

Abstract
AIM: To determine the outcome of orthotopic heart transplantation (OHT) in immunoglobulin light chain (AL) amyloidosis.
METHODS: The medical records of patients with AL who underwent orthotopic heart transplantation at the Mayo Clinic in Rochester Minnesota from 1992 to 2011 were reviewed. Patients met at least one of the following at: New York Heart Association class IV heart failure, ventricular thickness > 15 mm, ejection fraction < 40%. Selection guidelines for heart transplant included age < 60 years, absence of multiple myeloma and significant extra-cardiac organ involvement. Baseline characteristics including age, gender, organ involvement, and New York Heart Association functional class were recorded. Laboratory data, waiting time until heart transplant, and type of treatment of the underlying plasma cell disorder were recorded. Survival from the time of OHT was calculated using Kaplan-Meier survival curves. Survival of patients undergoing OHT for AL was compared to that of non-amyloid patients undergoing OHT during the same time period.
RESULTS: Twenty-three patients (median age 53 years) with AL received OHT. There were no deaths in the immediate perioperative period. Twenty patients have died post OHT. For the entire cohort, the median overall survival was 3.5 years (95%CI: 1.2, 8.2 years). The 1-year survival post OHT was 77%, the 2-year survival 65%, and the 5-year survival 43%. The 5-year survival for non-amyloid patients undergoing OHT during the same era was 85%. Progressive amyloidosis contributed to death in twelve patients. Of those without evidence of progressive amyloidosis, the cause of death included complications of autologous hematopoietic stem cell transplantation for 3 patients, post-transplant lymphoproliferative disorder for 2 patients; and for the remaining one death was related to each of the following causes: acute rejection; cardiac vasculopathy; metastatic melanoma; myelodysplastic syndrome; and unknown. Eight patients had rejection at a median of 1.8 mo post OHT (range 0.4 to 4.9 mo); only one patient died of rejection. Median survival of seven patients who achieved a complete hematologic response to either chemotherapy or autologous hematopoietic stem cell transplantation was 10.8 years.
CONCLUSION: Our data demonstrate that long term survival after heart transplant is feasible in AL patients with limited extra-cardiac involvement who achieve complete hematologic response.
Keywords: Heart transplantation, Autologous stem cell transplantation, Amyloidosis, Chemotherapy, Heart failure
Core tip: Heart failure due to immunoglobulin light chain (AL) amyloidosis is a devastating disease with poor prognosis. Orthotopic heart transplantation (OHT) is controversial. Twenty-three patients with AL amyloid underwent OHT at our institution over a twenty-year period. Median survival was 3.5 years following OHT. Median survival of seven patients who achieved a complete hematologic response to treatment for AL was almost 11 years. This study demonstrates that long term survival after heart transplant is feasible in AL patients with limited extra-cardiac involvement who achieve complete hematologic response.
INTRODUCTION
Immunoglobulin light chain (AL) amyloidosis is a systemic plasma cell disorder, characterized by the production of a kappa or lambda monoclonal light chain by a clonal population of bone marrow plasma cells[1]. The monoclonal light chain misfolds into an insoluble beta-pleated sheet conformation. The aberrant protein subsequently accumulates in tissues, leading to organ dysfunction.
Cardiac involvement occurs in 50% of patients with systemic AL amyloidosis and is the most important risk factor for adverse prognosis and death[2,3]. Amyloid fibrils infiltrate the extracellular space of the valves, atria and ventricles, as well as the perivascular spaces, resulting in biventricular wall thickening without ventricular dilation[4]. As a result, atrial pressure increases, and atrial dilation occurs despite amyloid infiltration. Clinical features of cardiac amyloidosis include restrictive physiology, heart failure, dysrhythmias, and sudden cardiac death[1,4,5]. In addition, there is evidence suggesting that the immunoglobulin free light chains are toxic to the myocardium[6].
Patients with advanced heart failure due to AL have an extremely poor prognosis and often do not survive long enough to benefit from therapy for amyloidosis. Orthotopic heart transplantation (OHT) in AL has been limited due to the risk of disease progression in other organs and recurrence of amyloid deposition in the transplanted heart[7,8]. Although earlier studies suggested inferior outcomes of OHT for compared with non-amyloid indications[8,9], more recent reports have found survival similar to other forms of heart failure[10]. Controversy regarding the role of OHT in AL remains[11,12] and many centers consider amyloidosis to be a contraindication to OHT. The purpose of the current study was to determine the long term outcome and predictors of survival in a large single-center cohort of patients undergoing OHT for AL.
MATERIALS AND METHODS
Study population
Patients were identified from our institutional database of cardiac transplantation recipients. The diagnosis of amyloidosis was confirmed by demonstrating Congo red positivity in tissue samples. AL amyloid was confirmed by laser dissection mass spectrometry in all but two patients, who had typing performed by immunohistochemistry. During the period from May 31, 1992 to December 31, 2011, 3117 patients with AL amyloidosis were seen at the Mayo Clinic. Twenty-one percent (668 patients) had overt congestive heart failure. One hundred and thirty-seven were referred for OHT evaluation, and 77 patients completed their evaluation for OHT. Of those completing the evaluation, 33 were deemed ineligible for OHT. Reasons for ineligibility included extensive amyloid in 29 and one instance of each of the following: Coexisting myeloma; coexisting lymphoma; improving cardiac status due to chemotherapy; and lack of financial approval. Forty-four patients (7% of patients with overt heart failure) completed evaluation and were listed for OHT, but only 23 were transplanted. Twenty-one were removed from the listing for the following reasons: Death (n = 12); further medical decline (n = 5); patient refusal (n = 2); myeloma (n = 1); and transplant elsewhere (n = 1). The median time to de-listing was 48 d (interquartile range 14, 111 d; range 0-341 d).
Throughout the 20-year period, all patients met at least one of the following at time of listing: New York Heart Association class IV heart failure, ventricular thickness > 15 mm, ejection fraction < 40%. In 1998, additional selection guidelines were added: Age < 60 years; combination of the urine light chain, serum monoclonal protein and bone marrow plasmacytosis that does not infer the presence of multiple myeloma or related disorders including low bone marrow plasma cell labeling index; absence of renal involvement as defined by a 24-h urine total protein excretion of < 500 mg and creatinine clearance > 50 mL/min per square meter unless combined renal transplant planned; absence of liver involvement - if elevation of alkaline phosphatase was thought to be due to heart failure, liver biopsy was to be done to exclude interstitial amyloid deposits. The presence of vascular deposits in a biopsy of the rectum, fat or viscera was not an exclusionary criterion. Assignment of organ involvement was according to the consensus criteria from the 10th International Symposium on Amyloid and Amyloidosis[13]. The modified body mass index (mBMI) was calculated as BMI multiplied by serum albumin level in gram per litre. For most patients the values used for listing and pre-operative BMI (and mBMI) were the same given the proximity of listing to OHT.
The autologous hematopoietic stem cell transplantation (AHSCT) protocol is as previously described, and 11 of the patients have been previously reported[14]. Demographic, clinical and laboratory data were collected from the Mayo Clinic Transplant Center database, the Robert A Kyle Dysproteinemia database, and all medical records were reviewed. Because most of these patients were treated before era of the serum immunoglobulin free light chain assay, the ability to assign a hematologic response was limited. The determination of hematologic response was a hybrid of the two consensus guidelines. If patients had serum immunoglobulin free light chains measured (n = 9), then the 2012 consensus response criteria were applied[15]; otherwise, the 10th consensus response criteria from the International Symposium on Amyloid and Amyloidosis were applied[13]. Two patients had measurable M-spikes, 8 had positive immunofixation of the serum or urine that could be followed, and 4 either had none of the aforementioned detected (or testing not performed prior to starting chemotherapy).
Immunosuppression
Post OHT, all patients received standard therapy for immunosuppression, according to our institutional protocol at the time of transplant. The first twenty-one patients received OKT3, cyclosporine, prednisone, and azathioprine or mycophenate mofetil. A gradual taper of cyclosporine was done over the first year to baseline immunosuppression. Surveillance endomyocardial biopsies to monitor for rejection were used to help guide prednisone taper. The last two patients received prednisone, mycophenate mofetil, and tacrolimus.
Statistical analysis
Medical records for the patients undergoing OHT for AL amyloidosis were reviewed. Survival from the time of OHT was calculated using Kaplan-Meier survival curves. Comparison of survival curves was done with the log-rank test. Baseline variables were tested for their impact on overall survival using Cox proportional modeling. The database was closed to follow up as of March 18, 2015. All statistics were calculated using JMP 10.0.0 (SAS, Carey, North Carolina).
RESULTS
Twenty-three patients with AL amyloidosis underwent OHT (Table 1). Fifty-two percent were female (n = 12), and all but two were Caucasian. Twenty-one patients had isolated cardiac involvement at baseline clinical evaluation; one patient (OHT#14) had mild peripheral nerve and gastrointestinal involvement, and one (OHT #15) had peripheral nerve involvement. Twenty-two patients had had a clonal lambda plasma cell disorder; one had a kappa clone. Three patients had renal transplantation, one simultaneous with the OHT and the others at 23 and 53 mo post OHT.
Table 1.
Demographics and orthotopic heart transplantation outcomes
| AL-OHT | M/F | Age at OHT | List to OHT (d) | Year OHT | PO FU (yr) | Major outcomes |
| 8 | F | 52 | 62 | 1997 | 16.5 | Alive, doing well |
| 22 | F | 53 | 1160 | 2011 | 3.9 | Alive, PTLD in remission, VGPR on bortezomib |
| 23 | M | 58 | 13 | 2011 | 3.1 | Alive, doing well |
| 1 | M | 45 | 86 | 1992 | 14.1 | Died, progressive amyloid; renal transplantation 53 mo post OHT |
| 17 | F | 51 | 94 | 2003 | 10.8 | Died, renal failure, debility; hematologic relapse and renal amyloid |
| 9 | M | 56 | 16 | 1998 | 8.6 | Died, metastatic melanoma |
| 2 | M | 44 | 126 | 1993 | 8.4 | Died, PTLD, sepsis, progressive amyloidosis |
| 12 | F | 57 | 44 | 1999 | 8.2 | Died, cardiogenic shock secondary to cardiac amyloid, required dialysis for renal amyloid post ASHCT#2 |
| 3 | M | 56 | 14 | 1994 | 7.5 | Died, progressive amyloid |
| 14 | M | 33 | 30 | 1999 | 6.3 | Died, cardiac allograft vasculopathy |
| 16 | F | 53 | 33 | 2000 | 5.4 | Died, progressive GI amyloid and stroke; renal transplant 23 mo post-op |
| 7 | M | 61 | 415 | 1997 | 3.5 | Died, progressive amyloid autonomic and peripheral neuropathy |
| 18 | M | 56 | 33 | 2004 | 3.1 | Died, complications of myelodysplastic syndrome |
| 5 | F | 47 | 68 | 1995 | 2.6 | Died, PTLD, progressive multifocal leukoencephalopathy |
| 10 | F | 54 | 5 | 1998 | 2.2 | Died, progressive amyloid |
| 19 | M | 62 | 18 | 2005 | 2.1 | Died, progressive amyloid peripheral neuropathy and GI involvement, recurrent pneumonia |
| 4 | M | 49 | 86 | 1994 | 1.2 | Died, progressive amyloid |
| 21 | F | 51 | 29 | 2007 | 1.2 | Died, progressive GI amyloid, right heart failure, renal failure, steroid myopathy vs amyloid neuropathy |
| 13 | F | 51 | 103 | 1999 | 0.9 | Died, sepsis, multiorgan failure after AHSCT |
| 11 | F | 48 | 31 | 1999 | 0.7 | Died, disseminated fungal infection after AHSCT |
| 15 | F | 60 | 33 | 1999 | 0.6 | Died, progressive amyloid peripheral and autonomic neuropathy |
| 20 | F | 52 | 33 | 2006 | 0.6 | Died, progressive amyloid and overwhelming infection |
| 6 | M | 56 | 99 | 1996 | 0.04 | Died, refractory rejection; had combined renal and cardiac transplant |
| Median (IQR) | 53 (33, 62) | 33 (29, 94) |
The first three rows are alive. M: Male; F: Female; PO FU: Post-operative follow-up; PTLD: Post-transplant lymphoproliferative disorder; GI: Gastrointestinal; VGPR: Very good partial response; IQR: Interquartile range.
Twenty patients have died post OHT (Table 1 and Figure 1A). The baseline disease burden is outlined in Table 2. For the entire cohort, the median overall survival was 3.5 years (95%CI: 1.2, 8.2 years). The 1-year survival post OHT was 77%, the 2-year survival 65%, and the 5-year survival 43% (Figure 1A). Progressive amyloidosis contributed to death in twelve patients. Of those without evidence of progressive amyloidosis, the cause of death included post-AHSCT complications for 3 patients, post-transplant lymphoproliferative disorder for 2 patients; and for the remaining there was one death related to each of the following causes: Acute rejection; cardiac vasculopathy; metastatic melanoma; myelodysplastic syndrome; and unknown. Eight patients had rejection at a median of 1.8 mo post OHT (range 0.4 to 4.9 mo); only one patient died of rejection).
Figure 1.
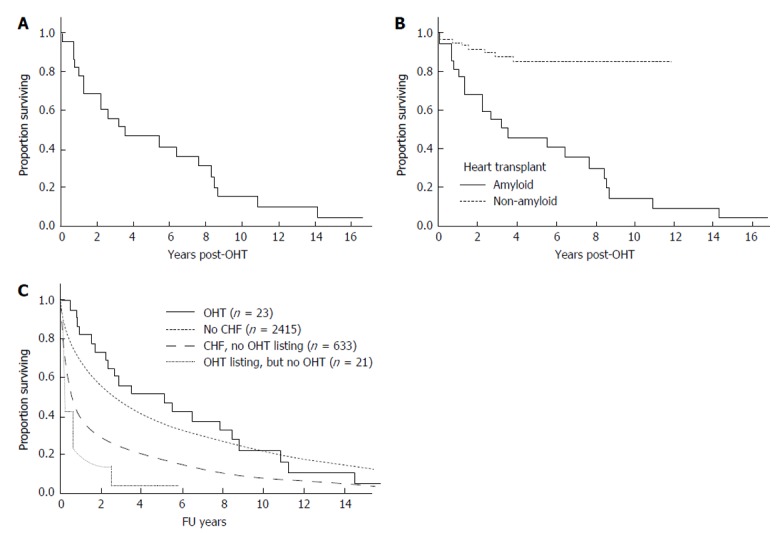
Overall survival. A: Overall survival from orthotopic heart transplant; B: Overall survival comparing OHT for AL amyloidosis to OHT from 1992 to 2011 for non-amyloid indications; C: Comparison of survival with non OHT subgroups. OHT: Orthotopic heart transplantation; CHF: Congestive heart failure; AL: Immunoglobulin light chain.
Table 2.
Baseline disease burden
| AL-OHT | IFE positive | g/dL | dFLC (mg/L) | Tx mBMI, kg g/L m2 | Creatinine (mg/dL) | Alkaline phosphatase (U/L) | IVS (mm) | EF (%) | BM PC (%) |
| 8 | Yes | 688 | 0.9 | 115 | 15 | 33 | 12 | ||
| 22 | Yes | 1.6 | 298 | 977 | 0.8 | 60 | 14 | 55 | 18 |
| 23 | NA | 0 | 1059 | 951 | 1.5 | 150 | 15 | 50 | 10 |
| 1 | Yes | 1120 | 1.4 | 112 | 16 | 40 | 8 | ||
| 17 | Yes | 0 | 70 | 913 | 0.9 | 302 | 16 | 64 | 12 |
| 9 | Yes | 0 | 271 | 822 | 1.7 | 334 | 12 | 78 | 8 |
| 2 | Yes | 1191 | 1.1 | 130 | 15 | 60 | 5 | ||
| 12 | Yes | 0 | 742 | 1.2 | 312 | 17 | 30 | 13 | |
| 3 | NA | 729 | 1.2 | 18 | 56 | 20 | |||
| 14 | Yes | 620 | 0.9 | 207 | 16 | 20 | 12 | ||
| 16 | No | 0 | 402 | 1.1 | 145 | 12 | 53 | 15 | |
| 7 | No | 1.2 | 132 | 15 | 57 | 6 | |||
| 18 | Yes | 0 | 245 | 935 | 1.2 | 90 | 18 | 56 | 17 |
| 5 | No | 599 | 0.9 | 428 | 22 | 40 | 4 | ||
| 10 | Yes | 0 | 629 | 0.7 | 110 | 16 | 44 | 5 | |
| 19 | Yes | 0 | 304 | 744 | 1.1 | 90 | 16 | 24 | 12 |
| 4 | NA | 693 | 1.1 | 14 | 20 | NA | |||
| 21 | Yes | 1.9 | 138 | 632 | 0.9 | 70 | 13 | 43 | 13 |
| 13 | Yes | 0.14 | 779 | 1.0 | 312 | 16 | 40 | 5 | |
| 11 | Yes | 0 | 279 | 513 | 0.7 | 156 | 21 | 30 | 12 |
| 15 | Yes | 788 | 1.2 | 118 | 17 | 50 | 9 | ||
| 20 | Yes | 0 | 87 | 865 | 1.5 | 112 | 14 | 40 | 7 |
| 6 | Yes | 894 | 1.1 | 229 | 13 | 35 | 19 | ||
| Median (IQR) | 27 (11-30) | 761 (631, 919) | 1.1 (0.9, 1.2) | 132 (111, 267) | 15 (14, 16) | 40 (34, 56) | 12 (7, 13) |
The first three rows are alive. IFE: Immunofixation of serum and/or urine; dFLC: Difference between involved and uninvolved immunoglobulin free light chains; Tx: Transplant; mBMI: Modified body mass index [albumin × weight/(height)2]; IVS: Interventricular septum; EF: Ejection fraction; BM PC: Bone marrow plasma cells; NA: Not available; IQR: Interquartile range.
Figure 1B shows a comparison of this cohort to patients undergoing isolated OHT at our center for non- amyloid indications, where 1 year overall survival is 94.8% ± 2.1% and 5-year survival 85.2% ± 4.4%. Given the small sample size, it is difficult to assess baseline factors that might predict for early death. Notably, no patient died within the immediate OHT peri-operative period. On univariate analysis none of the following factors were significant risk factors for poor overall survival: Age, gender, BMI or mBMI (at listing or at transplant), time from listing to OHT, serum creatinine, or bone marrow plasmacytosis.
Figure 1C shows the comparison of patients with AL who underwent OHT with AL patients without and with overt heart failure who did not undergo OHT, and with those who were listed but did undergo OHT.
Three patients had no therapy (chemotherapy or AHSCT) for amyloidosis and only four received treatment prior to OHT (Table 3). Reasons for no chemotherapy/AHSCT were: Inability to harvest stem cells for planned AHSCT; rejection two weeks after OHT; and death 7 mo after OHT. One patient received chemotherapy only prior to OHT, 4 received chemotherapy pre- and post-OHT, and 15 only post-OHT. The non-AHSCT first line therapies are shown in Table 3. Therapies beyond first line therapies included bortezomib, AHSCT, melphalan with corticosteroids, or an IMiD with dexamethasone; one patient received doxorubicin.
Table 3.
Chemotherapy and response
| AL-OHT | Rx relative to OHT | First amyloid directed therapy (Rx) | Response to1st Rx | Lines of Rx |
| 8 | Only Rx post-OHT | AHSCT1 | CR | 1 |
| 22 | Rx pre- and post-OHT | Mel-Dex | CR | ≥ 2 |
| 23 | Rx pre- and post-OHT | Bortezomib-Dex3 | VGPR | ≥ 2 |
| 1 | Only Rx post-OHT | Mel-Pred | CR | ≥ 2 |
| 17 | Only Rx post-OHT | AHSCT1 | CR | ≥ 2 |
| 9 | Only Rx post-OHT | AHSCT1 | CR | ≥ 2 |
| 2 | Only Rx pre-OHT | Mel-Pred | IFE positive | 1 |
| 12 | Only Rx post-OHT | AHSCT2 | PR | ≥ 2 |
| 3 | Only Rx post-OHT | AHSCT1 | CR | 1 |
| 14 | Only Rx post-OHT | AHSCT2 | IFE positive | 1 |
| 16 | Only Rx post-OHT | AHSCT1 | VGPR | ≥ 2 |
| 7 | Rx pre- and post-OHT | VBMCP | IFE positive | ≥ 2 |
| 18 | Only Rx post-OHT | AHSCT1 | VGPR | 1 |
| 5 | Only Rx post-OHT | Mel-Pred | NA | 1 |
| 10 | No treatment | None | NA | 0 |
| 19 | Only Rx post-OHT | AHSCT2 | PR | 1 |
| 4 | Rx pre- and post-OHT | Mel-Pred | IFE positive | ≥ 2 |
| 21 | Only Rx post-OHT | AHSCT2 | No response | ≥ 2 |
| 13 | Only Rx post-OHT | AHSCT1 | IFE positive | 1 |
| 11 | Only Rx post-OHT | AHSCT2 | IFE positive | 1 |
| 15 | No treatment | None | NA | 0 |
| 20 | Only Rx post-OHT | Dex | CR | 1 |
| 6 | No treatment | None | NA | 0 |
Melphalan conditioning 200 mg/m2;
Melphalan conditioning 140 mg/m2 in all but OHT #12 who got 150 mg/m2;
Patient had AHSCT as second line and received Melphalan conditioning 200 mg/m2. Amyloid directed therapy: AHSCT: Autologous hematopoietic stem cell transplant; Mel: Oral melphalan; Pred: Prednisone; Bortez: Bortezomib; Dex: Dexamethasone; VBMCP: Vincristine, BCNU, melphalan, cytoxan, prednisone; NA: Not available; CR: Complete hematologic response; PR: Partial response; VGPR: Very good partial response; IFE: Immunofixation.
After their first line therapy, seven patients achieved a complete hematologic response (CR), 3 a very good partial response, 2 a partial response, and 11 remained immunofixation positive or were not assessed before death (Table 3). As shown in Figure 2A, patients achieving a CR fared much better than those who did not, achieving a median survival of 10.8 years.
Figure 2.
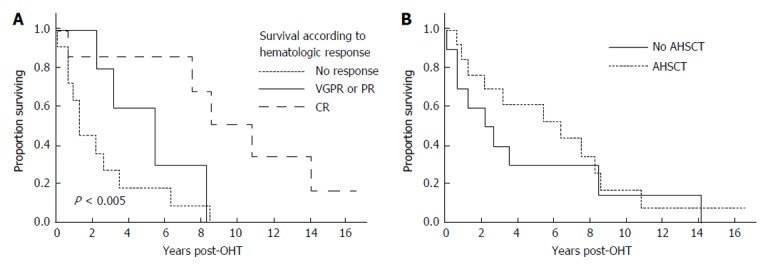
Influence of chemotherapy on overall survival. A: Overall survival (OS) based on hematologic response. The median OS for the 11 non-responders/non-evaluable patients was 1.2 years. The median OS for the 5 patients achieved VGPR or PR was 5.4 years. The median OS for the 7 patients achieving CR was 10.8 years; B: OS based on whether or not patient received AHSCT. The median OS for the 10 patients not undergoing AHSCT was 2.4 years; the median OS For the 13 patients undergoing AHSCT was 6.3 years. VGPR: Very good partial response; CR: Complete hematologic response; AHSCT: Autologous hematopoietic stem cell transplantation.
Thirteen patients underwent AHSCT, performed at a median of 8 mo (range 3-24 mo) post OHT with one patient having a second AHSCT 82 mo post OHT. In two patients AHSCT was planned but could not be performed due to inability to harvest stem cells. The median survival of those undergoing AHSCT was 6.3 years (95%CI: 1.2, 8.6 years). Figure 2B demonstrates survival outcomes of those who received AHSCT vs those who did not. Among the patients who underwent AHSCT, 8 received full dose melphalan conditioning (200 mg/m2), and 5 received attenuated doses. Two of the eight patients receiving full dose melphalan conditioning died within three years post AHSCT, and one is alive 33 mo at last follow-up. In contrast 3 of the 5 receiving attenuated melphalan conditioning died within 3-year post AHSCT. For the 2 who died within 100 d of AHSCT, the cause of death was disseminated fungal infection in one and sepsis leading to multi-organ failure in the other. Four of the AHSCT patients achieved a CR, 5 a very good partial response (PR) or PR, and 4 no significant response or not assessable.
DISCUSSION
Given the limited supply of donor hearts, OHT in AL amyloidosis remains controversial due to the risk of recurrent amyloidosis in the graft or progression of other organ involvement. This long term follow-up study reports the largest single center experience of OHT in AL. Our results support the use of OHT in AL amyloidosis patients with predominant cardiac involvement and no evidence of myeloma, especially if they have achieved (or are able to achieve) a complete hematologic response. Although the median overall survival of our cohort was only 3.5 years, those patients who achieved a complete hematologic response had a remarkably good overall median survival of more than 10 years.
Superior survival is reported in patients with AL amyloidosis and cardiac involvement who undergo OHT compared to patients who do not[14,16]. In a report of 14 patients from the United Kingdom, median overall survival was 7.5 years from OHT; in 8 patients who underwent AHSCT and OHT, survival was increased to 9.7 years[17]. These data are confounded by selection biases, but the fact remains that 30%-40% of patients with AL amyloidosis die within the first 6 mo of their diagnosis due to cardiac causes[18] making consideration of aggressive strategies imperative.
Most series of heart transplantation in AL have reasonable 1-year survival rates (Table 4). In our study there was no perioperative mortality. The major causes of death in our and other series are infection and progressive amyloidosis. If performed without chemotherapeutic support, 5-year survival is just 20%[19], Improved survival rates have been seen in patients who undergo AHSCT, with 1- and 5-year survival of 82% and 65% respectively in our earlier report[14]. Recent reports suggest improved short term outcomes with advances in chemotherapy and AHSCT, with the Stanford series reporting 1 year survival of 100%[20].
Table 4.
Orthotopic heart transplantation in patients with amyloidosis
| Ref. | n | AHSCT | Outcomes |
| Current series | 23 | 13 | 1-yr OS 77% |
| 5-yr OS 43% | |||
| MGH[10] | 18 | 14 | 5-yr OS 60% |
| United Kingdom 2004[19] | 171 | 3 | 1-yr 59% |
| 5-yr approximately 37% | |||
| United Kingdom 2010[17] | 141 | 8 | 1-yr OS 86% |
| 5-yr OS 45% | |||
| Spanish registry[28] | 13 | 3 | 1-yr OS 43% |
| 5-yr OS 36% | |||
| German group[29] | 12 | 5 | 1-yr OS 83% |
| 3-yr OS 83% | |||
| ISHLT Registry[8,30] | 102 | None | 1-yr 88% |
| 4-yr 38% | |||
| Maurer[16] | 10 | 8 | 1-yr 90% |
| Stanford[20] | 9 | 5 | 1-yr 100% |
| French registry[31] | 8 | 3 | 1-yr 89% |
Unclear how much overlap between these two groups. Intervals for Dubrey series was 1982-2002 and for Sattianayagam series, interval was 1984-2004, but there was no reference of which patients had been previously reported;
At least 8 were AL; unclear what other 2 were. ISHT: International Society for Heart Transplant; OS: Overall survival; AHSCT: Autologous hematopoietic stem cell transplantation.
In the MGH series of 18 AL patients undergoing OHT approximately 60% of patients were alive at 6 years[10], and, in contrast to earlier studies[8,9,21], was similar to that of non-amyloid patients. Although overall survival in our study was reduced compared with patients transplanted for non-amyloid indications, our series includes many early era patients who did not receive the benefit of current therapy for amyloidosis. Nevertheless, the long term survival of the patients in our series who achieved complete hematologic response was remarkably good.
Selecting patients with primarily cardiac involvement in AL is challenging. Subclinical extra-cardiac organ involvement may progress post heart transplant to clinically important disease. Perivascular intestinal amyloid is common and not viewed as a barrier to cardiac transplantation. However, in our experience, patients with significant mucosal intestinal involvement do poorly and are often not able to tolerate aggressive treatment for AL. Clearly not all patients with cardiac involvement will require heart transplant; there are patients with significant cardiac involvement who can have cardiac improvement with effective chemotherapy alone[22-25]. Perhaps cardiac biomarkers like ST-2 may lend insight to those with irreparable damage despite effective chemotherapy[26].
“Better selection” also means choosing those patients in whom the underlying plasma cell clone can be controlled, since in our study and others effective chemotherapy has resulted in the best outcomes post-OHT[10,16,20]. Most of the patients in our series did not receive chemotherapy prior to OHT because the only chemotherapies available at the time of their diagnosis were oral melphalan and prednisone and high dose melphalan with AHSCT. Newer treatment options[27], especially bortezomib containing regimens, are less myelosuppressive, making pre-OHT therapy a possibility. Furthermore, the improvement in chemotherapeutic regimens makes hematologic response more likely in the current era.
Achieving a hematologic response pre-OHT is not a simple matter. Time is of the essence in these patients. In our experience and others, approximately 40% of AL patients listed do not undergo OHT either due to death or deterioration[10]. This seems to be related to both delayed diagnosis, as well as inability to support these patients with traditional heart failure therapy and devices. In the MGH series, patients with amyloidosis had a mortality hazard ratio of 4.7 (95%CI: 2.8, 11.8) as compared to non-amyloidosis patients while on the waiting list[10]. The only predictive factor of survival to OHT in that study was BMI - patients with lower BMI fared better than those with higher BMI, although this was not confirmed in our study.
The number of AL amyloid patients transplanted at our institution in recent years has declined. This reduction is multifactorial, and reflects patients receiving earlier and more effective bone marrow directed treatment, more rigorous selection, the availability of OHT for AL at other medical centers, and our own reluctance to offer OHT after some discouraging outcomes. However, the excellent long term survival in this study of patients achieving CR , coupled with markedly improved short term survival recently reported[20] have prompted renewed enthusiasm for OHT in AL in highly selected patients.
We recognize that our study is limited by being a small series of highly selected patients, lacking currently available cardiac biomarkers and modern markers of clonal burden and access to current treatment regimens. Despite these limitations, in carefully selected patients, long term survival can be achieved. Moving forward the challenge will continue to be the selection of the appropriate patients. The patients likely to derive the most benefit are those who: (1) have plasma cells that are responsive to chemotherapy; (2) have clinically significant involvement of the heart only; and (3) are not demonstrating significant cardiac response despite effective chemotherapy. The current lack of effective short term cardiac support and the rapidly progressive nature of AL cardiac amyloidosis warrant consideration of revised guidelines for organ allocation in these patients.
ACKNOWLEDGMENTS
Robert A Kyle Hematologic Malignancies Program, the Predolin Foundation, and the Multiorgan Transplant Database from the William J Von Liebig Center for Transplantation and Clinical Regeneration, Mayo Clinic Rochester, MN, United States.
COMMENTS
Background
Cardiac involvement is present in approximately 50% of patients with immunoglobulin light chain (AL) amyloidosis amyloidosis and is associated with a dismal prognosis. Heart transplant for AL amyloid is controversial, due to concerns about amyloid deposition in the transplanted heart and the potential for increased morbidity and mortality from the underlying plasma cell disorder.
Research frontiers
The research goal was to review a single center experience with cardiac transplantation for AL amyloid and determine outcome.
Innovations and breakthroughs
This study demonstrates that long term survival is possible in highly selected patients with AL amyloid who undergo cardiac transplantation if the underlying plasma cell disorder can be controlled.
Applications
Patients with cardiac AL amyloid and limited extra cardiac involvement may be considered for cardiac transplantation. Long term survival is possible in those who achieve a complete hematologic response to chemotherapy or autologous stem cell transplantation.
Terminology
Immunoglobulin light chain AL is a plasma cell disorder which results in deposition of amyloid fibrils in the organs and tissues of the body. Autologous hematopoietic stem cell transplantation is a strategy to treat the underlying plasma cell disorder that causes AL amyloidosis.
Peer-review
The authors presented a good overview of patients with AL amyloidosis + advanced heart failure who received cardiac transplantation.
Footnotes
Institutional review board statement: The Mayo Foundation Institutional Review Board approved this study.
Informed consent statement: Patients were not required to give informed consent because of observational, retrospective nature of the study.
Conflict-of-interest statement: The authors of this manuscript have no conflicts of interest to disclose pertinent to this research.
Data sharing statement: No additional data available.
Open-Access: This article is an open-access article which was selected by an in-house editor and fully peer-reviewed by external reviewers. It is distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/
Peer-review started: January 27, 2016
First decision: February 29, 2016
Article in press: April 11, 2016
P- Reviewer: den Uil CA, Tang JM S- Editor: Gong XM L- Editor: A E- Editor: Liu SQ
References
- 1.Merlini G, Seldin DC, Gertz MA. Amyloidosis: pathogenesis and new therapeutic options. J Clin Oncol. 2011;29:1924–1933. doi: 10.1200/JCO.2010.32.2271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Dispenzieri A, Gertz MA, Kyle RA, Lacy MQ, Burritt MF, Therneau TM, McConnell JP, Litzow MR, Gastineau DA, Tefferi A, et al. Prognostication of survival using cardiac troponins and N-terminal pro-brain natriuretic peptide in patients with primary systemic amyloidosis undergoing peripheral blood stem cell transplantation. Blood. 2004;104:1881–1887. doi: 10.1182/blood-2004-01-0390. [DOI] [PubMed] [Google Scholar]
- 3.Dispenzieri A, Gertz MA, Kyle RA, Lacy MQ, Burritt MF, Therneau TM, Greipp PR, Witzig TE, Lust JA, Rajkumar SV, et al. Serum cardiac troponins and N-terminal pro-brain natriuretic peptide: a staging system for primary systemic amyloidosis. J Clin Oncol. 2004;22:3751–3757. doi: 10.1200/JCO.2004.03.029. [DOI] [PubMed] [Google Scholar]
- 4.Falk RH. Diagnosis and management of the cardiac amyloidoses. Circulation. 2005;112:2047–2060. doi: 10.1161/CIRCULATIONAHA.104.489187. [DOI] [PubMed] [Google Scholar]
- 5.Sharma N, Howlett J. Current state of cardiac amyloidosis. Curr Opin Cardiol. 2013;28:242–248. doi: 10.1097/HCO.0b013e32835dd165. [DOI] [PubMed] [Google Scholar]
- 6.Mishra S, Guan J, Plovie E, Seldin DC, Connors LH, Merlini G, Falk RH, MacRae CA, Liao R. Human amyloidogenic light chain proteins result in cardiac dysfunction, cell death, and early mortality in zebrafish. Am J Physiol Heart Circ Physiol. 2013;305:H95–103. doi: 10.1152/ajpheart.00186.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dubrey S, Simms RW, Skinner M, Falk RH. Recurrence of primary (AL) amyloidosis in a transplanted heart with four-year survival. Am J Cardiol. 1995;76:739–741. doi: 10.1016/s0002-9149(99)80214-8. [DOI] [PubMed] [Google Scholar]
- 8.Hosenpud JD, DeMarco T, Frazier OH, Griffith BP, Uretsky BF, Menkis AH, O’Connell JB, Olivari MT, Valantine HA. Progression of systemic disease and reduced long-term survival in patients with cardiac amyloidosis undergoing heart transplantation. Follow-up results of a multicenter survey. Circulation. 1991;84:III338–III343. [PubMed] [Google Scholar]
- 9.Kpodonu J, Massad MG, Caines A, Geha AS. Outcome of heart transplantation in patients with amyloid cardiomyopathy. J Heart Lung Transplant. 2005;24:1763–1765. doi: 10.1016/j.healun.2004.08.025. [DOI] [PubMed] [Google Scholar]
- 10.Gray Gilstrap L, Niehaus E, Malhotra R, Ton VK, Watts J, Seldin DC, Madsen JC, Semigran MJ. Predictors of survival to orthotopic heart transplant in patients with light chain amyloidosis. J Heart Lung Transplant. 2014;33:149–156. doi: 10.1016/j.healun.2013.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Nohria A. Should we avoid heart transplantation in cardiomyopathy due to radiotherapy/chemotherapy or amyloidosis? The devil is in the details. J Heart Lung Transplant. 2012;31:1253–1256. doi: 10.1016/j.healun.2012.10.014. [DOI] [PubMed] [Google Scholar]
- 12.DePasquale EC, Nasir K, Jacoby DL. Outcomes of adults with restrictive cardiomyopathy after heart transplantation. J Heart Lung Transplant. 2012;31:1269–1275. doi: 10.1016/j.healun.2012.09.018. [DOI] [PubMed] [Google Scholar]
- 13.Gertz MA, Comenzo R, Falk RH, Fermand JP, Hazenberg BP, Hawkins PN, Merlini G, Moreau P, Ronco P, Sanchorawala V, et al. Definition of organ involvement and treatment response in immunoglobulin light chain amyloidosis (AL): a consensus opinion from the 10th International Symposium on Amyloid and Amyloidosis, Tours, France, 18-22 April 2004. Am J Hematol. 2005;79:319–328. doi: 10.1002/ajh.20381. [DOI] [PubMed] [Google Scholar]
- 14.Lacy MQ, Dispenzieri A, Hayman SR, Kumar S, Kyle RA, Rajkumar SV, Edwards BS, Rodeheffer RJ, Frantz RP, Kushwaha SS, et al. Autologous stem cell transplant after heart transplant for light chain (Al) amyloid cardiomyopathy. J Heart Lung Transplant. 2008;27:823–829. doi: 10.1016/j.healun.2008.05.016. [DOI] [PubMed] [Google Scholar]
- 15.Palladini G, Dispenzieri A, Gertz MA, Kumar S, Wechalekar A, Hawkins PN, Schönland S, Hegenbart U, Comenzo R, Kastritis E, et al. New criteria for response to treatment in immunoglobulin light chain amyloidosis based on free light chain measurement and cardiac biomarkers: impact on survival outcomes. J Clin Oncol. 2012;30:4541–4549. doi: 10.1200/JCO.2011.37.7614. [DOI] [PubMed] [Google Scholar]
- 16.Maurer MS, Raina A, Hesdorffer C, Bijou R, Colombo P, Deng M, Drusin R, Haythe J, Horn E, Lee SH, et al. Cardiac transplantation using extended-donor criteria organs for systemic amyloidosis complicated by heart failure. Transplantation. 2007;83:539–545. doi: 10.1097/01.tp.0000255567.80203.bd. [DOI] [PubMed] [Google Scholar]
- 17.Sattianayagam PT, Gibbs SD, Pinney JH, Wechalekar AD, Lachmann HJ, Whelan CJ, Gilbertson JA, Hawkins PN, Gillmore JD. Solid organ transplantation in AL amyloidosis. Am J Transplant. 2010;10:2124–2131. doi: 10.1111/j.1600-6143.2010.03227.x. [DOI] [PubMed] [Google Scholar]
- 18.Kumar SK, Gertz MA, Lacy MQ, Dingli D, Hayman SR, Buadi FK, Short-Detweiler K, Zeldenrust SR, Leung N, Greipp PR, et al. Recent improvements in survival in primary systemic amyloidosis and the importance of an early mortality risk score. Mayo Clin Proc. 2011;86:12–18. doi: 10.4065/mcp.2010.0480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Dubrey SW, Burke MM, Hawkins PN, Banner NR. Cardiac transplantation for amyloid heart disease: the United Kingdom experience. J Heart Lung Transplant. 2004;23:1142–1153. doi: 10.1016/j.healun.2003.08.027. [DOI] [PubMed] [Google Scholar]
- 20.Davis MK, Kale P, Liedtke M, Schrier S, Arai S, Wheeler M, Lafayette R, Coakley T, Witteles RM. Outcomes after heart transplantation for amyloid cardiomyopathy in the modern era. Am J Transplant. 2015;15:650–658. doi: 10.1111/ajt.13025. [DOI] [PubMed] [Google Scholar]
- 21.Dubrey SW, Burke MM, Khaghani A, Hawkins PN, Yacoub MH, Banner NR. Long term results of heart transplantation in patients with amyloid heart disease. Heart. 2001;85:202–207. doi: 10.1136/heart.85.2.202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Grogan M, Gertz MA, Kyle RA, Tajik AJ. Five or more years of survival in patients with primary systemic amyloidosis and biopsy-proven cardiac involvement. Am J Cardiol. 2000;85:664–665, A11. doi: 10.1016/s0002-9149(99)00832-2. [DOI] [PubMed] [Google Scholar]
- 23.Madan S, Kumar SK, Dispenzieri A, Lacy MQ, Hayman SR, Buadi FK, Dingli D, Rajkumar SV, Hogan WJ, Leung N, et al. High-dose melphalan and peripheral blood stem cell transplantation for light-chain amyloidosis with cardiac involvement. Blood. 2012;119:1117–1122. doi: 10.1182/blood-2011-07-370031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Dispenzieri A, Gertz MA, Kumar SK, Lacy MQ, Kyle RA, Saenger AK, Grogan M, Zeldenrust SR, Hayman SR, Buadi F, et al. High sensitivity cardiac troponin T in patients with immunoglobulin light chain amyloidosis. Heart. 2014;100:383–388. doi: 10.1136/heartjnl-2013-304957. [DOI] [PubMed] [Google Scholar]
- 25.Palladini G, Lavatelli F, Russo P, Perlini S, Perfetti V, Bosoni T, Obici L, Bradwell AR, D’Eril GM, Fogari R, et al. Circulating amyloidogenic free light chains and serum N-terminal natriuretic peptide type B decrease simultaneously in association with improvement of survival in AL. Blood. 2006;107:3854–3858. doi: 10.1182/blood-2005-11-4385. [DOI] [PubMed] [Google Scholar]
- 26.Dispenzieri A, Gertz MA, Saenger A, Kumar SK, Lacy MQ, Buadi FK, Dingli D, Leung N, Zeldenrust S, Hayman SR, et al. Soluble suppression of tumorigenicity 2 (sST2), but not galactin-3, adds to prognostication in patients with systemic AL amyloidosis independent of NT-proBNP and troponin T. Am J Hematol. 2015;90:524–528. doi: 10.1002/ajh.24001. [DOI] [PubMed] [Google Scholar]
- 27.Gertz MA. Immunoglobulin light chain amyloidosis: 2013 update on diagnosis, prognosis, and treatment. Am J Hematol. 2013;88:416–425. doi: 10.1002/ajh.23400. [DOI] [PubMed] [Google Scholar]
- 28.Roig E, Almenar L, González-Vílchez F, Rábago G, Delgado J, Gómez-Bueno M, Crespo-Leiro MG, Arizón JM, de la Fuente L, Manito N. Outcomes of heart transplantation for cardiac amyloidosis: subanalysis of the spanish registry for heart transplantation. Am J Transplant. 2009;9:1414–1419. doi: 10.1111/j.1600-6143.2009.02643.x. [DOI] [PubMed] [Google Scholar]
- 29.Kristen AV, Sack FU, Schonland SO, Hegenbart U, Helmke BM, Koch A, Schnabel PA, Röcken C, Hardt S, Remppis A, et al. Staged heart transplantation and chemotherapy as a treatment option in patients with severe cardiac light-chain amyloidosis. Eur J Heart Fail. 2009;11:1014–1020. doi: 10.1093/eurjhf/hfp121. [DOI] [PubMed] [Google Scholar]
- 30.Hosenpud JD, Uretsky BF, Griffith BP, O’Connell JB, Olivari MT, Valantine HA. Successful intermediate-term outcome for patients with cardiac amyloidosis undergoing heart transplantation: results of a multicenter survey. J Heart Transplant. 1990;9:346–350. [PubMed] [Google Scholar]
- 31.Mignot A, Varnous S, Redonnet M, Jaccard A, Epailly E, Vermes E, Boissonnat P, Gandjbakhch I, Herpin D, Touchard G, et al. Heart transplantation in systemic (AL) amyloidosis: a retrospective study of eight French patients. Arch Cardiovasc Dis. 2008;101:523–532. doi: 10.1016/j.acvd.2008.06.018. [DOI] [PubMed] [Google Scholar]