

Abstract
Becker muscular dystrophy (BMD) is an X-linked recessive disorder involving mutations of the dystrophin gene. Cardiac involvement in BMD has been described and cardiomyopathy represents the number one cause of death in these patients. In this paper, the pathophysiology, clinical evaluations and management of cardiomyopathy in patients with BMD will be discussed.
Keywords: Becker muscular dystrophy, Cardiomyopathy, X-linked recessive disorder, Dystrophin
Core tip: Becker muscular dystrophy (BMD) is an X-linked recessive disorder involving mutations of the dystrophin gene. This condition is rare but not uncommon. However, there are limited articles on this topic. Patients with BMD can present with mental retardation and diffuse muscular dystrophy. Cardiomyopathy is the number one cause of death in BMD. This paper aims to provide a comprehensive overview of BMD pathophysiology and management. The paper will discuss both the established treatments as well as exciting new research on gene therapy.
INTRODUCTION
Becker muscular dystrophy (BMD), first described by Doctor Peter Emil Becker in 1955, is an X-linked recessive disorder involving mutations of the dystrophin gene. The dystrophin gene located on chromosome Xp21.1, codes for a large protein that serves as a scaffolding protein in both skeletal and cardiac muscle. In BMD, the mutations allow for expression of truncated but functional dystrophin or a reduced amount of dystrophin protein. BMD is characterized by progressive skeletal muscle weakness. It affects one in 18450 males with the prevalence of at least 2.4/100000[1]. Researchers started correlating BMD with cardiac involvement in 1960s[2]. BMD patients may live until the fifth or sixth decade of life and cardiomyopathy represents the number one cause of death in these patients[1,3].
CARDIOMYOPATHY IN BMD
The frequency of cardiac involvement in BMD is 60% to 75%[4]. The average age of onset of cardiac involvement is 28.7 ± 7.1 years[5]. Severe dilated cardiomyopathy (DCM) in patients less than 20-year-old is rare. The primary pathology of cardiomyopathy in BMD is thought to be due to diffuse degeneration and fibrosis in the ventricles especially the inferolateral region and the conduction tissue[4]. Myocardial damage preferentially in the inferolateral wall is presumed to be due to exaggerated mechanical stress and not due to limited distribution of dystrophin in this region.
There are different cardiac manifestations in BMD ranging from very subtle signs to severe cardiomyopathy requiring cardiac transplant[6]. Most of BMD patients have asymptomatic cardiac involvement. Only up to one third of patients develop DCM with symptoms of heart failure. Studies have shown that there appears to be no correlation between skeletal muscle involvement and the severity or time of onset of myocardial involvement[1,5]. The majority of BMD patients have skeletal muscle impairment before the onset of cardiac symptoms. However, there are rare cases in which cardiomyopathy may represent the initial manifestation. Ruiz-Cano et al[7] described a patient who was diagnosed with DCM and subsequently needed a heart transplant in less than 1 year. Eleven years after heart transplant, this patient developed lower extremities muscle weakness and was diagnosed with BMD based on muscle biopsy.
PATHOPHYSIOLOGY OF CARDIOMYOPATHY
The detailed molecular mechanism of the development of cardiomyopathy in BMD has not been well established. Currently, the mechanism is thought to be secondary to increase in intracellular calcium influx. Elevation of intracellular calcium results in mitochondrial deregulation, protease calpain-mediated necrosis and NF-κB activation. This leads to degradation of troponin I compromises the contraction of the cardiomyocyte and eventually results in cardiomyocyte death. The exact mechanism causing an increased intracellular calcium influx is subject to a debate. One proposed mechanism is that dysfunction of the sarcolemmal stretch activated channels causes an increased influx of calcium. Others believe that the absence of dystrophin causes cells to have a fragile membrane that is leaky and thus allowing intracellular calcium influx[8,9] (Figure 1).
Figure 1.
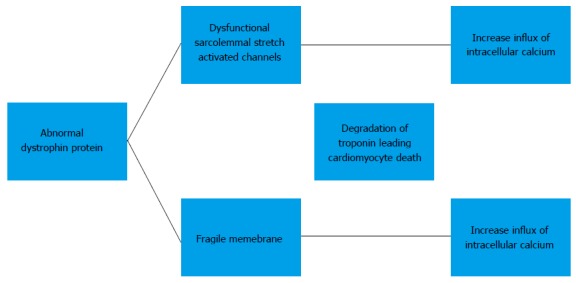
Proposed pathways leading to myocyte death.
GENOTYPE AND CARDIOMYOPATHY CORRELATION
The wide phenotypic variation in the severity of cardiomyopathy in BMD patients may be due to different mutations in the dystrophin gene. Deletions affecting the amino terminal domain or mutations resulting in disruption of spectin repeat in the rod domain of the dystrophin protein, and mutations involving exons 12 and 14 to 17 or 31 to 42 are associated with early onset of cardiomyopathy[1,10]. People with deletion mutations of exons 2 to 9 or exons 45 to 49 are at risk of developing DCM in the second and third decades of life respectively[11]. Specifically, deletion of the intron located between exon 48 and 49 is associated with cardiomyopathy. Deletion around exon 1 damages the expression and or function of dystrophin selectively in cardiac muscle. On the other hand, no cardiac abnormality is seen in patients with deletions on the 5’ side[12]. Further studies are needed to unify these findings. However, these findings suggest that BMD patients with certain mutations may have significant cardiac involvement and need more careful and regular cardiac evaluation.
Previous studies have shown that there is no correlation between the extent of cardiac and skeletal muscle disease in patients with BMD. A possible, straightforward explanation is that different genetic mutations lead to different phenotypes. However, recent findings suggest that there may be another explanation. Cardiac dystrophin may interact with proteins that are different from those of skeletal dystrophin. Johnson et al[13] using antibody-based immunoprecipitation, discovered that there is a different interaction between members of the dystrophin-associated protein complex (neuronal nitric oxide synthase and β2-syntrophin) with cardiac and skeletal dystrophin. Neuronal nitric oxide synthase does not interact with cardiac dystrophin while β2-syntrophin interacts with cardiac but not skeletal dystrophin. They also found that there is a unique interaction between cardiac dystrophin and Cavin-1 (polymerase I and transcript release factor), Ahnak1 (neuroblast differentiation-associated protein), Cypher (a PDZ-LIM domain Z-line protein), and CRYAB (crystalline, alpha B). The significance of these interactions remains to be determined.
CARDIOMYOPATHY IN FEMALE CARRIERS
Female carriers of dystrophin mutations may develop cardiomyopathy even without skeletal muscle disease. There are reports of electrocardiographic and echocardiographic evidence of cardiomyopathy among BMD carriers. However, the significance of cardiomyopathy in female carriers has been a source of debate, since it does not appear to affect life expectancy[14]. Hence, the benefit of routine cardiac surveillance in BMD carriers is unclear. Currently, there is no consensus on the need for regular cardiac surveillance in BMD carriers.
EKG FINDINGS
Typical EKG changes in BMD include an R:S ratio ≥ 1 in lead V1, tall R waves in the right precordial leads, deep Q waves in the inferolateral leads, short PR, and longer QTc interval. There are also conduction abnormalities including incomplete and complete right bundle branch block, incomplete and complete left bundle branch block, and infra-hisian block[1,2,12].
DEVICE THERAPY
BMD patients with cardiomyopathy can develop atrial and ventricular arrhythmias. The degree of arrhythmia is proportional to the severity of left ventricular dysfunction. The benefit of an implantable cardioverter-defibrillator (ICD) in BMD patients has not been established. Therefore, the same criteria is used for prophylactic ICD implantation in BMD patients as in other forms of nonischemic DCM[4,15]. Resynchronization therapy with biventricular pacing may also be considered to reduce heart failure symptoms[15-18].
CARDIAC TRANSPLANT
Although there were case reports dated back to the 1990s of patients with BMD who successfully underwent cardiac transplantation[19], inherited myopathies remained as a relative contraindication for heart transplant for a number of reasons. First, immunosuppression after transplant may cause progression of muscle impairment. Secondly, respiratory muscle dysfunction may make it difficult to wean off the ventilator post-operatively.
Wu et al[20] challenged these traditional concerns in a study comparing patients with muscular dystrophy to a matched cohort of patients with idiopathic DCM after heart transplant. The results showed that survival, rate of infection, cardiac rejection, and transplant vasculopathy post heart transplant were similar between the two groups. The limitation of this study was its small sample size and the possibility of selection bias. Nonetheless, the findings of this study suggest that BMD patients who have only mild muscular disability and no involvement of respiratory muscles may successfully undergo cardiac transplantation[7,20]. Patients with BMD may have a small additional risk of rhabdomyolysis and malignant hyperthermia reaction[21].
Cardiac rehabilitation after heart transplant in a patient with BMD has also been shown in a case report to improve cardiac function[6].
FUTURE THERAPEUTIC PERSPECTIVES
There are ongoing investigations looking at the introduction of a modified functional dystrophin gene via gene transfer as well as molecular correction of the mutated dystrophin gene. There are adeno-associated virus capsids that target cardiomyocytes specifically which allow gene expression in the heart even when the capsid is delivered via a peripheral vein. There are two types of synthetic dystrophin genes: Mini-dystrophin and micro-dystrophin. In mini-dystrophin, part of the rod domain is removed, while in micro-dystrophin, a significant portion of the rod and the C-terminal domain are removed. Mini-dystrophin transferred in a mouse model showed normalization of EKG and improved myocardial fibrosis and ejection fraction. Similarly, micro-dystrophin was able to restore normal heart rate, PR and QT interval, and cardiomyocyte integrity. The challenge of this gene therapy is the immune rejection of the viral vector or the newly expressed dystrophin protein[17].
Another gene therapy is exon skipping. In this method, antisense oligonucleotides (AONs) are used to remove mutated exons resulting in a truncated but functional protein. Applying this method in a mouse model with mutated dystrophin showed favorable echocardiographic changes[17,22-27]. Mendell et al[28] showed that AONs increased functional dystropin-positive fibers in an open-labeled human study. Unfortunately, subsequent Phase III trails failed to show clinical benefits[29]. However, Goyenvalle et al[25] recently showed a new class of AONs made up of tricyclo-DNA (tcDNA) might hold promise for future therapy. Using a mouse model, they showed tcDNA increases dystrophin expression in skeletal and cardiac muscles and improvement in cardio-respiratory function.
Lastly, there is sarcoplasmic reticulum calcium ATPase 2a (SERCA2a) gene therapy. The role of SERCA2a is to pump cytoplasmic calcium into the sarcoplasmic reticulum to restore calcium homeostatis and prevent cell death. Shin et al[26] found that increasing SERCA2a gene expression in mice using adeno-associated virus serotype-9 Lead to EKG improvement. This finding is especially encouraging because a Phase II trail by Jessup et al[27] showed that SERCA2a gene therapy improved heart failure symptoms, increased functional status and improved left ventricular end-systolic and end-diastolic volume in patients with end-stage heart failure.
IMAGING FINDINGS
The echocardiogram shows a dilated left ventricle with wall motion abnormality especially in the posterior and lateral wall. There is also impaired diastolic function even in those with normal systolic function. Mitral and tricuspid regurgitation are common findings[3].
Cardiovascular magnetic resonance imaging (CMR) is beginning to be accepted as a more sensitive modality than echocardiography in providing information on ventricular size and function, and detecting regional myocardial deformation. Contrast enhanced CMR (ceCMR), using late gadolinium enhancement as an indication of myocardial damage, allows for detection of even small areas of myocardial deformation[24]. Using ceCMR, Yilmaz et al[1] showed that myocardial damage in BMD begins in the subepicardium of the inferolateral wall. However, Soslow et al[22] showed recently that spatial mapping of the longitudinal relaxation time constant (T1) CMR might be superior to ceCMR in detecting early myocardial fibrosis. As such, more research is warranted to ascertain the best modality for detecting early fibrosis in BMD.
Previously, it has been recommended that BMD patients undergo a screening ECG and echocardiogram at the time of diagnosis and every five years thereafter if the findings are normal. However, as CMR becomes widely accepted, it is recommended it be initiated at diagnosis and then at least every two years even in the case of normal findings. This rigorous screening procedure is proposed with the hope of early cardiomyopathy detection so that effective treatment can be initiated to slow the progression of cardiac dysfunction (Table 1).
Table 1.
Summary of current diagnostic modalities
| Imaging modalities | Description |
| Echocardiogram | Evaluating for wall motion abnormality and cardiac function |
| Contrast enhanced cardiovascular magnetic resonance | Evaluating for early tissue fibrosis |
| Spatial mapping cardiovascular magnetic resonance | Evaluating for early tissue fibrosis |
OTHER ASSESSMENT METHODS
There are other methods that can either support the diagnosis or monitor left ventricular function. Chest X-ray may show cardiomegaly, pleural effusion, and pulmonary congestion. Cardiac troponin I is a marker for myocardial damage. Brain natriuretic peptide, released following ventricular overload and increased wall stress, has been proposed as a marker for monitoring of left ventricular dysfunction[30].
PHARMACOTHERAPY
Angiotensin-converting enzyme inhibitors (ACEIs) have been shown to delay the progression of LV dysfunction, improve left ventricular function, and confer a mortality benefit. However, there is no universal guideline on the best time for the initiation of ACEI in patients with BMD. Suggestions have been made for ACEI to be given when left ventricular ejection fraction is less than 55%[31,32].
β blockers are beneficial in patients with DCM. Therefore, β blockers may have positive effects on BMD patients with cardiomyopathy. A Japanese study comparing patients with different types of muscular dystrophies on ACEI alone vs ACEI plus β blocker showed that the combination of ACEI and β blocker provided a significant improvement on left ventricular fractional shortening[33]. Therefore, β blockers are recommended to be used in accordance with current heart failure guidelines. Clinically, hypotension may limit the use of a β-blocker.
Corticosteroids have been shown to improve muscle strength and function. Numerous studies implicated the role of steroid in prolonging ambulation and stabilization of pulmonary function. However, corticosteroids have many adverse effects, which include Cushing’s, hypertension, osteoporosis and hyperglycemia[9].
There has been no large trial examining the mortality benefit of angiotensin receptor blockers (ARBs) in BMD patients with cardiomyopathy. It is possible that ARBs are efficacious based on studies showing their benefit in other causes of heart failure. Diuretics and digoxin can be used as adjuncts for symptom reduction although no mortality benefit has been demonstrated. Aldosterone blockade can be added for patients with NYHA Class III or IV who are already on optimal doses of an ACEI and β blocker[15]. Calcium channel blockers such as diltiazem, flunarizine, and nifedipine have not been shown to be beneficial[34].
Current data suggested that there might be a role of eplerenone in treating BMD. Raman et al[23] showed that eplerenone in addition to ACEIs slow down the progression of left ventricular systolic function decline. The exact mechanism is unknown. But evidence from ceCMR suggested that it is likely secondary to eplerenone anti-inflammatory effect.
Ivabradine is a medication that selectively blocks the I(f) current in sinoatrial cells and slows heart rate. Unlike β blockers, ivabradine does not cause hypotension. Ivabradine may reverse cardiac remodeling thus providing a mortality benefit. A case report on Ivabradine in BMD cardiomyopathy has shown benefits. Randomized controlled trials are needed for further evaluation. Currently, this medication is not available in the United States[16].
CONCLUSION
There are still many unknowns regarding BMD cardiomyopathy. Imaging techniques need to be optimized further to allow for early diagnosis of CM. Different pharmacological and gene therapies currently being developed offer hope for patients with BMD cardiomyopathy.
Footnotes
Conflict-of-interest statement: The authors have no conflict of interest to report.
Open-Access: This article is an open-access article which was selected by an in-house editor and fully peer-reviewed by external reviewers. It is distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/
Peer-review started: December 28, 2015
First decision: February 2, 2016
Article in press: April 22, 2016
P- Reviewer: Amiya E, De Ponti R, Kettering K, Rodriguez-Cruz M, Sakabe K, Satoh H, Said SAM S- Editor: Qiu S L- Editor: A E- Editor: Jiao XK
References
- 1.Yilmaz A, Sechtem U. Cardiac involvement in muscular dystrophy: advances in diagnosis and therapy. Heart. 2012;98:420–429. doi: 10.1136/heartjnl-2011-300254. [DOI] [PubMed] [Google Scholar]
- 2.Steare SE, Dubowitz V, Benatar A. Subclinical cardiomyopathy in Becker muscular dystrophy. Br Heart J. 1992;68:304–308. doi: 10.1136/hrt.68.9.304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Connuck DM, Sleeper LA, Colan SD, Cox GF, Towbin JA, Lowe AM, Wilkinson JD, Orav EJ, Cuniberti L, Salbert BA, et al. Characteristics and outcomes of cardiomyopathy in children with Duchenne or Becker muscular dystrophy: a comparative study from the Pediatric Cardiomyopathy Registry. Am Heart J. 2008;155:998–1005. doi: 10.1016/j.ahj.2008.01.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Rajdev A, Groh WJ. Arrhythmias in the muscular dystrophies. Card Electrophysiol Clin. 2015;7:303–308. doi: 10.1016/j.ccep.2015.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Groh WJ. Arrhythmias in the muscular dystrophies. Heart Rhythm. 2012;9:1890–1895. doi: 10.1016/j.hrthm.2012.06.038. [DOI] [PubMed] [Google Scholar]
- 6.Srinivasan R, Hornyak JE, Badenhop DT, Koch LG. Cardiac rehabilitation after heart transplantation in a patient with Becker’s muscular dystrophy: a case report. Arch Phys Med Rehabil. 2005;86:2059–2061. doi: 10.1016/j.apmr.2005.03.036. [DOI] [PubMed] [Google Scholar]
- 7.Ruiz-Cano MJ, Delgado JF, Jiménez C, Jiménez S, Cea-Calvo L, Sánchez V, Escribano P, Gómez MA, Gil-Fraguas L, Sáenz de la Calzada C. Successful heart transplantation in patients with inherited myopathies associated with end-stage cardiomyopathy. Transplant Proc. 2003;35:1513–1515. doi: 10.1016/s0041-1345(03)00515-3. [DOI] [PubMed] [Google Scholar]
- 8.Kaspar RW, Allen HD, Montanaro F. Current understanding and management of dilated cardiomyopathy in Duchenne and Becker muscular dystrophy. J Am Acad Nurse Pract. 2009;21:241–249. doi: 10.1111/j.1745-7599.2009.00404.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.van Westering TL, Betts CA, Wood MJ. Current understanding of molecular pathology and treatment of cardiomyopathy in duchenne muscular dystrophy. Molecules. 2015;20:8823–8855. doi: 10.3390/molecules20058823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jefferies JL, Eidem BW, Belmont JW, Craigen WJ, Ware SM, Fernbach SD, Neish SR, Smith EO, Towbin JA. Genetic predictors and remodeling of dilated cardiomyopathy in muscular dystrophy. Circulation. 2005;112:2799–2804. doi: 10.1161/CIRCULATIONAHA.104.528281. [DOI] [PubMed] [Google Scholar]
- 11.Kaspar RW, Allen HD, Ray WC, Alvarez CE, Kissel JT, Pestronk A, Weiss RB, Flanigan KM, Mendell JR, Montanaro F. Analysis of dystrophin deletion mutations predicts age of cardiomyopathy onset in becker muscular dystrophy. Circ Cardiovasc Genet. 2009;2:544–551. doi: 10.1161/CIRCGENETICS.109.867242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Melacini P, Fanin M, Danieli GA, Fasoli G, Villanova C, Angelini C, Vitiello L, Miorelli M, Buja GF, Mostacciuolo ML. Cardiac involvement in Becker muscular dystrophy. J Am Coll Cardiol. 1993;22:1927–1934. doi: 10.1016/0735-1097(93)90781-u. [DOI] [PubMed] [Google Scholar]
- 13.Johnson EK, Zhang L, Adams ME, Phillips A, Freitas MA, Froehner SC, Green-Church KB, Montanaro F. Proteomic analysis reveals new cardiac-specific dystrophin-associated proteins. PLoS One. 2012;7:e43515. doi: 10.1371/journal.pone.0043515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Holloway SM, Wilcox DE, Wilcox A, Dean JC, Berg JN, Goudie DR, Denvir MA, Porteous ME. Life expectancy and death from cardiomyopathy amongst carriers of Duchenne and Becker muscular dystrophy in Scotland. Heart. 2008;94:633–636. doi: 10.1136/hrt.2007.125948. [DOI] [PubMed] [Google Scholar]
- 15.Romfh A, McNally EM. Cardiac assessment in duchenne and becker muscular dystrophies. Curr Heart Fail Rep. 2010;7:212–218. doi: 10.1007/s11897-010-0028-2. [DOI] [PubMed] [Google Scholar]
- 16.Finsterer J, Stöllberger C, Berger E. Beneficial effect of ivabradine in dilated cardiomyopathy from Becker muscular dystrophy. Herz. 2012;37:702–705. doi: 10.1007/s00059-012-3643-8. [DOI] [PubMed] [Google Scholar]
- 17.Lai Y, Duan D. Progress in gene therapy of dystrophic heart disease. Gene Ther. 2012;19:678–685. doi: 10.1038/gt.2012.10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Stöllberger C, Finsterer J. Left ventricular synchronization by biventricular pacing in Becker muscular dystrophy as assessed by tissue Doppler imaging. Heart Lung. 2005;34:317–320. doi: 10.1016/j.hrtlng.2005.03.003. [DOI] [PubMed] [Google Scholar]
- 19.Donofrio PD, Challa VR, Hackshaw BT, Mills SA, Cordell AR. Cardiac transplantation in a patient with muscular dystrophy and cardiomyopathy. Arch Neurol. 1989;46:705–707. doi: 10.1001/archneur.1989.00520420127038. [DOI] [PubMed] [Google Scholar]
- 20.Wu RS, Gupta S, Brown RN, Yancy CW, Wald JW, Kaiser P, Kirklin NM, Patel PC, Markham DW, Drazner MH, et al. Clinical outcomes after cardiac transplantation in muscular dystrophy patients. J Heart Lung Transplant. 2010;29:432–438. doi: 10.1016/j.healun.2009.08.030. [DOI] [PubMed] [Google Scholar]
- 21.Quinlivan RM, Dubowitz V. Cardiac transplantation in Becker muscular dystrophy. Neuromuscul Disord. 1992;2:165–167. doi: 10.1016/0960-8966(92)90002-n. [DOI] [PubMed] [Google Scholar]
- 22.Soslow JH, Damon BM, Saville BR, Lu Z, Burnette WB, Lawson MA, Parra DA, Sawyer DB, Markham LW. Evaluation of post-contrast myocardial t1 in duchenne muscular dystrophy using cardiac magnetic resonance imaging. Pediatr Cardiol. 2015;36:49–56. doi: 10.1007/s00246-014-0963-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Raman SV, Hor KN, Mazur W, Halnon NJ, Kissel JT, He X, Tran T, Smart S, McCarthy B, Taylor MD, et al. Eplerenone for early cardiomyopathy in Duchenne muscular dystrophy: a randomised, double-blind, placebo-controlled trial. Lancet Neurol. 2015;14:153–161. doi: 10.1016/S1474-4422(14)70318-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Dittrich S, Tuerk M, Haaker G, Greim V, Buchholz A, Burkhardt B, Fujak A, Trollmann R, Schmid A, Schroeder R. Cardiomyopathy in Duchenne Muscular Dystrophy: Current Value of Clinical, Electrophysiological and Imaging Findings in Children and Teenagers. Klin Padiatr. 2015;227:225–231. doi: 10.1055/s-0034-1398689. [DOI] [PubMed] [Google Scholar]
- 25.Goyenvalle A, Griffith G, Babbs A, El Andaloussi S, Ezzat K, Avril A, Dugovic B, Chaussenot R, Ferry A, Voit T, et al. Functional correction in mouse models of muscular dystrophy using exon-skipping tricyclo-DNA oligomers. Nat Med. 2015;21:270–275. doi: 10.1038/nm.3765. [DOI] [PubMed] [Google Scholar]
- 26.Shin JH, Bostick B, Yue Y, Hajjar R, Duan D. SERCA2a gene transfer improves electrocardiographic performance in aged mdx mice. J Transl Med. 2011;9:132. doi: 10.1186/1479-5876-9-132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Jessup M, Greenberg B, Mancini D, Cappola T, Pauly DF, Jaski B, Yaroshinsky A, Zsebo KM, Dittrich H, Hajjar RJ. Calcium Upregulation by Percutaneous Administration of Gene Therapy in Cardiac Disease (CUPID): a phase 2 trial of intracoronary gene therapy of sarcoplasmic reticulum Ca2+-ATPase in patients with advanced heart failure. Circulation. 2011;124:304–313. doi: 10.1161/CIRCULATIONAHA.111.022889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mendell JR, Rodino-Klapac LR, Sahenk Z, Roush K, Bird L, Lowes LP, Alfano L, Gomez AM, Lewis S, Kota J, et al. Eteplirsen for the treatment of Duchenne muscular dystrophy. Ann Neurol. 2013;74:637–647. doi: 10.1002/ana.23982. [DOI] [PubMed] [Google Scholar]
- 29.Lu QL, Cirak S, Partridge T. What Can We Learn From Clinical Trials of Exon Skipping for DMD? Mol Ther Nucleic Acids. 2014;3:e152. doi: 10.1038/mtna.2014.6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mori K, Manabe T, Nii M, Hayabuchi Y, Kuroda Y, Tatara K. Plasma levels of natriuretic peptide and echocardiographic parameters in patients with Duchenne’s progressive muscular dystrophy. Pediatr Cardiol. 2002;23:160–166. doi: 10.1007/s00246-001-0040-0. [DOI] [PubMed] [Google Scholar]
- 31.Bosser G, Lucron H, Lethor JP, Burger G, Beltramo F, Marie PY, Marçon F. Evidence of early impairments in both right and left ventricular inotropic reserves in children with Duchenne’s muscular dystrophy. Am J Cardiol. 2004;93:724–727. doi: 10.1016/j.amjcard.2003.12.005. [DOI] [PubMed] [Google Scholar]
- 32.Duboc D, Meune C, Lerebours G, Devaux JY, Vaksmann G, Bécane HM. Effect of perindopril on the onset and progression of left ventricular dysfunction in Duchenne muscular dystrophy. J Am Coll Cardiol. 2005;45:855–857. doi: 10.1016/j.jacc.2004.09.078. [DOI] [PubMed] [Google Scholar]
- 33.Kajimoto H, Ishigaki K, Okumura K, Tomimatsu H, Nakazawa M, Saito K, Osawa M, Nakanishi T. Beta-blocker therapy for cardiac dysfunction in patients with muscular dystrophy. Circ J. 2006;70:991–994. doi: 10.1253/circj.70.991. [DOI] [PubMed] [Google Scholar]
- 34.Toifl K, Presterl E, Graninger W. [Ineffectiveness of diltiazem in Duchenne muscular dystrophy: a placebo-controlled double-blind study] Wien Klin Wochenschr. 1991;103:232–235. [PubMed] [Google Scholar]