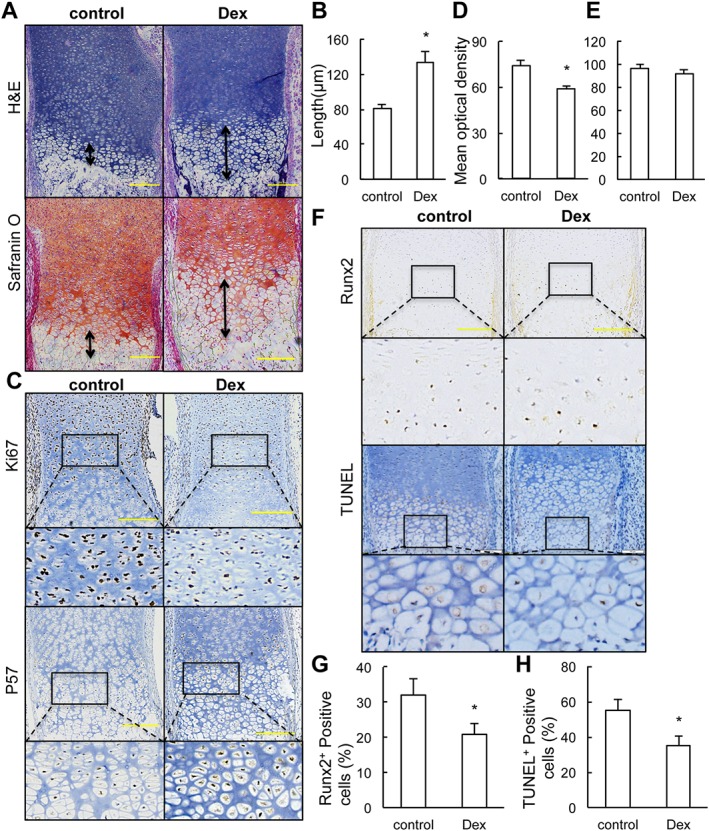
Prenatal dexamethasone treatment inhibits the terminal differentiation and apoptosis of hypertrophic chondrocytes in fetal long bone. (a) Representative H&E staining and safranin O staining images of distal femur from control and fetus with prenatal dexamethasone (Dex) treatment. (b) Quantification of the length of the hypertrophic zone. (c) Immunostaining of Ki67 and p57. (d, e) The staining intensity was determined by measuring the mean optical density obtained from six sections of different sample using Image Pro Plus System 6.0. (f) Immunostaining of Runx2 and TUNEL staining in sections of distal epiphysis femur from control and fetus with prenatal dexamethasone exposure. (g, h) Quantification of the mean optical density of Runx2‐positive (g) and TUNEL‐positive chondrocytes (h). The data are presented as the mean ± SEM, *P < 0.05 versus control; Student's t‐test; n = 6 per group. Scale bar = 200 μm.
The expansion of the hypertrophic zone in fetuses with dexamethasone treatment could be the result of increased proliferation or altered differentiation of the chondrocyte cell. To test the effect of prenatal dexamethasone treatment on chondrocyte proliferation and early differentiation, we performed immunostaining analysis of Ki67, a marker for replicating cells, and p57, a marker for cell cycle exit. We found reduced chondrocyte proliferation, but no modifications in early differentiation (Figure 1C–E). The above results further demonstrate that elongation of the hypertrophic zone is the major side effect of dexamethasone on fetal long bones.
Runx2 is an important transcription factor required for chondrocyte maturation and highly expressed in chondrocytes proceeding toward terminal differentiation (Ueta et al., 2001). Immunostaining of Runx2 revealed fewer mature chondrocytes in hypertrophic zone of long bone of prenatal dexamethasone‐treated fetuses compared with those of the control fetuses (Figure 1F, G). These results suggest that prenatal dexamethasone exposure delays chondrocyte maturation.
Hypertrophic chondrocytes undergo cell death and are gradually replaced by bone in the process of endochondral ossification. To investigate whether prenatal dexamethasone treatment affects terminal differentiation and apoptosis of chondrocyte, TUNEL staining was performed on fetal long bones. In femur sections from control fetuses, 55% of chondrocytes in the hypertrophic zone of epiphyseal cartilage were apoptotic. In contrast, only 35% of chondrocytes in femoral cartilage from dexamethasone‐treated fetuses were TUNEL positive (Figure 1F, H), suggesting that prenatal dexamethasone exposure delays terminal differentiation and apoptosis of the chondrocytes.
Prenatal dexamethasone exposure impairs bone formation in primary ossification centre
The metaphyseal bone is derived directly from the hypertrophic zone through a bone‐modelling process, leading to the formation of primary ossification centres. As shown in Figure 2A, von Kossa staining and H&E staining showed a striking shortened primary ossification centre in femur and significantly reduced long bone length after prenatal dexamethasone exposure, indicating that such exposure delayed primary ossification. For fetuses in the control group, the primary ossification centre was well developed with thick and long bone spicules in mineralized area. However, for fetuses after prenatal dexamethasone treatment, the mineralized area was much smaller, and the trabecular spicules were much thinner and shorter (Figure 2A). In addition, we observed a 32% reduction in the length of femur; this was due mainly to a striking 51% reduction in the length of the primary ossification centre (Figure 2B). This result indicates that the bone formation process in the fetus is delayed by prenatal dexamethasone exposure as well. To clarify the formation of mature osteoblasts, we therefore assayed for the mRNA expression of osteocalcin and BSP using qRT‐PCR. The results showed that prenatal dexamethasone treatment suppressed the expression of these bone marker genes in fetal bone (Figure 2C). These data demonstrate that prenatal dexamethasone exposure impairs bone formation in the primary ossification centres of fetal long bones.
Figure 2.
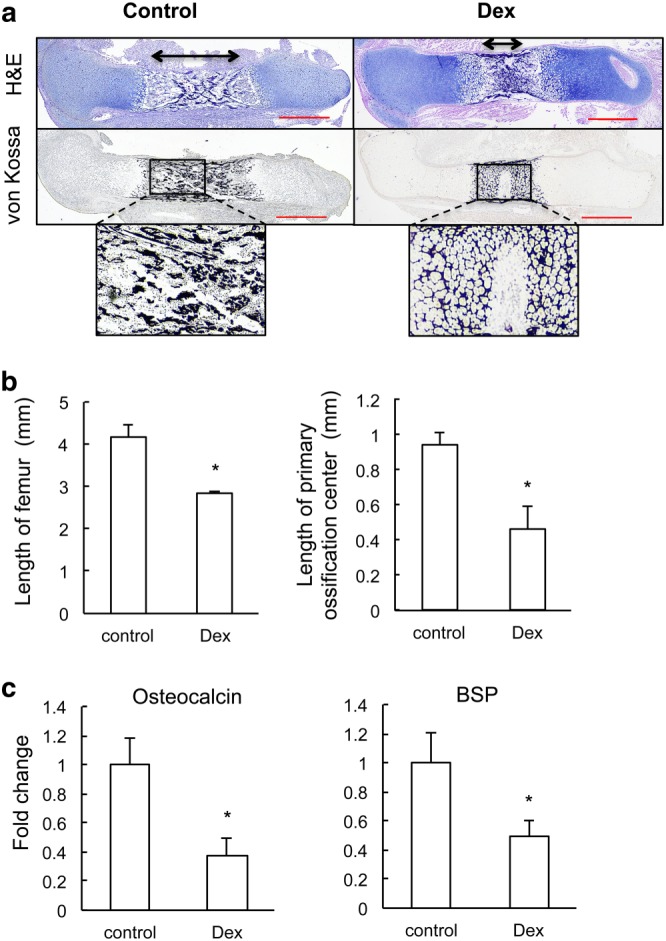
Prenatal dexamethasone (Dex) treatment delays endochondral bone formation. (a) Representative H&E staining and von Kossa staining images of full‐length femur from control and fetus with prenatal dexamethasone treatment. (b) Quantification of the whole length of femur and length of primary ossification centre. *P < 0.05 versus control; Student's t‐test; n = 6 per group. (c) qRT‐PCR analysis of gene expression of bone formation markers (osteocalcin and BSP). *P < 0.05 versus control; Mann–Whitney U‐test; n = 12 per group. Scale bar = 500 μm.
Prenatal dexamethasone exposure delays osteoclastogenesis
Genesis and recruitment of osteoclasts are key events in the primary ossification of the hypertrophic cartilage anlage. Osteoclast‐deficient mice also exhibit accumulation of hypertrophic chondrocytes and delayed endochondral ossification (Li et al., 2000). Formation of mature osteoclasts was evaluated by TRAP staining (Figure 3A). The results showed that the size of TRAP+ cells in fetal bone with prenatal dexamethasone treatment was much smaller than those in the control group. Quantification (Figure 3B) showed a significant decrease in the number of TRAP+ cells at the chondro‐osseous junction in prenatal dexamethasone‐treated fetus, compared with those in control fetuses. In primary ossification centre, the number of TRAP+ cells of the prenatal dexamethasone‐treated fetuses was also significantly reduced compared with that in control group. We further assessed the osteoclast differentiation in primary ossification centres by qRT‐PCR analyses of the expression of osteoclast‐specific function genes including those for cathepsin K(Ctsk), for the calcitonin receptor (Calcr) and for TRAP. Results showed that the expression of Ctsk was not influenced, whereas the expression of Calcr and TRAP was significantly reduced in fetuses with prenatal dexamethasone exposure (Figure 3C).
Figure 3.
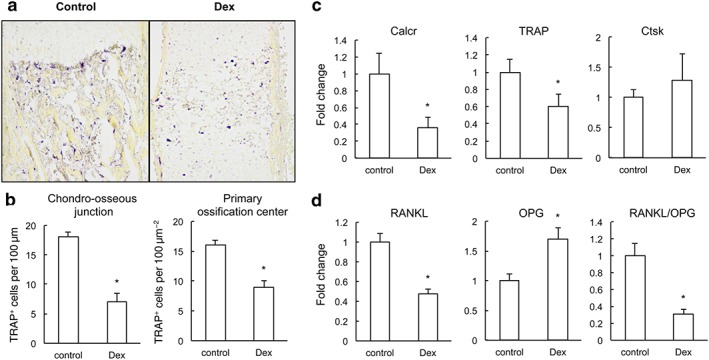
Prenatal dexamethasone (Dex) treatment suppresses osteoclast differentiation in fetal long bone. (a) TRAP staining (purple) of fetal femur. (b) The number of TRAP+ cells along the chondro‐osseous junction and marrow cavity was quantified microscopically. *P < 0.05 versus control; Student's t‐test; n = 6 per group. (c) qRT‐PCR measurement of mRNA expression of Calcr, TRAP and Ctsk in fetal bone from control and fetus treated with prenatal dexamethasone exposure. *P < 0.05 versus control; Mann–Whitney U‐test; n = 12 per group. (d) qRT‐PCR measurement of mRNA expression of RANKL and OPG in fetal bone from control and fetus treated with prenatal dexamethasone exposure. *P < 0.05 versus control; Mann–Whitney U‐test; , n = 22 per group.
To understand how prenatal dexamethasone treatment reduces the number of osteoclasts in the primary ossification centre, we measured the mRNA expression of two major determinants for osteoclastogenesis, RANKL and OPG, in the primary ossification centre using qRT‐PCR. There was a 52% reduction in RANKL expression and 1.7‐fold increment in OPG expression in the fetuses with prenatal dexamethasone exposure, which combined together resulted in an overall 69% decrease in the RANKL/OPG ratio in the primary ossification centre of the prenatal dexamethasone‐exposed fetuses (Figure 3D). These results suggest that prenatal dexamethasone treatment down‐regulates RANKL expression in bone, which in turn reduces the osteoclastogenesis.
Dexamethasone treatment regulates RANKL and OPG expression in primary chondrocytes and osteoblastic cells
As mature and hypertrophic chondrocytes, osteoblasts and osteocytes are the main cells that co‐express both RANKL and OPG (Silvestrini et al., 2005), we next treated primary cultures of chondrocytes with dexamethasone (10, 50, 250 and 1000 nM), to determine whether the down‐regulation of RANKL/OPG ratio is due to the direct effect of dexamethasone on these cells. The qRT‐PCR results revealed that dexamethasone significantly repressed the mRNA expression of RANKL, while enhancing the mRNA expression of OPG, resulting in decreased RANKL/OPG ratio after 48 h of treatment (Figure 4A). These results are consistent with the in vivo result we described above (Figure 3D).
Figure 4.

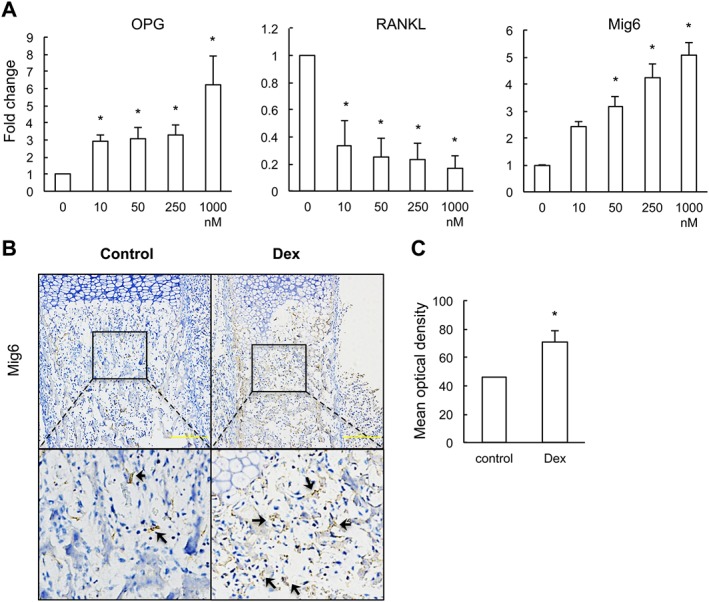
Dexamethasone (Dex) regulates the expression of RANKL and OPG partially through activating Mig6. (a) qRT‐PCR measurement of mRNA expression of RANKL and OPG in primary chondrocytes treated with various concentrations of dexamethasone (10, 50, 250 and 1000 nM) for 48 h (Kruskal–Wallis test). (b) qRT‐PCR measurements of mRNA expression of Mig6, EGFR, EGF, TGF‐α and heparin‐binding EGF (HB‐EGF) in fetal long bone. *P < 0.05 versus control; Mann–Whitney U‐test; n = 12 per group. (c) Immunostaining of Mig6 and phosphor‐EGFR in chondrocytes: black arrow heads show positive stained chondrocytes. (d, e) Quantification of the mean optical density of Mig6‐positive stained chondrocytes (d) and phosphorylated EGFR (e). (f) Dexamethasone dose‐dependently activates mRNA expression of Mig6 in primary chondrocytes (Kruskal–Wallis test). (g) Dexamethasone suppresses the phosphorylation of EGFR in chondrocytes through activating Mig6. Rat primary epiphyseal chondrocytes were treated with 250 nM dexamethasone with or without Mig6 siRNAs for 48 h. Cell lysates were collected for Western blotting of Mig6 and phospho‐EGFR. (h) Knockdown of Mig6 by siRNA completely blocks dexamethasone‐induced mRNA expression of OPG. Primary chondrocytes were transfected with either negative control or Mig6 siRNAs followed by dexamethasone treatment; 48 h later, the mRNA level of Mig6 was measured by qRT‐PCR. *P < 0.05 versus control; Kruskal–Wallis test; n = 7 per group. Scale bar = 200 μm.
EGFR signalling is important for expression of RANKL and OPG in chondrocytes (Zhang et al., 2011, 2013). To investigate whether dexamethasone affects the expression of RANKL and OPG by modulating EGFR signalling, we assessed the mRNA expression of EGFR and its ligands in long bone by qRT‐PCR. Results showed that dexamethasone did not alter the mRNA expression of EGFR and its ligands but it markedly increased the expression of Mig6 (Figure 4B). Immunohistochemical analysis of femur sections confirmed that the protein levels of Mig6 in the hypertrophic chondrocytes of fetuses with dexamethasone exposure were much higher than that of the control fetuses (Figure 4C, D). Mig6 blocks the formation of asymmetric EGFR dimers, thereby inhibiting the activities of EGFR tyrosine kinases (Zhang et al., 2007). We therefore investigated whether the up‐regulation of Mig6 induced by dexamethasone exposure further affected EGFR signalling, by analyzing EGFR phosphorylation in femur sections. Prenatal dexamethasone treatment induced a pronounced reduction in phosphorylation of the EGFR in the long bone of fetuses (Figure 4C, E). Consistent with the in vivo results mentioned above, dexamethasone treatment concentration‐dependently stimulated the mRNA expression of Mig6 in primary chondrocytes (Figure 4F). These results were further confirmed by the measurement of protein expression of Mig6, which showed that dexamethasone treatment induced significant increase in Mig6 expression and decrease in EGFR phosphorylation in chondrocytes. In contrast, knockdown of Mig6 by siRNA blocked the inhibitory effect of dexamethasone on EGFR phosphorylation (Figure 4G). These findings showed that prenatal dexamethasone treatment suppressed EGFR activity by up‐regulating Mig6 expression in fetal long bones.
To further understand whether Mig6 mediated the regulatory effect of dexamethasone on expression of RANKL and OPG in chondrocytes, we treated primary chondrocytes with dexamethasone (250 nM) and assayed gene expression using qRT‐PCR. The results revealed that siRNA knockdown of Mig6 completely abolished the up‐regulation of OPG expression in primary chondrocytes stimulated by dexamethasone (Figure 4H). This implies that prenatal dexamethasone exposure regulates osteoclastogenesis partially via up‐regulating Mig6 expression.
Mature osteoblasts also produce RANKL and OPG, which are the predominant factors regulating osteoclast differentiation in the bone cavity (Khosla, 2001). We further assessed the effect of dexamethasone on the expression of those genes in primary osteoblasts. qRT‐PCR results showed that dexamethasone treatment concentration‐dependently up‐regulated OPG expression and down‐regulated RANKL expression (Figure 5A). Interestingly, we also found that dexamethasone exposure could activate Mig6 expression in osteoblasts (Figure 5A). In addition, the immunohistochemistry results further demonstrated the up‐regulation of Mig6 protein in fetal bone with prenatal dexamethasone exposure (Figure 5B, C).
Figure 5.
Dexamethasone (Dex) regulates the expression of RANKL, OPG and Mig6 in osteoblastic cells. (a) qRT‐PCR measurements of mRNA expression of RANKL and OPG in primary osteoblastic cells treated with various concentrations of dexamethasone (10, 50, 250 and 1000 nM) for 48 h. *P < 0.05 versus control; Kruskal–Wallis test; n = 6 per group. (b) Immunostaining of Mig6 in bone marrow cavity: black arrow heads show positive stained osteoblasts. (b) The mean optical density of positive stained chondrocytes was quantified. *P < 0.05 versus control; Student's t‐test; n = 6 per group. Scale bar = 200 μm.
Low bone mass in adult offspring with prenatal dexamethasone treatment
Accumulating evidence from animal studies and clinical data has raised concerns regarding the long‐term consequences of prenatal dexamethasone treatment (Godfrey et al., 2011). Next, we investigated whether prenatal dexamethasone treatment had long‐term effect on longitudinal bone growth and bone mass in 12‐week‐old rat offspring. Results showed that there was no significant difference in the length of femur between offspring with and without prenatal dexamethasone treatment (data not shown). However, μCT imaging demonstrated that bone mass in 12‐week‐old rat offspring after prenatal dexamethasone treatment was less than that present in control offspring (Figure 6A). This was mainly due to a reduced trabecular number and an increased trabecular separation (Figure 6B). These data indicate that prenatal dexamethasone treatment may lead to less bone mass in the adult offspring.
Figure 6.
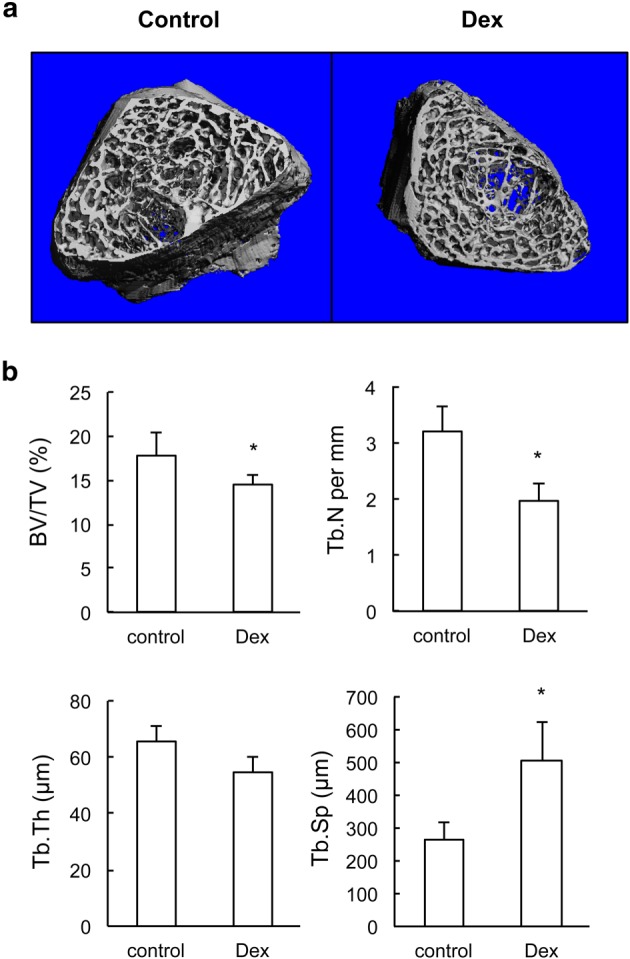
Twelve‐week‐old offspring with prenatal dexamethasone (Dex) treatment have reduced bone mass. (a) Representative μCT images of femur from 12‐week‐old offspring with prenatal saline or dexamethasone treatment. (b) Quantitative μCT analysis of trabecular bone microarchitecture. *P < 0.05 versus control; Student's t‐test; n = 12 per group. BV/TV, bone volume per tissue volume; Tb. N, trabecular number; Tb. Sp, trabecular separation; Tb. Th, trabecular thickness.
Discussion
Dexamethasone is used antenatally to accelerate the maturation of fetal respiratory and cardiovascular systems when there is a risk of preterm delivery, as well as to provide treatment for asthma of pregnant women and congenital adrenal hyperplasia of the fetus (Tan and Thomson, 2000; Maryniak et al., 2014). We undertook this study to examine the effect of prenatal dexamethasone exposure on fetal bone development and clarify the underlying mechanisms. The dose of dexamethasone used in human pregnancy varies from 0.02 mg·kg−1·day−1 for congenital adrenal hyperplasia (Meyer‐Bahlburg et al., 2004) to 0.1–0.5 mg·kg−1·day−1 for indicated preterm labour (Bloom et al., 2001; Peaceman et al., 2005). In animal experiments, the lower therapeutic dose of dexamethasone (0.03 mg·kg−1 every second day) induced loss of trabecular bone and retardation of bone growth in developmental piglets (Tomaszewska et al., 2013). Our previous work found that prenatal dexamethasone treatment could dose‐dependently increase intrauterine growth retardation rate in mice (Xu et al., 2011). Considering the dose conversion between rat and human (conversion factor is 0.16) (Reagen‐Shaw et al., 2007), prenatal treatment with dexamethasone in rats at 1 mg·kg−1 in this study is comparable with that prescribed for pregnant women (0.2–0.3 mg·kg−1) (Jobe and Soll, 2004). The present study indicated that prenatal dexamethasone treatment at this therapeutic dose (1 mg·kg−1·day−1) induced an adverse effect on long bone development with enlarged hypertrophic zones of growth plate in fetuses. Further, our data demonstrated that prenatal dexamethasone treatment can retard bone development and lead to the reduction of bone mass in later life. From a clinical point of view, it is important to use the proper dosage of dexamethasone.
Dexamethasone exerts different effects on bone growth at different developmental stages. In post‐natal development, dexamethasone treatment exerts growth‐inhibiting effects systemically through down‐regulation of the growth hormone/insulin‐like growth factor 1 axis (Nilsson et al., 2005) and inhibition of chondrocyte proliferation, mineralization and increment of apoptosis in growth plates (Nilsson et al., 2005; Chagin et al., 2010; Zaman et al., 2014). In prenatal stages, the negative effects of dexamethasone treatment on bone development of human are substantial in the last trimester of pregnancy when maximal fetal growth occurs (Weiler et al., 1997; So and Ng, 2005). Maternal treatment with dexamethasone either continuously or on alternate days induced retarded growth with an immature skeleton and shortened and less mineralized long bones in the fetus (Sliwa et al., 2010; Carbone et al., 2012). In line with these studies, we have demonstrated that prenatal dexamethasone exposure at middle and late gestational stages impaired skeleton development with shortened and less mineralized long bones.
In the embryonic stage, the long bones of vertebrate skeletons are developed through endochondral ossification, during which the mesenchymal cells initially undergo condensation and chondrogenesis to form cartilage anlagen, which is shaped like the prospective bone. Later, the cartilaginous anlagen is ossified, and the bone marrow is formed to give rise to mature bones (Amizuka et al., 2012). Bone growth along the longitudinal axis relies on chondrocyte proliferation, hypertrophy and production of an extracellular matrix. Accordingly, any interference in these processes would lead to the shortening of the bone length (Rossi et al., 2002). Our study demonstrated that prenatal dexamethasone exposure slightly inhibited the proliferation of chondrocytes, whereas it did not affect the expression level of p57, an early differentiation marker. This indicated that the delayed endochondral ossification induced by dexamethasone treatment was not derived from the suppressed early differentiation of chondrocytes. Further, we demonstrated that dexamethasone exposure significantly down‐regulated the expression of Runx2, a marker for chondrocyte maturation, and inhibited chondrocyte apoptosis. Because the chondrocyte‐specific activity of Runx2 is essential for endochondral ossification (Chen et al., 2011), our data strongly suggest that dexamethasone may delay endochondral ossification by suppressing the terminal differentiation and apoptosis of the hypertrophic chondrocytes.
Osteoclasts are critical and indispensable to the degradation of mineralized hypertrophic cartilage during endochondral bone formation (Touaitahuata et al., 2014). Mice lacking osteoclasts such as RANK knockout mice have extended hypertrophic cartilage zone (Li et al., 2000). Interestingly, our data showed a decreased number of osteoclasts at the chondro‐osseous junction of fetal bone after prenatal dexamethasone exposure, indicating that the expansion of the hypertrophic zone was due to the reduced osteoclastogenesis. Hypertrophic chondrocytes are one of the major sources of RANKL, controlling mineralized cartilage resorption at the growth plate (Xiong et al., 2011). Therefore, the enlarged hypertrophic zone we observed in long bones of fetuses after prenatal dexamethasone exposure is because of the reduced number of osteoclasts at the chondro‐osseous junction. Our in vivo and in vitro data further showed that dexamethasone exposure down‐regulated RANKL expression and up‐regulated OPG expression in chondrocytes, and this correlates well with the decreased number of osteoclasts at the chondro‐osseous junction. These results indicate that dexamethasone delays the endochondral ossification process by suppressing osteoclastogenesis.
In addition to reduced osteoclastogenesis at chondro‐osseous junctions, we also found that prenatal dexamethasone exposure reduced osteoeoclast numbers in the bone cavity, indicating decreased osteoclastogenesis in bone marrow. Osteoclasts have the unique ability to resorb mineralized bone and this process is an important part of bone modelling in early bone development stage (Crockett et al., 2011). In the bone cavity, osteoblast lineage cells produce RANKL and OPG to control osteoclastogenesis (Martin and Sims et al., 2015). We found that dexamethasone concentration‐dependently suppressed RANKL expression, whereas it activated OPG expression in primary osteoblasts, which may in turn delay the osteoclastogenesis. Our data also showed that the expression of bone formation marker osteocalcin and BSP was significantly suppressed, indicating that dexamethasone delayed bone modelling in the fetus.
EGFR and its ligands have previously been shown to be highly expressed in the chondrocytes and regulate endochondral ossification in growth plates (Zhang et al., 2011, 2013; Usmani et al., 2012). EGFR signalling supported the osteoclastogenesis at chondro‐osseous junctions and promoted the remodelling of growth plate cartilage extracellular matrix into bone during endochondral ossification (Zhang et al., 2011, 2013). Conversely, deficiency of Mig6 can induce abnormal endochondral ossification (Zhang et al., 2005; Shepard et al., 2013). EGFR‐specific tyrosine kinase inhibitors and EGFR knockdown impaired the RANKL‐mediated activation of osteoclastogenic signalling pathways and enlarged the growth plates with expanded hypertrophic zones (Yi et al., 2008; Zhang et al., 2011, 2013). Consistent with these reports, we found that dexamethasone induced accumulation of hypertrophic chondrocytes with delayed maturation and terminal differentiation; this phenotype is similar to that in EGFR‐deficient rat and mice (Zhang et al., 2011, 2013). Interestingly, dexamethasone has previously been shown to suppress EGFR signalling by up‐regulating Mig6 in fibroblasts (Xu et al., 2005). Our present study demonstrated that dexamethasone suppressed EGFR signalling in chondrocytes through Mig6. It is possible that dexamethasone treatment leads to the delayed osteoclastogenesis at chondro‐osseous junction by suppressing EGFR signalling. In supportof this possibility, we found a decreased number of osteoclasts adjacent to chondrocytes at the junction. In vitro study further showed that dexamethasone depressed the expression of RANKL, whereas it enhanced the expression of OPG partly by up‐regulating Mig6 in chondrocytes. Our results suggest that Mig6 is partly involved in mediating the toxic effect of dexamethasone on bone development in utero.
In conclusion, we have performed in vivo and in vitro experiments to clarify the mechanisms underlying bone developmental retardation in fetuses exposed to prenatal dexamethasone. The results demonstrated that prenatal dexamethasone exposure retarded endochondral ossification by delaying the terminal differentiation and apoptosis of hypertrophic chondrocytes and suppressing osteoclastogenesis. Mig6 in chondrocytes and osteoblast may be partly involved in the detrimental effect of dexamethasone on osteoclast differentiation. To our knowledge, the detailed mechanism of fetal bone growth retardation by maternal dexamethasone treatment was not reported previously in any other studies. This study will lead to a better understanding of the possible long‐term effect of dexamethasone on bone metabolism in later life. In addition, this study also indicates that long‐term prenatal dexamethasone treatment may have serious clinical consequences and the doses used should be optimized.
Author contributions
X.Z., Y.S.‐G., J.M., H.H. and L.W. performed the research; X.Z., H.W. and L.C. designed the research study; X.Z. and H.W. analysed the data; X.Z. and H.W. wrote and revised the paper; all authors approved the final manuscript.
Conflict of interest
The authors declare no conflicts of interest.
Declaration of transparency and scientific rigour
This Declaration acknowledges that this paper adheres to the principles for transparent reporting and scientific rigour of preclinical research recommended by funding agencies, publishers and other organisations engaged with supporting research.
Acknowledgements
This study was supported by grants from the National Science & Technology Pillar Program of China (no. 2013BAI12B01‐3), National Natural Science Foundation of China (nos. 81573515, 81220108026, 81430089 and 81371940) and Scientific Research Foundation for the Returned Overseas Chinese Scholars, State Education Ministry (to X.Z.).
Zhang, X. , Shang‐Guan, Y. , Ma, J. , Hu, H. , Wang, L. , Magdalou, J. , Chen, L. , and Wang, H. (2016) Mitogen‐inducible gene‐6 partly mediates the inhibitory effects of prenatal dexamethasone exposure on endochondral ossification in long bones of fetal rats. British Journal of Pharmacology, 173: 2250–2262. doi: 10.1111/bph.13506.
References
- Alexander SPH, Fabbro D, Kelly E, Marrion N, Peters JA, Benson HE et al. (2015a). The Concise Guide to PHARMACOLOGY 2015/16: Catalytic receptors. Br J Pharmacol 172: 5979–6023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Davenport AP, Kelly E, Marrion N, Peters JA, Benson HE et al. (2015b). The Concise Guide to PHARMACOLOGY 2015/16: G Protein‐Coupled Receptors. Br J Pharmacol 172: 5744–5869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Fabbro D, Kelly E, Marrion N, Peters JA, Benson HE et al. (2015c). The Concise Guide to PHARMACOLOGY 2015/16: Catalytic receptors. Br J Pharmacol 172: 5979–6023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amizuka N, Hasegawa T, Oda K, Luiz de Freitas PH, Hoshi K, Li M et al. (2012). Histology of epiphyseal cartilage calcification and endochondral ossification. Front Biosci (Elite Ed) 4: 2085–2100. [DOI] [PubMed] [Google Scholar]
- Anastasi S, Fiorentino L, Fiorini M, Fraioli R, Sala G, Castellani L et al. (2003). Feedback inhibition by RALT controls signal output by the ErbB network. Oncogene 22: 4221–4234. [DOI] [PubMed] [Google Scholar]
- Bloom SL, Sheffield JS, Mclntire DD, Leveno KJ (2001). Antenatal dexamethasone and decreased birth weight. Obstet Gynecol 97: 485–490. [DOI] [PubMed] [Google Scholar]
- Carbone DL, Zuloaga DG, Hiroi R, Foradori CD, Legare ME, Handa RJ (2012). Prenatal dexamethasone exposure potentiates diet‐induced hepatosteatosis and decreases plasma IGF‐1 in a sex‐specific fashion. Endocrinology 153: 295–306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chagin AS, Karimian E, Sundström K, Eriksson E, Sävendahl L (2010). Catch‐up growth after dexamethasone withdrawal occurs in cultured postnatal rat metatarsal bones. J Endocrinol 204: 21–29. [DOI] [PubMed] [Google Scholar]
- Chen H, Ghori‐Javed FY, Rashid H, Serra R, Gutierrez SE, Javed A (2011). Chondrocyte‐specific regulatory activity of Runx2 is essential for survival and skeletal development. Cells Tissues Organs 194: 161–165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng X, Chen JL, Ma ZL, Zhang ZL, Lv S, Mai DM et al. (2014). Biphasic influence of dexamethasone exposure on embryonic vertebrate skeleton development. Toxicol Appl Pharmacol 281: 19–29. [DOI] [PubMed] [Google Scholar]
- Citri A, Yarden Y (2006). EGF‐ERBB signaling: towards the systems level. Nat Rev Mol Cell Biol 7: 505–516. [DOI] [PubMed] [Google Scholar]
- Crockett JC, Rogers MJ, Coxon FP, Hocking LJ, Helfrich MH (2011). Bone remodeling at a glance. J Cell Sci 124: 991–998. [DOI] [PubMed] [Google Scholar]
- Crowther CA, McKinlay CJ, Middleton P, Harding JE (2011). Repeat doses of prenatal corticosteroids for women at risk of preterm birth for improving neonatal health outcomes. Cochrane Database Syst Rev 6: CD003935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Curtis MJ, Bond RA, Spina D, Ahluwalia A, Alexander SP, Giembycz MA et al. (2015). Experimental design and analysis and their reporting: new guidance for publication in BJP. Br J Pharmacol 172: 3461–3471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Godfrey KM, Inskip HM, Hanson MA (2011). The long‐term effects of prenatal development on growth and metabolism. Semin Reprod Med 29: 257–265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jobe AH, Soll RF (2004). Choice and dose of corticosteroid for antenatal treatments. Choice and dose of corticosteroid for antenatal treatments. Am J Obstet Gynecol 190: 878–881. [DOI] [PubMed] [Google Scholar]
- Khosla S (2001). Minireview: the OPG/RANKL/RANK system. Endocrinology 142: 5050–5055. [DOI] [PubMed] [Google Scholar]
- Kilkenny C, Browne W, Cuthill IC, Emerson M, Altman DG (2010). Animal research: reporting in vivo experiments: the ARRIVE guidelines. Br J Pharmacol 160: 1577–1579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J, Sarosi I, Yan XQ, Morony S, Capparelli C, Tan HL et al. (2000). RANK is the intrinsic hematopoietic cell surface receptor that controls osteoclastogenesis and regulation of bone mass and calcium metabolism. Proc Natl Acad Sci U S A 97: 1566–1571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin TJ, Sims NA (2015). RANKL/OPG: critical role in bone physiology. Rev Endocr Metab Disord 16: 131–139. [DOI] [PubMed] [Google Scholar]
- Maryniak A, Ginalska‐Malinowska M, Bielawska A, Ondruch A (2014). Cognitive and social function in girls with congenital adrenal hyperplasia—influence of prenatally administered dexamethasone. Child Neuropsychol 20: 60–70. [DOI] [PubMed] [Google Scholar]
- McGrath JC, Lilley E (2015). Implementing guidelines on reporting research using animals (ARRIVE etc.): new requirements for publication in BJP. Br J Pharmacol 172: 3189–3193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McMullen S, Mostyn A (2009). Animal models for the study of the developmental origins of health and disease. Proc Nutr Soc 68: 306–320. [DOI] [PubMed] [Google Scholar]
- Meyer‐Bahlburg HF, Dolezal C, Baker SW, Carlson AD, Obeid JS, New MI (2004). Cognitive and motor development of children with and without congenital adrenal hyperplasia after early‐prenatal dexamethasone. J Clin Endocrinol Metab 89: 610–614. [DOI] [PubMed] [Google Scholar]
- Morales WJ, Diebel ND, Lazar AJ, Zadrozny D (1986). The effect of antenatal dexamethasone administration on the prevention of respiratory distress syndrome in preterm gestations with premature rupture of membranes. Am J Obstet Gynecol 154: 591–595. [DOI] [PubMed] [Google Scholar]
- Murphy KE, Hannah ME, Willan AR, Hewson SA, Ohlsson A, Kelly EN et al. (2008). Multiple courses of antenatal corticosteroids for preterm birth (MACS): a randomized controlled trial. Lancet 372: 2143–2151. [DOI] [PubMed] [Google Scholar]
- Murphy KE, Willan AR, Hannah ME, Ohlsson A, Kelly EN, Matthews SG et al. (2012). Effect of antenatal corticosteroids on fetal growth and gestational age at birth. Obstet Gynecol 119: 917–923. [DOI] [PubMed] [Google Scholar]
- Nilsson O, Marino R, De Luca F, Phillip M, Baron J (2005). Endocrine regulation of the growth plate. Horm Res 64: 157–165. [DOI] [PubMed] [Google Scholar]
- Peaceman AM, Bajaj K, Kumar P, Grobman WA (2005). The interval between a single course of antenatal steroids and delivery and its association with neonatal outcomes. Am J Obstet Gynecol 193 (3Pt2): 1165–1169. [DOI] [PubMed] [Google Scholar]
- Pest MA, Russell BA, Zhang YW, Jeong JW, Beier F (2014). Disturbed cartilage and joint homeostasis resulting from a loss of mitogen‐inducible gene 6 in a mouse model of joint dysfunction. Arthritis Rheum 66: 2816–2827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reagan‐Shaw S, Nihal M, Ahmad N (2007). Dose translation from anima to human studies revisited. FASEB J 22: 659–661. [DOI] [PubMed] [Google Scholar]
- Rossi F, MacLean HE, Yuan W, Francis RO, Semenova E, Lin CS et al. (2002). p107 and p130 coordinately regulate proliferation, Cbfa1 expression, and hypertrophic differentiation during endochondral bone development. Dev Biol 247: 271–285. [DOI] [PubMed] [Google Scholar]
- Scheving LA, Buchanan R, Krause MA, Zhang X, Stevenson MC, Russell WE (2007). Dexamethasone modultes ErbB tyrosine kinase expression and signaling through multiple and redundant mechanisms in cultured rat hepatocytes. Am J Physiol Gastrointest Liver Physiol 293: G552–G559. [DOI] [PubMed] [Google Scholar]
- Schneider MR, Sibilia M, Erben RG (2009). The EGFR network in bone biology and pathology. Trends Endocrinol Metab 20: 517–524. [DOI] [PubMed] [Google Scholar]
- Shalhoub V, Conlon D, Tassinari M, Quinn C, Partridge N, Stein GS et al. (1992). Glucocorticoids promote development of the osteoblast phenotype by selectively modulating expression of cell growth and differentiation associated genes. J Cell Biochem 50: 425–440. [DOI] [PubMed] [Google Scholar]
- Shepard JB, Jeong JW, Maihle NJ, O'Brien S, Dealy CN (2013). Transient anabolic effects accompany epidermal growth factor receptor signal activation in articular cartilage in vivo. Arthritis Res Ther 15: R60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silvestrini G, Ballanti P, Patacchioli F, Leopizzi M, Gualtieri N, Monnazzi P et al. (2005). Detection of osteoprotegerin (OPG) and its ligand (RANKL) mRNA and protein in femur and tibia of the rat. J Mol Histol 36: 59–67. [DOI] [PubMed] [Google Scholar]
- Sliwa E, Dobrowolski P, Piersiak T (2010). Bone development of suckling piglets after prenatal, neonatal or perinatal treatment with dexamethasone. J Anim Physiol Anim Nutr (Berl) 94: 293–306. [DOI] [PubMed] [Google Scholar]
- So KW, Ng PC (2005). Treatment and prevention of neonatal osteopenia. Curr Paediatr 15: 106–113. [Google Scholar]
- Southan C, Sharman JL, Benson HE, Faccenda E, Pawson AJ, Alexander SPH et al. (2016). The IUPHAR/BPS Guide to PHARMACOLOGY in 2016: towards curated quantitative interactions between 1300 protein targets and 6000 ligands. Nucl Acids Res: 44 (Database Issue): D1054–D1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Staal B, Williams BO, Beier F, Vande Woude GF, Zhang YW (2014). Cartilage‐specific deletion of Mig‐6 results in osteoarthritis‐like disorder with excessive articular chondrocyte proliferation. Proc Natl Acad Sci U S A 111: 2590–2595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taher L, Collette NM, Murugesh D, Maxwell E, Ovcharenko I, Loots GG (2011). Global gene expression analysis of murine limb development. PLoS One 6: e28358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan KS, Thomson NC (2000). Asthma in pregnancy. Am J Med 109: 727–733. [DOI] [PubMed] [Google Scholar]
- Tomaszewska E, Dobrowolski P, Puzio I (2013). Morphological changes of the cartilage and bone in newborn piglets evoked by experimentally induced glucocorticoid excess during pregnancy. J Anim Physiol Anim Nutr (Berl) 97: 785–796. [DOI] [PubMed] [Google Scholar]
- Touaitahuata H, Cres G, de Rossi S, Vives V, Blangy A (2014). The mineral dissolution function of osteoclasts is dispensable for hypertrophic cartilage degradation during long bone development and growth. Dev Biol 393: 57–70. [DOI] [PubMed] [Google Scholar]
- Ueta C, Iwamoto M, Kanatani N, Yoshida C, Liu Y, Enomoto‐ Iwamoto M et al. (2001). Skeletal malformations caused by overexpression of Cbfa1 or its dominant negative form in chondrocytes. J Cell Biol 153: 87–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Usmani SE, Pest MA, Kim G, Ohora SN, Qin L, Beier F (2012). Transforming growth factor controls the transition from hypertrophic cartilage to bone during endochondral bone growth. Bone 51: 131–141. [DOI] [PubMed] [Google Scholar]
- Wang K, Yamamoto H, Chin JR, Werb Z, Vu TH (2004). Epidermal growth factor receptor‐deficient mice have delayed primary endochondral ossification because of defective osteoclast recruitment. J Biol Chem 279: 53848–53856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiler HA, Paes B, Shah JK, Atkinson SA (1997). Longitudinal assessment of growth and bone mineral accretion in prematurely born infants treated for chronic lung disease with dexamethasone. Early Hum Dev 47: 271–286. [DOI] [PubMed] [Google Scholar]
- Xiong J, Onal M, Jilka RL, Weinstein RS, Manolagas SC, O'Brien CA (2011). Matrix‐embedded cells control osteoclast formation. Nat Med 17: 1235–1241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu D, Chen M, Pan XL, Xia LP, Wang H (2011). Dexamethasone induces fetal developmental toxicity through affecting the placental glucocorticoid barrier and depressing fetal adrenal function. Environ Toxicol Pharmacol 32: 356–363. [DOI] [PubMed] [Google Scholar]
- Xu D, Makkinje A, Kyriakis JM (2005). Gene 33 is an endogenous inhibitor of epidermal growth factor receptor signaling and mediates dexamethasone‐induced suppression of EGF function. J Biol Chem 280: 2924–2933. [DOI] [PubMed] [Google Scholar]
- Yi T, Lee HL, Cha JH, Ko SI, Kim HJ, Shin HI et al. (2008). Epidermal growth factor receptor regulates osteoclast differentiation and survival through cross‐talking with RANK signaling. J Cell Physiol 217: 409–422. [DOI] [PubMed] [Google Scholar]
- Young BK, Klein SA, Katz M, Wilson SJ, Douglas GW (1980). Intravenous dexamethasone for prevention of neonatal respiratory distress: a prospective controlled study. Am J Obstet Gynecol 138: 203–209. [DOI] [PubMed] [Google Scholar]
- Zaman F, Chrysis D, Huntjens K, Chagin A, Takigawa M, Fadeel B et al. (2014). Dexamethasone differentially regulates Bcl‐2 family proteins in human proliferative chondrocytes: role of pro‐apoptotic bid. Toxicol Lett 224: 196–200. [DOI] [PubMed] [Google Scholar]
- Zhang X, Pickin KA, Bose R, Jura N, Cole PA, Kuriyan J (2007). Inhition of the EGF receptor by binding of MIG6 to an activating kinase domain interface. Nature 450: 741–744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, Sichari VA, Lan S, Zhu J, Koyama E, Dupuis HL et al. (2011). The critical role of the epidermal growth factor receptor in endochondral ossification. J Bone Miner Res 26: 2622–2633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, Zhu J, Li Y, Lin T, Siclari VA, Chandra A et al. (2013). Epidermal growth factor receptor signaling regulates epiphyseal cartilage development through β‐catenin‐dependent and ‐independent pathways. J Biol Chem 288: 32229–32240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang YW, Su Y, Lanning N, Swiatek PJ, Bronson RT, Sigler R et al. (2005). Targeted disruption of Mig‐6 in the mouse genome leads to early onset degenerative joint disease. Proc Natl Acad Sci U S A 102: 11740–11745. [DOI] [PMC free article] [PubMed] [Google Scholar]