

Abstract
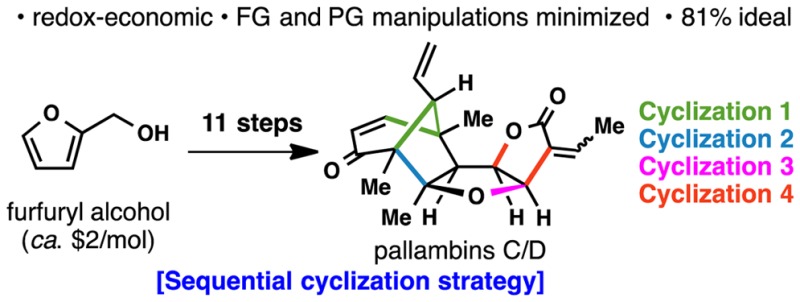
The structurally intriguing terpenes pallambins C and D have been assembled in only 11 steps from a cheap commodity chemical: furfuryl alcohol. This synthesis, which features a redox-economic approach free of protecting-group manipulations, assembles all four-ring systems via a sequential cyclization strategy. Of these four-ring constructing operations, two are classical (Robinson annulation and Mukaiyama aldol) and two are newly devised. During the course of this work a method for the difunctionalization of enol ethers was developed, and the scope of this transformation was explored.
Pallambins A–D (1–4, Figure 1), isolated from the epilithic liverwort Pallavicinia ambigua,1a exhibit no significant bioactivity, and their sole justification for study in the laboratory rests on their extraordinary chemical architectures. Indeed, with 4–6 rings, 7–10 contiguous stereocenters, and 2 quaternary stereocenters, these challenging terpenoid isolates are an ideal proving ground for the development of unique strategies and methods. Two elegant approaches to the synthesis of pallambins have appeared, one from the Wong group2 and the other from the Carreira3 group. The former route furnished 3 and 4 in 38 steps from the Wieland–Miescher ketone, and the latter afforded 1 and 2 in 23 steps from a fulvene. Ongoing interest in simplifying the synthesis of complex natural products by eliminating concession steps inspired our route to the pallambins.4 Here, an 11-step route to 3 and 4 is documented, wherein 9 of the 11 steps are strategic and protecting group manipulations are absent.5a−5c
Figure 1.
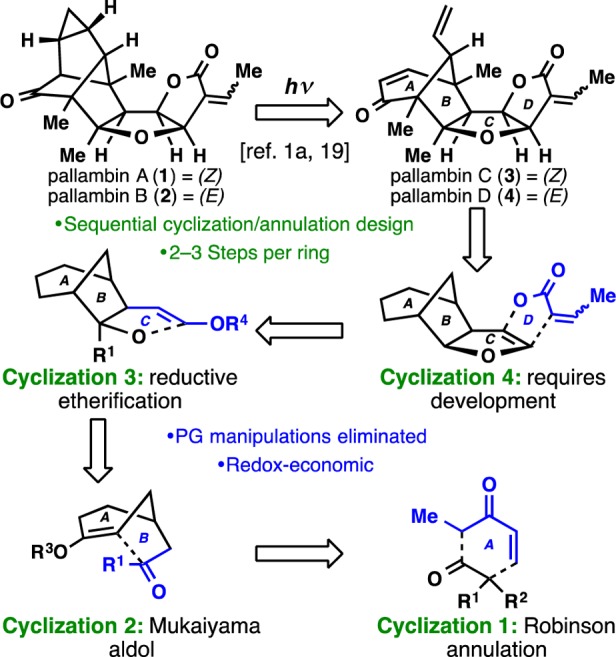
A sequential cyclization strategy enables an exceptionally concise pathway to pallambins C and D.
As outlined in Figure 1, the concise route to pallambins was enabled by a unique retrosynthetic design featuring four different cyclizations to forge the four different ring systems present in 3 and 4. Thus, sequential ring disconnections were made without considering precedent; maximizing strategic efficiency was the sole criterion. Cyclization 4 would append the D-ring onto a C-ring dihydrofuran. Since such a disconnection was not known in the forward sense, it presented an opportunity to bring such a reaction into being. Next, cyclization 3 would forge the C-ring by a reductive etherification process. The bicyclo[3.2.1]octane system could in principle arise by way of a Mukaiyama-type aldol reaction (cyclization 2). Notably, this type of cyclization bears resemblance to the proposed biosynthesis of the pallavicinia family of diterpenoids from a common labdane precursor.1b,6 Finally, the venerable Robinson annulation (cyclization 1) could be recruited at the outset to construct the A-ring.
The total syntheses of 3 and 4, as outlined in Scheme 1, commence with the abundant feedstock chemical furfuryl alcohol. Tandem Eschenmoser–Claisen rearrangement7 and reduction8 of the resulting amide was carried out in a single flask to produce aldehyde 5 in 75% yield after acidic workup. Although a seemingly trivial building block, the previously reported preparation of 5 (not scalable in our hands) required 3 steps and proceeded in <20% yield.9 Numerous approaches were evaluated (see SI for a summary) before an optimized solution was identified. Cyclization 1 (Robinson annulation) was then applied to aldehyde 5 and ethyl vinyl ketone (EVK) to afford 6 in 68% yield. Conjugate addition of a vinyl cuprate species followed by treatment with TMSCl delivered the latent nucleophile TMS-enol ether 7 as a single diastereomer in 75% yield.10 Chemoselective oxidative scission of the furan heterocycle11 could be accomplished using 1O2 to deliver the sensitive keto-aldehyde 8, which was used directly after photosensitizer removal with activated carbon. Attempts to purify 8 resulted in spontaneous nonstereoselective (1:1 dr at C-8) B-ring formation and thus 8 was carried forth in crude form. Cyclization 2 (Mukaiyama aldol), the pivotal bicyclo[3.2.1]octane forming step, was accomplished in good yield, and a remarkable effect of the Lewis acid and order of addition was empirically discovered (see SI for summary). For instance, if BF3·OEt2 was utilized, complete selectivity was observed for the undesired diastereomer at C-8 (structure verified by X-ray crystallography). If TiCl4 was added to a solution of 8, a 1:1 mixture of diastereomers was obtained. Ultimately, a reverse addition wherein the substrate was added slowly to a precooled solution of TiCl4 was required to obtain complete selectivity for the desired diastereomer 9 (structure verified by X-ray crystallography).12 In this cyclization, reaction with the less electrophilic methyl ketone is observed due to geometric constraints, thus preventing the enol ether from engaging the aldehyde.
Scheme 1. Total Synthesis of Pallambins C (3) and D (4).

Reagents and conditions: (1) furfuryl alcohol (1 equiv), 1,1-dimethoxy-N,N-dimethylpropan-1-amine (1.5 equiv), PhMe, 110 °C then TMDS (2 equiv), Ti(OiPr)4 (1.5 equiv), 50 °C (75%, one-pot); (2) ethyl vinyl ketone (1.5 equiv), Bu4NBr (10 mol %), 60% aq. KOH, PhMe, 23 °C (68%); (3) vinyl magnesium bromide (3.5 equiv), CuBr·DMS (20 mol %), HMPA (4 equiv), TMSCl (1.1 equiv), THF, −78 °C (75%); (4) O2, methylene blue, hν, DCM, −10 °C, then thiourea (1.5 equiv), 23 °C; (5) TiCl4 (1.5 equiv), Et2O, −78 °C (58%, 2 steps); (6) CH(OMe)3 (1.5 equiv), BF3·OEt2 (1.1 equiv), MgSO4 (25 equiv), DCM, 0 °C then AcBr (1 equiv) (57%); (7) Bu3SnH (1.5 equiv), AIBN (1 equiv), PhMe, 110 °C (90%); (8) LiHMDS (2 equiv), PhSeCl (2 equiv), THF, −78 °C then H2O2 (5.0 equiv), 0 °C (60%, one-pot); (9) PPTS (4 equiv), pyridine (4 equiv), PhCl, 130 °C; (10) dimethyl malonate (5 equiv), SnCl4 (5 equiv), DBU (5 equiv), I2 (1 equiv), DCM, 23 °C (87%, 2 steps); (11) 2 M NaOH, MeOH, 23 °C then Et3N (10.0 equiv), MeCN, 60 °C then LiHMDS (2.5 equiv), MeCHO (5 equiv), THF, −78 °C then Et3N (30 equiv), MsCl (5.0 equiv), DMAP, DCM, 23 °C (94%, one-pot); TMDS = 1,1,3,3-tetramethyldisiloxane, EVK = ethyl vinyl ketone, DMS = dimethyl sulfide, HMPA = hexamethylphosphoramide, MB = methylene blue, DCM = dichloromethane, AIBN = 2,2′-azobis(2-methylpropionitrile), DBU = 1,8-diazabicyclo[5.4.0]undec-7-ene, LiHMDS = lithium bis(trimethylsilyl)amide, PPTS = pyridinium p-toluenesulfonate, DMAP = 4-(dimethylamino)pyridine
Cyclization 3 required the seemingly simple conversion of enal 9 to a THF ring such as 12. This proved exceptionally challenging as all attempts to effect chemo- and stereoselective reduction of the C9–C11 olefin failed (see SI for a summary). Notably, Shenvi’s hydrogen-atom-transfer conditions were one of the only methods to provide the desired selectivity at C-9 in a simplified model substrate,13 however, these conditions were found to be unsuitable when applied to a system possessing the bridgehead vinyl group, despite extensive optimization efforts. Serendipitously, during our efforts to overcome this roadblock, an unusual reaction was discovered which ultimately circumvented the challenge. As an effort to address the chemoselectivity issue, conversion of enal 9 to acetal A was attempted using CH(OMe)3 in the presence of BF3·OEt2 (Figure 2). Surprisingly, this resulted in spontaneous C-ring formation, giving mixed acetal 10 with incorporation of a C-9 methyl ether in good yield. This transformation likely proceeds via generation of acetal A followed by Lewis acid promoted formation of a reactive oxocarbenium (C), which undergoes addition at C-9 with residual methanol to deliver fleeting intermediates B and D. For geometric reasons, only D can cyclize to afford the desired THF ring 10.
Figure 2.
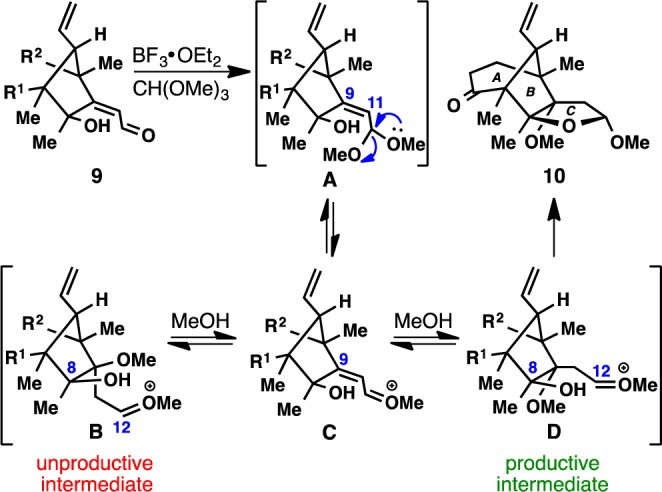
A mechanistic model for C-ring formation.
When attempts at direct conversion of 10 to 12 failed, intermediate 10 was treated with AcBr to furnish 11 in 57% overall yield (Scheme 1).14 Although this cyclization effectively installed the C-ring of the pallambins, it necessitated a nonstrategic reduction to replace the alkyl bromide with hydrogen (11 → 12, AIBN, SnBu3H, 90%). One-pot desaturation proceeded smoothly delivering enone 13 in 60% yield. Elimination of methanol from 13 could be accomplished using PPTS buffered with pyridine at 130 °C to afford the highly sensitive but thermally stable dihydrofuran 14, which was used directly after an aqueous wash.15
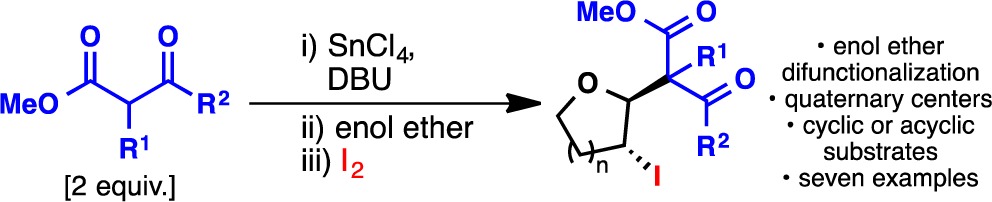
As anticipated, cyclization 4, the final ring-stitching operation of the synthesis, required invention. Although many clever strategies have been described to fashion the [3.3.0]furofuranone ring system,16 none were suitable for the task of selectively establishing ring-D on an enol ether such as 14. Well-trodden approaches such as epoxidation to introduce the C-11 hydroxyl group and downstream C–C bond formation failed in our hands due to chemo- and stereoselectivity issues (see SI for a summary). An annulation was thus designed to embrace the innate steric and electronic preferences of this ring system.17a−17c First, the tin-enolate of dimethyl malonate was generated using DBU in the presence of SnCl4 (DCM, 10 min, 23 °C). Next, enol ether 14 was added to this solution followed by I2. The desired iodo-diester 15 emerged from this protocol in 87% overall yield from 13 as a single diastereomer. The use of a tin enolate is essential as this nucleophile preferentially attacks the softer iodonium species initially formed upon addition of I2 to 14. Indeed, the use of sodium malonate leads exclusively to the iodinated malonate (quantitative yield) rather than the desired adduct 15 (for an optimization table, see SI). As shown in Table 1, this enol ether difunctionalization process appears to be general with seven additional examples. Notably, both cyclic and acyclic systems can be employed as well as alkyl malonates to generate quaternary centers.
Table 1. Scope of the Enol-Ether Difunctionalization Reaction.
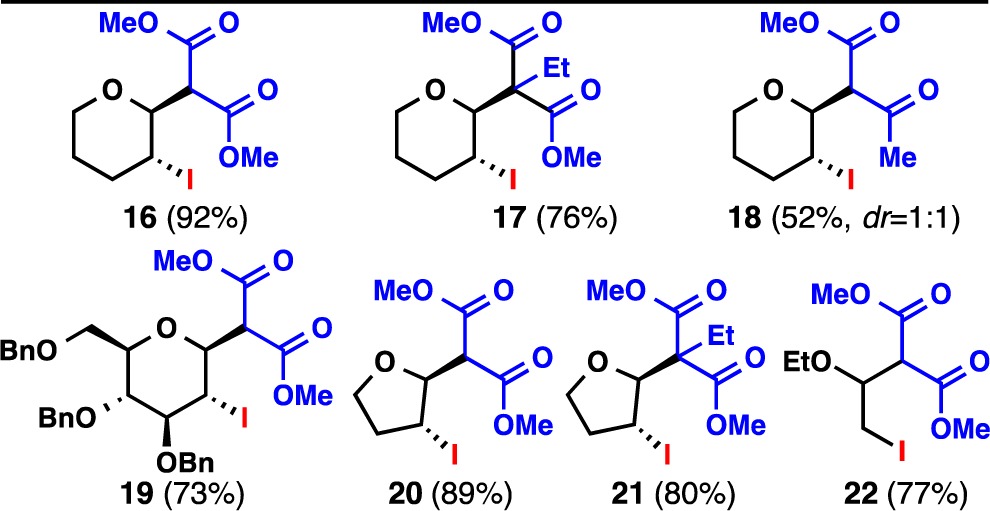
Cyclization 4 could be easily completed from 15 with concomitant formation of the natural isolates by engineering the following single-flask sequence: (1) alkaline hydrolysis of the esters (2 M NaOH) followed by addition of TMSCl to quench excess base; (2) Et3N-induced lactonization and decarboxylation (60 °C; (3) aldol addition to acetaldehyde; and (4) elimination with MsCl/Et3N. This one-pot set of four operations provided pallambins C (3) and D (4) in a 1:2 ratio in 94% isolated yield. The yield of this single step can be attributed to the near quantitative conversion of each individual transformation and the orthogonal nature of the conditions that do not necessitate individual isolation, purification, or workup; a simple evaporation suffices after operations 1–3. When directly compared with the multiflask sequence, this single-flask protocol saves 6 h of labor and eliminates three extractions and two chromatographic separations, providing a great example of how “pot economy” can simplify synthetic endeavors.18 At the outset of our synthetic campaign, we sought to examine and implement Lou’s proposed1a photoinduced interconversion of 3 and 4 to access 1 and 2 (Figure 1). During the course of our studies, Lou et al. demonstrated19 the photochemical interconversion of 3 and 4 to 1 and 2 is indeed operative providing formal access to 1 and 2 through the synthetic route detailed above.
A concise synthesis of the pallambins has thus been achieved. The four key ring systems found in 3 and 4 were generated by employing four sequential cyclizations, two of which are precedented and two of which required development. This design originated from a desire to eliminate extraneous redox-manipulations, functional group interconversions, and protecting group manipulations. In fact, of the 11 discrete steps of this synthesis, only two are nonstrategic (steps 7 and 9), making it 81% ideal.4a
Acknowledgments
Financial support for this work was provided by the NSF GRFP (predoctoral fellowship to L.P.M.), JSPS (postdoctoral fellowship S.U.), and the NIH (GM-097444). We thank Dr. D.-H. Huang and Dr. L. Pasternack for NMR spectroscopic assistance and Prof. A. L. Rheingold and Dr. C. E. Moore for X-ray crystallographic analysis.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/jacs.6b04816.
Author Present Address
† Department of Chemistry, Temple University, 1901 N. 13th Street, Philadelphia, PA 19122, United States
Author Contributions
§ These authors contributed equally to this paper.
The authors declare no competing financial interest.
Supplementary Material
References
- a Wang L.-N.; Zhang J.-Z.; Li X.; Wang X.-N.; Xie C.-F.; Zhou J.-C.; Lou H.-X. Org. Lett. 2012, 14, 1102. 10.1021/ol3000124. [DOI] [PubMed] [Google Scholar]; b Toyota M.; Saito T.; Asakawa Y. Chem. Pharm. Bull. 1998, 46, 178. 10.1248/cpb.46.178. [DOI] [Google Scholar]
- Xu X.-S.; Li Z.-W.; Zhang Y.-J.; Peng X.-S.; Wong H. N. C. Chem. Commun. 2012, 48, 8517. 10.1039/c2cc34310j. [DOI] [PubMed] [Google Scholar]
- Ebner C.; Carreira E. M. Angew. Chem. 2015, 127, 11379. 10.1002/ange.201505126. [DOI] [PubMed] [Google Scholar]
- a Gaich T.; Baran P. S. J. Org. Chem. 2010, 75, 4657. 10.1021/jo1006812. [DOI] [PubMed] [Google Scholar]; b Newhouse T.; Baran P. S.; Hoffmann R. W. Chem. Soc. Rev. 2009, 38, 3010. 10.1039/b821200g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Baran P. S.; Maimone T. J.; Richter J. M. Nature 2007, 446, 404. 10.1038/nature05569. [DOI] [PubMed] [Google Scholar]; b Young I. S.; Baran P. S. Nat. Chem. 2009, 1, 193. 10.1038/nchem.216. [DOI] [PubMed] [Google Scholar]; c Maimone T. J.; Ishihara Y.; Baran P. S. Tetrahedron 2015, 71, 3652. 10.1016/j.tet.2014.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Z.-J.; Lou H.-X.; Yu W.-T.; Fan P.-H.; Ren D.-M.; Ma B.; Mei J. Helv. Chim. Acta 2005, 88, 2637. 10.1002/hlca.200590204. [DOI] [Google Scholar]
- a Wick A. E.; Felix D.; Steen K.; Eschenmoser A. Helv. Chim. Acta 1964, 47, 2425. 10.1002/hlca.19640470835. [DOI] [Google Scholar]; b Ficini J.; Pouliquen J.; Normant H. C. R. Acad. Sci., Ser. IIc: Chim. 1969, 268, 1446. [Google Scholar]; c Feugeas C.; Olschwang D. Bull. Soc. Chim. Fr. 1968, 4985. [Google Scholar]
- a Bower S.; Kreutzer K.; Buchwald S. L. Angew. Chem., Int. Ed. Engl. 1996, 35, 1515. 10.1002/anie.199615151. [DOI] [Google Scholar]; b Laval S.; Dayoub W.; Favre-Reguillon A.; Demonchaux P.; Mignani G.; Lemaire M. Tetrahedron Lett. 2010, 51, 2092. 10.1016/j.tetlet.2010.02.008. [DOI] [Google Scholar]
- Kuznetsova S. V.; Anisimov A. V.; Viktorova E. A. Zh. Org. Khim. 1988, 24, 861. [Google Scholar]
- a Garnsey M. R.; Lim D.; Yost J. M.; Coltart D. M. Org. Lett. 2010, 12, 5234. 10.1021/ol1022728. [DOI] [PubMed] [Google Scholar]; b Jones T. K.; Denmark S. E. J. Org. Chem. 1985, 50, 4037. 10.1021/jo00221a015. [DOI] [Google Scholar]
- Iesce M. R.; Cermola F.; Temussi F. Curr. Org. Chem. 2005, 9, 109. 10.2174/1385272053369222. [DOI] [Google Scholar]
- a The observed difference in diastereoselectivity between BF3·Et2O and TiCl4 is likely attributable to the ability of TiCl4 to chelate the C-8 and C-12 carbonyl groups during the course of the reaction; b Upon addition of substrate to a stoichiometric solution of Lewis acid, the desired Mukaiyama aldol reaction takes place instantly. The alkoxide resulting from attack at the C-8 ketone likely forms a stoichiometric complex with the Lewis acid preventing a nonselective retro-aldol, aldol reaction sequence at C-8; c For a related phenomenon during the synthesis of ginkgolide B see:Desai M. C.; Ghosh A. K.; Houpis I. N. In Strategies and Tactics in Organic Synthesis; Lindberg T., Ed.; Academic Press: New York, 1991, Vol. 3, p 89. [Google Scholar]
- Iwasaki K.; Wan K. K.; Oppendisano A.; Crossley S. W. M.; Shenvi R. A. J. Am. Chem. Soc. 2014, 136, 1300. 10.1021/ja412342g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki Z.; Morita K. J. Org. Chem. 1967, 32, 31. 10.1021/jo01277a008. [DOI] [Google Scholar]
- Corey E. J.; Kang M. C.; Desai M. C.; Ghosh A. K.; Houpis I. N. J. Am. Chem. Soc. 1988, 110, 649. 10.1021/ja00210a083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng X.-S.; Ylagan R. M. P.; Siu Y. M.; Wong H. N. C. Chem. - Asian J. 2015, 10, 2070. 10.1002/asia.201500288. [DOI] [PubMed] [Google Scholar]
- a Kitagawa O.; Inoue T.; Taguchi T. Tetrahedron Lett. 1992, 33, 2167. 10.1016/0040-4039(92)88168-5. [DOI] [Google Scholar]; b Kitagawa O.; Fujiwara H.; Suzuki T.; Taguchi T.; Shiro M. J. Org. Chem. 2000, 65, 6819. 10.1021/jo000784z. [DOI] [PubMed] [Google Scholar]; c Yuan Y.; Shi Z. Synlett 2007, 20, 3219. 10.1055/s-2007-992388. [DOI] [Google Scholar]
- a Hayashi Y.; Umemiya S. Angew. Chem., Int. Ed. 2013, 52, 3450. 10.1002/anie.201209380. [DOI] [PubMed] [Google Scholar]; b Hayashi Y. Chem. Sci. 2016, 7, 866. 10.1039/C5SC02913A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J.-Z.; Zhu R.-X.; Li G.; Wang L.-N.; Sun B.; Chen W.-F.; Liu L.; Lou H.-X. Org. Lett. 2012, 14, 5624. 10.1021/ol302295a. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.