

Abstract
We present a high content multiwell plate cell-based assay approach to quantify protein interactions directly in cells using Förster resonance energy transfer (FRET) read out by automated fluorescence lifetime imaging (FLIM). Automated FLIM is implemented using wide-field time-gated detection, typically requiring only 10 s per field of view (FOV). Averaging over biological, thermal and shot noise with 100’s to 1000’s of FOV enables unbiased quantitative analysis with high statistical power. Plotting average donor lifetime vs. acceptor/donor intensity ratio clearly identifies protein interactions and fitting to double exponential donor decay models provides estimates of interacting population fractions that, with calibrated donor and acceptor fluorescence intensities, can yield dissociation constants. We demonstrate the application to identify binding partners of MST1 kinase and estimate interaction strength among the members of the RASSF protein family, which have important roles in apoptosis via the Hippo signalling pathway. KD values broadly agree with published biochemical measurements.
With increasing knowledge of intracellular signalling networks, it becomes more evident that molecules can be involved in processes occurring in multiple pathways. Understanding the complex interconnections between different pathways requires comprehensive identification of specific binding partners, and therefore it is important to develop higher throughput techniques to search for new interactions. Currently, biochemical methods are most often used to this end and provide high sensitivity. However, they require long separation procedures, during which the active molecules are isolated from their native environment and may present different reaction kinetics than in live cells where molecular crowding and high compartmentalisation could have an impact. Fluorescence microscopy – particularly exploiting genetically expressed fluorescent proteins – can be applied directly to map and quantify protein interactions in live or fixed cells and preserve information concerning the inhomogeneous cellular distribution of molecules, with typical spatial resolution below 0.5 μm. With the advent of superresolution microscopy, the prospect of sub-50 nm resolution could permit the study of the organisation and dynamics of molecules within organelles and large interacting complexes1,2. However, manual fluorescence microscopy experiments are subject to operator bias and it is impractical to undertake measurements on a sufficient number of cells to identify systematic errors and to average over “biological noise”. Large scale screening using automated fluorescence microscopes can provide higher throughput studies of signalling processes with improved statistical significance. To date, high content analysis platforms for cell imaging have been mostly based on fluorescence intensity readouts and have predominantly been applied to study the effects of inhibitors on signalling pathways3. Other fluorescence parameters may also be utilised to assay molecular environment (fluorescence lifetime) or fluorophore orientation (polarisation/anisotropy).
A widely used fluorescence technique to study bi-molecular interactions within cells is FRET (Förster resonant energy transfer), which utilises the non-radiative (dipole-dipole) energy transfer from a fluorescent donor to an acceptor that can take place only when the two fluorophores are situated at distances <~10 nm. In the case of two proteins labelled with donor and acceptor tags, this implies that FRET occurs only if and when the two proteins interact with each other. FRET has therefore been widely exploited to study protein interactions using fluorescence microscopes. However, its application for high content analysis in automated multiwell plate readers is much more limited. FRET can be read out using a wide range of techniques4 although most of these are not practical for rapid automated assays of multiwell plates where hundreds to thousands of fields of view are imaged in a single experiment.
One approach to detect FRET is to measure the fluorescence intensity ratio of the acceptor and the donor fluorophores observing the increase of the fluorescence intensity in the acceptor channel with the simultaneous decrease of the intensity in the donor channel. This spectral ratiometric imaging acquisition is fast but requires additional control samples to correct for spectral cross-talk between the fluorophores and to calibrate the spectral response of the specific optical set-up (instrument and sample corrections), making comparison between different samples difficult. Quantitation can be degraded by unknown variations in donor-acceptor stoichiometry and quantitative readouts of FRET efficiency and population fraction of FRETing donors are not possible without additional measurements of reference FRET constructs5,6.
It is also possible to utilise the depolarisation of the acceptor fluorescence as a FRET readout Polarisation-based measurements can achieve similar acquisition speeds as spectral ratiometric readouts and are highly sensitive to detect the occurrence of FRET, but it is again difficult to quantify FRET efficiencies and population fractions of interacting donors7,8. Polarisation has been applied as a first step to screen for possible interaction partners that were subsequently investigated using fluorescence lifetime9.
Fluorescence lifetime imaging (FLIM)10 provides a more robust approach to reading out FRET since only measurements of the donor fluorophores are required – negating the need for spectral calibration and providing readouts that can be directly compared between instruments and which can be translated from cell-based assays to animal models11. Compared to spectral or polarisation ratiometric techniques, FLIM requires more detected photons to achieve a given accuracy, so this is a slower modality for mapping and quantifying FRET in high content analysis. However, FLIM can also provide more quantitative readouts in a single spectral channel since the fluorescence decay profiles can be fitted to complex models in order to obtain the FRET efficiency and the interacting population fraction. Time-resolved measurements can be extended to analyse homoFRET and polarisation anisotropy decays using appropriate models. Fitting lifetime data to complex models typically requires 10,000’s of photons – compared to ~200 photons required to fit to a monoexponential decay model12 – and it is not possible to detect such high photon numbers per pixel from biological samples such as cells labelled with fluorescent proteins before photobleaching and phototoxicity ensue. However, global analysis techniques that fit data simultaneously from many pixels can overcome this limitation (subject to assumptions about spatial invariance of lifetime components across the data set) and enable FLIM data with only 100’s photons/pixel (i.e. compatible with live cell imaging) to be fitted to complex decay models. Quantitative information can also be directly obtained without fitting using the phasor analysis approach13,14.
In this paper we report the application of a prototype high content assay platform providing unsupervised FLIM FRET of multiwell plate arrays that can identify protein binding partners in their cellular context and quantify the dissociation constant, KD. In order to achieve the fast FLIM acquisition required for reading 96-well plates, we utilise wide-field time-gated imaging to realise a FLIM microscope that is able to automatically acquire wide-field or optically sectioned fluorescence lifetime images with a typical mean acquisition time of 10 seconds per field of view for cells expressing fluorescent proteins, including the time required to move from the previous field of view and to automatically focus the microscope. For the first time to our knowledge, we report the application of automated FLIM FRET to screen for protein binding partners within cells – here shown to identify interactions between the Ras-association domain family (RASSF) and mammalian sterile 20-like kinases (MST) – and the estimation of the KD for these interactions.
The RASSF family consists of ten members, RASSF1-10, which share a common Ras association domain. The role of this domain is not yet fully understood15, but the RASSF proteins are components of the MST/Hippo pathway, which is considered to restrict cell proliferation, thus they potentially play important roles in suppressing tumourigenesis16,17,18,19,20,21,22. This could oppose the Raf/Mek/Erk stimulation of cell growth/proliferation that also depends on Ras activation (Fig. 1). More recently, RASSF1 and MST1 have also been shown to influence the cardiac function in response to stress23, whilst RASSF5 and MST1 are involved in mediating TNFα- and TRAIL-induced apoptosis24.
Figure 1. Schematic of Ras-dependent pathways determining cell fate.
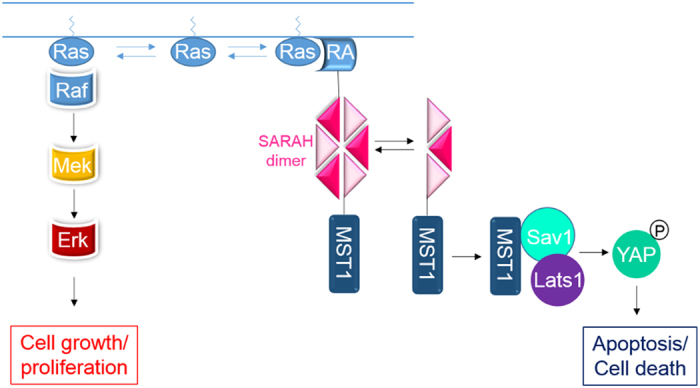
The C-terminal domains differ between the classical RASSF1-6 and the N-terminal RASSF7-10. The classical RASSF members have a common α-helical SARAH domain that is absent in the N-terminal RASSF members, which instead are predicted to have coiled-coil motifs at various positions towards their C-terminal region25. The SARAH domain, whose name is essentially derived from the three proteins that share this common feature at their extreme C-terminal region: Salvador/RASSF/Hippo16 is also found in the MST kinases. The mammalian homologues, WW45, RASSF and MST respectively, are components of the well conserved Hippo signalling pathway, which was first described in Drosophila melanogaster.
Recent studies have shown that the SARAH domain is able to dimerise in solution26,27,28, so it was hypothesized that dimerisation could represent a key mechanism of interaction between the MST kinases and RASSF proteins. It is thought that, through this dimerisation, RASSFs are involved in the regulation of the catalytic activity of MST kinases (Fig. 1). Being themselves devoid of enzymatic activity, RASSF proteins may act as scaffolds binding the MST kinases. Previous studies have shown interaction between several RASSF members and different Ras proteins15,29,30,31, leading to the hypothesis that Ras association localizes RASSFs and the MST kinases to the cell membrane, thus bringing the MST kinase domains into close proximity for trans-activating phosphorylation, driving the MST/Hippo pathway and cellular apoptosis17,18,32.
The SARAH dimer is formed via a head-to-tail interaction of the two helices in an antiparallel arrangement. Although different coiled-coil motifs have been described to form oligomers33,34, there is no evidence thus far to suggest that the predicted coiled-coils in the N-terminal RASSF7-10 are capable of associating with the SARAH domain of MST to promote their interactions. This study aimed to confirm at the cellular level that SARAH-mediated dimerisation is the mode of interaction by identifying the RASSF proteins that associate with MST1 kinase or its isolated SARAH domain. In addition, point mutations were also introduced within the SARAH domains of the more well-studied members, RASSF1 and 5, to study their effect on the dimerisation with the MST1-SARAH domain, which were assayed using FRET.
Results and Discussions
Figure 2 illustrates the fluorescent constructs that have been created to assay the RASSF-MST interactions using FRET. All ten RASSF proteins have been modified by attaching the fluorescent protein EGFP to their N-terminus to serve as a donor. Similarly, the MST1 kinase and its isolated SARAH domain (SARAHMST1) were labelled with mCherry at the N-terminus to provide the acceptor for the FRET assays. To evaluate the effect of possible non-specific interactions on the FRET readouts (e.g. arising from high local concentration of donor and acceptor), two negative controls were employed: the free fluorescent protein mCherry was expressed (without being linked to the MST1 or SARAHMST1 domain) and, as a more biologically relevant control, the kinase domain of the MST1 with a deletion of the SARAH domain (MST1ΔSARAH) was tagged with mCherry at its N-terminus.
Figure 2. Schematic representation of the fluorescent constructs used for the FRET assays.

The domain structure of the RASSF family members, of their possible interacting partners (MST1 kinase and its isolated SARAHMST1 domain) and of the negative controls are shown.
Figure 3 shows a diagram of the automated FLIM multiwell plate microscope that can be configured for wide-field imaging or for optical sectioning using a Nipkow spinning disk unit, the latter providing more quantitative readouts at the cost of increased complexity. FLIM is realised using a gated optical intensifier that acts as a fast (~100 ps rise time) electronic shutter synchronised with the laser pulses, opening at various delays after excitation to provide time-gated fluorescence intensity images for each time delay, integrated over a few seconds. From these images, the fluorescence decay profiles can be reconstructed and analysed by fitting to an appropriate exponential decay model.
Figure 3. Schematic of automated plate reader based on time-gated fluorescence lifetime imaging (FLIM).

(A) The pulsed excitation light is selected with an appropriate filter from the “white light” emitted by an ultrafast supercontinuum laser source and enters the microscope either in a wide-field configuration or via a Nipkow disk unit to provide optical sectioning. The fluorescence is detected via a gated optical intensifier (GOI) that acts as a fast (~100 ps rise time) electronic shutter synchronised with the laser pulses. The GOI opens at various delays after excitation (e.g. t1, t2, t3) and intensity images are acquired with a CCD camera at each time delay, integrating for a few seconds. (B) Lifetime determination. The time-gated images (t1, t2, t3) are used to reconstruct the fluorescence decay of the fluorophore, which is analysed by fitting exponential decay functions, discriminating between the lifetime of the donor only (D only) and the lifetime of the donor undergoing FRET in the presence of the acceptor (D + A).
To obtain reliable statistics, we automatically acquired FLIM images from 10 fields of view (FOV) per well, using 5 time gates to sample the fluorescence decay profiles with exposure times around 1 s per gate for the donor (EGFP) images. Intensity images of the acceptor (mCherry) were also obtained with direct excitation for the same fields of view. Approximately 800 FLIM images were thus acquired for each multiwell plate. Such large FRET data volumes require rapid automated analysis, for which we have developed an open source program called FLIMfit35 (available at http://www.openmicroscopy.org/site/products/partner/flimfit), based on the variable projection method and providing tools for segmentation of cells containing both donor and acceptor, monoexponential and global analysis of EGFP lifetime using convolution and background correction, as well as analysis of fluorescence intensity images.
For these assays, COS7 cells were transfected with donor only plasmids (EGFP-RASSF) and two different conditions of donor plus acceptor constructs: (i) EGFP-RASSF + mCherry-SARAHMST1 (interaction partner) and (ii) EGFP-RASSF + mCherry-MST1ΔSARAH as the negative control (Fig. 4A). Figure 4 shows the results of this screen, displaying the mean donor lifetime fitted to a monoexponential decay profile and averaged across 10 FOV per well. Although we expect to identify two EGFP lifetime components corresponding to free and SARAH-bound RASSF in co-transfected cells, here we show that a monoexponential fit (equation 1) provides a convenient average lifetime value (τ) per cell that can be used for qualitative readouts of the occurrence or absence of the protein-protein interaction.
Figure 4. Comparison of the RASSF family members in terms of dimerisation with the SARAHMST1 domain using FRET.

(A) Plate map showing average EGFP donor lifetimes (ps) calculated for 10 fields of view (FOV) per well using a monoexponential fit. (B) False-colour FLIM images of cells from a typical FOV in each well showing the EGFP lifetime (ps) per pixel. (C) Box plots showing median EGFP lifetimes, interquartile (box range), standard deviation (whisker), 1% and 99% percentile (×) and minimum/maximum values (−) calculated for individual cells averaged over 10 FOV per well using monoexponential analysis: green: EGFP-RASSF(1–10) only; red: EGFP-RASSF(1–10) + mCherry-SARAHMST1; blue: EGFP-RASSF(1–10) + mCherry-MST1ΔSARAH (see Supplementary material for a table of differences in mean fluorescence lifetime). (D) Acceptor/donor intensity ratios (ImCherry/IEGFP) averaged over each cell for all the conditions in the plate. The colour code is the same as in C). (E) Scattered plots of EGFP lifetimes versus acceptor/donor intensity ratios (ImCherry/IEGFP) calculated for individual cells (with same colour code as for C). FLIM data were acquired with wide-field imaging.
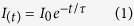 |
The average EGFP donor lifetimes calculated from the monoexponential pixel-wise fit of all FOV in each of the 96 wells of the RASSF-SARAH plate are displayed as a colour coded plate map (Fig. 4A). Box plots of lifetimes calculated per cell per condition are also shown (Fig. 4C). A montage of FLIM images showing one FOV per well is presented in Fig. 4B.
When cells co-transfected with the negative control (mCherry-MST1ΔSARAH) are compared with those transfected with the donor only, the average of the mean lifetime differences for each RASSF family member was 6 ± 8 ps, with a maximum change in mean fluorescence lifetime of 16 ps for RASSF3. These results represent the biological noise in our measurement. Therefore, to be conservative, we considered that a lifetime shift of at least 32 ps – i.e. twice the maximum difference observed between donor only and donor plus negative control – should be required for it to be considered significant.
In the case of RASSF and SARAHMST1 co-transfection, the average EGFP lifetime is reduced by 130–310 ps for RASSF1-6 (Table S.3, Supplementary informationm). This reduction is above our threshold for significance, as outlined above. For RASSF7-10, the reduction of the mean EGFP lifetime in cells co-transfected with SARAHMST1 was less than 32 ps, suggesting little or no donor-acceptor interaction (Table S.3, Supplementary information).
The donor and acceptor-labelled proteins used in these experiments were encoded using separate plasmids and therefore they will not be expressed in a 1:1 ratio in the cells (Fig. 4D). Variations in the donor/acceptor ratio do impact the magnitude of the FRET readout and could also impair the significance of the negative controls. For example, if more acceptor molecules are expressed in one cell compared to another, more donor molecules may be quenched and the average donor fluorescence lifetime per cell would then be shorter. In order to clarify this issue, 2D plots of EGFP donor lifetimes versus acceptor/donor intensity ratios have been constructed after segmenting individual cells in all FOVs (Fig. 4E). While a finite spread of the EGFP lifetimes is observed for each condition, the EGFP lifetime distributions for RASSF1-6/SARAHMST1 co-transfection only have a small overlap with the distribution of the negative control, the average lifetime being reduced even at low acceptor concentration, as would be expected for FRET. For RASSF7-10/SARAHMST1 co-transfection, the EGFP lifetime distributions are centred on similar values as for the negative control, even for high acceptor/donor ratios, indicating a lack of FRET and therefore a weak or no interaction. These 2D plots support the qualitative readouts provided by the average EGFP lifetime obtained from the monoexponential fit of intensity decays that are seen to be robust in the presence of variation in the donor/acceptor stoichiometry. Overall, these results indicate the specific interaction based on the dimerisation of the SARAH domains between RASSF1-6 and MST1, while there is very little or no interaction between the SARAH domain and the coiled-coil or unstructured regions at the C-terminal end of RASSF7-10.
The results of this intracellular FRET assay are supported by biochemical data15 and by the SARAH domain heterodimer structural models (depicted in Fig. S.1 in the Supplementary information) showing that most of the main interacting residues of all six SARAHRASSF monomers are well-conserved and aligned to heterodimerise with the SARAHMST1 monomer. The contact interface mainly involves the side chains and non-polar residues for all six heterodimers, with a small degree of polar or charged interaction between the acidic and basic residues (see also Table S.1 in the Supplementary information).
Based on these structural models, we selected three key non-polar residues in the main helix for further mutational studies. These are residues that align to L444, L448 and L451 in SARAHMST1 and they are highly conserved, as well as major contributors to dimer formation and stability. We applied our FRET screening technique to study the effect of three point mutations within the SARAH domains of RASSF1 and RASSF5C, which are the two best characterised RASSF members with published literature on their L308P and L224A mutants respectively23,36. All mutations involved the replacement of leucine residues with proline at the positions described above. We chose to perform these mutations since proline residues have been shown to introduce distortions (kinks) to α-helices37, in our case in the main helix facilitating the dimerisation with the SARAHMST1 domain26,28.
Figures 5 and 6 show the results of FRET assays of interaction between the wild type isolated SARAHMST1 domain and the point mutated RASSF1 and RASSF5 constructs. The average EGFP lifetimes obtained by fitting to a monoexponential decay model indicate that all three point mutations introduced within the SARAH domain of RASSF1 inhibit dimerisation with the isolated SARAHMST1 domain (Fig. 5A). The box plots in Fig. 5B indicate that the average values of the EGFP lifetimes show differences of less than 17 ps when co-expressed with the MST1ΔSARAH domain or with mCherry alone as negative controls. The distributions of the EGFP lifetimes versus the acceptor/donor intensity ratios of all RASSF1 mutants overlap with those of the negative control for all acceptor expression levels (Fig. 5D).
Figure 5. Effect of three different point mutations within the SARAH domain of RASSF1 on the dimerisation with the isolated SARAHMST1.

(A) Plate map showing the average EGFP lifetimes calculated for 10 fields of view per well when fitting to a monoexponential decay profile. The wild-type EGFP-RASSF1 assay shows that mCherry alone can serve as a negative control as well as the mCherry-MST1ΔSARAH. (B) Box plots showing median EGFP lifetimes, interquartile (box range), standard deviation (whisker), 1% and 99% percentile (×) and minimum/maximum values (−) for segmented cells in different conditions within the plate: green: EGFP-RASSF1 (wild type and mutants) only; red: EGFP-RASSF1 (wild type and mutants) + mCherry-SARAHMST1; blue: EGFP-RASSF1 (wild type and mutants) + mCherry (see Supplementary material for a table of differences in mean fluorescence lifetime). (C) Average acceptor/donor intensity ratios (ImCherry/IEGFP) for the segmented cells in different conditions within the plate (same colour code as in B). (D) 2D plots of acceptor/donor intensity ratios versus EGFP lifetimes for the segmented cells in different conditions within the plate (same colour code as in B). FLIM data were acquired with wide-field imaging.
Figure 6. The effect of three different point mutations within the SARAH domain of RASSF5C on the dimerisation with the isolated SARAHMST1.

(A) Plate map showing the average EGFP lifetimes calculated for 10 fields of view per well fitted to a monoexponential decay model. (B) Box plots showing median EGFP lifetimes, interquartile (box range), standard deviation (whisker), 1% and 99% percentile (×) and minimum/maximum values (−) for the segmented cells in different conditions within the plate: green: EGFP-RASSF5C (wild type and mutants) only; red: EGFP-RASSF5C (wild type and mutants) + mCherry-SARAHMST1; blue: EGFP-RASSF5C (wild type and mutants) + mCherry (see Supplementary material for a table of differences in mean fluorescence lifetime). (C) Average intensity ratios acceptor/donor (ImCherry/IEGFP) for the segmented cells in different conditions within the plate (same colour code as in B). (D) 2D plots of intensity ratios acceptor/donor versus EGFP lifetimes for the segmented cells in different conditions within the plate (same colour code as in B). FLIM data were acquired with wide-field imaging.
In the case of the three RASSF5C mutants, we observe a reduction in the average EGFP lifetimes compared to the negative control based on the box plots in Fig. 6B, suggesting that dimerisation of the SARAH domains still occurs. The distributions on the 2D plots in Fig. 6D are clearly different for the mCherry-SARAHMST1 domain co-transfection compared to the co-transfection with mCherry (negative control). However, the reduction in mean EGFP lifetime in individual cells expressing the mutants is smaller than that observed with the wild type RASSF5C, suggesting that the fraction of the bound molecules is reduced in the case of RASSF5C mutants compared to the wild-type protein, which could be due to a reduction in binding affinity.
These FRET screening data are also supported by biochemical assays, represented in Figs 7 and 8, where full length proteins were used to better mimic physiological conditions. For RASSF1 (Fig. 7), only the wild-type protein showed strong binding to MST1, whereas the signals from the mutants were significantly weaker. In the case of RASSF5 (Fig. 8), the mutants were still detected at a significant level, but reduced compared to the wild-type protein. In agreement with the FRET data, these biochemical assays indicate that, while binding still occurs, the affinities are reduced by the mutations.
Figure 7. Effects of mutations in the SARAHRASSF1 domain on dimerisation with full length MST1.

(A) The SARAH domain sequence of RASSF1. Main interacting non-polar (yellow), acidic (red) and basic (blue) residues are shown. The three positions in which mutations were introduced are marked by asterisks (*). (B) (i) Co-immunoprecipitation assay to show heterodimerisation between myc-MST1 K59R and wild-type (WT) EGFP-RASSF1 and its three mutants. The loading controls are shown below. (ii) Quantification of the bands in terms of relative intensity to the WT control (Mean ± SD. n = 3; *p < 0.05, **p < 0.01, ***p < 0.001). (C) Co-immunoprecipitation assay of the negative controls. A simultaneous negative control was performed using cell lysates containing only EGFP-RASSF1 or its mutants. The loading controls are shown below.
Figure 8. Effects of mutations in the SARAHRASSF5 domain on dimerization with full length MST1.

(A) The SARAH domain sequence of RASSF5. Main interacting non-polar (yellow), acidic (red) and basic (blue) residues are shown. The three positions in which mutations were introduced are marked by asterisks (*). (B) (i) Co-immunoprecipitation assay to show heterodimerisation between myc-MST1 K59R and wild-type (WT) EGFP-RASSF5 and its three mutants. The loading controls are shown below. (ii) Quantification of the bands in terms of relative intensity to the WT control (Mean ± SD. n = 3; *p < 0.05, **p < 0.01, ***p < 0.001). (C) Co-immunoprecipitation assay of the negative controls. A simultaneous negative control was performed using cell lysates containing only EGFP-RASSF5 or its mutants. The loading controls are shown below.
The different degrees of disruption to heterodimerisation caused by the SARAH mutations in RASSF1 and RASSF5 observed both in our FRET and biochemical data (Figs 5, 6, 7, 8) could be due to distinct biochemical and structural properties of the individual SARAH domains, such as local variations in the specific residues involved in the individual heterodimeric interfaces or in the residues surrounding the mutated sites. It is possible that the neighbouring residues could compensate for the effects of the mutation in RASSF5, but not in RASSF1. Similarly, the leucine residues and hydrophobic interactions may play a more critical role for RASSF1 heterodimerisation compared to RASSF5. Alternatively, the kinks introduced by the proline mutations could affect the secondary structure of SARAH as a whole. It has been shown that the MST1 binding interface increases due to these distortions28, so it is plausible that the introduction of proline into RASSF1-SARAH severely distorts its helical structure to the detriment of its ability to dimerise. This effect could be less severe in RASSF5, thus its mutants retain their ability to heterodimerise, albeit at a diminished level.
We also investigated the differences in the binding characteristics between the isolated SARAH domain and the full length MST1 when interacting with those RASSF proteins that are able to dimerise (RASSF1-6). Figure 9A shows the plate map for this experiment. Initially we fitted the donor fluorescence data to a monoexponential decay model, as for the previous assays. The plate map of EGFP donor lifetimes averaged over 10 FOV are shown colour-coded in Fig. 9A together with box plots of the lifetimes on a per cell basis in Fig. 9B. It is immediately apparent that the EGFP lifetimes are more reduced when the RASSF proteins 1–6 interact with the isolated SARAHMST1 domain compared to when they dimerise with the full length MST1. This is observed for all RASSF1-6, and is supported by the 2D plots of EGFP lifetimes vs. donor/acceptor ratios (Fig. 9D) even though the donor/acceptor ratios vary among the different conditions within the plate (Fig. 9C).
Figure 9. Comparison of the RASSF family members in terms of dimerisation with the isolated SARAHMST1 domain and the full length MST1 using FRET.

(A) Plate map showing average EGFP lifetimes (ps) calculated for 10 fields of view per well by fitting to a monoexponential decay model. (B) Box plots showing median EGFP lifetimes, interquartile (box range), standard deviation (whisker), 1% and 99% percentile (×) and minimum/maximum values (−) calculated for individual cells from 10 FOV per well; green: EGFP-RASSF(1–6) only; red: EGFP-RASSF(1–6) + mCherry-SARAHMST1; purple: EGFP-RASSF(1–6) + mCherry-MST1. (C) Average acceptor/donor intensity ratios (ImCherry/IEGFP) for all the conditions in the plate with the same colour code as in (C). (D) 2D scatter plots of acceptor/donor intensity ratios (ImCherry/IEGFP) versus EGFP lifetime calculated for individual cells with same colour code as (B). FLIM data were acquired with optical sectioning using Nipkow disc unit.
These data could be explained by a larger mean donor-acceptor FRET distance for the interaction of full length MST1 with RASSF1-6 compared to the interaction with the isolated SARAH domain, e.g. due to steric constraints. This would reduce the FRET efficiency, due to its dependence to the sixth power of the donor-acceptor distance. Alternatively, the fraction of bound molecules, e.g. due to a different binding affinity of RASSF1-6, could be different for the two interactions.
To understand more about the interactions producing the observed differences in the readout based on fitting to the monoexponential decay model, the data underlying Fig. 9 was fitted to a double exponential decay model (equation 2), using the global analysis capabilities of FLIMfit35.
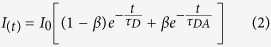 |
The two lifetime components contributing to the EGFP (donor) decay profiles arise from non-interacting RASSF molecules (unquenched donor, τD) and from RASSF molecules that are bound either to the isolated SARAH domain or to the full length MST1 kinase (donor quenched by FRET, τDA). The unquenched donor lifetime was determined using data from the cells transfected only with RASSF1-6 and this component was fixed during the global fitting. The donor lifetime quenched by FRET was allowed to vary, but was constrained to be spatially invariant across all cells within a given experimental condition. Thus, the fraction of interacting molecules (β term in equation 2) could be estimated and these results are presented in the box plots shown in Fig. 10A (the full list of parameters obtained from this analysis are presented in the Supplementary information). Owing to the challenges associated with quantifying FRET interactions between fluorescent proteins that are applicable to all such assays38, these values should be treated as estimates. Although they show relatively broad distributions, they indicate a lower fraction of bound molecules in the case of the RASSF interaction with the full length MST1 protein compared to the isolated SARAHMST1 domain.
Figure 10. Results of global fitting of the donor fluorescence decay data underlying Fig. 9 to a double exponential decay model.

(A) FRET population fractions for RASSF1-6 interacting with SARAHMST1 (red) and full length MST1 (purple). (B,C) EGFP and mCherry calibration of intensity versus fluorophore concentration. (D) Dissociation constants (KD) for RASSF1-6 interacting with SARAHMST1 (red) and full length MST1 (purple).
To understand if the results in Fig. 10A are indeed due to a difference in binding affinities, we need to estimate the dissociation constants KD for the interactions of MST1 with the different RASSF1-6 proteins. For this we can use the binding population fractions from the global FLIM analysis, but it is also necessary to estimate the concentrations of the MST1 and RASSF1-6 proteins from the EGFP and mCherry fluorescence intensities. To this end we used the Nipkow disc unit to implement optically sectioned FLIM in order to constrain the detected emission to a well-defined focal volume. The instrument was calibrated using solutions of purified fluorescent EGFP and mCherry at known concentrations in phosphate buffer pH 7.4. Figure 10B,C shows the linear relationship between the average detected fluorescence intensity per pixel and the fluorophore concentration.
Dissociation constants KD were calculated for each cell assuming a bi-molecular reaction (equation 3), where D is the donor-labelled partner, A is the acceptor-labelled partner and DA is the complex formed by their association:
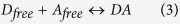 |
KD is then given by equation 4, which relates the concentrations of the binding partners to the complex:
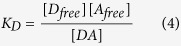 |
Using the fluorophore concentration calibration we can determine the total donor (Dtotal) and acceptor (Atotal) concentrations per cell, while the FRET fraction β obtained from the global analysis provides an estimate of the concentration of the DA complex via the bound fraction of the donor.
We can then write:
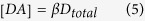 |
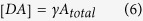 |
where γ is the bound fraction of the acceptor molecules within the complex. This fraction can be calculated from Eq. 5 and 6:
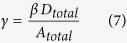 |
Knowing the bound D and A fractions, we can obtain the free fractions and re-write the KD expression:
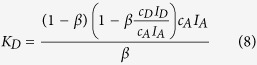 |
where ID and IA are the fluorescence intensities of the donor and acceptor respectively, which are linearly proportional to the concentrations via the proportionality constants cD and cA (Fig. 10B,C). A full derivation of this equation is presented in the Supplementary information. To estimate the donor (EGFP) concentration in the cells, the fitted initial intensity of the fluorescence decay (at t = 0) was used rather than the total fluorescence intensity because the FRETing and non-FRETing donors have different brightness due to their different lifetimes and quantum yields.
The KD values obtained for all the conditions in the plate are plotted in Fig. 10D and are in the same range as previously published values: C. Herrmann and colleagues39,40,41 determined a dissociation constant KD in the order of hundreds of nM for the RASSF5-MST1 complex in FRET experiments using stopped-flow fluorimetry, while the self-association constant for RASSF5 was found to be 5–10 μM, and that for MST1 was in the low nM range. In their case, KD was calculated as the ratio between the association rate constant kon and the dissociation rate constant koff. They measured a higher koff when full length RASSF5 dimerised with the isolated SARAH domain of MST1 than in the case when dimerisation was performed between the isolated SARAH domains of the two proteins, indicating that the full length proteins have a lower affinity than the isolated dimerising domains. Although in our experiments the KD values for the isolated SARAH domain and the full length MST1 are not clearly separated (Fig. 10D), there appears to be a trend towards higher average KD values for the RASSF-full length MST1 interaction, suggesting it has a lower interaction strength than the MST1 interaction with the isolated SARAH domain.
Conclusions
We have developed a high content assay utilising FLIM FRET to screen for binding partners of MST1 kinase among the RASSF protein family and to quantify the relative interaction affinities. Our custom automated FLIM multiwell plate microscope based on time gated detection is capable of rapid automated image acquisition and therefore facilitates systematic studies of bimolecular processes to provide statistically robust readouts that quickly highlight any systematic errors and effectively average over biological variations. We note that the results presented here and in our previous work11,35 highlight that the ability to apply global fitting over such large data sets enables us to take advantage of FRET assays with modest lifetime changes (100–200 ps).
We have demonstrated how a relatively simple wide-field FLIM plate microscope can be applied with fitting to monoexponential decay models to provide robust qualitative readouts of FRET, enabling protein interactions to be identified. This is of practical significance since fitting to monoexponential decay models is much less sensitive to system errors such as variations in the instrument response function, compared to fitting to more complex models and there is a wide range of software tools available to fit FLIM data to a monoexponential decay models on a pixel-wide basis. We also note the importance of plotting the ratio of acceptor to donor fluorescence intensities as a function of donor lifetime to elucidate the impact of relative concentrations, e.g. due to variations in transfection efficiency. For more quantitative measurements, the global fitting capabilities of software tools such as FLIMfit complement the capacity of the FLIM plate reader to acquire 100’s–1000’s of FOV and permit the population of FRETing donors to be estimated. We have shown that this can be extended to estimate the KD of protein interactions, which could be used to map systematically signalling networks, providing that the donor and acceptor fluorophore concentrations can be quantified and for this we implemented optical sectioning using a spinning Nipkow disc with our wide-field detection.
The variation in expression levels enabled us to overcome the impossibility of varying the concentrations of the interacting partners within cells in a controlled manner, as usually done when determining KD. By analysing a large number of cells resulting from segmenting hundreds of fields of view, it was possible to obtain data for a range of protein concentrations within a single experiment. We note that for the case of RASSF6, the statistics were less favourable due to relatively fewer cells surviving the transfection process - although the same conditions were applied as for the other RASSF proteins. Thus the data for RASSF6 should be interpreted with particular caution.
The values obtained for the KD are in reasonable agreement with those obtained in previous experiments utilising different biochemical techniques and report that the binding affinity is lower in the case of heterodimerisation between RASSF proteins and full length MST1 kinase compared to the heterodimerisation of RASSFs with the isolated SARAH domain from MST1. Our experiments thus illustrate the potential to apply automated high content FLIM FRET assays to screen for binding partners and estimate KD values in cells, which should offer advantage in convenience and biological relevance compared to in vitro measurements using purified proteins. To our knowledge, automated FRET-based assays to determine KD have previously been applied only in solution, either by intensity measurements42,43,44 or by time-resolved measurements of europium luminescence45. Previous reports on KD determination using FRET in cells are limited to intensity-based FRET46,47, although there is one report of using FLIM to detect FRET and calculate the KD48, but these measurements were not implemented in an automated platform to screen protein-protein interactions. Fluorescence correlation spectroscopy has also been used to determine KD49,50.
We believe that this automated FLIM FRET HCA approach provides a means to screen for protein interactions in their native context that could be scaled to screen large compound libraries. It could also be applied to map cell signalling networks. However, the quantification of the strength of specific interactions does rely on key simplifying assumptions. Below we point out some limitations of the current implementation:
The approach here using a simple donor/acceptor FRET pair is applicable to bimolecular interactions, including dimerisation, with a stoichiometry of 1:1. If more than two binding partners interact, e.g. to oligomerise or to form a complex, then FRET could take place between multiple donors and acceptors. The analysis and fitting model would have to be adapted and potentially more complex labelling schemes should be considered, as well as more sophisticated readouts including time-resolved fluorescence anisotropy or parallel measurements of acceptor as well as donor fluorescence. While this would be challenging, we note that three- or four-colour FRET schemes have been implemented using single molecule measurements51,52 or confocal/multiphoton fluorescence microscopy53,54. These approaches have been used to study conformational changes in RNA and DNA, multiple protein interactions55,56 and oligomerisation57, although KD values have not been obtained from such studies. Our current technique could be extended to read out multiple bimolecular interactions within the same or different signalling pathways using multiplexed FRET probes, as we and others have previously shown58,59.
Our approach provides information on the interaction strength between the expressed fluorescently-labelled proteins but one has to consider that, depending on the cell type, the corresponding unlabelled endogenous proteins could also be interacting with the labelled proteins and this would impact the estimates of KD50. Most cell-signalling components are expressed at relatively low levels (e.g. compared with housekeeping proteins) and for the COS7 cells used here, we expect the concentration of the endogenous proteins to be 5–10× lower than the corresponding over-expressed labelled protein. Nevertheless, further controls could be implemented in future studies that could include performing experiments in knockout cell lines for proteins of interest or depleting endogenous proteins to verify that this has no effect on KD estimates. Another approach to overcome this problem would be to label the endogenous proteins using gene editing techniques such as CRISPR/Cas and assay their interactions.
Estimations of KD based on FRET measurements using fluorescent proteins as donor and acceptor fluorophores can be subject to artefacts owing to the uncertainty in the average κ2 dipole orientation factor that arise from the fact that the fluorophores do not dynamically randomise their relative orientations during the fluorescence decay38, since the rotational correlation time of fluorescent proteins is typically large compared to the excited state lifetime60. This can lead to extended FRET efficiency probability distributions that could impact the estimation of the FRETing population fraction and therefore KD. Estimations of the FRETing population fraction can also be impacted by dark acceptor states38. These considerations impact all quantitative FRET measurements with fluorescent proteins yet such measurements are widely used and have provided a range of insights into biological processes. If these considerations can be addressed, e.g. by implementing FRET with smaller fluorophores that do result in dynamic averaging of dipole orientation, then the precision and reliability of KD estimation could be improved.
Our estimation of KD requires knowledge of the absolute concentration of donor and acceptor fluorophores, which we obtain by assuming that the quantum yield of the GFP and mCherry fluorescent proteins is the same in aqueous solution as it is in the cell and that it does not vary significantly throughout the cell. Previous measurements of EGFP report that it presents similar brightness in the cytoplasm and nucleus to what it presents in solution61.
The automated FLIM FRET assays reported in this work were undertaken with fixed cells, but could readily be applied to live cells for which similar performance is expected, in line with our previous work62. We are developing an open hardware approach to FLIM high content analysis and the latest versions of our open source software for data acquisition and analysis, together with and descriptions of hardware components is available on our website at http://www3.imperial.ac.uk/photonics/research/biomedical-imaging/openflimhca.
Materials and Methods
DNA constructs
Full length RASSF1-10 were cloned into the Gateway®-modified pEGFP-C1 vector (Clontech) to produce constructs with an N-terminus EGFP tag as described in ref. 15. MST1 (residues 1-487), MST1ΔSARAH (residues 1-431) and MST1-SARAH (residues 432-487) were cloned into pmCherry-C1 vector (Clontech) by restriction digest and ligation at the BglII and HindIII sites. The pTriEx6-MST1 K59R kinase-dead mutant construct used in the biochemical studies has been previously described in ref. 15. All RASSF5 constructs used in this study were derived from the RASSF5C isoform, which has an identical C-terminus region and SARAH domain to RASSF5A.
Mutagenesis was performed using the QuickChange® Site-Directed Mutagenesis Kit (Stratagene) following the manufacturer’s instructions. All mutant constructs were sequence verified.
Co-immunoprecipitation was performed using the Anti-c-Myc Immunoprecipitation kit (Sigma) using co-transfected cell lysates as described in ref. 15. All co-IP assays were repeated three times.
Western blots
Antibodies used for identification are anti-GFP (B2) (Santa Cruz), anti-GAPDH (Santa Cruz), anti-myc (in-house). Bands from Western blotting were quantified using ImageJ. The relative intensity of the WT control was set at 1 for each experiment and used as a reference point. The error bars are the standard deviations, p-values were calculated using the Student’s t-test and indicated as follows: p ≤ 0.05 (*), p ≤ 0.01 (**) and p ≤ 0.001 (***).
Protein modelling
Docking programs Hex (http://hexserver.loria.fr/index.php) and ClusPro (http://nrc.bu.edu/cluster) were used for rigid body docking and to run simulations of the heterodimers consisting of the monomer structures of MST1 (PDB: 2JO8) and the RASSF SARAH homology models from ref. 15. Each run generated 100 or more solutions that were ranked by cluster sizes and the top two ranked models were selected and analysed using naccess (http://www.bioinf.manchester.ac.uk/naccess/).
Cells
COS7 cells (ECACC) were grown in DMEM supplemented with 10% fetal calf serum, 2 mM glutamine, 1 mM sodium pyruvate and 1% penicillin-streptomycin and were used for all experiments. All fluorescent constructs were transfected via electroporation. Typically, 1–1.5 × 106 cells were suspended in 100 μl homemade electroporation buffer (140 mM KCl, 8 mM NaCl, 0.88 mM MgSO4, 2.97 mM Na2HPO4, 1.06 mM NaH2PO4 and 0.5% (w/v) bovine serumalbumin (pH = 7.4), filtered through a 0.2 μm membrane for sterilisation). 4.5 μg of plasmids in different combinations (indicated in the figures) were added to this suspension. Electroporation was performed using an Amaxa NucleofectorTM II (Lonza, Switzerland) using the manufacturer’s program for COS7 cells. Cells were then seeded in a 96-well plate (Greiner Bio-One) at a density of 30 000 cells/well and fixed the following day using 4% paraformaldehyde for 20 minutes at room temperature, washed 3 times in phosphate buffer saline (PBS) and imaged in PBS.
Automated FLIM multiwell plate reader
The instrument shown in Fig. 2 was constructed around a motorised Olympus IX 81-Z microscope with ZDC autofocus. The pulsed excitation radiation (60 MHz repetition rate) is selected from the output of a supercontinuum laser (SC 400-6, Fianium Ltd, UK) using band pass filters (Semrock) arranged in a motorised filter wheel.
For wide-field imaging the excitation is directed via a single-mode optical fibre to the back illumination port of the microscope after passing through a rotating diffuser wheel and relayed to the focal plane of the microscope to realise Köhler illumination. The samples arrayed in a 96-well plate were mounted on a motorised x-y stage (Märzhäuser Wetzlar GmbH, Germany) and imaged using a 20 × objective (Olympus UPlanFl 20×/0.5). Appropriate dichroic mirrors and emission filters in the motorised filter cube wheel (GFP: excitation 472/30 nm, dichroic 495 nm, emission 520/35 nm; mCherry: excitation 545/30 nm, dichroic 570 nm, emission 610/75 nm) provided automated selection of spectral channels. The emitted fluorescence light was imaged via the left-hand port of the microscope to a gated optical intensifier (GOI) (Kentech Instruments Ltd., UK) and the resulting gated images at the phosphorus screen were imaged to a cooled CCD camera (Orca ER II, Hamamatsu, Japan). The GOI gating voltage signal is synchronised and delayed with respect to the laser excitation pulses under computer control. For the work reported here, the GOI gate width was set to 1 ns and typically time-gated images of EGFP fluorescence were acquired at 5 different delays after excitation while only one time gated image (at the beginning of the decay) was acquired for the mCherry emission. The integration time of the CCD camera was set to 1–2 s per gate delay for EGFP and 5–6 s for the mCherry image acquisition such that the dynamic range of the CCD was utilised.
For the optically sectioned FLIM acquisitions used to provide the data for the KD calculations, the instrument was configured to incorporate a spinning Nipkow disk unit (CSU-X1 Yokogawa Electric Corporation, Japan), as described in refs 63, 64, 65, with a 40× air objective (Olympus, LUCPLFLN 40) with an NA of 0.6. The pulsed excitation was directed via a polarisation-preserving single mode optical fibre to the input of the spinning Nipkow disk unit and the fluorescence image was relayed onto the GOI where the time-gated images were acquired as for the wide-field configuration.
In addition to the time-gated FLIM FRET data, a FLIM acquisition of a scattering sample detected at the excitation wavelength was acquired to provide an instrument response function (IRF) for the data analysis. Time-gated FLIM was also applied to a well containing only PBS in order to determine the time varying background.
FLIM data acquisition and analysis
The instrument is controlled using a programme written in LabVIEW (National Instruments, USA). This controls the automatic movement of the stage, the autofocusing of each field of view, the automatic change of the excitation filters, of the filters and dichroics in the filter cube wheel, the objective lens change, the GOI gating and the CCD camera acquisition. A “prefind” scan was implemented to image the well plate using fluorescence intensity to identify and localise cells and to acquire donor and acceptor intensity images. Specific fields of view in various wells were selected for subsequent FLIM after applying an intensity threshold. FLIM data analysis was performed using the custom written open source software, FLIMfit, described in detail in ref. 35 and freely available at www.openmicroscopy.org/site/products/partner/flimfit. For the work reported here we utilised the following capabilities of FLIMfit: cell segmentation based on donor and/or acceptor intensity; calculation of average fluorescence intensity of donor and acceptor per cell; fitting the donor fluorescence intensity decays to monoexponential and to double exponential decay models (including instrument response function (IRF) and time-varying background correction); global fitting of donor fluorescence intensity decays across multiple fields of view and wells; visualisation of FLIM data (including rendering of plate maps showing mean EGFP decay times per well and images of one field of view per well). To utilise the relatively small changes in donor lifetime that we have obtained in the FLIM FRET assays reported here from fits to double exponential decay models, it is critical to minimise fluctuations in the IRF and to account for any residual variation. This was realised by acquiring FLIM data of a reference dye solution (rhodamine 6G) in some of the plate wells and fitting the measured decay data to a monoexponential model in order to precisely determine the relative excitation time (i.e. the start of the decay profile, t0), for each plate. This information was combined with the measurement of a scattering sample to construct the IRF that is convolved with the exponential decay model to provide the function to which the experimental FLIM data is fitted. Graphs of lifetime and intensity ratio parameters were plotted in Origin 8 (OriginLab, USA).
Additional Information
How to cite this article: Margineanu, A. et al. Screening for protein-protein interactions using Förster resonance energy transfer (FRET) and fluorescence lifetime imaging microscopy (FLIM). Sci. Rep. 6, 28186; doi: 10.1038/srep28186 (2016).
Supplementary Material
Acknowledgments
The authors gratefully acknowledge funding from the UK Biotechnology and Biological Sciences Research Council (BBSRC BB/E003621/1 and BB/H00713X/1), the UK Engineering and Physical Sciences Research Council (EPSRC Pathways to Impact grant) and the Wellcome Trust (WT 095931/Z/11/Z). DK and SW acknowledge PhD studentships from the Institute of Chemical Biology EPSRC funded Doctoral Training Centre.
Footnotes
Author Contributions A.M., J.J.C., D.J.K. and S.K. performed the experiments, A.M., D.J.K., S.C.W., C.W.D., M.K. and P.M.W.F. contributed to the analysis and interpretation of the data, S.K., D.J.K., S.C.W., C.W.D. and P.M.W.F. designed the instrumentation, D.F. performed structural protein models and calculations, A.M., C.W.D., M.K. and P.M.W.F. designed the study and A.M., J.J.C., D.J.K., S.C.W., C.W.D., M.K. and P.M.W.F. contributed to writing the manuscript.
References
- Eggeling C., Willig K. I., Sahl S. J. & Hell S. W. Lens-based fluorescence nanoscopy. Quart. Rev. Biophys. 48, 178–243 (2015). [DOI] [PubMed] [Google Scholar]
- Patterson G., Davidson M., Manley S. & Lippincott-Schwartz J. Superresolution imaging using single-molecule localization. Annu. Rev. Phys. Chem. 61, 345–367 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zanella F., Lorens J. B. & Link W. High content screening: seeing is believing. Trends Biotechnol. 28, 237–245 (2010). [DOI] [PubMed] [Google Scholar]
- Jares-Erijman E. A. & Jovin T. M. Imaging molecular interactions in living cells by FRET microscopy. Curr. Opin. Chem. Biol. 10, 409–416 (2006). [DOI] [PubMed] [Google Scholar]
- Hoppe A., Christensen K. & Swanson J. A. Fluorescence resonance energy transfer-based stoichiometry in living cells. Biophys. J. 83, 3652–3664 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen H., Puhl H. L. 3rd, Koushik S. V., Vogel S. S. & Ikeda S. R. Measurement of FRET efficiency and ratio of donor to acceptor concentration in living cells. Biophys, J. 91, L39–41 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bader A. N., Hofman E. G., van Bergen en Henegouwen P. M. P. & Gerritsen H. C. Imaging of protein cluster sizes by means of confocal time-gated fluorescence anisotropy microscopy. Opt. Express 15, 6934–6945 (2007). [DOI] [PubMed] [Google Scholar]
- Warren S. C., Margineanu A., Katan M., Dunsby C. & French P. M. W. Homo-FRET based biosensors and their application to multiplexed imaging of signalling events in live cells. Int. J. Mol. Sci. 16, 14695–14716 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matthews D. R. et al. A multi-functional imaging approach to high-content protein interaction screening. PLoS ONE 7, e33231 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fluorescence Lifetime Spectroscopy and Imaging: Principles and Applications Eds. Marcu L., French P. M. W., Elson D. S., CRC Press, pp. 1–322, (2015). [Google Scholar]
- Kumar S. et al. FLIM FRET technology for drug discovery: automated multiwell-plate high-content analysis, multiplexed readouts and application in situ. ChemPhysChem 12, 609–626 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Köllner M. & Wolfrum J. How many photons are necessary for fluorescence-lifetime measurements? Chem. Phys. Lett. 200, 199–203 (1992). [Google Scholar]
- Digman M., Caiolfa V. R., Zamai M. & Gratton E. The phasor approach to fluorescence lifetime imaging analysis. Biophys. J. 94, L14–16 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eichorst J. P., Teng K. W. & Clegg R. M. Polar plot representation of time-resolved fluorescence, in Fluorescence Spectroscopy and Microscopy: Methods and Protocols, Methods in Molecular Biology Eds. Engelborghs Y., Visser A. J. W. G. vol. 1076, Springer Science+Business Media, pp. 97–112 (2014). [DOI] [PubMed] [Google Scholar]
- Chan J. J. et al. Comparative analysis of interactions of RASSF1-10. Adv. Biol. Regul. 53, 190–201 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheel H. & Hofmann K. A novel inter action motif, SARAH, connects three classes of tumor suppressor. Curr. Biol. 13, R899–R900 (2003). [DOI] [PubMed] [Google Scholar]
- Oh H. J. et al. Role of the tumor suppressor RASSF1A in Mst1-mediated apoptosis. Cancer Res. 66, 2562–2569 (2006). [DOI] [PubMed] [Google Scholar]
- Praskova M., Khoklatchev A., Ortiz-Vega S. & Avruch J. Regulation of the MST1 kinase by autophosphorylation, by the growth inhibitory proteins, RASSF1 and NORE1, and by Ras. Biochem J. 381, 453–462 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khokhlatchev A. et al. Identification of a novel Ras-regulated proapoptotic pathway. Curr. Biol. 12, 253–265 (2002). [DOI] [PubMed] [Google Scholar]
- Guo C. et al. RASSF1A is part of a complex similar to the Drosophila Hippo/Salvador/Lats tumor-suppressor network. Curr. Biol. 17, 700–705 (2007). [DOI] [PubMed] [Google Scholar]
- Ikeda M. et al. Ras-association domain family protein 6 induces apoptosis via both caspase-dependent and caspase-independent pathways. Exp. Cell Res. 313, 1484–1495 (2007). [DOI] [PubMed] [Google Scholar]
- Ikeda M. et al. Hippo pathway-dependent and -independent roles of RASSF6. Sci. Signal 2, ra59 (2009). [DOI] [PubMed] [Google Scholar]
- Del Re D. P. et al. Proapoptotic Rassf1A/Mst1 signaling in cardiac fibroblasts is protective against pressure overload in mice. J. Clin. Invest. 120, 3555–67 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park J. et al. Tumor suppressor ras association domain family 5 (RASSF5/NORE1) mediates death receptor ligand-induced apoptosis. J. Biol. Chem. 285, 35029–38 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sherwood V., Recino A., Jeffries A., Ward A. & Chalmers A. D. The N-terminal RASSF family: a new group of Ras-association-domain containing proteins, with emerging links to cancer formation. Biochem. J. 425, 303–311 (2010). [DOI] [PubMed] [Google Scholar]
- Hwang E. et al. Structural insight into dimeric interaction of the SARAH domains from Mst1 and RASSF family proteins in the apoptosis pathway. Proc. Nat. Acad. Sci. USA 104, 9236–9241 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ni L. et al. Structural basis for autoactivation of human Mst2 kinase and its regulation by RASSF5. Structure 21, 1757–1768 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hwang E. et al. Structural basis of the heterodimerization of the MST and RASSF SARAH domains in the Hippo signalling pathway. Acta Crystallogr. D Biol. Crystallogr. 70, 1944–1953 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miertzschke M. et al. Characterization of interactions of adapter protein RAPL/Nore1B with RAP GTPases and their role in T cell migration. J. Biol. Chem. 282, 30629–30634 (2007). [DOI] [PubMed] [Google Scholar]
- Stieglitz B. et al. Novel type of Ras effector interaction established between tumour suppressor NORE1A and Ras switch II. EMBO J. 27, 1995–2005 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez-Viciana P., Sabatier C. & McCormick F. Signaling specificity by Ras family GTPases is determined by the full spectrum of effectors they regulate. Mol. Cell. Biol. 24, 4943–54 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Avruch J., Praskova M., Ortiz-Vega S., Liu M. & Zhang X. F. Nore1 and RASSF1 regulation of cell proliferation and of the MST1/2 kinases. Methods Enzymol. 407, 290–310 (2006). [DOI] [PubMed] [Google Scholar]
- Ciani B. et al. Molecular basis of coiled-coil oligomerization-state specificity. Proc. Natl. Acad. Sci. USA 107, 19850–19855 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moutevelis E. & Woolfson D. N. A periodic table of coiled-coil protein structures. J. Mol. Biol. 385, 726–32 (2009). [DOI] [PubMed] [Google Scholar]
- Warren S. C. et al. Rapid global fitting of large fluorescence lifetime imaging microscopy datasets. PLoS ONE 8, e70687 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raab M., Smith X., Matthess Y., Strebhardt K. & Rudd C. E. SKAP1 protein PH domain determines RapL membrane localization and Rap1 protein complex formation for T cell receptor (TCR) activation of LFA-1. J. Biol. Chem. 286, 29663–29670 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barlow D. J. & Thornton J. M. Helix geometry in proteins. J. Mol. Biol. 201, 601–619 (1988). [DOI] [PubMed] [Google Scholar]
- Vogel S. S., Nguyen T. A., van der Meer B. W. & Blank P. S. The impact of heterogeneity and dark acceptor states on FRET: Implications for using fluorescent protein donors and acceptors. PLoS ONE 7, e49593 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koturenkiene A. Molecular basis for apoptotic Ras signalling through Nore1-MST1 multi-protein complex. PhD thesis, University Bochum (2008).
- Constantinescu Aruxandei D., Makbul C., Koturenkiene A., Ludemann M. B. & Herrmann C. Dimerization-induced folding of MST1 SARAH and the influence of the intrinsically unstructured inhibitory domain: low thermodynamic stability of monomer. Biochemistry 50, 10990–1000 (2011). [DOI] [PubMed] [Google Scholar]
- Makbul C. et al. Structural and Thermodynamic Characterization of Nore1-SARAH: A small, helical module important in signal transduction networks. Biochemistry 52, 1045–1054 (2013). [DOI] [PubMed] [Google Scholar]
- Song Y., Madahar V. & Liao J. Development of FRET assay into quantitative and high-throughput screening technology platforms for protein–protein interactions. Ann. Biomed. Eng. 39, 1224–1234 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song Y., Rodgers V. G. J., Schultz J. S. & Liao J. Protein interaction affinity determination by quantitative FRET technology. Biotechnol. Bioeng. 109, 2875–2883 (2012). [DOI] [PubMed] [Google Scholar]
- Chakraborty S., Hu S.-Y., Wu S.-H., Karmenyan A. & Chiou A. The interaction affinity between vascular cell adhesion molecule-1 (VCAM-1) and very late antigen-4 (VLA-4) analyzed by quantitative FRET. PLoS One 10, e0121399 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du Y. et al. A time-resolved fluorescence resonance energy transfer assay for high-throughput screening of 14-3-3 protein–protein interaction inhibitors. Assay Drug Dev. Technol. 11, 367–381 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen H., Puhl H. L. III & Ikeda S. R. Estimating protein-protein interaction affinity in living cells using quantitative Förster resonance energy transfer measurements. J. Biomed. Optics 12, 054011 (2007). [DOI] [PubMed] [Google Scholar]
- Mehta K., Hoppe A. D., Kainkaryam R., Woolf P. J. & Linderman J. J. A computational approach to inferring cellular protein binding affinities from quantitative fluorescence resonance energy transfer imaging. Proteomics 9, 5371–5383 (2009). [DOI] [PubMed] [Google Scholar]
- Day R. N. Measuring protein interactions using Förster resonance energy transfer and fluorescence lifetime imaging microscopy. Methods 66, 200–207 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hom E. F. Y. & Verkman A. S. Analysis of coupled bimolecular reaction kinetics and diffusion by two-color fluorescence correlation spectroscopy: enhanced resolution of kinetics by resonance energy transfer. Biophys. J. 83, 533–546 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foo Y. H., Naredi-Rainer N., Lamb D. C., Ahmed S. & Wohland T. Factors affecting the quantification of biomolecular interactions by fluorescence cross-correlation spectroscopy. Biophys. J. 102, 1174–1183 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hohng S., Joo C. & Ha T. Single-Molecule three-color FRET. Biophys. J. 87, 1328–1337 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim H. et al. Protein-guided RNA dynamics during early ribosome assembly. Nature 506, 334–340 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee J. et al. Single-molecule four-color FRET. Angew. Chem. Int. Ed. Engl. 49, 9922–9925 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao M., Huang R. & Peng L. Quantitative multi-color FRET measurements by Fourier lifetime excitation-emission matrix. Optics Express 20, 26806–26827 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galperin E., Verkhusha V. V. & Sorkin A. Three-chromophore FRET microscopy to analyze multiprotein interactions in living cells. Nat. Methods 1, 209–217 (2004). [DOI] [PubMed] [Google Scholar]
- Wallrabe H., Sun Y., Fang X., Periasamy A. & Bloom G. Three-Color FRET expands the ability to quantify the interactions of several proteins involved in actin filament nucleation. Proc. SPIE 822, 82260J (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scott B. L. & Hoppe A. D. Three-dimensional reconstruction of three way FRET microscopy improves imaging of multiple protein-protein interactions. PLoS One 11, e0152401 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grant D. M. et al. Multiplexed FRET to image multiple signaling events in live cells. Biophys. J. 95, L69–L71 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao M., Wan X., Li Y., Zhou W. & Peng L. Multiplexed 3D FRET imaging in deep tissue of live embryos. Scientific Rep. 5, 13991 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarkar P., Vogel S. S., Gryczynski I. & Gryczynski Z. Photophysical properties of Cerulean and Venus fluorescent proteins. J. Biomed. Opt. 14, 034047 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y., Müller J. D., Ruan Q. & Gratton E. Molecular Brightness characterization of EGFP in vivo by fluorescence fluctuation spectroscopy. Biophys. J. 82, 133–144 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelly D. J. et al. An automated multiwell plate reading FLIM microscope for live cell autofluorescence lifetime assays. J. Innov. Opt. Health Sci. 7, 1450025 (2014). [Google Scholar]
- Grant D. M. et al. High speed optically sectioned fluorescence lifetime imaging permits study of live cell signaling events. Opt. Express 15, 15656–15673 (2007). [DOI] [PubMed] [Google Scholar]
- Talbot C. B. et al. High speed unsupervised fluorescence lifetime imaging confocal multiwell plate reader for high content analysis. J Biophotonics 1, 514–521 (2008). [DOI] [PubMed] [Google Scholar]
- Alibhai D. et al. Automated fluorescence lifetime imaging plate reader and its application to Förster resonant energy transfer readout of Gag protein aggregation. J. Biophotonics 6, 398–408 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.