

Abstract
Transcription factors have traditionally been viewed with skepticism as viable drug targets, but they offer the potential for completely novel mechanisms of action that could more effectively address the stem cell like properties, such as self-renewal and chemo-resistance, that lead to the failure of traditional chemotherapy approaches. Core binding factor is a heterodimeric transcription factor comprised of one of 3 RUNX proteins (RUNX1-3) and a CBFβ binding partner. CBFβ enhances DNA binding of RUNX subunits by relieving auto-inhibition. Both RUNX1 and CBFβ are frequently mutated in human leukemia. More recently, RUNX proteins have been shown to be key players in epithelial cancers, suggesting the targeting of this pathway could have broad utility. In order to test this, we developed small molecules which bind to CBFβ and inhibit its binding to RUNX. Treatment with these inhibitors reduces binding of RUNX1 to target genes, alters the expression of RUNX1 target genes, and impacts cell survival and differentiation. These inhibitors show efficacy against leukemia cells as well as basal-like (triple-negative) breast cancer cells. These inhibitors provide effective tools to probe the utility of targeting RUNX transcription factor function in other cancers.
Keywords: CBFβ, RUNX, PPI, Transcription factor inhibitor, Leukemia, Triple negative breast cancer
Graphical Abstract

Highlights
-
•
Small molecule inhibitors of CBFβ-RUNX protein-protein interaction developed.
-
•
Inhibitors alter occupancy of RUNX1 on target genes and alter their expression.
-
•
Inhibitors show efficacy against leukemia cell lines and basal-like (triple negative) breast cancer cell lines.
Transcription factors are proteins that bind to DNA and regulate how much of other proteins are made. We describe the development of inhibitors of the interaction between two transcription factors, CBFβ and RUNX. Both these transcription factors are the targets of alterations in human leukemia as well as in a number of solid tumors. The inhibitor changes the behavior of the RUNX transcription factor and alters the levels of proteins it regulates. We show these inhibitors may have potential utility for leukemia as well as one specific type of breast cancer which has a very poor prognosis.
1. Introduction
Core binding factor (CBF) is a heterodimeric transcription factor composed of a DNA-binding RUNX subunit (encoded by one of three genes: RUNX1, RUNX2, or RUNX3) and a non-DNA-binding CBFβ subunit which increases the affinity of RUNX proteins for DNA. All three RUNX proteins as well as CBFβ have been shown to be critical regulators of specific developmental pathways. RUNX1 and CBFβ are essential for definitive hematopoiesis, where they regulate expression of genes associated with proliferation, differentiation, and survival of stem and progenitor cells (FRIEDMAN, 2009, DE BRUIJN and SPECK, 2004, WANG et al., 2010, LINK et al., 2010). RUNX2 is essential for normal bone formation by way of transcriptional regulation of genes critical for bone development (KOMORI et al., 1997, OTTO et al., 1997). Both RUNX1 and RUNX3 play key roles in neuronal development.
Perhaps not surprisingly, based on their critical roles in normal development, RUNX proteins and CBFβ are targets of genetic alteration in a variety of cancers. Both RUNX1 and CBFB undergo chromosomal translocations in a subset of acute myeloid leukemia (AML) and acute lymphocytic leukemia (ALL) patients where the corresponding fusion proteins have clearly been shown to be drivers of disease (Blyth et al., 2005). For the fusion proteins AML1-ETO and TEL-AML1, the binding of the fusion proteins to CBFβ has been shown to be essential for transformation (Roudaia et al., 2009). RUNX1 is also mutated in a subset of AML and myelodysplastic syndrome (MDS) patients. In addition, RUNX1 has recently been implicated in a number of epithelial cancers (SCHEITZ et al., 2012, SCHEITZ and TUMBAR, 2013). Altered expression of RUNX2 has been implicated in breast and prostate cancers (Blyth et al., 2005). Silencing of RUNX3 by DNA methylation has been linked to intestinal and lung cancers (Lee et al., 2013). Due to the importance of these proteins for normal development as well as in a variety of cancers, small molecules which can modulate their activity are useful tools to address function and test new therapeutic approaches.
Small molecule inhibitors of protein-protein interactions, particularly in the context of transcription factors, is still a relatively nascent field, in part due to the long and widely held belief that this class of interactions is “undruggable”. With an increasing number of success stories of small molecule inhibitors modulating protein-protein interactions (ARKIN et al., 2014a, LARAIA et al., 2015, ARKIN and WHITTY, 2009), including transcription factors, this paradigm is clearly changing. Along this vein, we have developed tool compounds which bind to CBFβ and inhibit CBFβ binding to RUNX proteins as a probe for the role of this important protein-protein interaction in function as well as its potential therapeutic applications. The most potent compounds we have developed inhibit this protein-protein interaction at low micromolar concentrations, use an allosteric mechanism to achieve inhibition, displace CBFβ from RUNX1 in cells, change occupancy of RUNX1 on target genes, alter expression of RUNX1 target genes, and show clear effects on leukemia and basal-like breast cancer cells consistent with on-target activity on RUNX protein activity.
2. Materials and Methods
2.1. Chemical Synthesis
Details of the chemical synthesis and characterization of the compounds is provided in Supplemental Information.
2.2. FRET Assays
FRET assays were carried out as described previously (ILLENDULA et al., 2015, GORCZYNSKI et al., 2007) using 100 nM Cerulean-Runt domain and 100 nM Venus-CBFβ (1-141).
2.3. Pharmacokinetics Analysis of AI-12-126 and AI-14-91
Details of the pharmacokinetics analysis are provided in Supplemental Information.
2.4. GLIDE Docking
2.4.1. Ligand Preparation
Low energy 3D structures of compounds were produced using LigPrep 2.5. Epik 2.2 was used to generate ionization/tautomeric states of compounds. Minimum energy conformations 3 per ligand were generated using OPLS-2005 force field.
2.4.2. Protein Preparation
The CBFβ crystal structure (PDB code 1E50) was loaded from Protein Data Bank and prepared using Protein Preparation Wizard. The protein was pre-processed by assigning the bond orders, added hydrogen and filled in the missing loops and the side chains using Prime 3.0. Waters beyond 5 Å from hetero groups were removed, the protein is optimized and Impref-minimization was carried using the OPLS-2005 force field.
2.4.3. Docking
In Grid Generation, under docking tab we have used the site as a centroid of binding site residues in the protein. The active site residues were determined by chemical shift perturbations in 15N-1H and 13C-1H HSQC NMR experiments of protein binding to AI-4-57. The following residues were selected for grid generation: V86, L88, R90, E91, Y96, K98, A99, K111, G112, W113, M122, G123, C124. Docking was carried out using the Virtual Screening Workflow framework. All the compounds were docked flexibly and after docking 100% of best compounds with all good states were scored by MM-GBSA.
2.5. CBFβ Mutant Proteins
Wildtype CBFβ (1-141) and CBFβ (1-141) mutants R90E, K98E and K111E were expressed at 15 °C in 15N labeled minimal media. Proteins were purified using a Ni-NTA column, cleaved with rTev protease digestion overnight followed by size exclusion chromatography to remove the affinity tag and impurities. Protein samples at 150 μM were dialyzed, inserted into an NMR buffer, and titrated with 600 μM AI-4-57. All 15N-1H HSQCs were recorded on a Bruker 800 MHz NMR spectrometer equipped with a cryoprobe.
2.6. NMR Spectroscopy
All NMR-based experiments were acquired using CBFβ (1-141) solutions in buffer containing 50 mM KPi, 0.1 mM EDTA, 1 mM DTT, 0.01%(w/v) sodium azide, 5%(v/v) DMSO and 5%(v/v) D2O at a final pH of 7.5. All experiments were recorded on samples of uniformly labeled 15N CBFβ concentrated to 0.5 mM at 25 °C on a Bruker 18.8 T spectrometer equipped with a CryoProbe™. All NMR data were processed using NMRPipe. Samples containing compound were made by adding AI-4-57 in 100% DMSO to yield an equimolar protein-compound solution.
15N and 13C chemical shift perturbations were determined from 15N- and 13C-HSQC experiments, respectively, collected in the presence and absence of AI-4-57. Peaks were assigned using previously known resonances, and spectra were overlaid and compared using CcpNMR software suite (Vranken et al., 2005). Weighted chemical shift changes in parts per million were calculated by using the equation ∆ 15N + 1HN) = |∆ δHN | + (|∆ δN |/4.69) as described in Yuan et al., (2002). A resonance shift of 0.1 ppm or more was considered to be significant.
All 15N relaxation measurements were carried out with samples of CBFβ + AI-4-57 and CBFβ alone. 15N T1 experiments were conducted using relaxation delays of 10, 180, 300, 500, 1300, 1800, and 2300 ms. 15N T2 experiments used relaxation delays of 10, 25, 50, 75, 100, 150, 200, 225, and 250 ms. Peak intensities were fit to y = Ae− Bx, where B is the relaxation rate (R), using CcpNMR to determine T1 and T2 relaxation times. R1 and R2 relaxation rates, the inverses of T1 and T2, were used to calculate R1 ∗ R2 and R2/R1 values for protein with compound and protein with DMSO alone. Differences in R1, R2, R1 ∗ R2 and R2/R1 were calculated between the values for the CBFβ + AI-4-57 and CBFβ alone samples. Residues with changes in R1 ∗ R2 greater than two standard deviations above or below a trimmed mean consisting of the median 60% of the data were considered significantly different.
Saturation transfer difference NMR samples were composed of 200 μM CBFβ, 2 mM AI-4-57, 10% D2O, and 5% DMSO in 50 mM KPi, 1 mM DTT, 0.1 mM EDTA, 001% w/v NaN3, pH 7.5 in a final volume of 200 μl. All STD experiments were performed using a 600 MHz Bruker NMR spectrometer at 25 °C with saturation times of 500, 750, 1000, 1500, and 2000 ms. Samples were irradiated at 0.4 ppm (protein) and 30 ppm (off-resonance control) and the difference spectra calculated using MestReNova.
2.7. Co-immunoprecipitation Assays
4 × 106 SEM cells were treated with DMSO or 10 μM of AI-4-88, AI-10-47, AI-10-104, AI-12-126 and AI-14-91 for 6 h. Cells were lysed in modified RIPA buffer (50 mM Tris pH 7.5, 150 mM NaCl, 1% NP40, 0.25% sodium deoxycholate and 1 mM EDTA). RUNX1 was immunoprecipitated from cell lysates using anti-RUNX1 antibody (Active Motif, cat #39000,) and protein-A Agarose beads (Roche Applied Science) as follows: cell lysates were mixed with protein A agarose beads and 2 μg RUNX1 antibody in IP buffer I (50 mM Tris pH 7.5, 150 mM NaCl, 0.5% NP40, 0.25% sodium deoxycholate) and rotated at 10 rpm for 5 h. Agarose beads were washed twice with IP Buffer I followed by washing with IP buffer II (50 mM Tris pH 7.5, 0.1% NP40, 0.05% sodium deoxycholate). All lysis, immunoprecipitation, and washing steps included DMSO/corresponding inhibitor (10 μM). The beads were heated at 95 °C for 12 min in Western blot loading buffer (100 mM Tris-HCL pH 6.8, 200 mM DTT, 4% SDS, 0.2% Bromophenol-blue, 20% glycerol). The eluted protein was resolved in a 12% polyacrylamide gel. CBFβ was detected using anti-CBFβ antibody (provided by Nancy A. Speck). The membrane was re-probed with anti-RUNX1 antibody and detected using Clean-Blot IP Detection Reagents (Thermo Scientific). Relative band intensities were quantified using ImageJ software.
2.8. Effect of CBFβ Inhibitors on RUNX1 Occupancy and Target Gene Expression
Inducible Runx1 (iRunx) ES cells were maintained and differentiated essentially as described in Lichtinger et al. (2012).
2.8.1. ES Cell Maintenance
Inducible Runx1 (iRunx) ES cells were maintained on primary mouse embryonic fibroblasts in ES maintenance media: DMEM (high glucose from powder (Sigma D5648)), supplemented with 15% FCS, 1 mM Sodium pyruvate, 100 units/ml Penicillin and 100 μg/ml Streptomycin, 1 mM glutamine, 0.15 mM MTG, 25 mM HEPES buffer, 103 U/ml ESGRO® (Millipore), 1 × non-essential amino acids (Sigma). Prior to differentiation the ES cells were grown without feeder cells for two passages on gelatinised tissue culture treated plates.
2.8.2. ES cell differentiation
A single cell suspension of ES cells was transferred into IVD media on 15 cm low adherence bacteriological plates (Sterilin) at a concentration of 2.5 × 104/ml. IVD media - IMDM supplemented with 15% FCS, 100 units/ml Penicillin and 100 μg/ml Streptomycin, 1 mM glutamine, 0.15 mM MTG, 0.18 mg/ml Human transferrin (Roche 652,202) and 50 μg/ml Ascorbic acid. After 3.25 days the embryoid bodies were collected, briefly digested in Tryple Express (Life Technologies) and gently dissociated. To obtain a single cell suspension the cells were passed through a cell strainer and resuspended in IMDM + 20% FCS. Flk1 + ve (CD309) cells were isolated using a biotinylated Flk1 antibody (eBioscience 13-5821) at 5 μl per 107 cells for 15 min on ice, followed by 2 × wash with MACS buffer (PBS + 5% BSA and 2 mM EDTA). Cells bound by the antibody were then isolated using MACS anti-biotin beads and MACS LS columns (Miltenyi Biotec) according to the manufacturer's instructions. Isolated Flk1 + ve cells were plated in Blast media at a concentration of 9 × 103/cm2 on gelatinized tissue culture treated dishes. Blast media–IMDM supplemented with 10% FCS, 100 units/ml Penicillin and 100 μg/ml Streptomycin, 1 mM glutamine, 0.45 mM MTG, 0.18 mg/ml Human transferrin, 25 μg/ml Ascorbic acid, 20% D4T conditioned media, 5 μg/l mVEGF (Peprotech), 10 μg/l mIL-6 (Miltenyi Biotec). After approximately 40 h in blast culture, iRunx was induced by the addition of 0.1 μg/ml doxycycline (dox). At the same time as dox induction, the indicated concentrations of AI-14-91 or the control compound AI-4-88 were added. Adherent cells were washed and harvested by trypsinisation 24 h later and used for either FACS analysis, protein extract preparation, resuspended in TRIzol (Life Technologies) for RNA isolation or crosslinked for chromatin isolation.
2.8.3. mRNA expression analysis
RNA was extracted from cells using TRIzol® (Life Technologies) according to the manufacturer's instructions. DNase treatment was performed using Turbo DNase (Ambion) according to manufacturer's instructions and a further clean up step was performed using NucleoSpin RNA columns (Macherey Nagel). First strand cDNA synthesis was carried out using Superscript II (Life Technologies) according to manufacturer's instructions using 250 ng of RNA. Real Time PCR was carried out using ABI SYBR® green master mix with 2.5 μl of diluted cDNA and 0.25 μM primer per 10 μl reaction on an ABI 7900HT machine. Analysis was carried out according to the 2− ΔΔCt method, normalizing to GAPDH and using the + Dox sample as the calibrator, samples were measured in duplicate. Error bars represent standard deviation where n = 3. See Table 1 for gene expression primer sequences.
Table 1.
Primers for gene expression.
| Gene | Forward primer | Reverse primer |
|---|---|---|
| Runx1 | GCAGGCAACGATGAAAACTACTC | CAAACCTGAGGTCGTTGAATCTC |
| NFE2 | TCCTCAGCAGAACAGGAACAG | GGCTCAAAAGATGTCTCACTTGG |
| Gfi1 | GTGAGCCTGGAGCAACACAA | CTCTTGAAGCTCTTGCCACAGA |
| Itga2b (CD41) | AGAGGGCCATTCCTGTCTG | GTCGATTCCGCTTGAAGAAG |
| β-2-microglobulin | TTCTGGTGCTTGTCTCACTG | CAGTATGTTCGGCTTCCCATTC |
| Spi1 (Pu.1) | CCATAGCGATCACTACTGGGATTT | TGTGAAGTGGTTCTCAGGGAAGT |
| cebpa | GCAGGAGGAAGATACAGGAAGCT | ACACCTAAGTCCCTCCCCTCTAAA |
| cebpb | GTTTCGGGACTTGATGCAATC | CGCAGGAACATCTTTAAGTGAT |
2.8.4. FACS Analysis
Surface marker expression for CD41 and KIT (CD117) was assessed by FACS analysis using CD117-APC (BD Pharmingen 553356) and CD41 PE-Cy7 (e-Bioscience 25-0411) antibodies.
2.8.5. Western Blotting
Whole cell extracts were run on 4–20% TGX gels (Bio-Rad) and blotted using the Trans-Blot Turbo transfer system (Bio-Rad) according to the manufacturer's instructions. Blots were blocked with 4% milk powder in 0.05% TBS-Tween and incubated with anti-HA (1:1000 dilution, H6908 Sigma), anti-CBFβ (1:400, sc20693 Santa Cruz), and anti-GAPDH antibodies (1:8000 dilution, ab8245 Abcam). Proteins were visualized using Pierce SuperSignal West Pico Chemiluminescent substrate (Thermo Scientific).
2.8.6. Chromatin Immunoprecipitation
Chromatin immunoprecipitation was performed essentially as described previously (Lichtinger et al., 2012) with the exception that the material was crosslinked with Di(N-succinimidyl)-glutarate (DSG; Sigma) and 1% Formaldehyde (Thermo Fisher) before quenching with 1/10th volume 2 M glycine. Nuclei were prepared essentially as described previously (Lefevre et al., 2003), sonicated using a Bioruptor water bath in immunoprecipitation buffer I (25 mM Tris 1 M, pH 8.0, 150 mM NaCl, 2 mM EDTA, pH 8.0, 1% TritonX-100 and 0.25% SDS). After centrifugation the sheared 0.5–2 kb chromatin fragments (1–2 × 106 cells) were diluted with 2 volumes immunoprecipitation buffer II (25 mM Tris, pH 8.0, 150 mM NaCl, 2 mM EDTA, pH 8.0, 1% TritonX-100, 7.5% glycerol) and precipitation was carried out for 2–3 h at 4 °C using 2 μg anti-HA antibody (H6908) coupled to 15 μl Protein-G dynabeads. Beads were washed with low salt buffer (20 mM Tris, pH 8.0, 150 mM NaCl, 2 mM EDTA, pH 8.0, 1% TritonX-100, 0.1% SDS), high salt buffer (20 mM Tris, pH 8.0, 500 mM NaCl, 2 mM EDTA, pH 8.0, 1% TritonX-100, 0.1% SDS), LiCl buffer (10 mM Tris, pH 8.0, 250 mM lithium chloride, 1 mM EDTA, pH 8.0, 0.5% NP40, 0.5% sodium-deoxycholate) and TE pH 8.0 containing 50 mM sodium chloride. The immune complexes were eluted in 100 μl elution buffer (100 mM NaHCO3, 1% SDS) and after adding 4 μl 5 M sodium chloride and 0.5 μl proteinase K, the crosslinks were reversed at 65 °C overnight. DNA was extracted using the Ampure PCR purification kit (Beckman Coulter) according to the manufacturer's instructions and analyzed by qPCR. Primers used for ChIP analysis are shown in Table 2.
Table 2.
Primers for ChIP.
| Gene | Forward primer | Reverse primer |
|---|---|---|
| Pu.1 3′ enhancer | GCTGTTGGCGTTTTGCAAT | GGCCGGTGCCTGAGAAA |
| Oct4 promoter | TGGGCTGAAATACTGGGTTC | TTGAATGTTCGTGTGCCAAT |
| Itga2b (CD41) promoter | CTGTGAAAGTCCAGCCACCAT | AGTGAGCCAGGCAGCGAAT |
| NFE2 -3kb | TGTTTGGCAACAATGCTTGTG | CAACCCACCTCCACTACGTAT |
| Chr2 | AGGGATGCCCATGCAGTCT | CCTGTCATCAGTCCATTCTCCAT |
| Chr1 | TGCTCCACAGTGTCCATGTACA | AGCAATTTCATGGGTGAGAGAAG |
2.9. Differentiation of HPC-7 Cells
Effects of inhibitors on HPC-7 cell differentiation was carried out as described in Supplemental Information.
2.10. Viability of a Panel of Leukemia Cell Lines and Normal Cells With CBFβ Inhibitors
2.10.1. Cell Lines
Leukemia cell lines were purchased either from the American Type Culture Collection (ATCC), Dr. Chiosis Laboratory (MOLM-13, HEL and M0-91) or Dr. Lucio's Castilla's laboratory (ME-1). All cell lines have been authenticated. Cells were cultured in Iscove's Modified Dulbecco's Medium (IMDM; Life Technologies) supplemented with 10% fetal bovine serum (FBS) according to culture conditions indicated by ATCC and 1% penicillin/streptomycin (Pen/Strep; Life Technologies).
2.10.2. Cell Viability Assay
AML cell lines were cultured at 5 × 105 cells/ml in 96-well plates and treated for 48 h with the indicated compounds at different doses or with DMSO. Cells were stained with DAPI (Life Technologies). At least 2 × 104 events for cell lines per condition were characterized on a BD LSR II flow cytometer. Data analysis was performed using FlowJo 9.3 software for Mac OS X (TreeStar). Cells that were negative for DAPI were scored as viable, and the viability was represented as the percent relative to untreated controls.
2.10.3. Colony Assays With Normal Human Cord Blood Cells
Colony forming assay was performed as described (Guzman et al., 2007). Briefly, primary cord blood (CB) cells were obtained from the New York Blood Bank. Mononuclear cells (MNCs) were isolated by Ficoll. MNCs were cultured in the presence or absence of 10 μM or 20 μM of the indicated compounds for 48 h in serum free medium supplemented with cytokines (Hassane et al., 2010). Cells (25,000) were plated in MethoCult H4434 (Stemcell Technologies). Colonies were scored after 14 days of culture.
2.11. Runx1 Knockdown
Knockdown of RUNX1 (Wang et al., 2011) was determined by quantitative immunoblotting (Janes, 2015) with the following antibodies: RUNX1 (Cell Signaling #4336, 1:1000), RUNX2 (MBL International #D130-3, 1:1000), CBFβ (Santa Cruz Biotechnologies #SC-20693, 1:1000), vinculin (Millipore #05-386, 1:10,000), tubulin (Abcam #ab89984, 1:20,000), and GAPDH (Ambion #AM4300, 1:20,000).
2.12. Effects of CBFβ Inhibitors on Acini Formation in 3D Culture of MCF10A-5E Cells and on the Viability of the Basal-like Breast Cancer Cell Line HC1143 in 3D Culture
MCF10A-5E cells expressing shRUNX1 have been described previously (JANES et al., 2010, WANG et al., 2011). HCC1143 cells were obtained from ATCC and cultured in growth medium (RPMI + 10% FBS) according to the provider's recommendations. For 3D culture, cells were seeded at 5000 cells/well atop growth factor-reduced matrigel (BD) in assay medium (MCF10A-5E) or growth medium (HCC1143) + 5 ng/ml EGF (Peprotech) + 2% matrigel as described previously (Debnath et al., 2003). CBFβ inhibitors (1 μM) or 0.1% DMSO were added at the time of plating and replenished every four days with fresh assay or growth medium + EGF + matrigel.
3. Results
3.1. Initial Lead for Small Molecule Inhibitors of CBFβ-RUNX Binding Which Bind to CBFβ
We recently reported the 2-pyridyl benzimidazole AI-4-57 as a compound which binds to the CBFβ portion of the CBFβ-SMMHC fusion protein and inhibits its binding to the Runt domain of RUNX proteins (Illendula et al., 2015). Using our previously described FRET assay for binding of the amino acid 1-141 portion of CBFβ to the Runt domain (Gorczynski et al., 2007), we showed this compound is also a modest potency inhibitor of the binding of wildtype CBFβ to the RUNX1 Runt domain (see Fig. 1, Table 3). In order to develop more potent analogs for use as tool compounds to probe RUNX and CBFβ protein function, we synthesized a library of analogs of AI-4-57 and characterized their activity.
Fig. 1.

Identification of pharmacophore. A. FRET assay results for AI-4-57, AI-10-104, and AI-14-91. Cerulean-Runt domain and Venus-CBFβ concentration was 100 nM. X-axis indicates compound concentration and y-axis is ratio between the emission intensities at 525 and 474 nm. Experiments were performed in duplicate. Error bars represent average ± standard deviation. The average data points were fit using Origin. B. Schemes 1–3 illustrate the targets of modifications to AI-4-57 as well as compounds synthesized and assayed to delineate the active pharmacophore. C. Results of STD NMR experiment with AI-4-57 (2 mM) and CBFβ (200 μM). Bottom panel shows 1D NMR spectrum of AI-4-57. Upper 2 panels show STD difference spectra (off-resonance (70 ppm) saturation spectrum minus on-resonance (0.4 ppm) CBFβ saturation spectrum) with saturation times of 500 and 2000 ms.
Table 3.
Results of FRET assays for selected compounds. All compounds were measured in duplicate. Each dataset was fit to derive an IC50 value. IC50 values are reported as the average of these two independent fits ± the standard deviation.
| Compound ID | Structure | FRET IC50 (μM) |
|---|---|---|
| AI-4-57 | 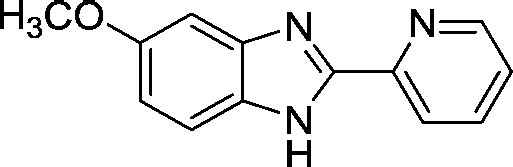 |
34.3 ± 0.3 |
| AI-10-47 | 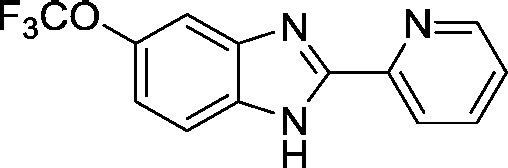 |
3.2 ± 0.5 |
| AI-10-104 (a) |
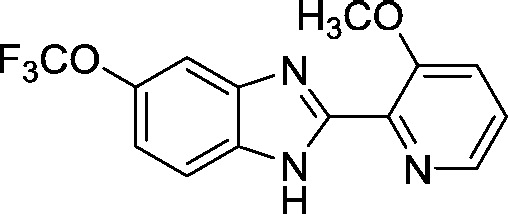 |
1.25 ± 0.06 |
| AI-12-16 (b) |
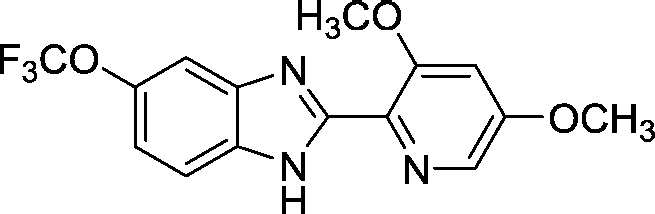 |
1.7 ± 0.2 |
| AI-14-55 (c) |
 |
4.8 ± 0.3 |
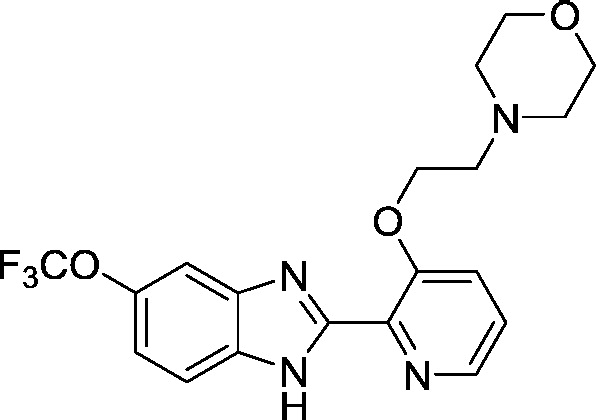
| AI-12-126 (d) |
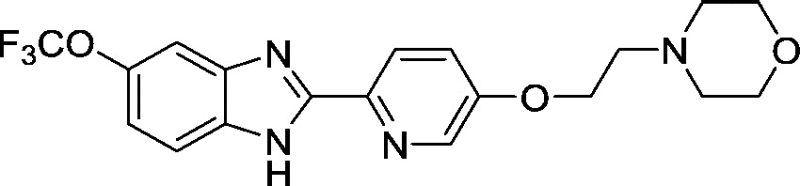 |
8.9 ± 1.3 |
| AI-14-91 (e) |
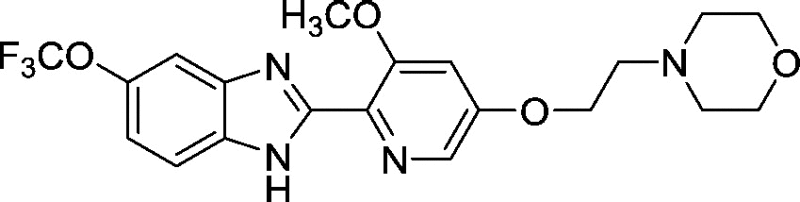 |
3.0 ± 0.5 |
| AI-14-18 (h) |
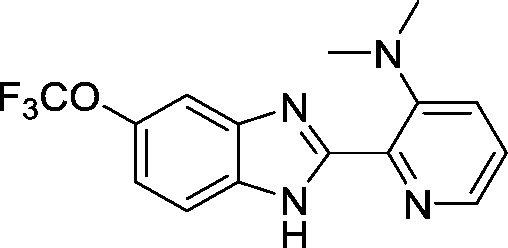 |
5.0 ± 0.3 |
| AI-14-72 (i) |
 |
1.7 ± 0.2 |
3.2. Identification of Pharmacophore and Exploration of Structure-Activity Relationships (SAR)
To get a better understanding of which functional groups of AI-4-57 are essential for its activity and to generate more effective inhibitors, a group of analogs were synthesized. All synthesized compounds were purified by flash chromatography and characterized by NMR and mass prior to evaluation in the FRET assay. As shown in Fig. 1b, Schemes 1–3 we probed several regions of AI-4-57 to define a pharmacophore for activity. Substitution of the pyridine ring with a phenyl ring (2a) or a furan (2b, Fig. 1b, Scheme 1) resulted in a complete loss of activity (Supplementary Table 1). Replacement of the methoxy functionality with a hydrogen (AI-4-88, 3a) also resulted in complete loss of activity. Fig. 1c shows the results of STD NMR analysis (MAYER and MEYER, 1999, MAYER and MEYER, 2001) of AI-4-57 binding to CBFβ. When short saturation times are used for this experiment, it can be effectively used to map epitopes on a molecule which are in close contact with the protein. With a short saturation time of 500 ms, we observe the signal for the methyl group of the methoxy functional group as well as the signal for the neighboring 4 hydrogen of the benzimidazole ring in the difference spectrum, consistent with this portion of the molecule being in close contact with the protein as well as with the effect of removing this substituent on activity. We further explored the 5-position by introducing fluorine (3c), triflouromethyl (3f), methyl (3d), and ethoxy (3b, Fig. 1b, Scheme 2) substitutions. Introduction of trifluoromethyl or methyl resulted in a loss of activity whereas introduction of fluorine or ethoxy resulted in compounds with similar activity (Supplementary Table 1). Moving the 5-methoxy to the 4-position (4a) resulted in reduced activity (Supplementary Table 1). Methyl (4h), methoxy (4g) and fluorine (4i) substitutions ortho to the pyridine nitrogen (Fig. 1b, Scheme 3) resulted in inactive compounds (Supplementary Table 1). As shown in Fig. 1c, short saturation time STD NMR identifies this hydrogen as being in close contact with the protein, providing a rationale for the loss of activity observed with substitutions at this position. This data established essential characteristics of the pharmacophore necessary for inhibition of CBFβ-Runt domain binding by this class of molecules. As shown in Supplementary Fig. 1, we subsequently prepared a series of compounds to further explore the SAR. FRET assay inhibition data for the most active compounds that emerged from this effort are shown in Table 3.
3.3. Inhibitors With Favorable ADMET Properties
Development of a useful tool compound which can be utilized for in vivo studies requires optimization not only of the activity of the compound but also its metabolic stability in vivo. In the context of our previous work on development of small molecule inhibitors that are specific for CBFβ-SMMHC (Illendula et al., 2015), we showed that AI-4-57 has a short half-life in mice with loss of the methyl group on the methoxy functionality being the resulting metabolite. Introduction of trifluoromethoxy abrogated the metabolic liability. Introduction of a trifluoromethoxy substitution into AI-4-57 yielded a compound (AI-10-47) with improved activity in the FRET assay (see Table 1) as well as in assays of cellular activity (see below). Introduction of this substitution into 7a (Supplementary Fig. 1), yielded the inhibitor AI-10-104 with IC50 = 1.25 μM (Table 1). However, administration of AI-10-104 to mice via intraperitoneal (IP) injection of a captisol formulation at 178 mg/kg resulted in significant sedative effects within 30 s, from which the mice recovered in approximately 1 h, whereas administration of a nanoparticle formulation at 200 mg/kg was lethal in approximately ~ 3.5 h. Hypothesizing that this effect is driven by an off-target activity, we have engineered additional analogs (AI-12-126, AI-14-55, and AI-14-91, see Table 1) where we have appended morpholine ring substituents to the pyridine ring, thereby altering the structure as well as polarity of the compounds. These compounds retain similar activity in the FRET assay to the parent compounds (see Table 3). Importantly, AI-12-126 and AI-14-91, when formulated as the HCl salts with captisol and administered IP at 100 mg/kg do not induce the sedative effects seen with AI-10-104 and are well-tolerated by mice. Measurements of the pharmacokinetic properties of these compounds in mice (see Supplementary Fig. 2) showed that at a dose of 100 mg/kg, we can achieve useful concentrations of the compounds with reasonable half-lives in vivo (AI-14-91, oral gavage, t1/2 = 203 min). These derivatives are, therefore, viable options for in vivo studies of CBFβ and RUNX function.
3.4. GLIDE Based Model of Inhibitor Binding to CBFβ
The structure of CBFβ was solved using NMR spectroscopy (Huang et al., 1999) and the crystallization of this very soluble protein has not been reported. Therefore, we have focused on using a combination of chemical shift perturbations in the NMR spectrum of CBFβ upon AI-4-57 binding combined with GLIDE based docking to gain insight into the binding mode. AI-4-57 induces chemical shift changes both in backbone NH resonances as well as in the sidechain CH resonances of the aromatic sidechains of Trp113 and Tyr96 in CBFβ (Illendula et al., 2015). These chemical shift changes provide key experimental information to identify the location of the binding site and guide docking using the program GLIDE. Interestingly, the chemical shift perturbation data indicate binding to a site that is spatially close to the Runt domain binding site on CBFβ but on the opposite side of the protein, i.e. the compounds act in an “allosteric” manner to inhibit the protein-protein interaction.
We used the residues identified as perturbed in the NMR data as constraints to dock AI-4-57 to CBFβ using GLIDE. We obtained two different docking poses with similar GLIDE docking scores, with the compound flipped by ~ 180° between the two (Fig. 2, Supplementary Fig. 3). In one pose (Supplementary Fig. 3), the pyridine ring is buried such that substitutions at the 5 position would sterically clash with the protein whereas in the other pose this position is not obstructed (Fig. 2). Substitutions at this position (compare for example AI-10-104 with AI-12-16 in Table 1) are well tolerated, indicating that the pose shown in Fig. 2 is the correct one.
Fig. 2.

GLIDE based docking of inhibitors to CBFβ. A. Surface representation of the binding pocket on CBFβ (grey) with AI-4-57 (blue) bound as determined using GLIDE, all overlayed on a ribbon representation of the structure of CBFβ. B. Surface representation of the binding pocket on CBFβ with AI-4-57 bound colored according to partial charge (red, negative; blue, positive; green, neutral) in same orientation as in A. C. Schematic showing the identity of the residues making contact with AI-4-57. D. Ribbon representation of the structure of CBFβ (blue) with AI-4-57 bound. The sidechains of R90 and K111 are displayed and colored red. E. Overlays of selected regions of 15N-1H HSQC spectra of CBFβ alone and CBFβ + AI-4-57 for wildtype, R90E, and K111E CBFβ proteins. Resonances for CBFβ alone are in blue and those for CBFβ + AI-4-57 are in red.
As shown in Fig. 2a, we see the compound binding into a relatively shallow pocket on the surface of the protein. The model shows rotation of the pyridine ring relative to the benzimidazole ring in the bound conformation, a result which is consistent with the increased activity of AI-10-104, with a 3-methoxy substituted pyridine ring which will force the pyridine ring out of planarity with the benzimidazole, relative to AI-10-47 (Table 3). Display of the partial charge distribution of the compounds and the protein (Fig. 2b) shows significant partial positive charge from the protein interacting favorably with partial negative charge from the compound. Fig. 2c identifies all the amino acids lining the binding pocket for the compound. We found the GLIDE based docking and scoring is able to distinguish inactive from active compounds, however its ability to quantitatively distinguish among the active compounds is limited.
In order to confirm the validity of this model, we have introduced 3 mutations into CBFβ to probe their effects on compound binding: Arg90- > Glu, Lys98- > Glu, and Lys111- > Glu. All of these residues are located in the binding pocket (Fig. 2d). The 15N-1H HSQC NMR spectrum of the Lys98Glu mutant protein showed wide-scale changes indicative of a disruption of the fold, so this mutation was not further analyzed. For the Arg90- > Glu and Lys111- > Glu mutant proteins, we have compared the observed chemical shift changes in an 15N-1H HSQC spectrum upon addition of AI-4-57 compared to the wildtype protein (Fig. 2e). For the Arg90 mutant we observe reduced chemical shift changes and for the Lys111 mutant, there is no chemical shift change, consistent with a loss of binding to the protein. This data experimentally validates the site of binding and supports the accuracy of the GLIDE model.
3.5. Molecular Basis for Allosteric Inhibition
As the CBFβ inhibitors bind at a site that is not on the binding interface for RUNX binding on CBFβ (see Fig. 3), they act via an allosteric mechanism. This could occur by way of structural changes induced in the protein by compound binding and/or by alteration of the dynamics of the protein upon compound binding. Significant chemical shift changes upon AI-4-57 binding are confined to the binding site for the compound (see Fig. 3). As shown in Fig. 3b and Supplementary Fig. 4, upon binding of AI-4-57 to CBFβ we do observe small chemical shift changes for amino acids located on the RUNX binding interface on CBFβ. As the magnitude of these chemical shift changes is quite small, it is clear that there is not any significant conformational change at the binding interface. We therefore hypothesized that there must be a change in dynamics associated with compound binding which mediates the reduced affinity. To test this idea, we have collected and analyzed 15N backbone R1 and R2 data and calculated the difference in R1 ∗ R2 between free CBFβ and CBFβ bound to AI-4-57 (Fig. 3c, Supplementary Fig. 5). Such an R1 ∗ R2 plot readily identifies residues with increased dynamics on either the μs-ms or ps-ns timescales (Kneller et al., 2002). Fig. 3c shows a plot of the difference between R1 ∗ R2 for CBFβ + AI-4-57 and CBFβ alone. In this plot, residues above zero indicate increased μs-ms timescale dynamics upon compound binding and residues below zero indicate increased ps-ns timescale dynamics upon compound binding. Residues which displayed R1 ∗ R2 difference values greater than two times the standard deviation from the trimmed mean were deemed to be significant. We observe changes in dynamics for residues located in the binding site for the inhibitor, resulting from both the binding and dissociation of compound as well as compound induced changes in protein dynamics (Fig. 3d). Interestingly, these changes occur both in the μs-ms timescale dynamics (principally near the benzimidazole ring) as well as on the ps-ns timescale (in the vicinity of the pyridine ring), with the latter perhaps a result of ring flipping of the pyridine ring. Importantly, we observe significant changes in dynamics for residues that are located at the binding interface for RUNX binding (Fig. 3e), including one residue that has been shown by previous mutagenesis studies to be an important contributor to the energetics of binding. Fig. 3e highlights two of these residues, Arg131 and Asn104. For Arg131, we observe increased μs-ms timescale dynamics with compound binding. Arg131 is a known contact residue with the Runt domain (Zhang et al., 2003). For Asn104, we observe an increase in ps-ns timescale dynamics with compound binding. Strikingly, our previous mutagenesis studies have shown that mutation of this residue has the largest effect of any of the CBFβ mutations we have characterized (Tang et al., 2000), i.e. Asn 104 is a hotspot residue for the binding of CBFβ to the Runt domain. This amino acid makes multiple contacts with residues in the Runt domain (Val159, Ala160, Thr161, Asp66) and mutation to Ala results in a 21-fold reduction in binding affinity (Tang et al., 2000).
Fig. 3.

Effects of CBFβ inhibitors on protein chemical shifts and backbone dynamics measured using NMR spectroscopy. A. Structure of the CBFβ (green) – Runt domain (magenta) heterodimer with a surface representation colored in red for the residues in the AI-4-57 binding pocket which undergo chemical shift changes upon binding. The orientation of AI-4-57 is that deduced from GLIDE docking. B. Structure of the CBFβ-Runt domain heterodimer with residues on the Runt domain binding interface which display chemical shift changes upon AI-4-57 binding having their surface colored in red. Residues in the Runt domain which make contact with these CBFβ residues are displayed as magenta colored surface representations. C. Plot of the difference in the measured 15N backbone R1 ∗ R2 values between CBFβ + AI-4-57 and CBFβ alone. D. Structure of the CBFβ-Runt domain heterodimer with the residues in the AI-4-57 binding pocket which display changes in R1 ∗ R2 upon AI-4-57 binding indicated as a surface representation. Residues showing increased ps-ns timescale motion are colored red and those showing increased μs-ms timescale motion are colored blue. E. Structure of the CBFβ-Runt domain heterodimer with residues on the Runt domain binding interface on CBFβ which show changes in R1 ∗ R2 upon AI-4-57 indicated as surface representations. Red indicates increased ps-ns timescale motion and blue indicates increased μs-ms timescale motion. Residues in the Runt domain which make contact with these residues are indicated as a surface representation and colored magenta. The identities of the residues involved are indicated in green for CBFβ and in magenta for the Runt domain.
3.6. Comparison to Ro5-3335
The small molecule Ro5-3335 was reported to be an inhibitor of CBFβ/RUNX function (Cunningham et al., 2012). This compound was reported to have been identified based on a hit from an alpha-screen for the CBFβ-Runt domain interaction, however the compound does not show significant effects in the alpha screen assay and does not show an effect on CBFβ-RUNX1 binding in cells (Cunningham et al., 2012). It also shows no effect on CBFβ-Runt domain binding in our FRET assay (Supplementary Fig. 6a). We also could not detect any interaction with either CBFβ or the RUNX1 Runt domain by NMR. The only target identification data presented (protein binding by UV measurement of adherence to nickel resin bound protein) indicated it binds to both CBFβ and RUNX1 (Cunningham et al., 2012). This seems unlikely from a chemical perspective, so the actual target of action is unclear. As this compound has structural similarity to known bromodomain inhibitors (SMITH et al., 2014, GALLENKAMP et al., 2014), we have screened this compound for activity against a panel of 32 bromodomains using a commercial screen (DiscoveRx BromoScan). This screen showed that at 1 μM, similar to the low μM concentrations where activity is reported for this compound in cellular assays, the SMARCA2 and WDR9(2) bromodomains were ~ 40% inhibited (Supplementary Fig. 6b). SMARCA2 is a member of the SWI/SNF family of proteins. The human SWI/SNF complex has been shown to associate with RUNX1 (Bakshi et al., 2010). In addition, SMARCA2 is listed as a common interaction partner of TBP and RUNX1 by the protein-protein interaction prediction program PIPS. Based on this, the lack of effect on CBFβ-RUNX binding, and the lack of clear target validation, we would suggest that the mechanism of action of Ro5-3335 may be based on inhibition of SMARCA2 rather than direct inhibition of CBFβ/RUNX, unlike the inhibitors we are reporting herein.
3.7. CBFβ Inhibitors Disrupt CBFβ-RUNX1 Binding in SEM Cells
To test whether the CBFβ inhibitors can disrupt the binding of full length CBFβ to full length RUNX1 in cells, we performed co-immunoprecipitation experiments in the SEM cell line. In order to limit effects on the viability of the cells, the treatment time was limited to 6 h. As shown in Fig. 4 (a replicate is shown in Supplementary Fig. 7; data for AI-4-57 in Supplementary Figure 8), we see a clear reduction in binding of CBFβ binding to RUNX1 in cells treated with 10 μM AI-10-104, AI-12-126, and AI-14-91. As AI-10-47 showed only a modest effect, we have measured the aqueous solubility of all 5 compounds tested in 0.25% DMSO by NMR spectroscopy, mimicking the conditions used for the coIP. Consistent with the coIP results, AI-10-104, AI-12-126, and AI-14-91 are significantly more soluble than AI-10-47, explaining the reduced activity seen for AI-10-47 in this assay. No effect was observed on the levels of CBFβ or RUNX1 upon treatment with inhibitors (Supplementary Figure 9).
Fig. 4.
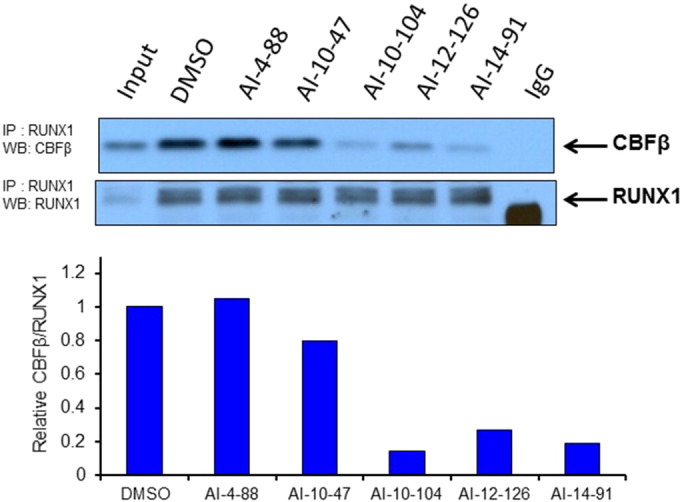
CBFβ inhibitors reduce CBFβ binding to RUNX1 in cells. Co-immunoprecipitation assays of lysates from acute myeloid leukemia SEM cells treated with AI-4-88, AI-10-47, AI-10-104, AI-12-126 and AI-14-91 at 10 μM for 6 h. RUNX1-bound CBFβ protein levels and immunoprecipitated RUNX1 are shown on the top panel. Quantification of RUNX1 bound CBFβ is shown on the bottom graph. Protein levels were normalized to RUNX1 and depicted relative to DMSO control.
3.8. CBFβ Inhibitors Reduce RUNX1 Occupancy on Target Genes and Alter Gene Expression
CBFβ increases the DNA binding activity of RUNX proteins by relieving auto-inhibition (Adya et al., 2000). Therefore, we would predict that inhibition of CBFβ-RUNX binding will reduce RUNX occupancy on target genes and alter their expression. As RUNX proteins are context-dependent regulators of gene expression, this could result in increases or decreases in concomitant gene expression depending on the target gene. To test this, we have used an inducible iRunx ES cell system which allows the expression of HA tagged Runx1 under the control of a doxycycline (dox) responsive promoter in a Runx1 −/− genetic background (Lichtinger et al., 2012). The cells were differentiated according to an established model of in vitro hematopoiesis in which ES cells are differentiated into hematopoietic progenitors (CHOI et al., 1998, SROCZYNSKA et al., 2009). This system provides a model which makes it possible to dissect the mechanisms that drive the differentiation process at different stages of hematopoietic development. In the absence of Runx1, cells are unable to progress past the hemogenic endothelium stage of development and generate hematopoietic progenitors (Fig. 5a). By inducing Runx1 and hence the assembly of activating (or repressive) complexes at Runx1-responsive loci its role in mediating the endothelial to hematopoietic transition can be examined.
Fig. 5.

Effects of CBFβ inhibitors on RUNX1 occupancy and target gene expression. A. Schematic diagram representing the inducible Runx1 (iRx) ES cell differentiation system. ES cells were allowed to form embryoid bodies (EB) in IVD culture media and hemangioblast Flk1 + ve cells were sorted and seeded into blast culture media and cultured for 40 h. In the absence of doxycycline (− dox) RUNX1 is absent and cells cannot progress from the HE1 stage of development. In the presence of doxycycline (+ dox) Runx1 is expressed and differentiation progresses, resulting in the formation of CD41 + ve/KIT + ve progenitors. B. Representative FACS histograms showing staining of the surface markers CD41 and KIT in cells treated with the compounds and dox as indicated. The percentage of cells expressing KIT and CD41 is reduced by treatment with AI-14-91 but not with the control compound AI-4-88. C. Gene expression analysis of inhibitor treated cells using RNA prepared from cells treated with and without dox and cells treated with 10 μM, 20 μM and 30 μM AI-14-91 or AI-4-88 in the presence of dox. Relative expression of genes is shown, normalized to GAPDH and using + dox as the calibrator. Error bars represent standard deviation where n = 3. D. ChIP analysis of HA-Runx1 binding following Runx1 induction by dox and treatment with inhibitor or control compound. ChIP with an anti-HA antibody recognizing HA tagged Runx1 was used to identify binding at selected amplicons and enrichment normalized to input and the Chr2 control is shown. Error bars represent standard deviation where n = 5. A one-way ANOVA test was used to analyze variance in HA enrichment values between the inhibitor and control compound, or between inhibitor and + dox treatment, * denotes p < 0.05 and ** denotes p < 0.01. E. Representative western blots showing levels of inducible Runx1 and CBFβ protein in the cell populations. Where indicated, cells were treated with 20 μM inhibitor or control compound.
ES cells were differentiated as described in the methods section and only adherent cells were harvested, since at this stage of differentiation very few cells had progressed to free floating progenitors. To examine the effect of CBFβ inhibition of RUNX1-dependent differentiation, we performed FACS analysis using antibodies against KIT, a growth factor receptor which is expressed on cells with haemopoietic potential, and CD41, a surface marker of newly committed multipotent progenitor cells. We found that treatment with the AI-14-91 inhibitor but not the inactive control compound (AI-4-88) resulted in a strong decrease in the percentage of cells expressing both these surface markers (Fig. 5b).
For gene expression analysis, we tested 4 genes that are activated by RUNX1 (increased by Dox induction) – Nfe2, Sfpi1 (Pu.1), Itga2b (CD41) and Gfi1. In each case, addition of the inhibitor (AI-14-91) but not the inactive control compound (AI-4-88) resulted in a dose dependent decrease in expression level (Fig. 5c).The expression level of Cebpa, an indirect target of RUNX, was also decreased. Cebpb which is not a direct Runx1 target was relatively unaffected by the inhibitor, as was β-2-microglobulin which was used as a negative control.
To test whether the disruption of RUNX1-CBFβ interaction abrogated chromatin binding of RUNX1, chromatin from differentiating cells was immunoprecipitated using an HA antibody which recognizes the HA tag on the iRUNX protein and examined RUNX1 binding to several well-known RUNX1 target regions (Fig. 5d) (Lichtinger et al., 2012). When normalized to input and a negative control region (Chr2), RUNX1 binding is increased upon dox induction at the Sfpi1 (Pu.1) 3′ enhancer (− 14kb 3′URE), the Itga2b (CD41) promoter, and the NFE2-3kb enhancer-element. Binding was reduced in the presence of the inhibitor (AI-14-91) but not the inactive control compound (AI-4-88). A control region of Chr1 and the Oct4 promoter, which is not active at this stage of differentiation, served as negative controls. Western blots demonstrated that cells under each of the dox induced conditions expressed comparable levels of RUNX1 and CBFβ protein (Fig. 5e).
3.9. CBFβ Inhibitors Induce Myeloid Differentiation of HPC-7 Cells
RUNX1 levels are high in hematopoietic stem cells and early progenitors, then decrease with differentiation into the myeloid lineage. We therefore hypothesized that CBFβ inhibitors would enhance differentiation into the myeloid lineage under conditions where that is preferred. To test this, we have cultured the HPC-7 cell line for 10 days under conditions where myeloid differentiation was favored. As shown in Supplementary Figure 10, addition of CBFβ inhibitors (AI-10-104, AI-14-91) to the culture results in a significant increase in differentiation as assessed by the appearance of the macrophage-specific surface marker CD11.
3.10. CBFβ Inhibitors Inhibit Growth of Leukemia Cell Lines
CBFβ/RUNX1 function is essential for normal hematopoiesis. Both CBFβ and RUNX1 are targets of chromosomal translocations in human leukemia. Recent studies showed that RUNX1 is required for the maintenance of leukemia cells expressing AML1-ETO, MLL-AF9, as well as T-ALL cells (BEN-AMI et al., 2013, GOYAMA et al., 2013, KWIATKOWSKI et al., 2014). Based on these findings, we hypothesized that CBFβ inhibitors may have utility for different types of leukemia. We therefore screened inhibitors against a panel of 11 leukemia cell lines with varying genotypes (see Fig. 6). We did not observe any activity for the inactive control compound AI-4-88 and only very weak activity for the modest potency AI-4-57. However, for all the more active inhibitors tested (AI-10-47, AI-10-104, and AI-14-91) we observed significant growth inhibition of the majority of the cell lines. Importantly, we observe only very modest effects, and no effect of AI-14-91 on the colony formation of normal human cord blood cells (Supplementary Figure 11), suggesting there could be a useful therapeutic window.
Fig. 6.

Effects of CBFβ inhibitors on the viability of a panel of leukemia cell lines. Percent viability for 11 leukemia cell lines after 48 h of treatment with the indicated compounds. The viability is represented as the percent DAPI negative cells relative to DMSO control. Each symbol/color represents an individual cell line. The symbol represents the mean of independent replicates and the error bars represent the S.E.M. The table indicates the morphology, type of leukemia, and known mutations for each of the 11 cell lines.
3.11. CBFβ Inhibitors Induce Increased Multiacinar Structures in Breast Cell 3D Culture and Block Growth of Basal-like Breast Cancer Cells in 3D Culture
RUNX-mediated gene expression is important for normal breast epithelial biology and breast cancer (LAU et al., 2006, PRATAP et al., 2006, PRATAP et al., 2009, SHORE, 2005, KADOTA et al., 2010, VAN BRAGT et al., 2014). To evaluate whether CBFβ inhibition would influence the multicellular organization of basal-like breast epithelia, we used 3D culture of MCF10A-5E cells (Janes et al., 2010). Knockdown of RUNX1 in these cells delays proliferation arrest and promotes the formation of non-spherical acini, indicating a role for RUNX1 during morphogenesis (Wang et al., 2011). When shRUNX1 or control cells were cultured with CBFβ inhibitors at 1 μM concentration, we observed a substantial increase in the formation of multiacinar structures compared to carrier control or inactive compound (Fig. 7a). The extent of multiacinus formation greatly exceeded that of RUNX1 knockdown, suggesting a phenotypic role for other RUNX-family members such as RUNX2, which is also expressed in these cells (Wang et al., 2011). The inhibitors appear to be initiating a nascent branching program that is reminiscent of when TGFβ-family signaling is impaired (Wang et al., 2014). RUNX transcription factors are well-recognized effectors of TGFβ-family ligands (Ito and Miyazono, 2003), which are naturally retained in the extracellular matrix (SCHNEYER et al., 2008, TAIPALE et al., 1996).
Fig. 7.

Effects of CBFβ inhibitors on acini formation by MCF10A-5E cells and viability of basal-like breast cancer cell line HC1143. CBFβ inhibitors alter acinar morphogenesis of basal-like breast epithelial cells and block survival of basal-like breast cancer cells in 3D organotypic culture. A. MCF10A-5E cells stably expressing shRUNX1 or shGFP control were grown in 3D culture for 10–11 days with the indicated compounds at 1 μM concentration and imaged by brightfield microscopy. The total number of multiacini per chamber was counted and scaled to the shGFP DMSO control. Data are shown as the mean ± s.e.m. of eight independent cultures. Quantitative immunoblotting for RUNX1, RUNX2, CBFβ, vinculin, tubulin, and GAPDH is shown to the right. B. HCC1143 cells were grown in 3D culture for 18 days with the indicated compounds at 1 μM concentration and imaged by brightfield microscopy. The total number of proliferating colonies per chamber was counted, and data are shown as the mean ± s.e.m. of four independent cultures. Scale bar is 200 μm.
RUNX and CBFβ mutations have been described in luminal breast cancer (BANERJI et al., 2012, ELLIS et al., 2012), but the role of RUNX in basal-like breast cancer is less clear (Janes, 2011). Increased levels of expression of RUNX1 and RUNX2 have been shown to be present in basal-like breast cancers and to correlate with poor prognosis (MCDONALD et al., 2014, FERRARI et al., 2014). We surveyed the Cancer Cell Line Encyclopedia and identified HCC1143 cells as a line in the Basal A subtype with high expression of RUNX transcripts as measured by microarray. When HCC1143 cells were grown under 3D culture conditions in the presence of active CBFβ inhibitors at 1 μM concentration, we observed a striking blockade in cell survival, with zero detectable colonies after 18 days (Fig. 7b). These results suggest that persistent RUNX-mediated gene expression is required in a subset of basal-like breast cancers, opening a potential therapeutic opportunity for CBFβ inhibitors.
4. Discussion
Transcription factors represent an important as well as challenging class of proteins for the development of small molecule inhibitors. Because of their function as crucial regulators of cell fate decisions in a normal and malignant context and as mediators of a wide swath of signaling pathways they are particularly attractive targets for cancer therapy. Moreover, it is now abundantly clear that tumorigenesis in multiple tissues involves the deregulation of gene expression and a subversion of the normal cell differentiation processes (Bonifer and Cockerill, 2011). However, transcription factors do not act alone, but function in the context of large multi-protein assemblies, which requires the targeting of intracellular protein-protein or protein-DNA interactions. This has proved to be more challenging than targeting enzymatic activity due to the nature of the interaction surfaces and the complexity of the assemblies, provoking the notion that this class of proteins is “undruggable”. Nevertheless, efforts in a number of labs have now demonstrated successful targeting of this class of proteins, opening up numerous avenues for novel modes of action to approach the treatment of cancer. Herein, we describe the development of a small molecule inhibitor which targets the heterodimeric core binding factor (CBF) transcription factor, specifically the protein-protein interaction between CBFβ and RUNX, the two subunits of CBF. We have optimized the activity as well as the ADMET properties based on an initial hit to generate tool compounds which we have shown bind to CBFβ and inhibit binding to RUNX1, resulting in decreased binding of RUNX1 to target sites in the genome and concomitant changes in the level of expression of RUNX1 target genes. While we have only experimentally demonstrated disruption of binding with RUNX1, all three RUNX proteins are known to bind CBFβ. In addition, the Runt domain of the RUNX proteins, to which CBFβ binds, is almost identical in sequence among the family members and no differences are found on the surface of the Runt domain which interacts with CBFβ. Based on this, it is our expectation that similar effects will be seen for these inhibitors on CBFβ-RUNX2 and CBFβ-RUNX3 binding. As such, the biological effects observed likely reflect activity against all the RUNX proteins present in a particular cell type. These compounds meet the criteria outlined by Frye for a high quality chemical probe (Frye, 2010), including a clear molecular profile of activity, mechanism of action, identity of active species, and proven utility.
Our inhibitors act via an allosteric mechanism, i.e. they do not bind at the protein-protein interface. Such a mode of action has been seen with numerous small molecule inhibitors of enzymes, but there have only been limited reports of such allosteric inhibitors of protein-protein interactions (Arkin et al., 2014b). However, this mechanism has clear advantages as the inhibitor does not have to directly compete with the partner protein. Our NMR relaxation data support a model for the activity of these compounds where induction of changes in the dynamics of critical hotspot binding residues on the RUNX binding interface on CBFβ mediate loss of binding. Such a dynamics based mechanism is consistent with numerous NMR studies of allosteric communication which have demonstrated altered dynamics as a mechanism.
The interaction between CBFβ and RUNX1 is essential for normal hematopoiesis. The CBFβ inhibitors block the induction of RUNX1-dependent genes during the endothelial-hematopoietic transition (EHT) and inhibit RUNX1 binding to its target genes. Moreover, this result highlights the crucial role of not only the presence of the complete CBF complex, but its actual binding to chromatin for the EHT. At later differentiation stages, RUNX1 inhibition enhances myeloid differentiation of the HPC-7 cell line, which is consistent with the reduction in expression of RUNX1 that occurs upon progressing from the hematopoietic stem cell (HSC) to the myeloid lineage. Based on these results, we explored the biological effects of the CBFβ inhibitors in a disease context. Since all three RUNX proteins (RUNX1, 2, 3) bind CBFβ, the biological effects of these inhibitors likely reflect the inhibition of all the RUNX proteins present in the cell lines used. We have previously shown that this interaction is essential for the transforming properties of the leukemia causing fusion proteins AML1-ETO and TEL-AML1 (Roudaia et al., 2009). Our data therefore suggest a potential therapeutic use of the CBFβ inhibitors in leukemia as they are effective in killing a variety of different leukemia cell lines, consistent with recent results indicating a critical role for both mutated and wild-type RUNX1 in such cells (BEN-AMI et al., 2013, GOYAMA et al., 2013), while having minimal impact on normal cord blood cells. However, the anti-tumor activity of the compounds is not restricted to hematological malignancies. CBFβ inhibitors increased multiacinar structures in breast cell 3D culture, confirming the crucial role of RUNX1 in this phenotype indicated by previous knock-down experiments, and completely inhibited the growth of a basal-like breast cancer cell line in 3D culture. Currently there is no effective therapy for basal-like breast cancer, so our results support targeting of the RUNX pathway for this very poor prognosis cancer. Our study clearly demonstrates the utility of these compounds for exploring the effects of CBFβ-RUNX inhibition across a broad spectrum of cancers.
Author Contributions
J.H.B., C.B., M.L.G., K.A.J., R.A.J., L.H.C., and M.A. designed the experiments. A.I., J.G., V.S.S.T., A.B., A.K., C.S., L.W., J.A.P., H.Z., C.K., A.P., Y.Z., Y.G., and L.D. performed the experiments and analyzed the data. J.H.B. wrote the manuscript.
Acknowledgements
This work was supported by grants from the National Cancer Institute (R01 CA140398) to J.H.B., L.H.C., and R.A.R., from the American Cancer Society (# 120668-RSG-11-047-01-DMC) and David and Lucile Packard Foundation (# 2009-34710) to K.A.J., from the National Institutes of Health (NIH) through the NIH Director's New Innovator Award Program (1 DP2 OD007399-01) to M.L.G., and from the Biotechnology and Biological Sciences Research Council (BB/I001220/2), Cancer Research UK and Leukaemia Lymphoma Research to C.B.
Footnotes
The authors have declared that no conflict of interest exists.
Supplementary data to this article can be found online at http://dx.doi.org/10.1016/j.ebiom.2016.04.032.
Appendix A. Supplementary Data
Mechanism of action of CBFß-RUNX inhibitors.
References
- Adya N., Castilla L.H., Liu P.P. Function of CBFbeta/Bro proteins. Semin. Cell Dev. Biol. 2000;11:361–368. doi: 10.1006/scdb.2000.0189. [DOI] [PubMed] [Google Scholar]
- Arkin M.R., Tang Y., Wells J.A. Small-molecule inhibitors of protein-protein interactions: progressing toward the reality. Chem. Biol. 2014;21:1102–1114. doi: 10.1016/j.chembiol.2014.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arkin M.R., Tang Y., Wells J.A. Small-molecule inhibitors of protein-protein interactions: progressing toward the reality. Chem. Biol. 2014;21:1102–1114. doi: 10.1016/j.chembiol.2014.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arkin M.R., Whitty A. The road less traveled: modulating signal transduction enzymes by inhibiting their protein-protein interactions. Curr. Opin. Chem. Biol. 2009;13:284–290. doi: 10.1016/j.cbpa.2009.05.125. [DOI] [PubMed] [Google Scholar]
- Bakshi R., Hassan M.Q., Pratap J., Lian J.B., Montecino M.A., Van Wijnen A.J., Stein J.L., Imbalzano A.N., Stein G.S. The human SWI/SNF complex associates with RUNX1 to control transcription of hematopoietic target genes. J. Cell. Physiol. 2010;225:569–576. doi: 10.1002/jcp.22240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banerji S., Cibulskis K., Rangel-Escareno C., Brown K.K., Carter S.L., Frederick A.M., Lawrence M.S., Sivachenko A.Y., Sougnez C., Zou L., Cortes M.L., Fernandez-Lopez J.C., Peng S., Ardlie K.G., Auclair D., Bautista-Pina V., Duke F., Francis J., Jung J., Maffuz-Aziz A., ONOFRIO R.C., Parkin M., Pho N.H., Quintanar-Jurado V., Ramos A.H., Rebollar-Vega R., Rodriguez-Cuevas S., Romero-Cordoba S.L., Schumacher S.E., Stransky N., Thompson K.M., Uribe-Figueroa L., Baselga J., Beroukhim R., Polyak K., Sgroi D.C., Richardson A.L., Jimenez-Sanchez G., Lander E.S., Gabriel S.B., Garraway L.A., Golub T.R., Melendez-Zajgla J., Toker A., Getz G., Hidalgo-MirandA A., Meyerson M. Sequence analysis of mutations and translocations across breast cancer subtypes. Nature. 2012;486:405–409. doi: 10.1038/nature11154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ben-Ami O., Friedman D., Leshkowitz D., Gs D., Orlovsky K., Pencovich N., Lotem J., Tanay A., Groner Y. Addiction of t(8;21) and inv(16) acute myeloid leukemia to native RUNX1. Cell Rep. 2013;4:1131–1143. doi: 10.1016/j.celrep.2013.08.020. [DOI] [PubMed] [Google Scholar]
- Blyth K., Cameron E.R., Neil J.C. The RUNX genes: gain or loss of function in cancer. Nat. Rev. Cancer. 2005;5:376–387. doi: 10.1038/nrc1607. [DOI] [PubMed] [Google Scholar]
- Bonifer C., Cockerill P.N. Chromatin mechanisms regulating gene expression in health and disease. Adv. Exp. Med. Biol. 2011;711:12–25. doi: 10.1007/978-1-4419-8216-2_2. [DOI] [PubMed] [Google Scholar]
- Choi K., Kennedy M., Kazarov A., Papadimitriou J.C., Keller G. A common precursor for hematopoietic and endothelial cells. Development. 1998;125:725–732. doi: 10.1242/dev.125.4.725. [DOI] [PubMed] [Google Scholar]
- Cunningham L., Finckbeiner S., Hyde R.K., Southall N., Marugan J., Yedavalli V.R., Dehdashti S.J., Reinhold W.C., Alemu L., Zhao L., Yeh J.R., Sood R., Pommier Y., Austin C.P., Jeang K.T., Zheng W., Liu P. Identification of benzodiazepine Ro5-3335 as an inhibitor of CBF leukemia through quantitative high throughput screen against RUNX1-CBFbeta interaction. Proc. Natl. Acad. Sci. U. S. A. 2012;109:14592–14597. doi: 10.1073/pnas.1200037109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Bruijn M.F., Speck N.A. Core-binding factors in hematopoiesis and immune function. Oncogene. 2004;23:4238–4248. doi: 10.1038/sj.onc.1207763. [DOI] [PubMed] [Google Scholar]
- Debnath J., Muthuswamy S.K., Brugge J.S. Morphogenesis and oncogenesis of MCF-10A mammary epithelial acini grown in three-dimensional basement membrane cultures. Methods. 2003;30:256–268. doi: 10.1016/s1046-2023(03)00032-x. [DOI] [PubMed] [Google Scholar]
- Ellis M.J., Ding L., Shen D., Luo J., Suman V.J., Wallis J.W., Van tine B.A., Hoog J., Goiffon R.J., Goldstein T.C., Ng S., Lin L., Crowder R., Snider J., Ballman K., Weber J., Chen K., Koboldt D.C., Kandoth C., Schierding W.S., Mcmichael J.F., Miller C.A., Lu C., Harris C.C., Mclellan M.D., Wendl M.C., Deschryver K., Allred D.C., Esserman L., Unzeitig G., Margenthaler J., Babiera G.V., Marcom P.K., Guenther J.M., Leitch M., Hunt K., Olson J., Tao Y., Maher C.A., Fulton L.L., Fulton R.S., Harrison M., Oberkfell B., Du F., Demeter R., Vickery T.L., Elhammali A., Piwnica-Worms H., Mcdonald S., Watson M., Dooling D.J., Ota D., Chang L.W., Bose R., Ley T.J., Piwnica-Worms D., Stuart J.M., Wilson R.K., Mardis E.R. Whole-genome analysis informs breast cancer response to aromatase inhibition. Nature. 2012;486:353–360. doi: 10.1038/nature11143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrari N., Mohammed Z.M., Nixon C., Mason S.M., Mallon E., Mcmillan D.C., Morris J.S., Cameron E.R., Edwards J., Blyth K. Expression of RUNX1 correlates with poor patient prognosis in triple negative breast cancer. PLoS One. 2014;9 doi: 10.1371/journal.pone.0100759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedman A.D. Cell cycle and developmental control of hematopoiesis by Runx1. J. Cell. Physiol. 2009;219:520–524. doi: 10.1002/jcp.21738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frye S.V. The art of the chemical probe. Nat. Chem. Biol. 2010;6:159–161. doi: 10.1038/nchembio.296. [DOI] [PubMed] [Google Scholar]
- Gallenkamp D., Gelato K.A., Haendler B., Weinmann H. Bromodomains and their pharmacological inhibitors. Chem. Med. Chem. 2014;9:438–464. doi: 10.1002/cmdc.201300434. [DOI] [PubMed] [Google Scholar]
- Gorczynski M.J., Grembecka J., Zhou Y., Kong Y., Roudaia L., Douvas M.G., Newman M., Bielnicka I., Baber G., Corpora T., Shi J., Sridharan M., Lilien R., Donald B.R., Speck N.A., Brown M.L., Bushweller J.H. Allosteric inhibition of the protein-protein interaction between the leukemia-associated proteins Runx1 and CBFbeta. Chem. Biol. 2007;14:1186–1197. doi: 10.1016/j.chembiol.2007.09.006. [DOI] [PubMed] [Google Scholar]
- Goyama S., Schibler J., Cunningham L., Zhang Y., Rao Y., Nishimoto N., Nakagawa M., Olsson A., Wunderlich M., Link K.A., Mizukawa B., Grimes H.L., Kurokawa M., Liu P.P., Huang G., Mulloy J.C. Transcription factor RUNX1 promotes survival of acute myeloid leukemia cells. J. Clin. Invest. 2013;123:3876–3888. doi: 10.1172/JCI68557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guzman M.L., Li X., Corbett C.A., Rossi R.M., Bushnell T., Liesveld J.L., Hebert J., Young F., Jordan C.T. Rapid and selective death of leukemia stem and progenitor cells induced by the compound 4-benzyl, 2-methyl, 1,2,4-thiadiazolidine, 3,5 dione (TDZD-8) Blood. 2007;110:4436–4444. doi: 10.1182/blood-2007-05-088815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hassane D.C., Sen S., Minhajuddin M., Rossi R.M., Corbett C.A., Balys M., Wei L., Crooks P.A., Guzman M.L., Jordan C.T. Chemical genomic screening reveals synergism between parthenolide and inhibitors of the PI-3 kinase and mTOR pathways. Blood. 2010;116:5983–5990. doi: 10.1182/blood-2010-04-278044. [DOI] [PubMed] [Google Scholar]
- Huang X., Peng J.W., Speck N.A., Bushweller J.H. Solution structure of core binding factor beta and map of the CBF alpha binding site. Nat. Struct. Biol. 1999;6:624–627. doi: 10.1038/10670. [DOI] [PubMed] [Google Scholar]
- Illendula A., Pulikkan J.A., Zong H., Grembecka J., Xue L., Sen S., Zhou Y., Boulton A., Kuntimaddi A., Gao Y., Rajewski R.A., Guzman M.L., Castilla L.H., Bushweller J.H. Chemical biology. A small-molecule inhibitor of the aberrant transcription factor CBFbeta-SMMHC delays leukemia in mice. Science. 2015;347:779–784. doi: 10.1126/science.aaa0314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito Y., Miyazono K. RUNX transcription factors as key targets of TGF-beta superfamily signaling. Curr. Opin. Genet. Dev. 2003;13:43–47. doi: 10.1016/s0959-437x(03)00007-8. [DOI] [PubMed] [Google Scholar]
- Janes K.A. RUNX1 and its understudied role in breast cancer. Cell Cycle. 2011;10:3461–3465. doi: 10.4161/cc.10.20.18029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janes K.A. An analysis of critical factors for quantitative immunoblotting. Sci. Signal. 2015;8 doi: 10.1126/scisignal.2005966. (rs2) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janes K.A., Wang C.C., Holmberg K.J., Cabral K., Brugge J.S. Identifying single-cell molecular programs by stochastic profiling. Nat. Methods. 2010;7:311–317. doi: 10.1038/nmeth.1442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kadota M., Yang H.H., Gomez B., Sato M., Clifford R.J., Meerzaman D., Dunn B.K., Wakefield L.M., Lee M.P. Delineating genetic alterations for tumor progression in the MCF10A series of breast cancer cell lines. PLoS One. 2010;5 doi: 10.1371/journal.pone.0009201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kneller J.M., Lu M., Bracken C. an effective method for the discrimination of motional anisotropy and chemical exchange. j. am. chem. soc. 2002;124:1852–1853. doi: 10.1021/ja017461k. [DOI] [PubMed] [Google Scholar]
- kom Ori t., YAGI H., Nomura S., Yamaguchi A., Sasaki K., Deguchi K., Shimizu Y., Bronson R.T., Gao Y.H., Inada M., Sato M., Okamoto R., Kitamura Y., Yoshiki S., Kishimoto T. Targeted disruption of Cbfa1 results in a complete lack of bone formation owing to maturational arrest of osteoblasts. Cell. 1997;89:755–764. doi: 10.1016/s0092-8674(00)80258-5. [DOI] [PubMed] [Google Scholar]
- Kwiatkowski N., Zhang T., Rahl P.B., Abraham B.J., Reddy J., Ficarro S.B., DASTUR A., AMZALLAG A., Ramaswamy S., Tesar B., Jenkins C.E., Hannett N.M., Mcmillin D., Sanda T., Sim T., Kim N.D., Look T., Mitsiades C.S., Weng A.P., Brown J.R., Benes C.H., Marto J.A., Young R.A., Gray N.S. Targeting transcription regulation in cancer with a covalent CDK7 inhibitor. Nature. 2014;511:616–620. doi: 10.1038/nature13393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laraia L., Mckenzie G., Spring D.R., Venkitaraman A.R., Huggins D.J. Overcoming chemical, biological, and computational challenges in the development of inhibitors targeting protein-protein interactions. Chem. Biol. 2015;22:689–703. doi: 10.1016/j.chembiol.2015.04.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lau Q.C., Raja E., Salto-Tellez M., Liu Q., Ito K., Inoue M., Putti T.C., Loh M., Ko T.K., Huang C., Bhalla K.N., Zhu T., Ito Y., Sukumar S. RUNX3 is frequently inactivated by dual mechanisms of protein mislocalization and promoter hypermethylation in breast cancer. Cancer Res. 2006;66:6512–6520. doi: 10.1158/0008-5472.CAN-06-0369. [DOI] [PubMed] [Google Scholar]
- Lee Y.S., Lee J.W., Jang J.W., Chi X.Z., Kim J.H., Li Y.H., Kim M.K., Kim D.M., Choi B.S., Kim E.G., Chung J.H., Lee O.J., Lee Y.M., Suh J.W., Chuang L.S., Ito Y., Bae S.C. Runx3 inactivation is a crucial early event in the development of lung adenocarcinoma. Cancer Cell. 2013;24:603–616. doi: 10.1016/j.ccr.2013.10.003. [DOI] [PubMed] [Google Scholar]
- Lefevre P., Melnik S., Wilson N., Riggs A.D., Bonifer C. Developmentally regulated recruitment of transcription factors and chromatin modification activities to chicken lysozyme cis-regulatory elements in vivo. Mol. Cell. Biol. 2003;23:4386–4400. doi: 10.1128/MCB.23.12.4386-4400.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lichtinger M., Ingram R., Hannah R., Muller D., Clarke D., Assi S.A., Lie A.L.M., Noailles L., Vijayabaskar M.S., Wu M., Tenen D.G., Westhead D.R., Kouskoff V., Lacaud G., Gottgens B., Bonifer C. RUNX1 reshapes the epigenetic landscape at the onset of haematopoiesis. EMBO J. 2012;31:4318–4333. doi: 10.1038/emboj.2012.275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Link K.A., Chou F.S., Mulloy J.C. Core binding factor at the crossroads: determining the fate of the HSC. J. Cell. Physiol. 2010;222:50–56. doi: 10.1002/jcp.21950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MAYER M., MEYER B. Characterization of ligand binding by saturation transfer difference NMR spectroscopy. Angewandte Chemie-International Edition. 1999;38:1784–1788. doi: 10.1002/(SICI)1521-3773(19990614)38:12<1784::AID-ANIE1784>3.0.CO;2-Q. [DOI] [PubMed] [Google Scholar]
- Mayer M., Meyer B. Group epitope mapping by saturation transfer difference NMR to identify segments of a ligand in direct contact with a protein receptor. J. Am. Chem. Soc. 2001;123:6108–6117. doi: 10.1021/ja0100120. [DOI] [PubMed] [Google Scholar]
- Mcdonald L., Ferrari N., Terry A., Bell M., Mohammed Z.M., Orange C., Jenkins A., Muller W.J., Gusterson B.A., Neil J.C., Edwards J., Morris J.S., Cameron E.R., Blyth K. RUNX2 correlates with subtype-specific breast cancer in a human tissue microarray, and ectopic expression of Runx2 perturbs differentiation in the mouse mammary gland. Dis. Model Mech. 2014;7:525–534. doi: 10.1242/dmm.015040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otto F., Thornell A.P., Crompton T., Denzel A., Gilmour K.C., Rosewell I.R., Stamp G.W., Beddington R.S., Mundlos S., Olsen B.R., Selby P.B., Owen M.J. Cbfa1, a candidate gene for cleidocranial dysplasia syndrome, is essential for osteoblast differentiation and bone development. Cell. 1997;89:765–771. doi: 10.1016/s0092-8674(00)80259-7. [DOI] [PubMed] [Google Scholar]
- Pratap J., Imbalzano K.M., Underwood J.M., Cohet N., Gokul K., Akech J., Van Wijnen A.J., Stein J.L., Imbalzano A.N., Nickerson J.A., Lian J.B., Stein G.S. Ectopic runx2 expression in mammary epithelial cells disrupts formation of normal acini structure: implications for breast cancer progression. Cancer Res. 2009;69:6807–6814. doi: 10.1158/0008-5472.CAN-09-1471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pratap J., Lian J.B., Javed A., Barnes G.L., Van Wijnen A.J., Stein J.L., Stein G.S. Regulatory roles of Runx2 in metastatic tumor and cancer cell interactions with bone. Cancer Metastasis Rev. 2006;25:589–600. doi: 10.1007/s10555-006-9032-0. [DOI] [PubMed] [Google Scholar]
- Roudaia L., Cheney M.D., Manuylova E., Chen W., Morrow M., Park S., Lee C.T., Kaur P., Williams O., Bushweller J.H., Speck N.A. CBFbeta is critical for AML1-ETO and TEL-AML1 activity. Blood. 2009;113:3070–3079. doi: 10.1182/blood-2008-03-147207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheitz C.J., Lee T.S., Mcdermitt D.J., Tumbar T. Defining a tissue stem cell-driven Runx1/Stat3 signalling axis in epithelial cancer. EMBO J. 2012;31:4124–4139. doi: 10.1038/emboj.2012.270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheitz C.J., Tumbar T. New insights into the role of Runx1 in epithelial stem cell biology and pathology. J. Cell. Biochem. 2013;114:985–993. doi: 10.1002/jcb.24453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneyer A.L., Sidis Y., Gulati A., Sun J.L., Keutmann H., Krasney P.A. Differential antagonism of activin, myostatin and growth and differentiation factor 11 by wild-type and mutant follistatin. Endocrinology. 2008;149:4589–4595. doi: 10.1210/en.2008-0259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shore P. A role for Runx2 in normal mammary gland and breast cancer bone metastasis. J. Cell. Biochem. 2005;96:484–489. doi: 10.1002/jcb.20557. [DOI] [PubMed] [Google Scholar]
- Smith S.G., Sanchez R., Zhou M.M. Privileged diazepine compounds and their emergence as bromodomain inhibitors. Chem. Biol. 2014;21:573–583. doi: 10.1016/j.chembiol.2014.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sroczynska P., Lancrin C., Pearson S., Kouskoff V., Lacaud G. In vitro differentiation of mouse embryonic stem cells as a model of early hematopoietic development. Methods Mol. Biol. 2009;538:317–334. doi: 10.1007/978-1-59745-418-6_16. [DOI] [PubMed] [Google Scholar]
- Taipale J., Saharinen J., Hedman K., Keski-OJA J. Latent transforming growth factor-beta 1 and its binding protein are components of extracellular matrix microfibrils. J. Histochem. Cytochem. 1996;44:875–889. doi: 10.1177/44.8.8756760. [DOI] [PubMed] [Google Scholar]
- Tang Y.Y., Shi J., Zhang L., Davis A., Bravo J., Warren A.J., Speck N.A., Bushweller J.H. Energetic and functional contribution of residues in the core binding factor beta (CBFbeta) subunit to heterodimerization with CBFalpha. J. Biol. Chem. 2000;275:39579–39588. doi: 10.1074/jbc.M007350200. [DOI] [PubMed] [Google Scholar]
- Van Bragt M.P., Hu X., Xie Y., Li Z. RUNX1, a transcription factor mutated in breast cancer, controls the fate of ER-positive mammary luminal cells. Elife. 2014;3 doi: 10.7554/eLife.03881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vranken W.F., Boucher W., Stevens T.J., Fogh R.H., pajon A., Llinas M., Ulrich E.L., Markley J.L., Ionides J., Laue E.D. The CCPN data model for NMR spectroscopy: development of a software pipeline. Proteins. 2005;59:687–696. doi: 10.1002/prot.20449. [DOI] [PubMed] [Google Scholar]
- Wang C.C., Bajikar S.S., Jamal L., Atkins K.A., Janes K.A. A time- and matrix-dependent TGFBR3-JUND-KRT5 regulatory circuit in single breast epithelial cells and basal-like premalignancies. Nat. Cell Biol. 2014;16:345–356. doi: 10.1038/ncb2930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang C.Q., Jacob B., Nah G.S., Osato M. Runx family genes, niche, and stem cell quiescence. Blood Cells Mol. Dis. 2010;44:275–286. doi: 10.1016/j.bcmd.2010.01.006. [DOI] [PubMed] [Google Scholar]
- Wang L., Brugge J.S., Janes K.A. Intersection of FOXO- and RUNX1-mediated gene expression programs in single breast epithelial cells during morphogenesis and tumor progression. Proc. Natl. Acad. Sci. U. S. A. 2011;108:E803–E812. doi: 10.1073/pnas.1103423108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan X., Davydova N., Conte M.R., Curry S., Matthews S. Chemical shift mapping of RNA interactions with the polypyrimidine tract binding protein. Nucleic Acids Res. 2002;30:456–462. doi: 10.1093/nar/30.2.456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang L., Li Z., Yan J., Pradhan P., Corpora T., Cheney M.D., Bravo J., Warren A.J., Bushweller J.H., Speck N.A. Mutagenesis of the Runt domain defines two energetic hot spots for heterodimerization with the core binding factor beta subunit. J. Biol. Chem. 2003;278:33097–33104. doi: 10.1074/jbc.M303972200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Mechanism of action of CBFß-RUNX inhibitors.