

Abstract

The nucleation of crystals in liquids is one of nature’s most ubiquitous phenomena, playing an important role in areas such as climate change and the production of drugs. As the early stages of nucleation involve exceedingly small time and length scales, atomistic computer simulations can provide unique insights into the microscopic aspects of crystallization. In this review, we take stock of the numerous molecular dynamics simulations that, in the past few decades, have unraveled crucial aspects of crystal nucleation in liquids. We put into context the theoretical framework of classical nucleation theory and the state-of-the-art computational methods by reviewing simulations of such processes as ice nucleation and the crystallization of molecules in solutions. We shall see that molecular dynamics simulations have provided key insights into diverse nucleation scenarios, ranging from colloidal particles to natural gas hydrates, and that, as a result, the general applicability of classical nucleation theory has been repeatedly called into question. We have attempted to identify the most pressing open questions in the field. We believe that, by improving (i) existing interatomic potentials and (ii) currently available enhanced sampling methods, the community can move toward accurate investigations of realistic systems of practical interest, thus bringing simulations a step closer to experiments.
1. Introduction
Crystal nucleation in liquids has countless practical consequences in science and technology, and it also affects our everyday experience. One obvious example is the formation of ice, which influences global phenomena such as climate change,1,2 as well as processes happening at the nanoscale, such as intracellular freezing.3,4 On the other hand, controlling nucleation of molecular crystals from solutions is of great importance to pharmaceuticals, particularly in the context of drug design and production, as the early stages of crystallization impact the crystal polymorph obtained.5,6 Even the multibillion-dollar oil industry is affected by the nucleation of hydrocarbon clathrates, which can form inside pipelines, endangering extraction.7,8 Finally, crystal nucleation is involved in many processes spontaneously occurring in living beings, from the growth of the beautiful Nautilus shells9 to the dreadful formation in our own brains of amyloid fibrils, which are thought to be responsible for many neurodegenerative disorders such as Alzheimer’s disease.10,11
Each of the above scenarios starts from a liquid below its melting temperature. This supercooled liquid(12) is doomed, according to thermodynamics, to face a first-order phase transition, leading to a crystal.13,14 Before this can happen, however, a sufficiently large cluster of crystalline atoms (or molecules or particles) must form within the liquid, such that the free energy cost of creating an interface between the liquid and the crystalline phase will be overcome by the free energy gain of having a certain volume of crystal. This event stands at the heart of crystal nucleation, and how this process has been, is, and will be modeled by means of computer simulations is the subject of this review.
The past few decades have witnessed an impressive body of experimental work devoted to crystal nucleation. For instance, thanks to novel techniques such as transmission electron microscopy at very low temperatures (cryo-TEM), we are now able to peek in real time into the early stages of crystallization.15 A substantial effort has also been made to understand which materials, in the form of impurities within the liquid phase, can either promote or inhibit nucleation events,16 a common scenario known as heterogeneous nucleation. However, our understanding of crystal nucleation is far from being complete. This is because the molecular (or atomistic) details of the process are largely unknown because of the very small length scale involved (nanometers), which is exceptionally challenging to probe in real time even with state-of-the-art measurements. Hence, there is a need for computer simulations, and particularly molecular dynamics (MD) simulations, where the temporal evolution of the liquid into the crystal is more or less faithfully reproduced. Unfortunately, crystal nucleation is a rare event that can occur on time scales of seconds, far beyond the reach of any conventional MD framework. In addition, a number of approximations within the computational models, algorithms, and theoretical framework used have been severely questioned for several decades. Although the rush for computational methods able to overcome this time-scale problem is now more competitive than ever, we are almost always forced to base our conclusions on the ancient grounds of classical nucleation theory (CNT), a powerful theoretical tool that nonetheless dates back 90 years to Volmer and Weber.17
In fact, these are exciting times for the crystal nucleation community, as demonstrated by the many reviews covering several aspects of this diverse field.18−24 This particular review is focused almost exclusively on MD simulations of crystal nucleation of supercooled liquids and supersaturated solutions. We take into account several systems, from colloidal liquids to natural gas hydrates, highlighting long-standing issues as well as recent advances. Although we review a substantial fraction of the theoretical efforts in the field, mainly from the past decade, our goal is not to discuss in detail every contribution. Instead, we try to pinpoint the most pressing issues that still prevent us from furthering our understanding of nucleation.
This article is structured in three parts. In the first part, we introduce the theoretical framework of CNT (section 1.1), the state-of-the-art experimental techniques (section 1.2), and the MD-based simulation methods (section 1.3) that in the past few decades have provided insight into nucleation. In section 2, we put such computational approaches into context, describing both achievements and open questions concerning the molecular details of nucleation for different types of systems, namely, colloids (section 2.1), Lennard-Jones (LJ) liquids (section 2.2), atomic liquids (section 2.3), water (section 2.4), nucleation from solution (section 2.5), and natural gas hydrates (section 2.6). In the third and last part of the article (section 3), we highlight future perspectives and open challenges in the field.
1.1. Theoretical Framework
1.1.1. Classical Nucleation Theory
Almost every computer simulation of crystal nucleation in liquids invokes some elements25 of classical nucleation theory (CNT). This theory has been discussed in great detail elsewhere,26−28 and we describe it here for the sake of completeness and also to introduce various terms used throughout the review. Nonetheless, readers familiar with CNT can skip to section 1.2.
CNT was formulated 90 years ago
through the contributions of Volmer and Weber,17 Farkas,29,30 Becker and Döring,31 and Zeldovich,32 on
the basis of the pioneering ideas of none other than Gibbs himself.33 CNT was created to describe the condensation
of supersaturated vapors into the liquid phase, but most of the concepts
can also be applied to the crystallization of supercooled liquids
and supersaturated solutions. According to CNT, clusters of crystalline
atoms (or particles or molecules) of any size are treated as macroscopic
objects, that is, homogeneous chunks of crystalline phase separated
from the surrounding liquid by a vanishingly thin interface. This
apparently trivial assumption is known as the capillarity approximation,
which encompasses most of the strengths and weaknesses of the theory.
According to the capillarity approximation, the interplay between
the interfacial free energy, 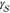 , and the difference in
free energy between
the liquid and the crystal,
, and the difference in
free energy between
the liquid and the crystal,  , fully describes the thermodynamics of
crystal nucleation. In three dimensions,34 the free energy of formation,
, fully describes the thermodynamics of
crystal nucleation. In three dimensions,34 the free energy of formation,  , for a spherical crystalline nucleus of
radius r can thus be written as the sum of a surface
term and a volume term
, for a spherical crystalline nucleus of
radius r can thus be written as the sum of a surface
term and a volume term
| 1 |
This function, sketched in Figure 1, displays a maximum corresponding to the so-called critical nucleus size n*
| 2 |
where 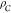 is the number density of the crystalline
phase. The critical nucleus size represents the number of atoms that
must be included in the crystalline cluster for the free energy difference,
is the number density of the crystalline
phase. The critical nucleus size represents the number of atoms that
must be included in the crystalline cluster for the free energy difference, 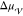 , to match the free energy cost due to the
formation of the solid–liquid interface. Clusters of crystalline
atoms occur within the supercooled liquid by spontaneous, infrequent
fluctuations, which eventually lead the system to overcome the free
energy barrier for nucleation
, to match the free energy cost due to the
formation of the solid–liquid interface. Clusters of crystalline
atoms occur within the supercooled liquid by spontaneous, infrequent
fluctuations, which eventually lead the system to overcome the free
energy barrier for nucleation
| 3 |
triggering the actual crystal growth (see Figure 1).
Figure 1.

Sketch of the free energy
difference,  , as a function of the crystalline nucleus
size n. A free energy barrier for nucleation,
, as a function of the crystalline nucleus
size n. A free energy barrier for nucleation, 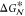 , must
be overcome to proceed from the (metastable)
supercooled liquid state to the thermodynamically stable crystalline
phase through homogeneous nucleation (purple). Heterogeneous nucleation
(green) can be characterized by a lower free energy barrier,
, must
be overcome to proceed from the (metastable)
supercooled liquid state to the thermodynamically stable crystalline
phase through homogeneous nucleation (purple). Heterogeneous nucleation
(green) can be characterized by a lower free energy barrier, 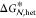 , and a smaller critical nucleus size, nhet*, whereas
in the case of spinodal decomposition (orange), the supercooled
liquid is unstable with respect to the crystalline phase, and the
transformation to the crystal proceeds in a barrierless fashion. The
three snapshots depict a crystalline cluster nucleating within the
supercooled liquid phase (homogeneous nucleation) or as a result of
the presence of a foreign impurity (heterogeneous nucleation), as
well as the simultaneous occurrence of multiple crystalline clusters
in the unstable liquid. This scenario is often labeled as spinodal
decomposition, although the existence of a genuine spinodal decomposition
from the supercooled liquid to the crystalline phase has been debated
(see text).
, and a smaller critical nucleus size, nhet*, whereas
in the case of spinodal decomposition (orange), the supercooled
liquid is unstable with respect to the crystalline phase, and the
transformation to the crystal proceeds in a barrierless fashion. The
three snapshots depict a crystalline cluster nucleating within the
supercooled liquid phase (homogeneous nucleation) or as a result of
the presence of a foreign impurity (heterogeneous nucleation), as
well as the simultaneous occurrence of multiple crystalline clusters
in the unstable liquid. This scenario is often labeled as spinodal
decomposition, although the existence of a genuine spinodal decomposition
from the supercooled liquid to the crystalline phase has been debated
(see text).
The kinetics of crystal
nucleation is typically addressed by assuming
that no correlation exists between successive events increasing or
decreasing the number of constituents of the crystalline nucleus.
In other words, the time evolution of the nucleus size is presumed
to be a Markov process, in which atoms in the liquid either order
themselves one by one in a crystalline fashion or dissolve one by
one into the liquid phase. In addition, we state that every crystalline
nucleus lucky enough to overcome the critical size n* quickly grows to macroscopic dimensions on a time scale much smaller
than the long time required for that fortunate fluctuation to come
about. If these conditions are met,35 the
nucleation rate, that is, the probability per unit time per unit volume
of forming a critical nucleus does not depend on time, leading to
the following formulation of the so-called steady-state nucleation
rate 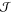
| 4 |
where kB is the
Boltzmann constant and  is a prefactor that we discuss
later. The
steady-state nucleation rate is the central quantity in the description
of crystallization kinetics, as much as the notion of critical nucleus
size captures most of the thermodynamics of nucleation.
is a prefactor that we discuss
later. The
steady-state nucleation rate is the central quantity in the description
of crystallization kinetics, as much as the notion of critical nucleus
size captures most of the thermodynamics of nucleation.
All
quantities specified up to now depend on pressure and most
notably temperature. In most cases, the interfacial free energy, 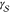 , is assumed to be linearly
dependent on
temperature, whereas the free energy difference between the liquid
and solid phases,
, is assumed to be linearly
dependent on
temperature, whereas the free energy difference between the liquid
and solid phases, 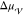 , is proportional to the supercooling,
, is proportional to the supercooling,  (or the supersaturation). Several approximations
exist to treat the temperature dependence of
(or the supersaturation). Several approximations
exist to treat the temperature dependence of 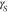 (36) and
(36) and 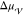 ,37 which can
vary substantially for different supercooled liquids.38 In any case, it follows from eq 3 that the free energy barrier for nucleation,
,37 which can
vary substantially for different supercooled liquids.38 In any case, it follows from eq 3 that the free energy barrier for nucleation, 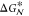 decreases with supercooling. In other words,
the farther one is from the melting temperature
decreases with supercooling. In other words,
the farther one is from the melting temperature  , the larger the thermodynamic driving force
for nucleation is.
, the larger the thermodynamic driving force
for nucleation is.
Interestingly, in the case of supercooled
liquids, kinetics goes
the other way, as the dynamics of the liquid slow down with supercooling,
thus hindering the occurrence of nucleation events. In fact, although
a conclusive expression for the prefactor the latter is still lacking,39,40 it is usually written within CNT
as27
it is usually written within CNT
as27
| 5 |
where  is the number of possible
nucleation sites
per unit volume,
is the number of possible
nucleation sites
per unit volume,  is the Zeldovich
factor27,41 (accounting for the fact that several postcritical
clusters might
still shrink without growing into the crystalline phase), and
is the Zeldovich
factor27,41 (accounting for the fact that several postcritical
clusters might
still shrink without growing into the crystalline phase), and  is a kinetic prefactor.39 The latter should represent the attachment
rate, that is, the frequency with which the particles in
the liquid phase
reach the cluster rearranging themselves in a crystalline fashion.
However, in a dense supercooled liquid,
is a kinetic prefactor.39 The latter should represent the attachment
rate, that is, the frequency with which the particles in
the liquid phase
reach the cluster rearranging themselves in a crystalline fashion.
However, in a dense supercooled liquid, 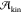 also
quantifies the ease with which the
system explores configurational space, effectively regulating the
amplitude of the fluctuations possibly leading to the formation of
a crystalline nucleus. In short, we can safely say that
also
quantifies the ease with which the
system explores configurational space, effectively regulating the
amplitude of the fluctuations possibly leading to the formation of
a crystalline nucleus. In short, we can safely say that  involves the atomic or molecular
mobility
of the liquid phase, more often than not quantified in terms of the
self-diffusion coefficient
involves the atomic or molecular
mobility
of the liquid phase, more often than not quantified in terms of the
self-diffusion coefficient 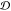 ,27 which obviously
decreases with supercooling. Thus, for a supercooled liquid, the competing
trends of
,27 which obviously
decreases with supercooling. Thus, for a supercooled liquid, the competing
trends of 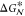 and
and  lead, in the case of diffusion-limited
nucleation,42 to a maximum in the nucleation
rate, as depicted in Figure 2. The same arguments apply when dealing with processes such
as the solidification of metallic alloys.43,44 In the case of nucleation from solutions,
lead, in the case of diffusion-limited
nucleation,42 to a maximum in the nucleation
rate, as depicted in Figure 2. The same arguments apply when dealing with processes such
as the solidification of metallic alloys.43,44 In the case of nucleation from solutions,  and
and 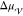 depend mainly on supersaturation. However,
the dependence of the kinetic prefactor on supersaturation is much
weaker than the temperature dependence of
depend mainly on supersaturation. However,
the dependence of the kinetic prefactor on supersaturation is much
weaker than the temperature dependence of 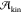 characteristic
of supercooled liquids.
As a result, there is usually no maximum in the nucleation rate as
a function of supersaturation for nucleation from solutions.45
characteristic
of supercooled liquids.
As a result, there is usually no maximum in the nucleation rate as
a function of supersaturation for nucleation from solutions.45
Figure 2.

Illustration of how certain quantities from CNT vary as
a function
of supercooling, ΔT, for supercooled liquids.
The free energy difference between the liquid and the solid phase  , the interfacial free energy
, the interfacial free energy 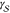 , and the kinetic prefactor
, and the kinetic prefactor  are reported as functions of ΔT in a generic case of diffusion-limited nucleation, characterized
by a maximum in the steady-state nucleation rate
are reported as functions of ΔT in a generic case of diffusion-limited nucleation, characterized
by a maximum in the steady-state nucleation rate  .
. 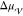 is zero at the melting temperature
is zero at the melting temperature 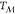 , and
, and 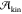 is vanishingly
small at the glass transition
temperature
is vanishingly
small at the glass transition
temperature  .
.
Although 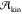 is supposed to play a minor role
compared
to the exponential term in eq 4, the kinetic prefactor has been repeatedly blamed for the
quantitative disagreement between experimental measurements and computed
crystal nucleation rates.39,46 Atomistic simulations
could, in principle, help to clarify the temperature dependence as
well as the microscopic origin of
is supposed to play a minor role
compared
to the exponential term in eq 4, the kinetic prefactor has been repeatedly blamed for the
quantitative disagreement between experimental measurements and computed
crystal nucleation rates.39,46 Atomistic simulations
could, in principle, help to clarify the temperature dependence as
well as the microscopic origin of 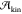 and also
of the thermodynamic ingredients
involved in the formulation of CNT. However, quantities such as
and also
of the thermodynamic ingredients
involved in the formulation of CNT. However, quantities such as 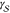 are not only infamously
difficult to converge
within decent levels of accuracy47,48 but can even
be ill-defined in many situations. For instance, it remains to be
seen whether
are not only infamously
difficult to converge
within decent levels of accuracy47,48 but can even
be ill-defined in many situations. For instance, it remains to be
seen whether 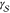 , which, in principle,
refers to a planar
interface under equilibrium conditions, can be safely defined when
dealing with small crystalline clusters of irregular shapes. In fact,
the early stages of the nucleation process often involve crystalline
nuclei whose size and morphology fluctuate on a time scale shorter
than the structural relaxation time of the surrounding liquid. Moreover,
the dimensions of such nuclei can be of the same order as the diffuse
interface between the liquid and the solid phases, thus rendering
the notion of a well-defined
, which, in principle,
refers to a planar
interface under equilibrium conditions, can be safely defined when
dealing with small crystalline clusters of irregular shapes. In fact,
the early stages of the nucleation process often involve crystalline
nuclei whose size and morphology fluctuate on a time scale shorter
than the structural relaxation time of the surrounding liquid. Moreover,
the dimensions of such nuclei can be of the same order as the diffuse
interface between the liquid and the solid phases, thus rendering
the notion of a well-defined 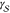 value quite dangerous.
As an example, Joswiak
et al.49 recently showed that, for liquid
water droplets,
value quite dangerous.
As an example, Joswiak
et al.49 recently showed that, for liquid
water droplets, 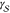 can strongly depend on
the curvature of
the droplet. The mismatch between the macroscopic interfacial free
energy and its curvature-dependent value can spectacularly affect
water-droplet nucleation, as reported by atomistic simulations of
droplets characterized by radii on the order of ∼0.5–1.5
nm. Some other quantities, such as the size of the critical cluster,
depend in many cases rather strongly on the degree of supercooling.
This is the case, for example, for the critical nucleus size n*, which can easily span 2 orders of magnitude in just
10 °C of supercooling.50,51
can strongly depend on
the curvature of
the droplet. The mismatch between the macroscopic interfacial free
energy and its curvature-dependent value can spectacularly affect
water-droplet nucleation, as reported by atomistic simulations of
droplets characterized by radii on the order of ∼0.5–1.5
nm. Some other quantities, such as the size of the critical cluster,
depend in many cases rather strongly on the degree of supercooling.
This is the case, for example, for the critical nucleus size n*, which can easily span 2 orders of magnitude in just
10 °C of supercooling.50,51
1.1.2. Two-Step Nucleation
Given the old age of CNT, it is no surprise that substantial efforts have been devoted to extend and/or improve its original theoretical framework. The most relevant modifications possibly concern the issue of two-step nucleation. Many excellent works have reviewed this subject extensively (see, e.g., refs (18, 24, 52, and 53)), so that we provide only the essential concepts here.
In the original formulation of CNT, the system has to overcome a single free energy barrier, corresponding to a crystalline nucleus of a certain critical size, as depicted in Figure 3. When dealing with crystal nucleation from the melt, it is rather common to consider the number of crystalline particles within the largest connected cluster, n, as the natural reaction coordinate describing the whole nucleation process. In many cases, the melt is dense enough that local density fluctuations are indeed not particularly relevant and the slow degree of freedom is in fact the crystalline ordering of the particles within the liquid network. However, one can easily imagine that, in the case of crystal nucleation of molecules in solutions, for example, the situation can be quite different. Specifically, in a realistically supersaturated solution, a consistent fluctuation of the solute density (concentration) could be required just to bring a number nρ of solute molecules close enough to form a connected cluster. Assuming that the molecules involved in such a density fluctuation will also order themselves in a crystalline fashion on exactly the same time scale is rather counterintuitive.
Figure 3.

Schematic comparison of one-step versus two-step nucleation for a generic supersaturated solution. (a) Sketch of the free energy difference ΔGn,nρ as a function of the number of solute molecules in the largest “connected” cluster (they can be ordered in a crystalline fashion or not) (nρ) and of the number of crystalline molecules within the largest connected cluster (n). The one-step mechanism predicted by CNT (purple) is characterized by a single free energy barrier for nucleation, ΔGn,nρ,one-step*. In contrast, the two-step nucleation requires a free energy barrier, ΔGnρ,two-step, to be overcome through a local density fluctuation of the solution, leading to a dense, but not crystalline-like, precursor. The latter can be unstable (green) or stable (orange) with respect to the liquid phase, being characterized by a higher (green) or lower (orange) free energy basin. Once this dense precursor has been obtained, the second step consists of climbing a second free energy barrier, ΔGn,two-step*, corresponding to the ordering of the solute molecules within the precursor from a disordered state to the crystalline phase. (b) One-step (purple) and two-step (green and orange) nucleation mechanisms visualized in the density (nρ)–ordering (n) plane. The one-step mechanism proceeds along the diagonal, as both nρ and n increase at the same time in such a way that a single free energy barrier has to be overcome. In this scenario, the supersaturated solution transforms continuously into the crystalline phase. On the other hand, within a two-step nucleation scenario, the system has to experience a favorable density fluctuation along nρ first, forming a disordered precursor that, in a second step, orders itself in a crystalline fashion, moving along the (n) coordinate and ultimately leading to the crystal.
In fact, the formation of crystals from molecules in solution often occurs according to a two-step nucleation mechanism that has no place in the original formulation of CNT. In the prototypical scenario depicted in Figure 3, a first free energy barrier, ΔGnρ,two-step*, has to be overcome by means of a density fluctuation of the solute, such that a cluster of connected molecules of size nρ is formed. This object does not yet have any sort of crystalline order, and depending on the system under consideration, it can be either unstable or stable with respect to the supersaturated solution (see Figure 3). Subsequently, the system has to climb a second free energy barrier, ΔGn,two-step*, to order the molecules within the dense cluster in a crystalline-like fashion. A variety of different nucleation scenarios have been loosely labeled as two-step, from crystal nucleation in colloids (see section 2.1) or Lennard-Jones liquids (see section 2.2) to the formation of crystals of urea or NaCl (see section 2.5), not to mention biomineralization (see, e.g., refs (18 and 53)) and protein crystallization (see, e.g., refs (54 and 55)).
In all of these cases, CNT as it is formulated is simply not capable of dealing with two-step nucleation. This is why, in the past few decades, a number of extensions and/or modifications of CNT have been proposed and indeed successfully applied to account for the existence of a two-step mechanism. Here, we mention the phenomenological theory of Pan et al.,54 who wrote an expression for the nucleation rate assuming a free energy profile similar to the one sketched in Figure 3, where dense metastable states are involved as intermediates on the path toward the final crystalline structure. The emergence of so-called prenucleation clusters (PNCs), namely, stable states within supersaturated solutions, which are known to play a very important role in the crystallization of biominerals, for example, was also recently fit into the framework of CNT by Hu et al.56 They proposed a modified expression for the excess free energy of the nucleus that takes into account the shape, size and free energy of the PNCs as well as the possibility for the PNCs to be either metastable or stable with respect to the solution. A comprehensive review of the subject is offered by the work of Gebauer et al.18 It is worth noticing that these extensions of CNT are mostly quite recent, as they were triggered by overwhelming experimental evidence for two-step nucleation mechanisms.
1.1.3. Heterogeneous Nucleation
CNT is
also the tool of the trade for heterogeneous crystal nucleation, that
is, nucleation that occurs on account of the presence of a foreign
phase (see Figure 1). In fact, nucleation in liquids occurs heterogeneously more often
than not, as in some cases, the presence of foreign substances in
contact with the liquid can significantly lower the free energy barrier 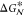 . A typical
example is given by the formation
of ice: As we shall see in sections 2.4.1 and 2.4.2, it
is surprisingly difficult to freeze pure water, which invariably takes
advantage of a diverse portfolio of impurities, from clay minerals
to bacterial fragments,16 to facilitate
the formation of ice nuclei.
. A typical
example is given by the formation
of ice: As we shall see in sections 2.4.1 and 2.4.2, it
is surprisingly difficult to freeze pure water, which invariably takes
advantage of a diverse portfolio of impurities, from clay minerals
to bacterial fragments,16 to facilitate
the formation of ice nuclei.
Heterogeneous nucleation is customarily formulated within the CNT framework in terms of geometric arguments.27 Specifically
| 6 |
where f(θ) ≤
1 is the shape factor, a quantity that accounts for
the fact that three different interfacial free energies must be balanced: 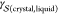 ,
, 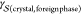 , and
, and 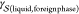 . For instance, considering a supercooled
liquid nucleating on top of an ideal planar surface offered by the
foreign phase, we obtain the so-called Young’s relation
. For instance, considering a supercooled
liquid nucleating on top of an ideal planar surface offered by the
foreign phase, we obtain the so-called Young’s relation
where θ is the contact angle, namely,
a measure of the extent to which the crystalline nucleus wets the foreign surface. Thus, the contact angle determines whether
and how much it could be easier for a critical nucleus to form in
an heterogeneous fashion, as for 0 ≤ θ < π,
the volume-to-surface energy ratio 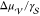 is larger for
the spherical cap nucleating
on the foreign surface than for the sphere nucleating in the liquid.
This simple formulation is clearly only a rough approximation of what
happens in reality. At first, the contact angle is basically a macroscopic
quantity, of which the microscopic equivalent is in most cases ill-defined
on the typical length scales involved in the heterogeneous nucleation
process.57 In addition, in most cases,
the nucleus will not be shaped like a spherical cap, and to make things
more complicated, many different nucleation sites with different morphologies
typically exist on the same impurity. Finally, the kinetic prefactor,
is larger for
the spherical cap nucleating
on the foreign surface than for the sphere nucleating in the liquid.
This simple formulation is clearly only a rough approximation of what
happens in reality. At first, the contact angle is basically a macroscopic
quantity, of which the microscopic equivalent is in most cases ill-defined
on the typical length scales involved in the heterogeneous nucleation
process.57 In addition, in most cases,
the nucleus will not be shaped like a spherical cap, and to make things
more complicated, many different nucleation sites with different morphologies
typically exist on the same impurity. Finally, the kinetic prefactor, 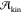 , becomes even more obscure in
heterogeneous
nucleation, as it is plausible that the foreign phase will affect
the dynamical properties of the supercooled liquid.
, becomes even more obscure in
heterogeneous
nucleation, as it is plausible that the foreign phase will affect
the dynamical properties of the supercooled liquid.
1.1.4. Nucleation at Strong Supercooling
Moving toward strong
supercooling, several things can happen to the
supercooled liquid phase. Whether one can avoid the glass transition
largely depends on the specific liquid under consideration and on
the cooling rate (see, e.g., ref (58)). Assuming that the system can be cooled sufficiently
slowly, hence avoiding both the glass transition and crystal nucleation,
one can, in principle, enter a supercooled regime in which the liquid
becomes unstable with respect to the crystalline phase. This region
of the phase diagram is known as the spinodal region, where the tiniest perturbation, for example, of the local density
or the degree of ordering leads the system toward the crystalline
phase without paying anything in terms of free energy (see Figure 1). In fact, below
a certain critical temperature,  , the free energy barrier for nucleation
is zero, and the liquid transforms spontaneously into the crystal
on very short time scales. The same picture holds for molecules in
solution, as nicely discussed by Gebauer et al.,18 and it cannot, by definition, be described by conventional
CNT, according to which a small
, the free energy barrier for nucleation
is zero, and the liquid transforms spontaneously into the crystal
on very short time scales. The same picture holds for molecules in
solution, as nicely discussed by Gebauer et al.,18 and it cannot, by definition, be described by conventional
CNT, according to which a small  value
persists even at the strongest supercoolings.59
value
persists even at the strongest supercoolings.59
Although spinodal regimes have been observed
in a variety of scenarios,60 the existence
of a proper spinodal decomposition from the supercooled liquid to
the crystalline phase has been debated (see, e.g., ref (61)). Enhanced-sampling MD
simulations,62 which we discuss in section 2.2, have suggested
that barrierless crystal nucleation is possible at very strong supercooling,
whereas other works claim that this is not the case (see, e.g., ref (63)). Here, we simply note
that, at strong supercooling—not necessarily within the presumed
spinodal regime—a number of assumptions on which CNT relies
become, if not erroneous, ill-defined. The list is long, and in fact,
a number of nucleation theories27 able
to at least take into account the emergence of a spinodal decomposition
exist, although they have mostly been formulated for condensation
problems. In any case, the capillarity approximation is most likely
to fail at strong supercoolings, as the size of the critical nucleus
becomes exceedingly small, down to losing its meaning in the event
of a proper spinodal decomposition. Moreover, we shall see, for instance,
in section 2.2, that
the shape of the crystalline clusters is anything but spherical at
strong supercooling and, at the same time, the kinetic prefactor assumes
a role of great importance. In fact, nucleation at strong supercooling
might very well be dominated by 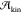 , as the mobility of the supercooled
liquid
is what really matters when the free energy barrier for nucleation
approaches vanishingly small values. Strong supercooling is important
because this is the regime in which most computational studies have
been performed. Large values of ΔT imply high
nucleation rates and smaller critical nuclei, although as one moves
away from
, as the mobility of the supercooled
liquid
is what really matters when the free energy barrier for nucleation
approaches vanishingly small values. Strong supercooling is important
because this is the regime in which most computational studies have
been performed. Large values of ΔT imply high
nucleation rates and smaller critical nuclei, although as one moves
away from 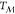 , most of the assumptions of CNT
are progressively
invalidated.
, most of the assumptions of CNT
are progressively
invalidated.
At this point, given the substantial approximations of CNT64 and especially its old age, the reader might be waiting for us to introduce the much more elegant, accurate, and comprehensive theories that experiments and simulations surely embrace today. Sadly, this is not the case. Countless flavors of nucleation theories exist. Many of them, such as dynamical nucleation theory,65 mean-field kinetic nucleation theory,66 and coupled flux theory,67−70 are mainly limited to condensation problems, and some others have only rarely been applied, for example, to crystallization in glasses,26 such as diffuse interface theory.71,72 Several improvements on CNT have been proposed, targeting specific aspects such as the shape of the crystalline nuclei73 or the finite size of the nonsharp crystal–liquid interface.49 Nucleation theories largely unrelated to CNT can also be found, such as classical density functional theory (cDFT)74−77 (classical, not to be confused with the celebrated quantum mechanical framework of Hohenberg and Kohn78). A fairly complete inventory of nucleation theories, together with an excellent review of nucleation in condensed matter, can be found elsewhere.79 Here, we do not discuss the details of any of these approaches, as indeed none of them has been consistently used to model crystal nucleation in liquids. This is because CNT, despite having many shortcomings, is a simple yet powerful theory that is able to capture at least qualitatively the thermodynamics and kinetics of nucleation for very different systems, from liquid metals to organic crystals. It has been extended to include heterogeneous nucleation, and it is fairly easy to modify it to take into consideration multicomponent systems such as binary mixtures as well.27,79
1.2. Experimental Methods
Several different experimental approaches have been employed to understand the thermodynamics and kinetics of crystal nucleation in liquids. Although this review discusses theory and simulations almost exclusively, we present in this section a concise overview of the state-of-the-art experimental techniques to highlight their capabilities as well as their limitations.
A schematic synopsis focusing on both spatial and temporal resolutions is sketched in Figure 4, and an inventory of notable applications is reported in Table 1. As already stated, nucleation is a dynamical process usually occurring on very small time and length scales (nanoseconds and nanometers, respectively). Thus, obtaining the necessary spatial and temporal resolutions is a tough technical challenge.
Figure 4.

Overview of some of the experimental methods that have been applied to characterize nucleation. Ranges of the spatial and temporal resolutions typical of each approach are reported on the x and y axes, respectively.
Table 1. Selection of Experimental Approaches That Have Been Employed to Study Nucleation Phenomena, along with Some Examples of Systems Examined.
| method | example(s) |
|---|---|
| confocal scanning microscopy | colloids,83,84 oogenesis in Xenopus(136) |
| AFM | glucose isomerase85 |
| SMRT-TEM, HREM | organic crystals,137,138 metal phosphate88 |
| cryo-TEM | CaCO3,86,87 magnetide,89 MCM41139 |
| femtosecond X-ray scattering | ice90,91 |
| high-speed visible or IR imaging | ice140 |
| analytical ultracentrifugation | CaCO3128 |
| powder diffraction | colloids,94,95 ice96 |
| FTIRS | ice,123−125 glycine,126 paracetamol127,141 |
| optical microscopy | colloids,81,82 ice92,93 |
| ambient-pressure XPS | ice142,143 |
| DSC | glass fibers,117 hydrates,118 ice,119−121 metal alloy122 |
| environmental SEM | CaP,144 ice145 |
| flow chamber | ice,129−131n-penthanol132 |
| cloud chamber | ice2,133−135 |
Indeed, true microscopic80 insight has rarely been achieved. For instance, colloids offer a playground where simple microscopy can image the particles involved in the nucleation events, which occur on such long time scales (seconds) that a full characterization in time of the process has been achieved.81,82 Specifically, confocal microscopy has led to three-dimensional imaging of colloidal systems, unraveling invaluable information about the critical nucleus size, for example.83,84
In a similar fashion, Sleutel et al. achieved molecular resolution of the formation of two-dimensional glucose isomerase crystals by means of atomic force microscopy.85 This particular investigation featured actual movies showing both crystal growth and the dissolution of precritical clusters, as well as providing information about the influence of the substrate. In addition, cryo-TEM techniques have recently provided two-dimensional snapshots of nucleation events at very low temperatures. In selected cases, where the time scales involved are again on the order of seconds, dynamical details have been obtained, as in the cases of CaCO3,86,87 metal phosphate,88 and magnetite.89
However, more often than not, crystal nucleation in liquids takes place within time windows too small (nanoseconds) to allow for a sequence of snapshots to be taken with high-spatial-resolution instruments. In these cases, microscopic insights cannot be obtained, and much more macroscopic measurements have to be performed.
In this context, several experimental approaches aim at examining a large number of independent nucleation events for a whole set of rather small configurations of the system, basically performing an ensemble average. For example, in droplet experiments, nucleation is characterized as a function of time or temperature. Freezing is identified for each nucleation event within the ensemble of available configurations by techniques such as femtosecond X-ray scattering,90,91 optical microscopy,81,82,92,93 and powder X-ray diffraction.94−96 From these data, the nucleation rate is often reconstructed by measuring either metastable zone widths97−104 or induction times105−110 (several examples are listed in, e.g., refs (111−115)), thus providing a solid connection to theoretical frameworks such as CNT (see section 1.1). An essential technical detail within this class of measurements is that the volume available for each nucleation event has to be as small as possible to reduce the occurrence of multiple nucleation events within the same configuration. High-throughput devices such as the lab-on-a-chip116 can significantly improve the statistics of the nucleation events, thus enhancing the capabilities of these approaches.
Another line of action focuses on the study of large, macroscopic systems. Freezing is detected by techniques such as differential scanning calorimetry,117−122 Fourier transform infrared spectroscopy (FTIRS),123−127 and analytical ultracentrifugation128 or by some flavor of chamber experiments.2,129−135 In this case, the frozen fraction of the overall system and/or the nucleation temperatures can be obtained, and in some cases, nucleation rates have been extracted (see Table 1).
Finally, experimental methods that can detect nucleation and the formation of the crystal (predominantly by means of optical microscopy) but do not provide any microscopic detail have helped to shed light on issues such as the role of the solvent or impurities. This is usually possible by examining the amount of crystalline phase obtained along with its structure.
Even though there are a large number of powerful experimental techniques and new ones emerging (e.g., ultrafast X-ray90), it is still incredibly challenging to obtain microscopic-level insight into nucleation from experiments. As we shall see now, MD simulations provide a powerful complement to experiments.
1.3. Molecular Dynamics Simulations
1.3.1. Brute-Force Simulations
When dealing
with crystal nucleation in liquids, atomistic simulations should provide
a detailed picture of the formation of the critical nucleus. The simplest
way to achieve this is by so-called brute-force MD simulations, which
involve cooling the system to below the freezing temperature and then
following its time evolution until nucleation is observed. Brute-force
simulations are the antagonist of enhanced-sampling simulations, where
specific computational techniques are used to alter the dynamics of
the system so as to observe nucleation on a much shorter time scale.
Monte Carlo (MC) techniques, although typically coupled with enhanced
sampling techniques, can be used to recover  ,146−148 but the calculation of
,146−148 but the calculation of 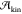 requires
other methods, such as kinetic
Monte Carlo (KMC).39 The natural choice
to simulate nucleation events is instead MD simulations, which directly
provide the temporal evolution of the system.
requires
other methods, such as kinetic
Monte Carlo (KMC).39 The natural choice
to simulate nucleation events is instead MD simulations, which directly
provide the temporal evolution of the system.
MD simulations
aimed at investigating nucleation are usually performed in the isothermal–isobaric
ensemble (NPT), where P (usually
ambient pressure) and T <  are kept constant by means of a barostat
and a thermostat, respectively. Such computational tweaking is a double-edged
sword. In fact, nucleation and most notably crystal growth are exothermic
processes,149 and within the length scale
probed by conventional atomistic simulations (1–104 nm), it is necessary to keep the system at constant temperature.
On the other hand, in this way, dynamical and structural effects in
both the liquid and the crystalline phases due to the heat developed
during the nucleation events are basically neglected.150−152 Although the actual extent of these effects is not yet clear, forcing
the sampling of the canonical ensemble is expected to be especially
dangerous when dealing with very small systems affected by substantial
finite-size effects. More importantly, thermostats and barostats affect
the dynamics of the system. Small coupling constants and clever approaches
(e.g., stochastic thermostats153) can be
employed to limit the effects of the thermostats, but in general,
care must be taken. The same reasoning applies for P and barostats as well. A density change of the system is usually
associated with nucleation,154 the crystalline
phase being more (or less, in the case of, e.g., water) dense than
the liquid parent phase.
are kept constant by means of a barostat
and a thermostat, respectively. Such computational tweaking is a double-edged
sword. In fact, nucleation and most notably crystal growth are exothermic
processes,149 and within the length scale
probed by conventional atomistic simulations (1–104 nm), it is necessary to keep the system at constant temperature.
On the other hand, in this way, dynamical and structural effects in
both the liquid and the crystalline phases due to the heat developed
during the nucleation events are basically neglected.150−152 Although the actual extent of these effects is not yet clear, forcing
the sampling of the canonical ensemble is expected to be especially
dangerous when dealing with very small systems affected by substantial
finite-size effects. More importantly, thermostats and barostats affect
the dynamics of the system. Small coupling constants and clever approaches
(e.g., stochastic thermostats153) can be
employed to limit the effects of the thermostats, but in general,
care must be taken. The same reasoning applies for P and barostats as well. A density change of the system is usually
associated with nucleation,154 the crystalline
phase being more (or less, in the case of, e.g., water) dense than
the liquid parent phase.
Three conditions must be fulfilled
to extract 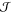 from brute-force
MD simulations:
from brute-force
MD simulations:
-
(1)
The system must be allowed to evolve in time until spontaneous fluctuations lead to a nucleation event.
-
(2)
The system size must be significantly larger than the critical nucleus.
-
(3)
Significant statistics of nucleation events must be collected.
Each of these conditions is surprisingly difficult to fulfill. The most daunting obstacle is probably the first one because of the so-called time-scale problem.155,156 In most cases, nucleation is a rare event, meaning that it usually occurs on a very long time scale; precisely how long depends strongly on ΔT. A rough estimate of the number of simulation steps required to observe a nucleation event within a molecular dynamics run is reported in Figure 5. Under the fairly optimistic assumption that classical MD simulations can cope with up to ∼105 molecules on a time scale of nano-/microseconds, there is only a very narrow set of conditions for which brute-force classical MD simulations could be used to investigate nucleation, usually only at strong supercooling. Time scales typical of first-principles simulations, also reported in Figure 5 assuming up to ∼102 molecules, indicate that unbiased ab initio simulations of nucleation events are unfeasible.
Figure 5.

Nucleation rate 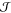 as a
function of the simulation time needed
within an MD simulation to observe a single nucleation event. The
blue shaded region highlights the approximate simulation times currently
affordable by classical MD simulations; clearly, only very fast nucleation
processes can be simulated with brute-force MD. For homogeneous ice
nucleation,
as a
function of the simulation time needed
within an MD simulation to observe a single nucleation event. The
blue shaded region highlights the approximate simulation times currently
affordable by classical MD simulations; clearly, only very fast nucleation
processes can be simulated with brute-force MD. For homogeneous ice
nucleation, 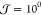 and
and 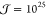 can typically be observed for ΔT = 30 K and
ΔT = 80 K, respectively.
In the derivations of classical and ab initio simulation times, 105 and 102 molecules, respectively, were considered,
together with the number density of a generic supercooled liquid,
can typically be observed for ΔT = 30 K and
ΔT = 80 K, respectively.
In the derivations of classical and ab initio simulation times, 105 and 102 molecules, respectively, were considered,
together with the number density of a generic supercooled liquid, 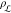 = 0.01 molecules·Å–3.
= 0.01 molecules·Å–3.
The second important condition is the size of the system. The number of atoms (or molecules) in the system defines the time scale accessible to the simulation and, thus, the severity of the time-scale problem. The reason large simulation boxes, significantly larger than the size of the critical nucleus, are needed is because periodic boundary conditions will strongly affect nucleation (and growth) if even the precritical nuclei are allowed to interact with themselves. This usually leads to unrealistically high nucleation rates. This issue worsens at mild supercooling, where the critical nucleus size rapidly increases toward dimensions not accessible by MD simulations.
Third, it is not sufficient to collect information on just one nucleation event. Nucleation is a stochastic event following a Poisson distribution (at least ideally; see section 1.1), and so to obtain the nucleation rate, one needs to accumulate decent statistics.
Taking these issues
into consideration, various approaches for
obtaining 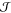 have emerged.
One approach, known as the
Yasuoka–Matsumoto method,157 involves
simulating a very large system, so that different nucleation events
can be observed within a single run. In this case, large simulation
boxes are needed to collect sufficient statistics and to avoid spurious
interactions between different nuclei. Another family of methods involves
running many different simulations using much smaller systems, which
is usually computationally cheaper. Once a collection of nucleation
events has been obtained, several methods for extracting
have emerged.
One approach, known as the
Yasuoka–Matsumoto method,157 involves
simulating a very large system, so that different nucleation events
can be observed within a single run. In this case, large simulation
boxes are needed to collect sufficient statistics and to avoid spurious
interactions between different nuclei. Another family of methods involves
running many different simulations using much smaller systems, which
is usually computationally cheaper. Once a collection of nucleation
events has been obtained, several methods for extracting 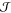 can be employed.
The simplest ones (mean
lifetime158 and survival probability159,160 methods) involve the fitting of the nucleation times to Poisson
statistics. A more in-depth technique, the so-called mean first-passage
method,161 allows for a detailed analysis
of the nucleus population but requires a probability distribution
in terms of nucleus size.
can be employed.
The simplest ones (mean
lifetime158 and survival probability159,160 methods) involve the fitting of the nucleation times to Poisson
statistics. A more in-depth technique, the so-called mean first-passage
method,161 allows for a detailed analysis
of the nucleus population but requires a probability distribution
in terms of nucleus size.
The literature offers a notable number of works in which brute-force MD simulations have been successfully applied. Most of them rely on one approach to circumvent the above-mentioned issues, particularly the time-scale problem. As we shall see in section 2, to simulate nucleation events, one almost always has to either choose a very simple system or increase the level of approximation sometimes dramatically, for instance, by coarse-graining the interatomic potential used.
1.3.2. Enhanced-Sampling Simulations
In the previous section, we introduced the time-scale problem, the main reason brute-force MD simulations are generally not feasible when studying crystal nucleation. Enhanced sampling methods alter how the system explores its configurational space, so that nucleation events can be observed within a reasonable amount of computational time. Broadly speaking, one can distinguish between free-energy methods and path-sampling methods, both of which have been extensively discussed elsewhere (see, e.g., refs (156 and 162−164)). Thus, only the briefest of introductions is needed here.
Of the many enhanced sampling methods, only a handful have been successfully
used to compute crystal nucleation rates. This is because information
is needed about both the thermodynamics of the system (the free energy
barrier for nucleation  ) and
the kinetics of the nucleation process
(the kinetic prefactor
) and
the kinetics of the nucleation process
(the kinetic prefactor 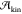 ). When dealing with crystal nucleation
in supercooled liquids, free-energy-based methods are rather common,
such as umbrella sampling (US)165−167 and metadynamics.168−170 In both cases, and indeed in almost all enhanced sampling methods
currently available, the free energy surface of the actual system
is coarse-grained by means of one or more order parameters or collective
variables. The choice of the order parameter is not trivial and can
have dramatic consequences. An external bias is then applied to the
system, leading to a modified sampling of the configurational space
that allows for the reconstruction of the free energy profile with
respect to the chosen order parameter and, thus, for the computation
of the free energy barrier. This approach has been successful in a
number of cases. However, there is a price to be paid: Upon introduction
of an extra term into the system Hamiltonian, the actual dynamics
of the system is to some extent hampered, and much of the insight
into the nucleation mechanism is lost. Moreover,
). When dealing with crystal nucleation
in supercooled liquids, free-energy-based methods are rather common,
such as umbrella sampling (US)165−167 and metadynamics.168−170 In both cases, and indeed in almost all enhanced sampling methods
currently available, the free energy surface of the actual system
is coarse-grained by means of one or more order parameters or collective
variables. The choice of the order parameter is not trivial and can
have dramatic consequences. An external bias is then applied to the
system, leading to a modified sampling of the configurational space
that allows for the reconstruction of the free energy profile with
respect to the chosen order parameter and, thus, for the computation
of the free energy barrier. This approach has been successful in a
number of cases. However, there is a price to be paid: Upon introduction
of an extra term into the system Hamiltonian, the actual dynamics
of the system is to some extent hampered, and much of the insight
into the nucleation mechanism is lost. Moreover, 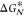 is only half of the story. To obtain
is only half of the story. To obtain 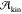 , one
needs complementary methods, usually
aimed at estimating the probability for the system on top of the nucleation
barrier—in the space of the selected order parameter—to
get back to the liquid phase or to evolve into the crystal. Most frequently,
such methods are based on some flavor of transition state theory,171−174 such as the Bennett–Chandler formulation,175,176 and require a massive set of MD or KMC simulations to be performed.
, one
needs complementary methods, usually
aimed at estimating the probability for the system on top of the nucleation
barrier—in the space of the selected order parameter—to
get back to the liquid phase or to evolve into the crystal. Most frequently,
such methods are based on some flavor of transition state theory,171−174 such as the Bennett–Chandler formulation,175,176 and require a massive set of MD or KMC simulations to be performed.
On the other hand, the ever-growing family of path-sampling methods can provide direct access to the kinetics of the nucleation process. These approaches again rely on the definition of an order parameter, but instead of applying an external bias potential, an importance sampling is performed so as to enhance the naturally occurring fluctuations of the system. Within the majority of the path-sampling approaches currently used, including transition interface sampling177−179 (TIS) and forward flux sampling180,181 (FFS), the ensemble of paths connecting the liquid and the crystal is divided into a series of interfaces according to different values of the order parameter. By sampling the probability with which the system crosses each of these interfaces, a cumulative probability directly related to the nucleation rate can be extracted. Other path-sampling techniques such as transition path sampling182,183 (TPS) rely instead on the sampling of the full ensemble of the reactive trajectories. In both cases, by means of additional simulations involving, for example, committor analysis distribution184 and thermodynamic integration,185 one can subsequently extract the size of the critical nucleus and the free energy difference between the solid and liquid phases, respectively. Many different path-sampling methods are available, but to our knowledge, only TPS, TIS, and most prominently FFS have allowed for estimates of crystal nucleation rates. Under certain conditions, path-sampling methods do not alter the dynamics of the system, allowing for invaluable insight into the nucleation mechanism. However, they are particularly sensitive to the slow dynamics of strongly supercooled systems, which hinder the sampling of the paths and makes them exceptionally expensive computationally. Although the past few decades have taught us that enhanced sampling techniques are effective in tackling crystal nucleation of colloids (see section 2.1), Lennard-Jones melts (see section 2.2), and other atomic liquids (see section 2.3), only recently have these techniques been applied to more complex systems.
One challenging scenario for simulations of nucleation is provided by the formation of crystals from solutions characterized by very low solute concentration. Although this occurrence is often encountered in real systems of practical interest, it is clearly extremely difficult for MD simulations, even if aided by conventional enhanced sampling techniques, to deal with just a few solute molecules dissolved within 103–106 solvent molecules. In these cases, the diffusion of the solute plays a role of great relevance, and the interaction between solvent and solute can enter the nucleation mechanism itself. Thus, obtaining information about the thermodynamics, let alone the kinetics, of nucleation at very low solute concentrations is presently a formidable task. However, efforts have been devoted to further our understanding of solute migration and solute-nuclei association, for example, as demonstrated by the pioneering works of Gavezzotti and co-workers186,187 and more recently by Kawska and co-workers.188,189 In the latter work, the authors illustrate an approach that relies on the modeling of the subsequent growth step, where solute particles (often ions) are progressively added to the (crystalline or not) cluster. After each of these growth steps, a structural optimization of the cluster and the solvent by means of MD simulations is performed. Although this method cannot provide quantitative results in terms of the thermodynamics and/or the kinetics of nucleation, it can, in principle, provide valuable insight into the very early stages of crystal nucleation when dealing with solutions characterized by very low solute concentrations.
On a final note, we mention seeded MD simulations. This technique relies on simulations in which a crystalline nucleus of a certain size is inserted into the system at the beginning of the simulation. Although useful information about critical nucleus size can be obtained in this way,51,190−192 the method does not usually allow for a direct calculation of the nucleation rate. However, seeded MD simulations are one of the very few methods by which it is currently possible to investigate solute precipitate nucleation (see, e.g., Knott et al.193). In this case, the exceedingly low attachment rate of the solute often prevents both free-energy- and transition-path-sampling-enhanced sampling methods from being applied effectively.
As we shall see in the next few sections, the daunting computational costs, together with the delicate choice of order parameter and the underlying framework of CNT, still make enhanced-sampling simulations of crystal nucleation in liquids an intimidating challenge.
2. Selected Systems
We have chosen to review different classes of systems, which we present in order of increasing complexity. We start in sections 2.1 and 2.2 with colloids and Lennard-Jones liquids, respectively. These systems are described by simple interatomic potentials that allow large-scale MD simulations, and thus with them, many aspects of CNT can be investigated and nucleation rates calculated. In some cases, the latter can can be directly compared to experimental results. As such, colloids and Lennard-Jones liquids represent a sort of benchmark for MD simulations of crystal nucleation in liquids, although we shall see that our understanding of crystal nucleation is far from satisfactory even within these relatively easy playgrounds. In section 2.3, we discuss selected atomic liquids of technological interest such as liquid metals, supercooled liquid silicon, and phase-change materials for which nucleation occurs on very small time scales. As the first example of a molecular system, we then focus on the most important liquid of them all, water. We review the body of computational work devoted to unraveling both the homogeneous (section 2.4.1) and heterogeneous (section 2.4.2) formation of ice, offering a historical perspective guiding the reader through the many advances that have furthered our understanding of ice nucleation in the past decades.
Next, we present an overview of nucleation from solution (section 2.5), where simulations have to deal with solute and solvent. We take into account systems of great practical relevance such as urea molecular crystals, highlighting the complexity of the nucleation mechanism, which is very different from what CNT predicts. Finally, section 2.6 is devoted to the formation of gas hydrates.
As a general rule, increasing the complexity of the system raises
more questions about the validity of the assumptions underpinning
CNT. The reader will surely notice that simulations have revealed
many drawbacks of CNT along the way and that reaching decent agreement
for the nucleation rate 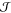 between experiments
and simulations still
remains a formidable task.
between experiments
and simulations still
remains a formidable task.
2.1. Colloids
Hard-sphere model systems take a special place in nucleation studies. One reason for this is the simplicity of the interatomic potential customarily used to model them: The only interaction a hard-sphere particle experiences comes from elastic collisions with other particles. Because there is no attractive force between particles, a hard-sphere system is entirely driven by entropy. As a consequence, the phase diagram is very simple and can be entirely described with one single parameter, the volume fraction Φ. Only two different phases are possible: a fluid and a crystal. At volume fractions Φ < 0.494, the system is in its fluid state; at 0.492 < Φ < 0.545, the system will be a mixture of fluid and crystalline states; and at Φ > 0.545, the thermodynamically most stable phase is the crystal. The transformation from fluid to crystal occurs through a first-order phase transition.194 Despite their simplicity, systems behaving like hard spheres can be prepared experimentally. Colloids made of polymers are commonly used for this purpose, the most prominent example being poly(methyl methacrylate) (PMMA) spheres coated with a layer of poly(12-hydroxystearic acid). After the spheres have been synthesized, they are dissolved in a mixture of cis-decaline and tetraline, which enables the use of a wide range of powerful optical techniques to investigate nucleation.195,196 The possibility of using these large hard spheres in nucleation experiments has two major advantages: First, a particle size larger than the wavelength used in microscopy experiments makes it possible to track the particle trajectories in real space. In additional, nucleation occurs in a matter of seconds, which allows experimentalists to follow the complete nucleation process in detail. Compared to other systems, it is therefore possible to observe the critical nucleus directly, for example, by confocal microscopy (see section 1.2), which is of crucial importance for understanding nucleation. These qualities of hard-sphere systems make them ideal candidates to advance our understanding of nucleation. As such, it is not surprising that the freezing of hard spheres is better characterized than any other nucleation scenario, and in fact, a number of excellent reviews in this field already exist.197−202 Our aim here is thus not to give a detailed overview of the field but to highlight some of the milestones and key discoveries and connect them to other nucleation studies. To keep the discussion reasonably brief, we limit the latter to neutral and perfectly spherical hard-sphere systems. However, we note that a sizable amount of work has been devoted to a diverse range of colloidal systems, such as nonspherical particles,203−208 charged particles, and mixtures of different colloidal particles,209−214 to name just a few.
Readers interested in the state of the
art in about 2000 are referred to other reviews.197,198 In the early 2000s, two major advances in the field were made, one
on the theoretical side and the other experimentally. In 2001, Auer
and Frenkel39 computed absolute nucleation
rates of a hard-sphere system using KMC simulations. They did so by
calculating Pcrit, the probability of
forming a critical nucleus spontaneously, and 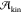 , the
kinetic prefactor. This made a direct
comparison between simulations and experiments possible. The outcome
was surprising and worrisome. They found that the experimental and
theoretical nucleation rates disagreed by several orders of magnitude.
This was surprising, because simulations did really well in describing
all sorts of properties of hard spheres before. It was worrisome because
only very few sound approximations were made by Auer and Frenkel to
obtain their nucleation rates. Their theoretical approach seemed to
be as good as it gets. The authors’ suggestion, that the problem
lay in experiments or, more precisely, in the interpretation of experiments,
showed a possible way to resolve the discrepancy. In the same year,
Gasser et al.83 conducted ground-breaking
experiments, imaging the nucleation of a colloidal suspension in real
space using confocal microscopy. Four snapshots of their system containing
approximately 4000 particles are shown in Figure 6. This was a significant step, because nucleation
had previously been investigated indirectly, using the structure factor
obtained from light-scattering experiments, for example. In their
study, they were able to directly measure the size of a critical nucleus
for the first time. Achieving sufficient temporal and spatial resolution
at the same time is possible thus far only for colloidal systems (for
more details about experimental techniques, see section 1.2). They found that the nucleus
was rather aspherical with a rough surface; both of these effects
are completely neglected in CNT. Note that aspherical nuclei also
appear in LJ systems, for example (see section 2.2.1). In addition, a random hexagonal close-packed
(rhcp) structure for the hard spheres was observed, in good agreement
with Auer and Frenkel.39 This is interesting,
because slightly different systems such as soft spheres and Lennard-Jones
particles seem to favor body-centered-cubic (bcc) stacking. However,
Gasser et al.’s study did not resolve the discrepancy between
experimental and simulated nucleation rates, as their results were
in agreement with earlier small-angle light-scattering experiments.215
, the
kinetic prefactor. This made a direct
comparison between simulations and experiments possible. The outcome
was surprising and worrisome. They found that the experimental and
theoretical nucleation rates disagreed by several orders of magnitude.
This was surprising, because simulations did really well in describing
all sorts of properties of hard spheres before. It was worrisome because
only very few sound approximations were made by Auer and Frenkel to
obtain their nucleation rates. Their theoretical approach seemed to
be as good as it gets. The authors’ suggestion, that the problem
lay in experiments or, more precisely, in the interpretation of experiments,
showed a possible way to resolve the discrepancy. In the same year,
Gasser et al.83 conducted ground-breaking
experiments, imaging the nucleation of a colloidal suspension in real
space using confocal microscopy. Four snapshots of their system containing
approximately 4000 particles are shown in Figure 6. This was a significant step, because nucleation
had previously been investigated indirectly, using the structure factor
obtained from light-scattering experiments, for example. In their
study, they were able to directly measure the size of a critical nucleus
for the first time. Achieving sufficient temporal and spatial resolution
at the same time is possible thus far only for colloidal systems (for
more details about experimental techniques, see section 1.2). They found that the nucleus
was rather aspherical with a rough surface; both of these effects
are completely neglected in CNT. Note that aspherical nuclei also
appear in LJ systems, for example (see section 2.2.1). In addition, a random hexagonal close-packed
(rhcp) structure for the hard spheres was observed, in good agreement
with Auer and Frenkel.39 This is interesting,
because slightly different systems such as soft spheres and Lennard-Jones
particles seem to favor body-centered-cubic (bcc) stacking. However,
Gasser et al.’s study did not resolve the discrepancy between
experimental and simulated nucleation rates, as their results were
in agreement with earlier small-angle light-scattering experiments.215
Figure 6.

Crystallization of PMMA with Φ = 0.45 observed by confocal microscopy. Red (large) and blue (small) spheres show crystal- and liquid-like particles, respectively. The size of the observed volume is 58 μm by 55 μm by 20 μm, containing about 4000 particles. After shear melting of the sample, snapshots were taken after (A) 20, (B) 43, (C) 66, and (D) 89 min. The time series shows how an aspherical nucleus forms and grows over time. Reprinted with permission from ref (83). Copyright 2001 American Association for the Advancement of Science.
Much of the subsequent work focused on trying to resolve this discrepancy between experiments and simulation. A step forward was made in 2006 and 2007.216,217 Schöpe et al. found experimental evidence supporting a two-step crystallization process (see section 1.1.2) in hard-sphere systems. Other systems such as proteins and molecules in solution (see section 2.5) were well-known at that time to crystallize through a more complex mechanism than that assumed by CNT. Even for hard-sphere systems, two-step nucleation processes were reported before 2006;218−220 the occurrence of this mechanism was attributed to details of the polydispersity of the hard spheres, however. The new insight provided by Schöpe et al. in 2006 and 2007 was that the two-step nucleation process is general, and as such, it does not depend on either polydispersity or volume fraction. In 2010, simulations performed by Schilling et al.221 supported these experimental findings. Using unbiased MC simulations, Schilling et al. were able to reproduce the evolution of the structure factor from previous experiments. Not even the simplest model system seemed to follow the traditional picture assumed in CNT. Could this two-step mechanism explain why the computational rates39 disagreed with experiments? At first, it seems like a tempting explanation, because Auer and Frenkel39 had to introduce order parameters to calculate absolute nucleation rates. Such a conclusion, however, automatically presupposes a reaction pathway, which might not necessarily match the nucleation pathway taken in experiments. Filion et al.222 showed in the same year, however, that very different computational approaches [brute-force MD, US, and FFS, which we described earlier (see section 1.3.2)] led to the same nucleation rates, all in agreement with Auer and Frenkel.39 They therefore concluded that the discrepancy between simulations and experiments did not lie in the computational approach employed by Auer and Frenkel. They offered two possible explanations, one being that hydrodynamic effects, completely neglected in the simulations, might play a role and the other being possible difficulties in interpreting the experiments. Schilling et al.147 tried to address one of the key issues when comparing experiments with simulations: uncertainties and error estimation. Whereas the determination of the most characteristic quantity in hard-sphere systems, the volume fractions, is straightforward for simulations, experimentalists are confronted with a more difficult task in this case. The typical error in determining the volume fraction experimentally is about ±0.004, which translates into an uncertainty in the nucleation rate of about an order of magnitude. Upon taking these considerations into account, the authors concluded that the discrepancy can be explained by statistical errors and uncertainties.
Does this mean that the past 10 years of research tried to explain a discrepancy that is actually not there? Filion et al.223 rightfully pointed out that, whereas the rates between experiments and simulations coincide at high volume fraction, they still clearly disagree in the low-volume-fraction regime. No simple rescaling justified by statistical uncertainty could possibly resolve that discrepancy. In their article, they also addressed a different issue. In a computational study in 2010, Kawasaki and Tanaka224 obtained, by means of Brownian dynamics,225 nucleation rates in good agreement with experiments, contrary to the nucleation rates computed by Auer and Frenkel using brute-force MD.39 It should be noted that Kawasaki and Tanaka did not use a pure hard-sphere potential, but used a Weeks–Chandler–Andersen potential instead. Was the approximation of a hard-sphere system, something that can never be fully realized in experiments, the problem all the time? What Filion et al. showed is that different computational approaches (brute-force MD, US, and FSS) all lead to the same nucleation rates, all of them in disagreement with what Kawasaki and Tanaka found. Through a detailed evaluation of their approach and that of Kawasaki and Tanaka, they concluded that their rates are more reliable. The discrepancy was back on the table, where it still remains and is as large as ever.
For a detailed comparison between experimental and computational rates, the reader is referred to ref (202). The message we want to convey here is that the disagreement between simulations and experiments in the simplest system still persists today. It is worth mentioning that this fundamental disagreement between simulations and experiments is not unique to colloids. Other systems such as water (sections 2.4.1 and 2.4.2) and molecules in solution (section 2.5) also show discrepancies of several orders of magnitude in nucleation rates. This long-standing debate is of great relevance to all investigations dealing with systems modeled using any flavor of hard-sphere potential. A notable example in this context is the crystallization of proteins, which are usually treated as hard spheres. Despite basically neglecting most of the complexity of these systems, this substantial approximation has allowed for a number of computational studies226−236 that, although outside the scope of this review, certainly contributed to furthering our understanding of the self-assembly of biological particles.
2.2. Lennard-Jones Liquids
Beyond hard spheres, the first step toward more realistic systems involves the inclusion of attractive interactions. The Lennard-Jones liquid is a widely studied model system that does just that. It can be seen as the natural extension of the hard-sphere model, to which it becomes equivalent when the strength of the attractive interactions goes to zero. LJ liquids were first introduced in 1924,237 and since then, they have been the subject of countless computational studies. LJ potentials allow for exceedingly fast MD simulations, and a wide range of thermodynamic information is available for them, such as the phase diagram238−242 and the interfacial free energy.243−245
The stable structure of the LJ system up to 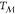 is a face-centered-cubic (fcc)
crystal;
slightly less stable in free energy is a hexagonal-close-packed (hcp)
structure, which, in turn, is significantly more stable then a third
body-centered-cubic (bcc) phase.246,247 With his
study of liquid argon in 1964, Rahman reported what is probably the
first LJ MD simulation.248 His findings
showed good agreement with experimental data for the pair distribution
function and the self-diffusion coefficient, thus demonstrating that
LJ potentials can properly describe noble elements in their liquid
form at ambient pressure. This conclusion was validated later by Verlet249 and McGinty.250 To
the best of our knowledge, nucleation of LJ liquids was investigated
for the first time in 1969 by de Wette et al.240 and in 1976 by Mandell et al.251 for two-dimensional and three-dimensional systems, respectively.
is a face-centered-cubic (fcc)
crystal;
slightly less stable in free energy is a hexagonal-close-packed (hcp)
structure, which, in turn, is significantly more stable then a third
body-centered-cubic (bcc) phase.246,247 With his
study of liquid argon in 1964, Rahman reported what is probably the
first LJ MD simulation.248 His findings
showed good agreement with experimental data for the pair distribution
function and the self-diffusion coefficient, thus demonstrating that
LJ potentials can properly describe noble elements in their liquid
form at ambient pressure. This conclusion was validated later by Verlet249 and McGinty.250 To
the best of our knowledge, nucleation of LJ liquids was investigated
for the first time in 1969 by de Wette et al.240 and in 1976 by Mandell et al.251 for two-dimensional and three-dimensional systems, respectively.
2.2.1. Nonspherical Nuclei
Early simulations252,253 investigating the condensation of LJ vapors into a liquid already indicated a substantial discrepancy with CNT rates. It is worth noticing that the order parameter for crystal-like particles presented by ten Wolde et al.252 fostered a considerable amount of later work devoted to improving the order parameters customarily used to describe crystal nucleation from the liquid phase (see, e.g., ref (254)). In 2008, Kalikmanov et al.255 compared CNT and cDFT (see section 1.1) simulations with condensation data for argon. They found that CNT spectacularly failed to reproduce experimental condensation rates, underestimating them by up to 26 orders of magnitude. This disagreement triggered a number of computational studies aimed at clarifying the assumption of the sphericity of the critical nucleus within the freezing of LJ liquids. By embedding pre-existing spherical clusters into supercooled LJ liquids, Bai and Li256,257 found values of the critical nucleus size in excellent agreement with CNT within a broad range of temperatures. However, these results have been disputed by the umbrella sampling simulations of Wang et al.,258 for example, as well as the path-sampling investigation of Moroni et al.259 In both cases, the nuclei became less spherical with increasing ΔT. In addition, Moroni et al. pointed out that the critical nucleus size is determined by a nontrivial interplay between the shape, the size, and the degree of crystallinity of the cluster. Such a scenario is clearly much more complex than the usual CNT picture, as it violates the capillarity approximation (see 1.1.1). Nonspherical nuclei were also observed by Trudu et al.,62 who extended the conventional CNT formula to account for ellipsoidal nuclei. Such a tweak gave much better estimations of both the critical nucleus size and the nucleation barrier. Recall that the shape of the critical nuclei can be observed experimentally in very few cases (see sections 1.2 and 2.1).
However, at
very strong supercooling, things fell apart because of the emergence
of spinodal effects (see section 1.1). Note that CNT fails at strong supercooling even
without the occurrence of spinodal effects, as the time lag (transient
time) needed for structural relaxation into the steady-state regime
results in a time-dependent nucleation rate.19 For instance, Huitema et al.260 showed
that incorporating the time dependence into the kinetic prefactor
yields an improved estimate of nucleation rates. In fact, by embedding
extensions to the original CNT framework, one can, in some cases,
recover a reasonable agreement between simulations and experiments
even at strong supercooling. As an example, Peng et al.261 also showed that including enthalpy-based terms
in the formulation of the temperature dependence of 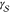 substantially improves
the outcomes of
CNT.
substantially improves
the outcomes of
CNT.
2.2.2. Polymorphism
Another aspect that
has been thoroughly addressed within the crystal nucleation of LJ
liquids is the structure of the crystalline clusters involved. The
mean-field theory approach of Klein and Leyvraz262 suggests a decrease of the nucleus density as well as an
increase of the bcc character when moving toward the spinodal region.
These findings were confirmed by the umbrella sampling approach of
ten Wolde et al.,252,263,264 who reported a bcc shell surrounding fcc cores. Furthermore, Wang
et al.258 showed that the distinction between
the crystalline clusters and the surrounding liquid phase falls off
as a function of ΔT. In fact, the free energy
barrier for nucleation, computed by means of umbrella sampling simulations
(see section 1.3.2), was found to be on the order of kBT at ΔT = 52%. In addition,
the nuclei undergo substantial structural changes toward nonsymmetric
shapes, a finding validated by the metadynamics simulations of Trudu
et al.62 The same authors investigated
the nucleation mechanism close to the critical temperature for spinodal
decomposition, 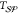 (see section 1.1.4), where the free energy basin corresponding
to the liquid phase turned out to be ill-defined, that is, already
overlapping with the free energy basin of the crystal. Such a finding
suggested that, below
(see section 1.1.4), where the free energy basin corresponding
to the liquid phase turned out to be ill-defined, that is, already
overlapping with the free energy basin of the crystal. Such a finding
suggested that, below 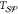 , there is no free energy barrier for nucleation,
indicating that the liquid is unstable rather than metastable and
that the crystallization mechanism has changed from nucleation toward
the more collective process of spinodal decomposition (see section 1.1.4 and Figure 1).
, there is no free energy barrier for nucleation,
indicating that the liquid is unstable rather than metastable and
that the crystallization mechanism has changed from nucleation toward
the more collective process of spinodal decomposition (see section 1.1.4 and Figure 1).
Insights into the interplay between nucleation and polymorphism have been provided by the simulations of ten Wolde et al.,252,263,264 among others, suggesting that, within the early stages of the nucleation process, the crystalline clusters are bcc-like, later turning into fcc crystalline kernels surrounded by bcc shells. These findings were validated by Desgranges and Delhommelle265 and Wang et al.258
More recently, Wang et al.266 performed a cDFT study to determine the difference between the free energy barrier for nucleation required for the creation of an fcc or bcc critical nucleus. In addition, the difficulty for nucleation of the three different crystal orientations for fcc was ranked (100) > (110) > (111). These studies confirm the presence of a two-step mechanism (see section 1.1.2) and the validity of Ostwald’s step rule267 for the LJ model. As we will see later (e.g., homogeneous ice nucleation, section 2.4.1), nucleation through metastable phases has also been observed for more complicated liquids. Important contributions regarding polymorph control during crystallization were made by Desgranges and Delhommelle,247,265,268 who investigated nucleation under different thermodynamic conditions. By keeping the temperature constant and altering the pressure, they were able to influence the amount of bcc particles. This reached up to a point where the nucleus was almost purely bcc-like. Calculation of the bcc–liquid line in the phase diagram showed that these nucleation events occurred in the bcc existence domain. Additionally, the transformation from fcc to hcp during crystal growth, well after the critical nucleus size has been reached, was studied by changing the temperature at constant pressure. As depicted in Figure 7, at ΔT = 10%, a small number of hcp atoms were observed surrounding the fcc core, whereas at ΔT = 22%, much larger hcp domains formed within the crystallite, suggesting that the conversion from hcp to fcc is hindered at higher temperatures. On a final note, we emphasize that many findings related to polymorphism are often quite dependent on the choice of the order parameters employed. This issue is not limited to LJ systems, and it is especially important when dealing with similarly dense liquid and crystalline phases (e.g., metallic liquids), where order parameters usually struggle to properly distinguish the different crystalline phases from the liquid.268 In particular, it remains to be seen whether the fractional bcc, fcc, and hcp contents of the LJ nuclei that we have discussed will stand the test of the last generation of order parameters.
Figure 7.

Cross section of postcritical crystalline clusters of 5000 LJ particles for ΔT = (a) 10% and (b) 22%. fcc-, hcp-, and bcc-like particles are depicted in gray, yellow, and red, respectively. At ΔT = 22% substantial hcp domains form within the crystallite, whereas at ΔT = 10%, hcp particles can be found almost exclusively on the surface of the fcc core. Reprinted with permission from ref (247). Copyright 2007 American Physical Society.
2.2.3. Heterogeneous Nucleation
Heterogeneous
crystal nucleation has also been investigated for a variety of LJ
systems. For instance, Wang et al.258 used
umbrella-sampling simulations (see section 1.3.2) to calculate the free energy barrier
for heterogeneous nucleation of an LJ liquid on top of an ideal impurity,
represented by a single fcc (111) layer of LJ particles. By explicitly
varying the lattice spacing of the substrate, asub, they calculated 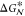 as a
function of asub – aequi, where aequi is the lattice
spacing of the equilibrium
crystalline phase.269 They found that
as a
function of asub – aequi, where aequi is the lattice
spacing of the equilibrium
crystalline phase.269 They found that  displays
a minimum for asub – aequi = 0, whereas
for large values of asub – aequi, nucleation proceeds within the bulk of
the supercooled liquid phase. These findings support the early argument
of the zero lattice mismatch introduced by Turnbull and Vonnegut270 to justify the striking effectiveness of AgI
crystals in promoting heterogeneous ice nucleation. In fact, in several
situations, one can define a disregistry or lattice mismatch δ
as
displays
a minimum for asub – aequi = 0, whereas
for large values of asub – aequi, nucleation proceeds within the bulk of
the supercooled liquid phase. These findings support the early argument
of the zero lattice mismatch introduced by Turnbull and Vonnegut270 to justify the striking effectiveness of AgI
crystals in promoting heterogeneous ice nucleation. In fact, in several
situations, one can define a disregistry or lattice mismatch δ
as
| 7 |
Values of δ close to or even
equal to
zero have often been celebrated as the main ingredient that makes
a crystalline impurity particularly effective in promoting heterogeneous
nucleation. However, the universality of this concept has been severely
questioned in the past few decades, as we shall see in section 2.4.2 for heterogeneous
ice nucleation. Nonetheless, it seems that the argument regarding
zero lattice mismatch can hold for certain simple cases, as demonstrated
by Mithen and Sear,271 who studied heterogeneous
nucleation of LJ liquids on the (111) and (100) faces of an fcc crystal
by means of FFS simulations (see section 1.3.2). They reported a maximum in the heterogeneous
nucleation rate for a small, albeit nonzero, value of δ (see Figure 8). The difference
between their study and that of Wang et al.258 is simply that many more values of δ were taken into account
by Mithen and Sear,271 thus allowing the
maximum of 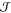 to be determined
more precisely. On a different
note, Dellago et al.272 performed TIS simulations
(see section 1.3.2) to investigate heterogeneous crystal nucleation of LJ supercooled
liquids on very small crystalline impurities. They found that even
tiny crystalline clusters of just ∼10 LJ particles can actively
promote nucleation and that the morphology of the substrate can play
a role as well. Specifically, whereas fcc-like clusters were rather
effective in enhancing nucleation rates, no substantial promotion
was observed for icosahedrally ordered seeds.
to be determined
more precisely. On a different
note, Dellago et al.272 performed TIS simulations
(see section 1.3.2) to investigate heterogeneous crystal nucleation of LJ supercooled
liquids on very small crystalline impurities. They found that even
tiny crystalline clusters of just ∼10 LJ particles can actively
promote nucleation and that the morphology of the substrate can play
a role as well. Specifically, whereas fcc-like clusters were rather
effective in enhancing nucleation rates, no substantial promotion
was observed for icosahedrally ordered seeds.
Figure 8.

Nucleation rates computed with the FFS method for a rigid hexagonal surface of LJ atoms in contact with a LJ liquid. Potentials 1 and 2 describe the interaction between substrate and liquid and differ only slightly by the value of σ they use. Error bars are standard deviations from five FFS runs. These results show that the maximum in the nucleation rate occurs at nonzero values of the lattice mismatch δ. Reprinted with permission from ref (271). Copyright 2014 AIP Publishing LLC.
MC simulations performed by Page
and Sear273 have demonstrated that confinement
effects can be of great relevance
as well. They computed heterogeneous nucleation rates for a LJ liquid
walled inbetween two flat crystalline planes characterized by a certain
angle θsub. A maximum of 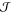 was
found for a specific value of θsub, boosting the
nucleation rate by several orders of magnitude
with respect to the promoting effect of a flat crystalline surface.
In addition, different values of θsub led to the
formation of different crystalline polymorphs.
was
found for a specific value of θsub, boosting the
nucleation rate by several orders of magnitude
with respect to the promoting effect of a flat crystalline surface.
In addition, different values of θsub led to the
formation of different crystalline polymorphs.
Finally, Zhang et al.274 recently probed the influence of structured and structureless LJ potential walls or nucleation rates. Both types of wall were found to increase the temperature at which nucleation occurs. However, this effect became negligible when moving toward vanishingly small liquid–wall interaction strengths. We shall see in section 2.4.2 that the interplay between the morphology of the substrate and the strength of the liquid–substrate interaction can lead to a diverse range of nucleation behavior.
2.2.4. Finite-Size Effects
MD simulations
of LJ liquids are computationally cheap, making them the perfect candidates
to examine how finite-size effects impact crystal nucleation. The
seminal work of Honeycutt and Andersen275 took into account up to 1300 LJ particles at 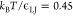 , which turned out to be too
few particles
to completely rule out the effects of periodic boundary conditions.
In fact, the authors suggested that extra care had to be taken because
of the diffuseness of the interface between the supercooled liquid
phase and the crystalline nucleus, which can induce an artificial
long-range order in the system, leading to a nonphysically high nucleation
rate. These findings are particularly relevant, as the critical nucleus
size at this ΔT value is on the order of just
a few tens of particles, representing a tiny fraction of the whole
system. Only a few years later, Swope and Andersen276 investigated the same effects by taking into account up
to 106 LJ particles subjected to the same strong supercooling
as probed by Honeycutt and Andersen.275 According to their large-scale MD simulations, 15000 particles seem
to be sufficient to avoid finite-size effects. This outcome must be
carefully pondered, as currently, the vast majority of simulations
dealing with crystallization of realistic systems cannot afford to
take into account system sizes 3 orders of magnitude larger than the
size of the critical nucleus. Consistent with Honeycutt and Andersen,275 Huitema et al.260 examined the nucleation of an LJ liquid in a wide range of temperatures
(70–140 K). Although nonphysical instantaneous crystallization
was observed for systems on the order of ∼500 particles, simulation
boxes containing about 10000 particles seemed to be free from finite-size
effects.
, which turned out to be too
few particles
to completely rule out the effects of periodic boundary conditions.
In fact, the authors suggested that extra care had to be taken because
of the diffuseness of the interface between the supercooled liquid
phase and the crystalline nucleus, which can induce an artificial
long-range order in the system, leading to a nonphysically high nucleation
rate. These findings are particularly relevant, as the critical nucleus
size at this ΔT value is on the order of just
a few tens of particles, representing a tiny fraction of the whole
system. Only a few years later, Swope and Andersen276 investigated the same effects by taking into account up
to 106 LJ particles subjected to the same strong supercooling
as probed by Honeycutt and Andersen.275 According to their large-scale MD simulations, 15000 particles seem
to be sufficient to avoid finite-size effects. This outcome must be
carefully pondered, as currently, the vast majority of simulations
dealing with crystallization of realistic systems cannot afford to
take into account system sizes 3 orders of magnitude larger than the
size of the critical nucleus. Consistent with Honeycutt and Andersen,275 Huitema et al.260 examined the nucleation of an LJ liquid in a wide range of temperatures
(70–140 K). Although nonphysical instantaneous crystallization
was observed for systems on the order of ∼500 particles, simulation
boxes containing about 10000 particles seemed to be free from finite-size
effects.
It is also worth pointing out that Peng et al.261 recently described a novel class of finite-size
effects unrelated to periodic boundary conditions. In fact, they showed
that the equilibrium density of critical nuclei, 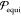 ,277 can effectively
influence the absolute value of nucleation rates. Specifically, at
very strong supercooling, the critical nuclei will on average form
very shortly after the transient time, whereas at mild ΔT, the stochastic nature of nucleation will lead to a consistent
scatter of the nucleation times. In other words, in the latter scenario,
either exceedingly large systems must be taken into account, or a
sizable number of independent simulations must be performed to deal
with the long tails of the distribution of nucleation times.
,277 can effectively
influence the absolute value of nucleation rates. Specifically, at
very strong supercooling, the critical nuclei will on average form
very shortly after the transient time, whereas at mild ΔT, the stochastic nature of nucleation will lead to a consistent
scatter of the nucleation times. In other words, in the latter scenario,
either exceedingly large systems must be taken into account, or a
sizable number of independent simulations must be performed to deal
with the long tails of the distribution of nucleation times.
2.3. Atomic Liquids
Various interatomic potentials have been developed to deal with atomic liquids. Examples include the Sutton–Chen potentials278 for several metals and the Tosi–Fumi potential279 for molten salts such as NaCl. Terms accounting for the directionality of covalent bonds have been included, for example, in the Stillinger–Weber potential280 for Si; the bond order potentials of Tersoff281,282 for Si, GaAs, and Ge; and the reactive potential of Brenner283 for carbon-based systems. Another class of interatomic potential is based on the concept of local electronic density and includes, for instance, the Finnis–Sinclair potentials278,284 for metallic systems, the whole family of the embedded-atom-method (EAM) potentials,285 and the glue potential286,287 for Au and Al.
Many of these potentials are still incredibly
cheap in terms of computer time, thus allowing for large-scale, unbiased
MD simulations. Recently, massively parallel MD runs succeeded in
nucleating supercooled liquid Al288 and
Fe289 using an EAM potential and a Finnis–Sinclair
potential, respectively. As up to 106 atoms were taken
into account, actual grain boundaries were observed, providing unprecedented
insight in to the crystal growth process. The nucleation of bcc Fe
crystallites and the evolution of the resulting grain boundaries at
different temperatures can be appreciated in Figure 9a. The sizable dimension of the simulation
boxes (∼50 nm) allowed nucleation events to be observed within
hundreds of picoseconds, and grain coarsening (i.e., the process by
which small crystallites end up incorporated into larger ones) is
also clearly visible. Mere visual inspection of the nucleation trajectories
depicted in Figure 9a suggests different nucleation regimes as a function of temperature.
In fact, the same authors calculated a temperature profile for the
nucleation rate, shown in Figure 9b, that demonstrates the emergence of a maximum of 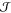 value characteristic
of diffusion-limited
nucleation (see section 1.1.1).
value characteristic
of diffusion-limited
nucleation (see section 1.1.1).
Figure 9.

Crystal nucleation of supercooled Fe by means of large-scale MD simulations. (a) Snapshots of trajectories at different temperatures. Crystalline (bcc) atoms are depicted in purple. Yellow circles highlight small crystalline grains doomed to be incorporated into the larger ones later on because of grain coarsening. (b) Nucleation rate as a function of temperature. Reprinted with permission from ref (290). Copyright 2015 Nature Publishing Group.
A field that has greatly benefited from MD simulations is the crystallization of metal clusters, as nicely reviewed by Aguado and Jarrold.291 For instance, it is possible to probe the interplay between the size of the clusters and the cooling rate upon crystal nucleation and growth. In this context, Shibuta292 reported three different outcomes for supercooled liquid Mo nanoparticles modeled by means of a Finnis–Sinclair potential, namely, the formation of a bcc single crystal, a glassy state, or a polycrystalline phase. In some cases, nucleation rates obtained from simulations were consistent with CNT, as in the case of Ni nanodroplets,293 for which nucleation events were again observed by means of brute-force MD simulations using the Sutton–Chen potential. The influence of the redox potential on the nucleation process has also been investigated. Milek and Zahn294 employed an enhanced flavor of the EAM potential to study the nucleation of Ag nanoparticles from solution. They established that the outcome of nucleation events is strongly influenced by the strength of the redox potential, able to foster either a rather regular fcc phase or a multitwinned polycrystal. Similar to what was done for LJ liquids, the effects of confinement were assessed for Au nanodomains modeled using the glue potential by Pan and Shou.295 According to their findings, smaller domains facilitate crystal nucleation. Lü and Chen296 instead investigated surface-layering-induced crystallization of Ni–Si nanodroplets using a modified EAM potential. It seems that, for this particular system, atoms proximal to the free surface of the droplet assume a crystalline-like ordering on very short time scales, thus triggering crystallization in the inner regions of the system. No such effect has been reported in the case of surface-induced crystallization in supercooled tetrahedral liquids such as Si and Ge, as investigated by Li et al.297 through FFS simulations employing both Tersoff and Stillinger–Weber potentials. The presence of the free surface facilitates crystal nucleation for this class of systems as well, but surface layering was not observed. Instead, the authors claimed that the surface reduces the free energy barrier for nucleation as it introduces a pressure-dependent term in the volume free energy change expected for the formation of the crystalline clusters. The situation is quite different for surface-induced ice nucleation, at least according to the coarse-grained mW model of Molinero and Moore.298 In fact, Haji-Akbari et al.299 recently investigated ice nucleation in free-standing films of supercooled mW water using both FFS and US, finding that, in these systems, crystallization is inhibited in the proximity of the vapor–liquid interface. Very recently, Gianetti et al.300 extended the investigation of Haji-Akbari et al.299 to the crystallization of a whole family of modified Stillinger–Weber liquids with different degrees of tetrahedrality λ, locating a crossover from surface-enhanced to bulk-dominated crystallization in free-standing films as a function of λ. Another seminal study by Li et al.,301 again using FFS, focused on homogeneous ice nucleation within supercooled mW water nanodroplets, where nucleation rates turned out to be strongly size dependent and in general consistently smaller (by several orders of magnitude) than the bulk case. FFS was also applied by Li et al.302 to examine homogeneous nucleation of supercooled Si. FFS has also been successful in predicting homogeneous crystal nucleation rates in molten NaCl, modeled using a Tosi–Fumi potential by Valeriani et al.303 Large discrepancies between their results and experimental nucleation rates can be appreciated when CNT is used to extrapolate the calculations to the milder supercooling probed by the actual measurements. Given that the authors obtained consistent results using two different enhanced sampling methods, this study hints again at the many pitfalls of CNT.
2.3.1. Phase-Change Materials
A unique
example of a class of materials for which nucleation can be effectively
addressed by brute-force MD simulations is given by so-called phase-change
materials.304,305 These systems are of great technological
interest as they are widely employed in optical memories (e.g., DVD-RW)
and in a promising class of nonvolatile memories known as phase-change
memory,306 based on the fast and reversible
transition from the amorphous to the crystalline phase. Although crystal
nucleation in amorphous systems, especially metallic and covalent
glasses, is beyond the scope of this review, we refer the reader to
the excellent work of Kelton and Greer26 for a detailed introduction. Here we just note that in phase change
memories the amorphous phase is often heated above the glass transition
temperature, so that crystal nucleation occurs within the supercooled
liquid phase. Phase-change materials used in optical and electronic
devices are typically tellurium-based chalcogenide alloys (see ref (305)). The family of the pseudobinary
compounds (GeTe)x(Sb2Te3)y represents a prototypical system.
Although both the structure and dynamics of these systems are far
from trivial, nucleation from the melt takes place on the nanosecond
time scale for a wide range of supercooling.304−306 Thus, with phase-change materials, we have a great opportunity to
investigate nucleation in a complex system by means of brute-force
MD simulations. We note that the crystallization of these systems
has been extensively characterized by different experimental techniques
[particularly TEM and AFM (see section 1.2); the crystallization kinetics has also
been recently investigated by means of ultrafast-heating calorimetry307 and ultrafast X-ray imagining308], but because of the exceptionally high nucleation rates,
it is difficult to extract information about the early stages of the
nucleation process. Thus, in this scenario, simulations could play
an important role. Unfortunately, phase-change materials require ab
initio methods or sophisticated interatomic potentials with first-principles
accuracy. In fact, several attempts have been made to study nucleation
in phase-change materials by ab initio MD in very small systems.309,310 Although these studies provided useful insights into the nucleation
mechanism, severe finite-size effects prevented the full characterization
of the crystallization process. The limited length and time scales
typical of first-principles calculations were recently outstripped
in the case of the prototypical phase-change material GeTe by the
capabilities of a neural network interatomic potential.311 Such potentials allow for a computational speedup
of several orders of magnitude compared to conventional ab initio
methods while retaining an accuracy close to that of the latter.312 Although nucleation rates have not yet been
calculated, detailed investigations of homogeneous and heterogeneous
nucleation have already been reported.313,314 For instance,
as shown in Figure 10, a single-crystalline nucleus formed in a 4000-atom model of supercooled
liquid GeTe in the 625–675 K temperature regime within a few
hundred picoseconds. On the same timescale, several nuclei appeared
below 600 K, suggesting that the free energy barrier for nucleation
is vanishingly small for this class of materials just above the glass
transition temperature. This is because of the fragility315 of the supercooled liquid, which displays a
substantial atomic mobility even at large supercoolings.316 Thus, in this particular case, the kinetic
prefactor 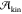 (see eq 5) is not hindered that much by the strong
supercooling,
whereas the free energy difference between the liquid and the crystal
(see eq 5) is not hindered that much by the strong
supercooling,
whereas the free energy difference between the liquid and the crystal  (see eq 1) skyrockets as expected, leading to the exceedingly
high nucleation rates characteristic of these materials.
(see eq 1) skyrockets as expected, leading to the exceedingly
high nucleation rates characteristic of these materials.
Figure 10.

Fast crystallization of supercooled GeTe by means of MD simulations with neural-network-derived potentials. The number of crystalline nuclei larger than 29 atoms at different temperatures in the supercooled liquid phase is reported as a function of time (notice the exceedingly small time scale at strong supercooling). Two snapshots at the highest and lowest temperatures showing only the crystalline atoms are also reported. At high temperature, a single nucleus is present, whereas several nuclei (each one depicted in a different color) appear at low temperature. The number of nuclei first increases and then decreases due to coalescence. Reprinted with permission from ref (313). Copyright 2013 American Chemical Society.
In conclusion, whereas MD simulations have by no means exhausted the field of crystal nucleation of atomic liquids, they have certainly provided insight into a number of interesting systems and paved the way for the study of more complex systems, as we shall see in the following sections.
2.4. Water
2.4.1. Homogeneous Ice Nucleation
Ice nucleation impacts many different areas, ranging from aviation317,318 to biological cells319 and Earth’s climate.320,321 It is therefore not surprising that a considerable body of work has been carried out to understand this fundamental process. We cannot cover it all here; instead, we give a general overview of the field, starting with a discussion of nucleation rates. This allows us to directly compare experiments and simulations and to identify strengths and weaknesses of different approaches. We then discuss insights into the nucleation mechanism. The heterogeneous formation of ice is presented in section 2.4.2.
Nucleation Rates
An important goal for both experiments and simulations is to extract nucleation rates. Experimental nucleation rates have been measured over a broad range of temperatures, most often with micrometer-sized water droplets so as to avoid heterogeneous nucleation. In Figure 11, we bring together nucleation rates obtained from various experiments, along with computed nucleation rates.
Figure 11.

Compilation of homogeneous nucleation rates for water, obtained by experiments and simulations. The x axis shows the supercooling with respect to the melting point of different water models or 273.15 K for experiment. The y axis shows the logarithm of the nucleation rate in m–3 s–1. Rates obtained with computational approaches are shown as solid symbols; experimental rates are shown as crossed symbols. For each computational study, the computational approach and the water force field used are specified. The nucleation study of Sanz et al.51 is not included in this graph, because their study was conducted at a small supercooling (20 K), which resulted in a very low estimated nucleation rate far outside this plot (it would correspond to −83 on the y axis). Taborek329 performed measurements with different setups, namely, using sorbitan tristearate (STS) and sorbitan trioleate (STO) as surfactants. Data for the graph were taken from refs (124, 299, 301, and 322−335).
Accessing nucleation rates from MD simulations became feasible only in the past few years as a result of advances in force fields (such as the coarse-grained mW298 potential) and enhanced sampling techniques described earlier (see section 1.3.2). These methods have therefore been widely used for studies of not only homogeneous but also heterogeneous nucleation (see section 2.4.2). From the comparison of experimental and computational nucleation rates reported in Figure 11, a few conclusions are apparent. First, nucleation rates vary hugely with supercooling, by a factor of more than 1035. Second, nucleation rates differ substantially (approximately 10 orders of magnitude) between simulations (solid symbols) and experiments (crossed symbols) at relatively small supercoolings (∼30–50 K). At larger supercoolings, the agreement appears to be slightly better, even though very few simulations have been reported at very strong supercooling. The third striking feature is that, whereas the experimental results agree well with each other (within 1–2 orders of magnitude), the computational rates differ from each other by a factor of approximately 1010.
What is the cause of disagreement between different computational approaches? Part of the reason is certainly that different water models lead to different rates; see, for example, Espinosa et al.326 Yet, even if the same water model is employed, the rates do not agree with each other very well. A neat example is offered by nucleation rates obtained using the mW model. An early study by Moore and Molinero336 succeeded in calculating the Avrami exponent337,338 for the crystallization kinetics of ice from brute-force MD simulations at very strong supercooling, obtaining results remarkably similar to experiment.339,340 However, mW nucleation rates turned out to be far less encouraging. In fact, Li et al.322 and Reinhardt and Doye324 both performed simulations using the mW model, obtaining nucleation rates that differed by about 5 orders of magnitude. The only major difference was the enhanced sampling technique employed, FFS by Li et al. and US by Reinhardt and Doye. The statistical uncertainties of the two approaches (1–2 orders of magnitude) are much smaller than the 5-orders-of-magnitude discrepancy between the two studies. It was also shown that the two methods agree very well with each other for colloids,222 for example (see section 2.1). The use of different computational approaches therefore also seems to be unlikely as the source of the disagreement. What the cause is remains elusive.
Because we cannot cover all of
the work shown in Figure 11 in detail here, we now discuss
just two studies. First, that of Sanz et al.,51 which agrees best with the experimental rates. The authors used
the TIP4P/2005 and TIP4P/Ice water models in combination with seeded
MD simulations (see section 1.3.2. For more details, the reader is referred to the original
article51). Seeding involves considerably
more assumptions than, for example, US or FFS. In particular, the
approach assumes a CNT-like free energy profile, although it does
not usually employ the macroscopic interfacial free energy. Furthermore,
the temperature dependence of key quantities such as 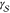 and
and  (see section 1.1.1) is approximated. Nevertheless, the
agreement between their nucleation rates and experiment seemingly
outperformed other approaches. In a more recent work, Espinosa et
al.326 obtained nucleation rates for a
few other water models as well. However, it should be noted that the
good agreement between the nucleation rates reported in refs (51 and 326) and the experimental data could originate from error cancellation.
In fact, whereas the rather conservative definition of crystalline
nucleus adopted in these works will lead to small nucleation barriers
(and thus to higher nucleation rates), the TIP4P family of water models
is characterized by small thermodynamic driving forces to nucleation,327 which, in turn, results in smaller nucleation
rates.
(see section 1.1.1) is approximated. Nevertheless, the
agreement between their nucleation rates and experiment seemingly
outperformed other approaches. In a more recent work, Espinosa et
al.326 obtained nucleation rates for a
few other water models as well. However, it should be noted that the
good agreement between the nucleation rates reported in refs (51 and 326) and the experimental data could originate from error cancellation.
In fact, whereas the rather conservative definition of crystalline
nucleus adopted in these works will lead to small nucleation barriers
(and thus to higher nucleation rates), the TIP4P family of water models
is characterized by small thermodynamic driving forces to nucleation,327 which, in turn, results in smaller nucleation
rates.
The second work we briefly discuss here is the very recent
study
(2015) of Haji-Akbari and Debenedetti.327 The authors directly calculated the nucleation rate at 230 K of
an all-atom model of water (TIP4P/ICE) using a novel FFS sampling
approach.327 This was a tour de force,
but strikingly, their rates differed from experiment by about 11 orders
of magnitude. The authors noted that this might be as close as one
can actually get to experiment with current classical water models.
This is because of the extreme sensitivity of nucleation rates to
thermodynamic properties such as 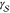 and
and 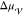 , which, according to CNT, enter exponentially
(section 1.1.1)
in the definition of
, which, according to CNT, enter exponentially
(section 1.1.1)
in the definition of  . For instance,
an uncertainty of only 6–7%
for
. For instance,
an uncertainty of only 6–7%
for 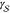 at 235 K leads to an
error of about 9 orders
of magnitude in
at 235 K leads to an
error of about 9 orders
of magnitude in  .322 Experimental
estimates for γ range from 25 to 35 mN/m;342 computational estimates range from about 20343 to 35 mN/m.344 As
another example, Haji-Akbari and Debenedetti327 explicitly quantified the extent to which the TIP4P/Ice model underestimates
the free energy difference
.322 Experimental
estimates for γ range from 25 to 35 mN/m;342 computational estimates range from about 20343 to 35 mN/m.344 As
another example, Haji-Akbari and Debenedetti327 explicitly quantified the extent to which the TIP4P/Ice model underestimates
the free energy difference 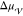 between the crystalline and liquid phases
and found that the mismatch between
between the crystalline and liquid phases
and found that the mismatch between  and
and 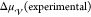 alone
leads to an overestimation of the
free energy barrier for nucleation of about 60%, which translates
into nucleation rates up to 9 orders of magnitude larger. In fact,
taking into account such a discrepancy brings the results of Haji-Akbari
and Debenedetti within the confidence interval of the experimental
data. Thus, it is clear that we simply do not know some key quantities
accurately enough to expect perfect agreement between simulations
and experiments.
alone
leads to an overestimation of the
free energy barrier for nucleation of about 60%, which translates
into nucleation rates up to 9 orders of magnitude larger. In fact,
taking into account such a discrepancy brings the results of Haji-Akbari
and Debenedetti within the confidence interval of the experimental
data. Thus, it is clear that we simply do not know some key quantities
accurately enough to expect perfect agreement between simulations
and experiments.
In addition to issues of modeling water/ice accurately, finite-size effects can be expected to also play a role [as they do with Lennard-Jones systems (section 2.2) and molecules in solution (section 2.5)]. Only recently was this issue addressed explicitly for ice nucleation by English and Tse345 in unbiased simulations with the mW model. They were able to simulate systems containing nearly 10 million water molecules on a microsecond time scale and found that larger systems favor the formation of crystallization precursors compared to smaller ones. Interestingly, lifetimes of the precursors were found to be less sensitive to system size. A quantitative understanding of finite-size effects on nucleation rates remains elusive nevertheless.
In summary, it can be said that, in terms of accurate nucleation rates, experiments are still clearly superior to simulations. However, the advantage of simulations is that the nucleation mechanism can also be obtained, which, at present, is not possible with experiments, although femtosecond X-ray laser spectroscopy might be able to partially overcome this limitation in the near future.90
Nucleation Mechanism
In 2002, Matsumoto et al.346 were the first to report a nucleation event in an unbiased simulation based on an all-atom model of water (TIP4P). Their landmark work opened the doors to the study of ice nucleation at an atomistic level. They found that nucleation took place once a sufficient number of long-lived hydrogen bonds were formed with a nucleus of ice. Recent evidence suggests that, most likely, their nucleation trajectory was driven by finite-size effects.51 Together with the simulations of Vrbka and Jungwirth,347 also affected by severe finite-size effects,51 the work of Matsumoto et al. remains, to date, the only seemingly unbiased MD simulation observing homogeneous ice nucleation with an all-atom force field.
What really enabled the community to investigate ice formation at a molecular level was the development of the coarse-grained mW potential for water298 in the early 2010s. Using unbiased MD simulations based on the mW force field, Moore and Molinero in 2011323 provided evidence that, in the supercooled regime around the homogenous nucleation temperature, Th, the fraction of 4-fold-coordinated water molecules increases sharply prior to a nucleation event. In a separate work, the same authors suggested348 that, at very strong supercooling, the critical nucleus is mostly made of cubic ice, which subsequently evolves into a mixture of stacking-disordered cubic and hexagonal ice layers. In the same year, Li et al.322 identified another structural motif that might play a role in ice nucleation. They consistently observed a topological defect structure in growing ice nuclei in their FFS simulations based on the mW representation of water. This defect, depicted in Figure 12a, can be described as a twin boundary with 5-fold symmetry, and it has also been observed302 in nucleation simulations of tetrahedral liquids simulated with the Stillinger–Weber potential, on which the mW coarse-grained model was built.
Figure 12.

(a) Formation of a topological defect with 5-fold symmetry during homogeneous ice nucleation. The snapshots (i–iv) show the time evolution of the defect structure, indicated by black dashed lines. Ic and Ih water molecules are shown in blue and red, respectively. Reprinted with permission from ref (322) (Copyright 2011 Royal Society of Chemistry), in which Li et al. performed FFS simulations of models containing about 4000 mW water molecules. (b) Nucleation of an ice cluster forming homogeneously from I0-rich precritical nuclei. Water molecules belonging to Ic, Ih, a clathrate-like phase, and I0 are depicted in yellow, green, orange, and magenta, respectively. (i,ii) A critical nucleus forms in an I0-rich region. (iii) The crystalline cluster evolves in a postcritical nucleus, formed by an Ic-rich core surrounded by an I0-rich shell. (iv) The same postcritical nucleus as depicted in iii, but only particles with 12 or more connections (among ice-like particles) are shown. The color map refers to the order parameter Q12 specified in ref (325), from which this image was reprinted with permission (Copyright 2014 Nature Publishing Group). The unbiased MD simulations on which the analysis is based feature 10000 mW molecules.
In 2012, another significant leap in understanding the nucleation mechanism of ice from a structural point of view was made by combining experimental and computational techniques.343 Specifically, Malkin et al. showed that ice forming homogeneously is stacking-disordered (the corresponding ice structure was called Isd), meaning that it is made out of cubic and hexagonal ice layers stacked in a random fashion.
In 2014, two studies substantiated the potential relevance of precursor structures prior to ice formation. Palmer et al. provided evidence for a liquid–liquid transition in supercooled water in a molecular model of water (ST2).350 In their study, the authors sampled the energy landscape of supercooled water and found two metastable liquid basins corresponding to low-density (LDL) and high-density (HDL) water. The appealing idea behind the transition from HDL to LDL prior to ice nucleation is that LDL is structurally closer to ice than HDL. Note that the existence of two metastable liquid basins was not a general finding: The mW model does not have a basin for LDL, for example.323 Indeed, the presence of this liquid–liquid phase transition is a highly debated issue.351,352
Another conceptually similar idea is ice formation through ice 0 (I0), proposed by Russo et al.325 Instead of a liquid–liquid phase transition that transforms water into another liquid state prior to nucleation, the authors proposed a new ice polymorph (I0) to bridge the gap between supercooled water and ice. I0 is a metastable ice polymorph and is structurally similar to the supercooled liquid. It has a low interfacial energy with both liquid water and ice Ic/Ih. Russo et al. therefore proposed I0 to bridge liquid water to crystalline Ic/Ih. Indeed, the authors found I0 at the surface of growing ice nuclei in MD simulations; we show part of a nucleation trajectory in Figure 12b. Furthermore, they showed that the shape of the nucleation barrier is much better described by a core–shell-like model (Ic/Ih core surrounded by I0) compared to the classical nucleation model. This is important, because it suggests that models that are based solely on CNT assumptions might not be appropriate for describing homogeneous ice nucleation.
However, the emergence of I0 has not yet been reported by any other nucleation study, including the recent work of Haji-Akbari and Debenedetti327 that we previously mentioned in the context of nucleation rates. In that work,327 the authors performed a topological analysis of the nuclei, validated by the substantial statistics provided by the FFS simulations. As depicted in Figure 13, the majority of nuclei that reached the critical nucleus size contained a large amount of double-diamond cages (DDCs, the building blocks of Ic), whereas nuclei rich in hexagonal cages (HCs, the building blocks of Ih) had a very low probability to overcome the free energy barrier for nucleation. In addition, even postcritical nuclei had a high content of DDCs, whereas HCs did not show any preference to appear within the core of the postcritical nuclei. This evidence is consistent with the findings reported in ref (323) and in contrast with the widely invoked scenario in which a kernel of thermodynamically stable polymorph (in this case, Ih) is surrounded by a shell of a less stable crystalline structure (in this case, Ic).
Figure 13.

(Left) A typical double-diamond cage (DDC, blue) and a hexagonal cage (HC, red), the building blocks of Ic and Ih, respectively. (Right) Temporal evolution of an ice nucleus from (i,ii) the early stages of nucleations up to (v,vi) postcritical dimensions, as observed in the FFS simulations of Haji-Akbari and Debenedetti.327 About 4000 water molecules, modeled with the TIP4P/Ice potential,349 were considered in the NPT ensemble at ΔT ≈ 40 K. One can clearly notice the abundance of DDCs throughout the whole temporal evolution. In contrast, HC-rich nuclei have only a marginal probability to cross the nucleation barrier (see text). Reprinted with permission from ref (327). Copyright 2015 National Academy of Sciences.
In the past few years, the understanding of homogeneous ice nucleation has improved dramatically. We now have a good understanding of the structure of ice that forms through homogeneous nucleation, stacking-disordered ice. Furthermore, there is very good agreement (within 2 orders of magnitude) between experimental nucleation rates in a certain temperature range. Computational methods face the problem of being very sensitive to some key thermodynamic properties; the nucleation rates they predict are therefore less accurate. On the other hand, they allow us to study conditions that are very challenging to probe experimentally, and they also provide insight into the molecular mechanisms involved in the crystallization process.
2.4.2. Heterogeneous Ice Nucleation
As mentioned in the previous section, homogeneous ice nucleation becomes extremely slow at moderate supercooling. This seems at odds with our everyday experience—we do not, for example, have to wait for temperatures to reach −30 °C before we have to use a deicer on our car windows. In fact, the formation of ice in nature occurs almost exclusively heterogeneously, thanks to the presence of foreign particles. These ice-nucleating agents facilitate the formation of ice by lowering the free energy barrier for nucleation (see Figure 1). Indeed, the work of Sanz et al.,51 in which homogeneous ice nucleation was studied using seeded MD (see section 1.3.1), found rates so low at temperatures above ΔT = 20 K that they concluded that all ice nucleation above this temperature must occur heterogeneously. Homogeneous nucleation is still of great importance in atmospheric processes and climate modeling, as under certain conditions, both heterogeneous and homogeneous nucleation are feasible routes toward the formation of ice in clouds, as reported in ref (354), for example.
In addition to the challenges (both computational and experimental) faced when investigating homogeneous ice nucleation, one also has to consider the structure of the water–surface interface and how this impacts the nucleation rate. Generally, the experimental data for the rates and characterization of the interfacial structure come from two different communities: Climate scientists have provided much information on how various particles, often dust particles or biological matter such as pollen, affect ice nucleation (as depicted in Figure 14), whereas surface scientists have invested a great deal of effort in trying to understand, at the molecular level, how water interacts with and assembles itself at surfaces (see, e.g., ref (355)). This means that there is a huge gap in our understanding, as the surfaces of the particles used to obtain rates are often not characterized, whereas surface science experiments are generally carried out at pristine, often metallic, surfaces under ultrahigh-vacuum conditions. We will see in this section that computational studies have gone some way toward bridging this gap, although there is still much work to be done should we wish to quantitatively predict a material’s ice-nucleating efficacy.
Figure 14.

Potential immersion-mode ice nucleus concentrations, Nice, a measure of the efficiency of a given substance to boost heterogeneous ice nucleation, as a function of temperature for a range of atmospheric aerosol species. Note the wide range of nucleating capability for materials as diverse as soot and bacterial fragments over a very broad range of temperatures. Reprinted with permission from ref (356). Copyright 2012 Royal Society of Chemistry.
Water on Crystalline Surfaces
From a computational perspective, it is the surface science experiments that lend themselves most readily to modeling. In fact, even relatively expensive computational methods such as density functional theory (DFT), which have not featured much in this article, have proven indispensable in furthering our understanding of how water behaves at surfaces, especially when used in conjunction with experiments (see, e.g., refs (355 and 357) for an overview). As such, early computational studies focused on understanding how the surface affected the first few layers of water, especially with respect to the concept of lattice mismatch (see section 2.2), where a surface that has a structure commensurate with ice acts as a template for the crystal. Nutt and co-workers358−360 investigated the adsorption structures of water at a model hexagonal surface and at BaF2(111) using interaction potentials derived from ab initio calculations. Although the surfaces under investigation had structures that matched the basal face of ice well, they found disordered structures of water to be more favorable than ice-like overlayers. Using DFT, Hu and Michaelides investigated the adsorption of water on the (001) face of the clay mineral kaolinite,361,362 a known ice-nucleating agent in the atmosphere. The (001) surface of kaolinite exposes a pseudohexagonal arrangement of OH groups that were proposed to be the cause of its good ice-nucleating ability.363 Although they found that a stable ice-like layer could form at the surface, the amphoteric nature of the kaolinite surface, depicted in Figure 15, meant that all of water molecules could participate in four hydrogen bonds, making further growth on top of the ice-like layer unfavorable. Croteau et al.364,365 investigated adsorption of water on kaolinite using the CLAYFF and SPC/E potentials366,367 and grand canonical Monte Carlo (GCMC). Although some hexagonal patches of water were seen in the contact layer, the overall structure was mostly disordered, and the hexagonal structures that did form were strained relative to those found in ice. Also using GCMC, Cox et al.368 investigated the role of lattice mismatch using model hexagonal surfaces and TIP4P water.369 They found that, for atomically flat surfaces, a nominally zero lattice mismatch produced disordered contact layers comprising smaller-sized rings (i.e., pentagons and squares) and observed hexagonal ice-like layers only for surfaces with larger lattice constants.
Figure 15.

The amphoteric nature of kaolinite is important to its ice-nucleating ability. (Left) Ice-like contact layers at the kaolinite surface, with the (a,c) basal and (b,d) prism faces of ice adsorbed on kaolinite, as viewed from the (a,b) side and (c,d) top. (Right) Adsorption energy of ice on kaolinite when bound through either its basal face (red data) or its prism face (blue data) for varying numbers of layers of ice. (Open and solid symbols indicate data obtained with a classical force field and with DFT, respectively.) When only the contact layer is present, the basal face structure is more stable than the prism face structure, but as soon as more layers are present, the prism face structure becomes more stable. This can be understood by the ability of the prism face to donate hydrogen bonds to the surface, and to the water molecules above, through the “dangling” hydrogen bonds seen in b and d. Reprinted with permission from ref (371). Copyright 2013 Royal Society of Chemistry.
Prior to ca. 2010, the above types of study were the state of the art for simulations of heterogeneous ice nucleation. Although they provided evidence that properties such as lattice match alone are insufficient to explain a material’s ice-nucleating ability, because ice nucleation itself was not directly observed, only inferences could be drawn about how certain properties might actually affect ice nucleation. Yan and Patey370 investigated the effects of electric fields on ice nucleation using brute-force molecular dynamics (the electric fields were externally applied and were not due to an explicit surface). They found that the electric field needed to act over only a small range (e.g., 10 Å) and that the ice that formed near the “surface” was ferroelectric cubic ice, although the rest of the ice that formed above was not. Cox et al. performed simulations of heterogeneous ice nucleation371 in which the atomistic natures of both the water and the surface were simulated explicitly, using TIP4P/2005 water372 and CLAYFF366 to describe kaolinite. Despite the fact that the simulations were affected by finite-size effects, the simulations revealed that the amphoteric nature of the kaolinite361,362 is important to ice nucleation. In the liquid, a strongly bound contact layer was observed, and for ice nucleation to occur, significant rearrangement in the above water layers was required. It was found that ice nucleated with its prism face, rather than its basal face, bound to the kaolinite, which was unexpected based on the theory that the pseudohexagonal arrangement of OH groups at the surface was responsible for templating the basal face. Cox et al. rationalized the formation of the prism of ice at the kaolinite as being due to its ability to donate hydrogen bonds both to the surface and to the water molecules above (see Figure 15), whereas the basal face maximizes hydrogen bonding to the surface only.361,362 More recent simulation studies, employing rigid and constrained models of kaolinite, have also found the amphoteric nature of the kaolinite surface to be important.373 However, the heterogeneous nucleation mechanism of water on clays is yet to be validated by unconstrained simulations unaffected by substantial finite-size effects.
Hydrophobicity and Surface Morphology
As in the case of simulations of homogeneous ice nucleation, the use of the coarse-grained mW potential298 has seen the emergence of computational studies that actually quantify the ice-nucleating efficiencies of different surfaces. Recently, Lupi et al.374 investigated ice nucleation at carbonaceous surfaces (both smooth graphitic and rough amorphous surfaces) using cooling ramps to measure nonequilibrium freezing temperatures ΔTf ≡ Tf – Tfhomo, where Tf is the temperature at which ice nucleates in the presence of a surface and Tf = 201 ± 1 K is the temperature at which homogeneous ice nucleation occurs. It was found that the rough amorphous surface did not enhance ice nucleation (ΔTf = 0 K), whereas the smooth graphitic surfaces promoted ice nucleation (ΔTf = 11–13 K). This was attributed to the fact that the smooth graphitic surface induced a layering in the density profile of water above the surface, whereas the rough amorphous surface did not. Lupi and Molinero quantified the extent of layering as
where ρ(z) is the density of water at a height z above the surface and ρ0 ≡ ρ(zbulk), where zbulk is a height where the density profile is bulk-like. In a subsequent work using the same methodology, Lupi and Molinero375 investigated how the hydrophilicity of graphitic surfaces affected ice nucleation. The hydrophilicity of the surface was modified in two different ways: first, by uniformly modifying the water–surface interaction strength and, second, by introducing hydrophilic species at the surface. It was found that the two ways produced qualitatively different results: Uniformly modifying the interaction potential led to enhanced ice nucleation, whereas increasing the density of hydrophilic species was detrimental to ice nucleation (although the surfaces still enhanced nucleation relative to homogeneous nucleation). It was concluded that hydrophilicity is not a good indicator of the ice-nucleating ability of graphitic surfaces. As for the difference between increasing the hydrophilicity by uniformly modifying of the interaction potential and by introducing hydrophilic species, Lupi and Molinero again saw that the extent of layering in water’s density profile above the surface correlated well with the ice-nucleating efficacy. The general applicability of the layering mechanism, however, was left as an open question.
Cox et al.376,377 addressed the question of the general applicability of the layering mechanism by investigating ice nucleation rates over a wider range of hydrophilicities (by uniformly changing the interaction strength) on two surfaces with different morphologies: (i) the (111) surface of a face-centered-cubic LJ crystal (fcc-111) that provided distinct adsorption sites for the water molecules and (ii) a graphitic surface, similar to that of Lupi et al.374 Although it was found that the layering mechanism (albeit with a slight modification to the definition used by Lupi et al.) could describe the ice-nucleating behavior of the graphitic surface, at the fcc-111 surface, no beneficial effects of layering were observed. This was attributed to fact that the fcc-111 surface also affected the structure of the water molecules in the second layer above the surface, in a manner detrimental to ice nucleation. It was concluded that layering of water above the surface can be beneficial to ice nucleation, but only if the surface presents a relatively smooth potential energy surface to the water molecules.
The studies at the carbonaceous and fcc-111 surfaces374−377 discussed above hinted that the heterogeneous nucleation mechanism could be very different at different types of surfaces. Although there is experimental evidence that, for example, different carbon nanomaterials are capable of boosting ice nucleation (see, e.g., ref (378)), most experiments can only quantify the ice-nucleating ability of the substrates (see section 1.2). However, the structure of the water–substrate interface and any insight into the morphology of the nuclei are typically not available, making simulations essential to complement the experimental picture. In this respect, Zhang et al.379 assessed that the (regular) pattering of a generic crystalline surface at the nanoscale can strongly affect ice formation. More generally, the interplay between hydrophobicity and surface morphology was recently elucidated by Fitzner et al.380 Brute-force MD simulations of heterogeneous ice nucleation were performed for the mW water model on top of several crystalline faces of a generic fcc crystal, taking into account different values of the water–surface interaction strength, as well as different values of the lattice parameter. The latter is involved in the rather dated270 concept of zero lattice mismatch, which we introduced in section 2.2 (see eq 7) and which has been often quoted as the main requirement of an effective ice-nucleating agent. However, a surprisingly nontrivial interplay between hydrophobicity and morphology was observed, as depicted in Figure 16. Clearly, neither the layering nor the lattice mismatch alone are sufficient to explain such a diverse scenario. In fact, the authors proposed three additional microscopic factors that can effectively aid heterogeneous ice nucleation on crystalline surfaces: (i) an in-plane templating of the first water overlayer on top of the crystalline surface; (ii) a first overlayer buckled in an ice-like fashion; and (iii) enhanced nucleation in regions of the liquid beyond the first two overlayers, possibly aided by dynamical effects and/or structural templating effects of the substrate extending past the surface water interface. In addition, it turned out that different lattice parameters can lead to the nucleation and growth of up to three different faces of ice [basal, prismatic, and secondary prismatic ({1120})] on top of the very same surface, adding a layer of complexity to the nucleation scenario. Insights into the interplay between hydrophobicity and morphology were also very recently obtained by Bi et al.,381 who investigated heterogeneous ice nucleation on top of graphitic surfaces by means of FFS simulations using the mW model. Among their findings, the authors suggested that the efficiency of ice-nucleating agents can be a function not only of surface chemistry and surface crystallinity but of the elasticity of the substrate as well.
Figure 16.

Interplay
between surface morphology and water–surface interaction
on the heterogeneous ice nucleation rate. (a) Heat maps representing
the values of ice nucleation rates on top of four different fcc surfaces
[(111), (100), (110), (211)], plotted as a function of the adsorption
energy, Eads, and the lattice parameter, afcc. The lattice mismatch δ with respect
to ice on (111) is indicated below the corresponding graph in panel
a. The values of the nucleation rate,  , are
reported as log10(J/J0), where J0 refers to the
homogeneous nucleation rate at the same
temperature. (b) Sketches of the different regions (white areas) in
(Eads,afcc) space in which a significant enhancement of the nucleation rate
is observed. Each region is labeled according to the face of Ih nucleating and growing on top of the surface [basal, prismatic,
or (11/200), together with an indication of what
it is that enhances the nucleation, where “temp”, “buck”,
and “highE” refer to the in-plane template
of the first overlayer, the ice-like buckling of the contact layer,
and the nucleation for high adsorption energies on compact surfaces,
respectively. Reprinted with permission from ref (380). Copyright 2015 American
Chemical Society.
, are
reported as log10(J/J0), where J0 refers to the
homogeneous nucleation rate at the same
temperature. (b) Sketches of the different regions (white areas) in
(Eads,afcc) space in which a significant enhancement of the nucleation rate
is observed. Each region is labeled according to the face of Ih nucleating and growing on top of the surface [basal, prismatic,
or (11/200), together with an indication of what
it is that enhances the nucleation, where “temp”, “buck”,
and “highE” refer to the in-plane template
of the first overlayer, the ice-like buckling of the contact layer,
and the nucleation for high adsorption energies on compact surfaces,
respectively. Reprinted with permission from ref (380). Copyright 2015 American
Chemical Society.
Computational Methods and Models
Enhanced sampling
techniques have also been used to investigate heterogeneous ice nucleation.
Reinhardt and Doye382 used umbrella sampling
with the mW model to investigate nucleation at a smooth planar interface
and at an ice-like surface. They found that the flat planar interface
did not help nucleate ice and that homogeneous nucleation was the
preferred pathway. One explanation given for this finding was that,
as the density of liquid water is higher than that of ice, an attractive
surface favors the liquid phase. It was also noted that the mW potential
imposes an energy penalty for nontetrahedral triplets, that removing
neighbors at the surface decreases this energetic penalty, and that
this reduction in tetrahedrality favors the liquid phase. Cabriolu
and Li recently studied ice nucleation at graphitic surfaces using
forward flux sampling,383 again with the
mW model. Under the assumptions that 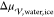 depends linearly on ΔT and
that
depends linearly on ΔT and
that 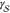 does not depend on ΔT, Cabriolu and Li also extracted the values of the contact
angle
at different temperatures, which, along with the free energy barrier,
turned out to be consistent with CNT for heterogeneous nucleation
(see section 1.1.3). Although intriguing, the generality of this finding to surfaces
that include strong and localized chemical interactions remains an
open question.
does not depend on ΔT, Cabriolu and Li also extracted the values of the contact
angle
at different temperatures, which, along with the free energy barrier,
turned out to be consistent with CNT for heterogeneous nucleation
(see section 1.1.3). Although intriguing, the generality of this finding to surfaces
that include strong and localized chemical interactions remains an
open question.
We have seen that, for both homogeneous and heterogeneous nucleation, using the coarse-grained mW model has greatly enhanced our ability to perform quantitative, systematic simulation studies of ice nucleation. We must face the fact, however, that this approach will further our understanding of heterogeneous ice nucleation only so far. As discussed for kaolinite,361,362,371,373 an explicit treatment of the hydrogen bonds is essential in describing heterogeneous ice nucleation. In addition, the mW model (as well as the majority of fully atomistic water models) cannot take into account surface-charge effects. Surfaces can polarize water molecules in the proximity of the substrate, alter their protonation state, and even play a role in determining the equilibrium structure of the liquid at the interface. In light of recent studies,370,384 it seems that these effects can heavily affect nucleation rates of many different systems. How, then, do we proceed? The answer is not clear. As discussed, enhanced sampling techniques such as umbrella sampling382 and forward flux sampling383 have been applied to heterogeneous ice nucleation with the mW model, and we have seen the latter applied successfully to homogeneous nucleation with an all-atom model of water;327 the computational cost, however, was huge. Although the presence of an ice-nucleating agent should help reduce this cost, the parameter space that we wish to study is large, and systematically studying how the various properties of a surface affect ice nucleation requires the investigation of many different surfaces.
There is another computational issue that also requires attention. Simulating heterogeneous ice nucleation under realistic conditions does not mean just mild supercooling; we also need realistic models of the surfaces that we wish to study! Most studies of kaolinite have considered only the planar interface, even though, in nature, kaolinite crystals have many step edges and defects. Ice nucleation at AgI was also recently studied,385,386 although bulk truncated structures for the exposed crystal faces were used. In the case of AgI(0001), this is problematic, as the wurtzite structure of the crystal means that this basal face is polar and likely to undergo reconstruction.387 Furthermore, AgI is photosensitive, and it has been shown experimentally that exposure to light enhances its ice-nucleating efficacy,388 suggesting that structural motifs at the surface very different from those expected from the bulk crystal structure are important. The development of computational techniques to determine surface structures, along with accurate force fields to describe the interaction with water, will be essential if we are to fully understand heterogeneous ice nucleation.
2.5. Nucleation from Solution
Understanding crystal nucleation from solution is a problem of great practical interest, influencing, for instance, pharmaceutical, chemical, and food processing companies. Being able to obtain a microscopic description of nucleation and growth would allow the selection of specific crystalline polymorphs, which, in turn, can have an enormous impact on the final product.389 An (in)famous case illustrating the importance of this issue is the drug Ritonavir,390,391 originally marketed as solid capsules to treat HIV. This compound has at least two polymorphs: the marketed and thoroughly tested polymorph (PI) and a second more stable crystalline phase PII that appeared after PI went to market. PII is basically nonactive as a drug because of a much lower solubility than PI. As such and, most importantly, because of the fact that PII had never been properly tested, Ritonavir was withdrawn from the market in favor of a much safer alternative in the form of gel capsules. Many other examples392 could be listed, as various environmental factors (such as the temperature, the degree of supersaturation, the type of solvent, and the presence of impurities) can play a role in determining the final polymorph of many classes of molecular crystals. Thus, it is highly desirable to pinpoint a priori the conditions leading to the formation of a specific polymorph possessing the optimal physical/chemical properties for the application of interest.
The term nucleation from solution encompasses a whole range of systems, from small molecules in aqueous or organic solvents to proteins, peptides, and other macromolecular systems in their natural environment. These systems are very diverse, and a universal nucleation framework is probably not applicable to all of these cases. The review by Dadey et al.393 discusses the role of the solvent in determining the final crystal. Many aspects of the nucleation of solute precipitates from solution were recently reviewed by Agarwal and Peters.394 In this section, we limit the discussion to small molecules in solution.
A central issue with MD simulations of nucleation from solution is the choice of order parameters able to distinguish different polymorphs. Many of these collective variables have been used in enhanced-sampling simulations (see section 1.3.2). Several examples can be found in refs (214 and 395−399). MD simulations of nucleation from solution are particularly challenging because of finite-size effects due to the nature of the solute/solvent system.152,400 In the NVT and NPT ensembles, where MD simulations of nucleation are usually performed, the total number of solute molecules is constant. However, the ratio between the numbers of solute molecules in the crystalline phase and in the solution varies during the nucleation events, leading to a change in the chemical potential of the system. This occurrence has negligible effects in the thermodynamic limit,401 but it can substantially affect the outcomes of, for example, free-energy-based enhanced-sampling simulations. Simulations of models containing a large number (103–105) of molecules can alleviate the problem,402 although this is not always the case.394,403,404 An analytic correction to the free energy for NPT simulations of nucleation of molecules from solution was proposed in refs (394 and 404) on the basis of a number of previous works (see, e.g., refs (152, 400, and 405)) and applied later in ref (406) as well. Alternative approaches include seeded MD simulations193,402 (see section 1.3.1) and simulations mimicking the grand canonical ensemble (μVT),407,408 where the number of constituents is not a constant and the number of molecules in—in this case—the solution is allowed to evolve in time. It is worth noticing that nucleation of molecules in solution is a challenging playground for experiments as well. For instance, quantitative data about nucleation of ionic solutions are amazingly hard to find within the current literature. This is in stark contrast with the vast amount of data covering, for example, ice nucleation (as illustrated in section 2.4).
2.5.1. Organic Crystals
Among the countless organic compounds, urea molecules can be regarded as a benchmark for MD simulation of nucleation from solution. This is because urea is a system of great practical importance that (i) displays fast nucleation kinetics and (ii) has only one experimentally characterized polymorph. Early studies by Piana and co-workers409,410 focused on the growth rate of urea crystals, which turned out to be consistent with experimental results. Years later, the inhibition of urea crystal growth by additives was investigated by Salvalaglio et al.411,412 The investigation of the early stages of nucleation was tackled only recently by Salvalaglio et al.413 for urea molecules in aqueous and organic (ethanol, methanol, and acetonitrile) solvents. In these studies, the authors employed metadynamics along with the generalized Amber force field.414,415 The resulting free energies, modified for finite-size effects related to the solvent,413 suggested that different solvents lead to different nucleation mechanisms. Whereas a single-step nucleation process is favored in methanol and ethanol, a two-step mechanism (see section 1.1.2) emerges for urea molecules in acetonitrile and water, as depicted in Figure 17. In this case, the initial formation of an amorphous—albeit dense—cluster is followed by evolution into a crystalline nucleus. Note that, according to the free energy surface reported in Figure 17a, the amorphous clusters (configurations 2 and 3 in Figure 17a,b) are unstable with respect to the liquid phase; that is, they are not metastable states having their own free energy bases, but rather, they originate from fluctuations within the liquid phase. This evidence, together with the fact that the transition state (configuration 4 in Figure 17a,b) displays a fully crystalline core, prompts the following, long-standing question: If the critical nucleus is mostly crystalline and the amorphous precursors are unstable with respect to the liquid phase, can we truly talk about a two-step mechanism? Reference (394) suggests the terms ripening regime two-step nucleation when dealing with stable amorphous precursors and crystallization-limited two-step nucleation when the amorphous clusters are unstable and the limiting step is the formation of a crystalline core within the clusters. Salvalaglio et al.413 also observed two polymorphs (PI and PII) in the early stages of the nucleation process. PI corresponds to the experimental crystal structure and is the most stable structure in the limit of an infinite crystal.416 PII, however, is more stable for small crystalline clusters. In agreement with the Ostwald rule (see section 2.2), the small crystalline clusters that initially form in solution are of the PII type, and the subsequent conversion from PII to PI seems to be an almost-barrierless process.
Figure 17.

(a) Free-energy surface (FES) associated with the early stages of nucleation of urea in aqueous solution, as obtained by Salvalaglio et al.406 from a well-tempered metadynamics simulation of 300 urea molecules and 3173 water molecules, within an isothermal–isobaric ensemble at p = 1 bar and T = 300 K (simulation S2 in ref (406) with a correction term to the free-energy included to represent the case of a constant supersaturation of 2.5). The contour plot of the FES is reported as a function of the number of molecules belonging to the largest connected cluster (n, along the ordinate) and the number of molecules in a crystal-like configuration within the largest cluster (no, along the abscissa). Note that n ≥ no by definition and that CNT would prescribe that the evolution of the largest cluster in the simulation box is such that n = no (i.e., only the diagonal of the contour plot is populated). The presence of an off-diagonal basin provides evidence of a two-step nucleation of urea crystals from aqueous solutions. This is further supported by the representative states sampled during the nucleation process, shown in panel b. Urea molecules are represented as blue spheres, and red connections are drawn between urea molecules falling within a cutoff distance of 0.6 nm of each other. Reprinted with permission from ref (406). Copyright 2015 National Academy of Sciences.
An approach similar to that employed in ref (413) was used to investigate crystal nucleation of 1,3,5-tris(4-bromophenyl)benzene molecules in water and methanol. These simulations showed the emergence of prenucleation clusters, consistent with recent experimental results137 based on single-molecule real-time transmission electron microscopy (SMRT-TEM; see section 1.2). The formation of prenucleation clusters in the early stages of nucleation from solution has been observed in several other cases.18,24,137,393,417 This is of great relevance, as CNT is not able to account for two- (or multi-) step nucleation. MD simulations have been of help in several cases, validating or supporting a particular mechanism. For instance, MD simulations provided evidence for two-step nucleation in aqueous solutions of α-glycine418 and n-octane (or n-octanol) solutions of d-/l-norleucine.419
2.5.2. Sodium Chloride
Sodium chloride (NaCl) nucleation from supersaturated brines represents an interesting challenge for simulations, as the system is relatively easy to model and experimental nucleation rates are available.
The first simulations of NaCl nucleation date back to the early 1990s, when Ohtaki and Fukushima420 performed brute-force MD simulations using very small systems (448 molecules including water molecules and ions) and exceedingly short simulation times (∼10 ps). Thus, the formation of small crystalline clusters that they observed was most likely a consequence of finite-size effects. More recently, the TPS simulations of Zahn421 suggested that the centers of stability for NaCl aggregates consist of nonhydrated Na+ ions octahedrally coordinated with Cl– ions, although the results were related to very small simulations boxes (containing 310 molecules in total).
Tentative insight into the structure of the crystalline clusters came with the work of Nahtigal et al.,422 featuring simulations of 4132 molecules (4000 water molecules and 132 ions) in the 673–1073 K range for supercritical water at different densities (0.17–0.34 g/cm3). They reported a strong dependence of the crystalline cluster size distribution on the system density, with larger clusters formed at lower densities. Moreover, the clusters appeared to be amorphous. The emergence of amorphous precursors was also reported in the work of Chakraborty and Patey,423,424 who performed large-scale MD simulations featuring 56000 water molecules and 4000 ion pairs in the NPT ensemble. The SPC/E model425 was used for water, and the ion parameters were those used in the OPLS426,427 force field. Their findings provided strong evidence for a two-step mechanism of nucleation, where a dense but unstructured NaCl nucleus is formed first, followed by a rearrangement into the rock salt structure, as depicted in Figure 18a. On a similar note, metadynamics simulations performed by Giberti et al.428 using the GROMOS429 force field for the ions and the SPC/E425 model for water suggested the emergence of a wurtzite-like polymorph in the early stages of nucleation. This precursor could be an intermediate state along the path from brine to the NaCl crystal. However, Alejandre and Hansen430 pointed out a strong sensitivity of the nucleation mechanism on the choice of the force field.
Figure 18.

(a) Snapshots
from an MD simulation of crystal nucleation of NaCl
from aqueous solution. The simulations, carried out by Chakraborty
and Patey,423 involved 56000 water molecules
and 4000 ion pairs (concentration of 3.97 m) in the NPT ensemble. All Na+ (black) and Cl– (yellow) ions within 2 nm of a reference Na+ ion (larger
and blue) are shown, together with water molecules (oxygen and hydrogen
atoms in red and white, respectively) within 0.4 nm from each ion.
From the relatively homogeneous solution (3 ns), an amorphous cluster
of ions emerges (5 ns). This fluctuation in the concentration of the
ions leads to a subsequent ordering of the disordered cluster (10
ns) in a crystalline fashion (30 ns), consistently with a two-step
nucleation mechanism. Reprinted with permission from ref (423). Copyright 2013 American
Chemical Society. (b) Comparison of NaCl nucleation rates,  , as a function
of the driving force for
nucleation, reported as 1/(Δμ/kBT)2. Red points and blue and gray (continuous)
lines were estimated by three different approaches in the simulations
of Zimmermann et al.402 Experimental data
obtained employing an electrodynamic levitator trap (Na et al.436), an efflorescence chamber (Gao et al.437), and microcapillaries (Desarnaud et al.438) are also reported, together with a tentative
fit (γfitexp, dotted line). Note the substantial (up to about 30 orders of magnitude)
discrepancy between experiments and simulations. Reprinted with permission
from ref (402). Copyright
2015 American Chemical Society.
, as a function
of the driving force for
nucleation, reported as 1/(Δμ/kBT)2. Red points and blue and gray (continuous)
lines were estimated by three different approaches in the simulations
of Zimmermann et al.402 Experimental data
obtained employing an electrodynamic levitator trap (Na et al.436), an efflorescence chamber (Gao et al.437), and microcapillaries (Desarnaud et al.438) are also reported, together with a tentative
fit (γfitexp, dotted line). Note the substantial (up to about 30 orders of magnitude)
discrepancy between experiments and simulations. Reprinted with permission
from ref (402). Copyright
2015 American Chemical Society.
In fact, very recent simulations by Zimmermann et al.402 demonstrated that the GROMOS force field overestimates the stability of the wurtzite-like polymorph. The authors employed a seeding approach within an NVT setup for which the absence of depletion effects was explicitly verified.152 The force fields used were those developed by Joung and Cheatham431 for Na+ and Cl– and SPC/E425 for water, which provide reliable solubilities and accurate chemical potential driving force.432 Using a methodology introduced in ref (193), the interfacial free energy and the attachment frequency δn were deduced. A thorough investigation of the latter demonstrated that the limiting factor for δn, which, in turn, strongly affects the kinetics of nucleation (see section 1.1.1), is not the diffusion of the ions within the solution but is instead the desolvation process needed for the ions to get rid of the solvent and join the crystalline clusters. Moreover, Zimmermann et al.402 evaluated the nucleation free energy barrier as well as the nucleation rate as a function of supersaturation, providing three estimates using different approaches. The results are compared with experiments in Figure 18b, showing a substantial discrepancy as large as 30 orders of magnitude. Interestingly, experimental nucleation rates are much smaller than what is observed in simulations, contrary to what has been observed for colloids, for example (see section 2.1). We stress that the work of Zimmermann et al. employed state-of-the-art computational techniques and explored NaCl nucleation under different conditions using a variety of approaches. The fact that these tour de force simulations yielded nucleation rates that differed significantly from experiments casts yet another doubt on the possibility of effectively comparing experiments and simulations. However, it must be noted that Zimmermann et al.402 assumed a value of about 5.0 molNaCl/kgH2O for the NaCl solubility in water, as proposed in ref (432). This differs substantially from the values independently obtained by Moucka et al.433 (3.64 molNaCl/kgH2O) and more recently by Mester and Panagiotopoulos434 (3.71 molNaCl/kgH2O). This discrepancy can explain the enormous mismatch reported by Zimmermann et al.,402 once again demonstrating the severe sensitivity of nucleation rates to any of the ingredients involved in their calculations.
On a final note, we stress that many other examples of molecular dynamics simulations looking at specific aspects of crystal nucleation from solution exist in the literature. For instance, a recent study by Anwar et al.435 describes secondary crystal nucleation, where crystalline seeds are already present within the solution. The authors suggest, for a generic solution represented by Lennard-Jones particles, a (secondary) nucleation mechanism enhanced by the existence of PNCs (see section 1.1.2). Kawska et al.189 stressed instead the importance of proton transfer within the early stages of nucleation of zinc oxide nanoclusters from an ethanol solution. The emergence of similar ripening processes, selecting specific crystalline polymorphs, for example, according to the effect of different solvents is still fairly unexplored but bound to be of great relevance in the future. Finally, several computational studies have dealt with the crystallization of calcium carbonate, which was recently reviewed extensively in ref (18) and thus, together with the broad topic of crystal nucleation of biominerals, is not discussed in here.
2.6. Natural Gas Hydrates
Natural gas hydrates are crystalline compounds in which small gas molecules are caged (or enclathrated) in a host framework of water molecules. As natural gas molecules (e.g., methane, ethane, propane) are hydrophobic, gas hydrates are favored by conditions of high pressure and low temperature, and are found to occur naturally in the ocean bed and in permafrost regions.439 With exceptionally high gas storage capabilities and the fact that it is believed that gas hydrates exceed conventional gas reserves by at least an order of magnitude,440 there is interest in trying to exploit gas hydrates as a future energy resource. Although gas hydrates might potentially play a positive role in the energy industry’s future, they are currently considered a hindrance: If mixed phases of water and natural gas are allowed to cool in an oil pipeline, then a hydrate can form and block the line, causing production to stall. Understanding the mechanism(s) by which gas hydrates nucleate is likely to play an important role in the rational design of more effective hydrate inhibitors.
2.6.1. Hydrate Structures
There are two main types of natural gas hydrates: structure I (sI), which has a cubic structure (space group Pm3̅n), and structure II (sII), which also has a cubic structure (space group Fd3̅m). (There is also a third, less common type, sH, which has a hexagonal crystal structure, but we do not discuss this structure any further here.) Structurally, the water frameworks of both sI and sII hydrates are similar to that of ice Ih, with each water molecule finding itself in an approximately tetrahedral environment with its nearest neighbors. Unlike ice Ih, however, the water framework consists of cages, with cavities large enough to accommodate a gas molecule.
Between the sI and sII hydrates, there exist three types of cages, which are denoted 5p6h depending on the numbers of five- and six-sided faces that make up the cage. For example, common to both the sI and sII hydrates is the 512 cage, where the water molecules sit on the vertices of a pentagonal dodecahedron. Along with 512 cages, the sI hydrate also consists of a 51262 cages, which have two six-sided faces and 12 five-sided faces: There are two 512 cages and six 51262 cages in the unit cell. The sII hydrate, on the other hand, has a unit cell made up of 16 512 cages and eight 51264 cages. Because of the larger size of the 51264 cage, the sII structure forms in the presence of larger guest molecules such as propane, whereas small guest molecules such as methane favor the sI hydrate. (This is not to say that small guest molecules are not present in sII, just that the presence of larger guest molecules is necessary to stabilize the larger cavities.) The sI, sII, and sH crystals structures are shown in Figure 19, along with the individual cage structures. Further details regarding the crystal structures of natural gas hydrates can be found in ref (439).
Figure 19.

Crystal structures of the sI, sII and sH gas hydrates, along with the corresponding cage structures. Only the water molecule positions are shown, as spheres connected by lines. Reprinted with permission from ref (441). Copyright 2007 John Wiley & Sons.
2.6.2. Homogeneous Nucleation
Historically, two main molecular mechanisms for hydrate nucleation have been proposed. First, Sloan and co-workers442,443 proposed the labile cluster hypothesis (LCH), which essentially describes the nucleation process as the formation of isolated hydrate cages that then agglomerate to form a critical hydrate nucleus. Second, the local structure hypothesis (LSH) was proposed after umbrella sampling simulations by Radhakrishnan and Trout444 suggested that the guest molecules first arrange themselves in a structure similar to the hydrate phase, which is accompanied by a perturbation (relative to the bulk mixture) of the water molecules around the locally ordered guest molecules. For the same reasons as already outlined elsewhere (see section 1.2), it is experimentally challenging the verify which, if either, of these two nucleation mechanisms is correct. What we will see in this section is how computer simulations of gas hydrate nucleation have been used to help shed light on this process.
Although not the first computer simulation study of natural gas hydrate formation (see, e.g., refs (444−447)), one the most influential simulation works on gas hydrate formation is that of Walsh et al.,448 in which methane hydrate formation was directly simulated under conditions of 250 K and 500 bar. It was found that nucleation proceeded through the cooperative organization of two methane and five water molecules into a stable structure, with the methane molecules adsorbed on opposite sides of a pentagonal ring of water molecules. This initial structure allowed the growth of more water faces and adsorbed methane, until a 512 cage formed. This process took on the order of 50–100 ns to complete. After persisting for ∼30 ns, this 512 cage opened when two new water molecules were inserted into the only face without an adsorbed methane molecule, on the side opposite to that where several new full cages were completed. This opening of the original 512 cage was then followed by the relatively fast growth of methane hydrate. The early stages of hydrate nucleation are shown in Figure 20. After ∼240 ns, the original 512 cage transformed into a 51263 cage, a structure not found in any equilibrium hydrate structure. Walsh et al. also found that 512 cages dominated, in terms of abundance, during the early stages of nucleation. 51262 cages (which along with the 512 cages comprise the sI hydrate) were the second most abundant, although their formation occurred approximately 100 ns after that of the initial 512 cages. A significant amount of the larger 51264 cages that are found in the sII hydrate was also observed, which was rationalized by the large number of face-sharing 512 cages providing an appropriate pattern. The 51263 cages were also observed in an abundance close to that of the 51262 cages. The final structure can be summarized as a mixture of sI and sII motifs, linked by 51263 cages. A similar structure had previously been reported as a result of hydrate growth simulations.449,450
Figure 20.

Early stages of hydrate nucleation observed by Walsh et al.:448 (A–C) A pair of methane molecules is adsorbed on either side of a single pentagonal face of water molecules. Partial cages form around this pair, near the eventual central violet methane molecule, only to dissociate over several nanoseconds. (D,E) A small cage forms around the violet methane, and other methane molecules adsorb at 11 of the 12 pentagonal faces of the cage, creating the bowl-like pattern shown. (F,G) The initial central cage opens on the end opposite to the formation of a network of face-sharing cages, and rapid hydrate growth follows. (H) A snapshot of the system after hydrate growth shows the fates of those methane molecules that made up the initial bowl-like structure (other cages not shown). Reprinted with permission from ref (448). Copyright 2009 American Association for the Advancement of Science.
Even though the work of Walsh et al.448 provided useful insight into the hydrate nucleation mechanism, the conclusions were based on only two independent nucleation trajectories. Soon after the publication by Walsh et al., Jacobsen et al.451 reported a set of 12 simulations using a methane–water model452 based on mW water under conditions of 210 K and 500 atm (the melting point of the model is approximately 300 K). Owing to the reduced computational cost of the coarse-grained model, they were also able to study a much larger system size than Walsh et al. (8000 water and 1153 guest molecules451 vs 2944 water and 512 guest molecules448). In agreement with Walsh et al., the initial stages of the nucleation mechanism were also dominated by 512 cages, and a mixture of sI and sII motifs connected by 51263 cages was observed. It was also observed that solvent-separated pairs of guest molecules were stabilized by greater numbers of guest molecules in the cluster. As gas hydrates are composed of solvent-separated pairs of guest molecules as opposed to contact pairs, this suggests a resemblance to the LSH, where the local ordering of guest molecules drives the nucleation of the hydrate. Jacobsen et al., however, also found a likeness to the LCH: Clusters of guest molecules and their surrounding water molecules formed long-lived blobs that slowly diffused in solution. These blobs could be considered large analogues of the labile clusters proposed in the LCH. Through analysis of their simulation data, Jacobsen et al. concluded that the blob is a guest-rich precursor in the nucleation pathway of gas hydrates with small guest molecules (such as methane). Note that the distinction between blobs and the amorphous clathrate is that the water molecules have yet to be locked into the clathrate hydrate cages in the former. The overall nucleation mechanism is depicted in Figure 21.
Figure 21.

Sketch of the nucleation mechanism of methane hydrates proposed in ref (451). Clusters of guest molecules aggregate in blobs, which transform into amorphous clathrates as soon as the water molecules arrange themselves in the cages characteristic of crystalline clathrate, which eventually form upon the reordering of the guest molecules—and thus of the cages—in a crystalline fashion. Note that the difference between the blob and the amorphous clathrate is that the water molecules have yet to be locked into clathrate hydrate cages in the former. Reprinted with permission from ref (451). Copyright 2010 American Chemical Society.
Both the work of Walsh et al. and that of Jacobson et al. suggest that amorphous hydrate structures are involved in the nucleation mechanism, although both studies were carried out under high driving forces. In ref (453), Jacobsen and Molinero addressed the following two questions raised by the above studies: How could amorphous nuclei grow into a crystalline form? Are amorphous nuclei precursors intermediates for clathrate hydrates under less forcing conditions? By considering the size-dependent melting temperature of spherical particles using the Gibbs–Thomson equation, Jacobson and Molinero found for all temperatures that the size of the crystalline critical nucleus was always smaller than that of the amorphous critical nucleus, with the two becoming virtually indistinguishable in terms of stability for very small nuclei of ∼15 guest molecules (i.e., under very forcing conditions). From a thermodynamic perspective, this would suggest that nucleation would always proceed through a crystalline nucleus. The observation of amorphous nuclei,445,448,451,454,455 even at temperatures as high as 20% supercooling, hints that their formation might be favored for kinetic reasons. Employing the CNT expression for the free energy barrier suggested that the amorphous nuclei could be kinetically favored up to 17% supercooling if γa/γx = 0.5, where γa and γx are the surface tensions of the liquid-amorphous and liquid-crystal structures, respectively. Jacobson and Molinero estimated γx ≈ 36 mJ/m2 and 16 < γa < 32 mJ/m2, so it is certainly plausible that amorphous precursors are intermediates for clathrate hydrates under certain conditions. The growth of clathrate hydrates from amorphous and crystalline seeds was also studied, where it was found that crystalline clathrate can grow from amorphous nuclei. As the simulation led to fast mass transport, the growth of postcritical nuclei was relatively quick, and the amorphous seed became encapsulated by a (poly)crystalline shell. Under conditions where an amorphous nucleus forms first because of a smaller free energy barrier but diffusion of the guest species becomes a limiting factor, it is likely that small nuclei would have long enough to anneal to structures of greater crystallinity before growing to the macroscopic crystal phase.
It thus appears that gas hydrates might exhibit a multistep nucleation process involving amorphous precursors for reasonably forcing conditions, but for temperatures close to coexistence, it seems that nucleation should proceed through a single crystalline nucleus. By assuming a CNT expression for the free energy (as well as the total rate), Knott et al.456 used the seeding technique (see section 1.3.1) to compute the nucleation rate for sI methane hydrate with relatively mild supersaturation of methane, in a manner similar to that of Espinosa et al.326 for homogeneous ice nucleation as discussed in section 2.4.1. They found vanishingly small homogeneous nucleation rates of 10–111 nuclei cm–3 s–1, meaning that, even with all of Earth’s ocean waters, the induction time to form one crystal nucleus homogeneously would be ∼1080 years! Knott et al. therefore concluded that, under mild conditions, hydrate nucleation must occur heterogeneously.
2.6.3. Heterogeneous Nucleation
Compared to homogeneous nucleation, the heterogeneous nucleation of gas hydrates has been little studied. Liang et al.454 investigated the steady-state growth of a H2S hydrate crystal in the presence of silica surfaces, finding that the crystal preferentially grew in the bulk solution rather than at the interface with the solid. They also observed that, in one simulation, local gas density fluctuations of the dissolved guest led to the spontaneous formation of a gas bubble from solution, which was located at the silica interface. This had two effects on the observed growth: (i) the bubble depleted most of the gas from solution, leading to an overall decrease of the crystal growth rate, and (ii) because of the location of the guest bubble, the silica surface effectively acted like a source of gas, promoting growth of the crystal closer to the interface relative to the bulk.
Bai et al. investigated the heterogeneous nucleation of CO2 hydrate in the presence of a fully hydroxylated silica surface, first in a two-phase system where the water and CO2 were well-mixed457 and then in a three-phase system where the CO2 and water were initially phase-separated.458 For the two-phase system, the authors reported the formation of an ice-like layer at the silica surface, above which a layer composed of semi-512 cage-like structures mediated the structural mismatch between the ice-like contact layer and the sI hydrate structure above. In the three-phase system, nucleation was observed at the three-phase contact line, along which the crystal nucleus also grew. This was attributed to the stabilizing effect of the silica on the hydrate cages, plus the requirement for the availability of both water and CO2. In a later work, Bai et al.459 investigated the effects of surface hydrophilicity (by decreasing the percentage of surface hydroxyl groups) and crystallinity on the nucleation of CO2 hydrate. They found that, in the case of decreased hydrophilicity, the ice-like layer at the crystalline surface vanished, replaced instead by a single liquid-like layer upon which the hydrate directly nucleated. Whereas shorter induction times to nucleation at the less hydrophilic surfaces were reported, little dependence on the crystallinity of the surface was observed. Although certainly an interesting observation, as only a single trajectory was performed for each system, studies in which multiple trajectories are used to obtain a distribution of induction times would be desirable, and as the hydrate actually appears to form away from the surface in all cases, a full comparison of the heterogeneous and homogeneous rates would also be a worthwhile pursuit.
There have also been a number of studies investigating the potential role of ice in the nucleation of gas hydrates. Pirzadeh and Kusalik460 performed MD simulations of methane hydrate nucleation in the presence of ice surfaces and reported that an increased density of methane at the interface induced structural defects (coupled 5–8 rings) in the ice that facilitated the formation of hydrate cages. Nguyen et al.461 used MD simulations to directly investigate the interface between a gas hydrate and ice and found the existence of an interfacial transition layer (ITL) between the two crystal structures. The water molecules in the ITL, which was found to be disordered and two to three layers of water in thickness, had a tetrahedrality and potential energy intermediate between those of either of the crystal structures and liquid water. The authors suggested that the ITL could assist the heterogeneous nucleation of gas hydrates from ice by providing a lower surface free energy than either of the ice–liquid and hydrate–liquid interfaces. Differential scanning calorimetry experiments by Zhang et al.462 found ice and hydrate formation to occur simultaneously (on the experimental time scale), which was attributed to the heterogeneous nucleation of ice, which, in turn, facilitated hydrate formation. Poon and Peters463 provide a possible explanation for ice acting as a heterogeneous nucleating agent for gas hydrates, aside from the structural considerations of refs (460 and 462): At a growing ice front, the local supersaturation of methane can be dramatically increased, to the extent that induction times to nucleation are reduced by as much as a factor 10100.
Computer simulations of hydrate nucleation have certainly contributed to our understanding of the underlying mechanisms, especially in the case of homogeneous nucleation. One fairly consistent observation across many simulation studies (e.g., refs (444, 445, 447, 448, 451, 453, and 464)) suggests that some type of ordering of dissolved guest molecules precedes the formation of hydrate cages. Another is that amorphous nuclei, consisting of structural elements of both sI and sII hydrates form when conditions are forcing enough. Nevertheless, open questions still remain. In particular, the prediction that homogeneous nucleation rates are vanishingly small under mild conditions456 emphasizes the need to better understand heterogeneous nucleation. To this end, enhanced sampling techniques such as FFS, which was recently applied to methane hydrate nucleation at 220 K and 500 bar,464 are likely to be useful, although directly simulating nucleation under mild conditions is still likely to be a daunting task. Another complicating factor is that, aside from the presence of solid particles, the conditions from which natural gas hydrates form are often highly complex; for example, in an oil or gas line, there is fluid flow, and understanding how this effects the methane distribution in water is likely to be an important factor in determining how fast gas hydrates form.465 In this respect, the formation of natural gas hydrates is a truly multiscale phenomenon.
3. Future Perspectives
We have described only a fraction of the many computer simulation studies of crystal nucleation in supercooled liquids and solutions. Still, we have learned that MD simulations have dramatically improved our fundamental understanding of nucleation. For instance, several studies on colloidal particles (see section 2.1) provided evidence for two-step nucleation mechanisms, and the investigation of LJ liquids yielded valuable insights into the effects of confinement (see section 2.2). In addition, the investigation of more realistic systems has provided outcomes directly related to problems of great relevance. For example, the influence of different solvents on the early stages of urea crystallization (see section 2.5) has important consequences in fine chemistry and in the fertilizer industry, and the molecular details of clathrate nucleation (see section 2.5) could help to rationalize and prevent hydrate formation in oil or natural gas pipelines. Thus, it is fair to say that MD simulations have been and will remain a powerful complement to experiments.
However, simulations are presently affected by several shortcomings, which hinder a reliable comparison with experimental nucleation rates and limit nucleation studies to systems and/or conditions often far from those investigated experimentally. These weaknesses can be classified in two main categories: (i) limitations related to the accuracy of the computational model used to represent the system and (ii) shortcomings due to the computational techniques employed to simulate nucleation events.
-
(i)
In an ideal world, ab initio calculations would be the tool of the trade. Unfortunately, in all but a handful of cases such as the phase-change materials presented in section 2.3, the time-scale problem makes ab initio simulations of crystal nucleation unfeasible (see Figure 5). As this will be the status quo for the next few decades, we are forced to focus our efforts on improving the current classical force fields and on developing novel classical interatomic potentials. This is a fundamental issue that affects computer simulations of materials as a whole. Although this is not really an issue for nucleation of simple systems such as colloids (section 2.1), things start to fall apart when dealing with more realistic systems (see, e.g., sections 2.5 and 2.6) and become even worse in the case of heterogeneous nucleation (see, e.g., section 2.4.2), as the description of the interface requires extremely transferable and reliable force fields. Machine learning techniques466 such as neural network potentials (see section 2.3 and refs (467 and 468)) are emerging as possible candidates to allow for classical MD simulations with an accuracy closer to first-principles calculations, but the field is constantly looking for other options that are capable of bringing simulations closer to reality.
-
(ii)
The limitations of the computational techniques currently employed to study crystal nucleation are those characteristic of rare-events sampling. Brute-force MD simulations (see section 1.3.1) allow for an unbiased investigation of nucleation events, but the time-scale problem limits this approach to very few systems, typically very distant from realistic materials (see, e.g., sections 2.1 and 2.2)—although notable exceptions exist (see section 2.3). It is also worth noticing that, whereas brute-force MD is not able to provide a full characterization of the nucleation process, useful insight can still be gained, for example, into prenucleation events.18,469 Enhanced sampling techniques (see section 1.3.2) are rapidly evolving and have the potential to take the field to the next level. However, free energy methods as they are do not give access to nucleation kinetics and, in the case of complex systems (see, e.g., sections 2.4.1 and 2.5), are strongly dependent on the choice of the order parameter. On the other hand, in light of the body of work reviewed, it seems that path-sampling methods can provide a more comprehensive picture of crystal nucleation. However, at the moment, these techniques are computationally expensive, and a general implementation is not available yet, although consistent efforts have recently been put in place. We believe that the development of efficient enhanced sampling methods specific to crystal nucleation is one of the crucial challenges ahead.
At the moment, simulations of crystal nucleation of complex liquids are restricted to small systems (102–104 particles), most often under idealized conditions. For instance, it is presently very difficult to take into account impurities or, in the case of heterogeneous nucleation, defects of the substrate. Indeed, defects seem to be ubiquitous in many different systems, such as ice, hard-sphere crystals, LJ crystals, and organic crystals as well. Defects are also often associated with polymorphism, but possibly because of the inherent difficulties in modeling them (or in characterizing them experimentally), they are under-represented in the current literature. These are important aspects that almost always impact experimental measurements and that should thus be included in simulations as well. In general, simulations of nucleation should allow us not only to provide microscopic insight but also to make useful predictions and/or to provide a general understanding to be applied to a variety of systems. These two ambitious goals are particularly challenging for simulations of heterogeneous nucleation. In light of the literature we have reviewed in this work, we believe that much of the effort in the future has to be devoted to (i) enabling atomistic simulations of heterogeneous nucleation dealing with increasingly realistic interfaces and (ii) obtaining general, maybe non-material-specific trends able to point the community into the right direction, even at the cost of sacrificing accuracy to a certain extent. On the other hand, we hope that the body of work reviewed here will inspire future experiments targeting cleaner, well-defined systems by means of novel techniques, possibly characterized by better temporal and spatial resolution. Improving on the current limitations of the computational models and techniques would enable simulations of much larger systems over much longer time scales, with a degree of accuracy that would allow a fruitful comparison with experiments. We think this should be the long-term objective for the field. Up to now, the only way to connect simulations and experiments has been through the comparison of crystal nucleation rates, which even now still exhibit substantial discrepancies for every single class of systems we have reviewed. This is true not only for complex liquids such as water (see section 2.4.1) but even for model systems such as colloids (section 2.1). This, together with the fact that, in some cases, even experimental data are scattered across several orders of magnitude, suggests that we are dealing with crystal nucleation in liquids within a flawed theoretical framework.
In fact, CNT is now 90 years old. It is thus no wonder that every aspect of this battered theory has been criticized at some point. However, some aspects have been questioned more frequently than others. For instance, the emergence of two-step (or even multistep) mechanisms for nucleation has been reported for many different systems (see sections 2.1, 2.2, 2.5, and 2.6) and cannot be easily embedded in CNT as it is, although several improvements on the original CNT formulation have appeared within the past decade (see section 1.1.2). Nonetheless, CNT is basically the only theory invoked by both experiments and simulations when dealing with crystal nucleation from the liquid phase. CNT is widely used because it offers a simple and unified picture for nucleation and it is often very useful. However, as demonstrated by both experiments and simulations, even the basic rules governing the formation of the critical nucleus can change dramatically from one system to another. Thus, we believe that any sort of theoretical universal approach, a brand new CNT, so to say, will be unlikely to significantly further the field. Indeed, we fear that the same reasoning will hold for the computational methods required. We cannot think of a single enhanced sampling technique capable of tackling the complexity of crystal nucleation as a whole. The interesting but uncomfortable truth is that each class of supercooled liquids often exhibits unique behavior, which, in turn, results in specific features ruling the crystal nucleation process. Thus, it is very much possible that different systems under different conditions could require different, ad hoc flavors of CNT. Although the latter have been evolving for decades, we believe that a sizable fraction of the new developments in the field should aim at producing particular flavors of CNT, specifically tailored to the problem at hand.
In conclusion, it is clear that MD simulations have proven themselves to be of the utmost importance in unraveling the microscopic details of crystal nucleation in liquids. We have reviewed important advances that have provided valuable insights into fundamental issues and diverse nucleation scenarios, complementing experiments and furthering our understanding of nucleation as a whole. Whether CNT can be effectively improved in a universal fashion is unclear. We feel that the ultimate goal for simulations should be to get substantially closer to the reality probed by experiments and that, to do so, we have to sharpen our computational and possibly theoretical tools. In particular, we believe that the community should invest in improving the classical interatomic potentials available as well as the enhanced sampling techniques currently used, enabling accurate simulations of crystal nucleation for systems of practical relevance.
Acknowledgments
This work was supported by the European Research Council under the European Union’s Seventh Framework Programme (FP/2007-2013)/ERC Grant Agreement 616121 (HeteroIce project). A.M. was also supported by the Royal Society through a Royal Society Wolfson Research Merit Award. We gratefully acknowledge Dr. Matteo Salvalaglio, Dr. Gareth Tribello, Dr. Richard Sear, and Prof. Daan Frenkel for insightful discussions and for reading an earlier version of the manuscript.
Glossary
Abbreviations
- bcc
body-centered cubic
- cDFT
classical density functional theory
- CNT
classical nucleation theory
- DFT
density functional theory
- DSC
differential scanning calorimetry
- fcc
face-centered cubic
- FFS
forward flux sampling
- FTIRS
Fourier transform infrared spectroscopy
- HDL
high-density liquid
- LCH
labile cluster hypothesis
- LDL
low-density liquid
- LSH
local structure hypothesis
- MD
molecular dynamics
- MetaD
metadynamics
- PNC
prenucleation cluster
- rhcp
random hexagonal close packed
- SEM
scanning electron microscopy
- sH
structure H
- sI
structure I
- sII
structure II
- SMRT-TEM
single-molecule-real-time TEM
- TEM
transmission electron microscopy
- TIS
transition interface sampling
- UMD
unbiased molecular dynamics
- US
umbrella sampling
- XPS
X-ray photoeletron spectroscopy
Biographies
Gabriele C. Sosso obtained his Ph.D. in Nanostructures and Nanotechnologies from The University of Milano-Bicocca (Milan, Italy) in 2012, under the supervision of Prof. Marco Bernasconi. He then moved to ETH Zurich (USI Campus Lugano, Lugano, Switzerland) as a Postdoctoral Researcher within the group of Prof. Michele Parrinello. He joined the group of Prof. Angelos Michaelides at University College London in 2015 as a Postdoctoral Research Associate. His research deals chiefly with computer simulations of disordered systems and phase transitions, particularly crystal nucleation and growth.
Ji Chen graduated from University of Science and Technology of China with a B.Sc. degree in physics in 2009. Under the supervision of Prof. Enge Wang and Prof. Xinzheng Li, he completed his Ph.D. degree in 2014 at Peking University. He is currently a Postdoctoral Research Associate in the group of Prof. Angelos Michaelides at University College London, working on two-dimensional water and ice. His research interests include surface chemical physics and materials under extreme conditions.
Stephen J. Cox graduated from University of Cambridge in 2010 with a B.A. in Natural Sciences, where he undertook research projects with Prof. Daan Frenkel and Prof. Michiel Sprik. Under the supervision of Prof. Angelos Michaelides at University College London, he completed his Ph.D. in computational chemistry in 2014, in which he primarily investigated heterogeneous ice nucleation. He is currently a Postdoctoral Research Fellow in the Chemical Sciences Division at Lawrence Berkeley National Laboratory, where he investigates the theory of self-assembly and ion solvation.
Martin Fitzner graduated in 2012 from Friedrich-Schiller-University Jena (Jena, Germany) with a first-class B.Sc. in physics. After completing his Ms.C. in 2014, focused on the electronic structure of topological insulators, he joined the group of Angelos Michaelides. He is currently working toward his Ph.D., employing computer simulations to further our molecular understanding of heterogeneous ice nucleation.
Philipp Pedevilla obtained his B.Sc. from Leopold Franzens University of Innsbruck (Innsbruck, Austria) in 2011. After spending a year at University College London as an Erasmus student, he obtained his M.Sc. from Leopold Franzens University of Innsbruck in 2013. He is currently working toward his Ph.D. degree at University College London in the group of Prof. Angelos Michaelides. His interests are ab initio and classical molecular dynamics simulations of aqueous interfaces, mainly aimed at understanding ice nucleation at the molecular level.
Andrea Zen holds a Ph.D. in statistical and biological physics from the International School for Advanced Studies (Trieste, Italy, 2009). Afterward, he was awarded a Research Fellowship from the University of Rome La Sapienza, and since 2014, he has worked as a Research Associate at the London Centre for Nanotechnology, University College London, in the group of Prof. Angelos Michaelides. His research focuses on statistical mechanics and electronic structure calculations, mostly using density functional theory and quantum Monte Carlo approaches.
Angelos Michaelides obtained a Ph.D. in Theoretical Chemistry in 2000 from The Queen’s University of Belfast. Following this, he worked as a Postdoctoral Research Associate and Junior Research Fellow at the University of Cambridge and then at the Fritz Haber Institute (Berlin, Germany) as an Alexander von Humboldt Research Fellow. Subsequently, he was promoted to Staff Scientist and Research Group Leader at the Fritz Haber Institute. In 2006, he moved to University College London, where, since 2009, he has been Professor of Theoretical Chemistry. Research in his group (www.chem.ucl.ac.uk/ice) involves computer simulations of catalytic and environmental interfaces, aiming at reaching a fundamental new understanding of elementary processes at such interfaces. Nucleation and water are major focuses of his work.
Author Present Address
† Chemical Sciences Division, Lawrence Berkeley National Laboratory, Berkeley, CA 94720, USA.
The authors declare no competing financial interest.
References
- Bartels-Rausch T. Chemistry: Ten Things We Need to Know about Ice and Snow. Nature 2013, 494, 27–29. 10.1038/494027a. [DOI] [PubMed] [Google Scholar]
- Murray B. J.; Wilson T. W.; Dobbie S.; Cui Z.; Al-Jumur S. M. R. K.; Möhler O.; Schnaiter M.; Wagner R.; Benz S.; Niemand M.; et al. Heterogeneous Nucleation of Ice Particles on Glassy Aerosols under Cirrus Conditions. Nat. Geosci. 2010, 3, 233–237. 10.1038/ngeo817. [DOI] [Google Scholar]
- Mazur P. Cryobiology: The Freezing of Biological Systems. Science 1970, 168, 939–949. 10.1126/science.168.3934.939. [DOI] [PubMed] [Google Scholar]
- Lintunen A.; Hölttä T.; Kulmala M. Anatomical Regulation of Ice Nucleation and Cavitation Helps Trees to Survive Freezing and Drought Stress. Sci. Rep. 2013, 3, 2031. 10.1038/srep02031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erdemir D.; Lee A. Y.; Myerson A. S. Polymorph Selection: The Role of Nucleation, Crystal Growth and Molecular Modeling. Curr. Opin. Drug. Discovery Dev. 2007, 10, 746–755. [PubMed] [Google Scholar]
- Cox J. R.; Ferris L. A.; Thalladi V. R. Selective Growth of a Stable Drug Polymorph by Suppressing the Nucleation of Corresponding Metastable Polymorphs. Angew. Chem., Int. Ed. 2007, 46, 4333–4336. 10.1002/anie.200605257. [DOI] [PubMed] [Google Scholar]
- Sloan E. D. Fundamental Principles and Applications of Natural Gas Hydrates. Nature 2003, 426, 353–363. 10.1038/nature02135. [DOI] [PubMed] [Google Scholar]
- Hammerschmidt E. G. Formation of Gas Hydrates in Natural Gas Transmission Lines. Ind. Eng. Chem. 1934, 26, 851–855. 10.1021/ie50296a010. [DOI] [Google Scholar]
- Velazquez-Castillo R.; Reyes-Gasga J.; Garcia-Gutierrez D. I.; Jose-Yacaman M. Nanoscale Characterization of Nautilus Shell Structure: An Example of Natural Self-Assembly. J. Mater. Res. 2006, 21, 1484–1489. 10.1557/jmr.2006.0190. [DOI] [Google Scholar]
- Harper J. D.; Lieber C. M.; Lansbury P. T. Jr Atomic Force Microscopic Imaging of Seeded Fibril Formation and Fibril Branching by the Alzheimer’s Disease Amyloid-β Protein. Chem. Biol. 1997, 4, 951–959. 10.1016/S1074-5521(97)90303-3. [DOI] [PubMed] [Google Scholar]
- Walsh D. M.; Lomakin A.; Benedek G. B.; Condron M. M.; Teplow D. B. Amyloid β-Protein Fibrillogenesis Detection of a Protofibrillar Intermediate. J. Biol. Chem. 1997, 272, 22364–22372. 10.1074/jbc.272.35.22364. [DOI] [PubMed] [Google Scholar]
- Debenedetti P. G.Metastable Liquids: Concepts and Principles; Princeton University Press: Princeton, NJ, 1996; pp 105–121. [Google Scholar]
- Stevenson J. D.; Wolynes P. G. The Ultimate Fate of Supercooled Liquids. J. Phys. Chem. A 2011, 115, 3713–3719. 10.1021/jp1060057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Many supercooled liquids under certain conditions take a more chaotic path toward amorphous systems.470 However, this topic, involving the uncanny phenomenon of the glass transition,315 lies outside the scope of this work.
- Habraken W. J. E. M.; Tao J.; Brylka L. J.; Friedrich H.; Bertinetti L.; Schenk A. S.; Verch A.; Dmitrovic V.; Bomans P. H. H.; Frederik P. M.; et al. Ion-Association Complexes Unite Classical and Non-Classical Theories for the Biomimetic Nucleation of Calcium Phosphate. Nat. Commun. 2013, 4, 1507. 10.1038/ncomms2490. [DOI] [PubMed] [Google Scholar]
- Murray B. J.; O’Sullivan D.; Atkinson J. D.; Webb M. E. Ice Nucleation by Particles Immersed in Supercooled Cloud Droplets. Chem. Soc. Rev. 2012, 41, 6519–6554. 10.1039/c2cs35200a. [DOI] [PubMed] [Google Scholar]
- Volmer M.; Weber A. Germ-Formation in Oversaturated Figures. Z. Phys. Chem. 1926, 119, 277–301. [Google Scholar]
- Gebauer D.; Kellermeier M.; Gale J. D.; Bergström L.; Cölfen H. Pre-Nucleation Clusters as Solute Precursors in Crystallisation. Chem. Soc. Rev. 2014, 43, 2348–2371. 10.1039/c3cs60451a. [DOI] [PubMed] [Google Scholar]
- Sear R. P. Quantitative Studies of Crystal Nucleation at Constant Supersaturation: Experimental Data and Models. CrystEngComm 2014, 16, 6506–6522. 10.1039/C4CE00344F. [DOI] [Google Scholar]
- Vekilov P. G. Nucleation_Vekilov. Cryst. Growth Des. 2010, 10, 5007–5019. 10.1021/cg1011633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu X.; Ting C. L.; Kusaka I.; Wang Z.-G. Nucleation in Polymers and Soft Matter. Annu. Rev. Phys. Chem. 2014, 65, 449–475. 10.1146/annurev-physchem-032511-143750. [DOI] [PubMed] [Google Scholar]
- Yi P.; Rutledge G. C. Molecular Origins of Homogeneous Crystal Nucleation. Annu. Rev. Chem. Biomol. Eng. 2012, 3, 157–182. 10.1146/annurev-chembioeng-062011-081029. [DOI] [PubMed] [Google Scholar]
- Anwar J.; Zahn D. Uncovering Molecular Processes in Crystal Nucleation and Growth by Using Molecular Simulation. Angew. Chem., Int. Ed. 2011, 50, 1996–2013. 10.1002/anie.201000463. [DOI] [PubMed] [Google Scholar]
- Zahn D. Thermodynamics and Kinetics of Prenucleation Clusters, Classical and Non-Classical Nucleation. ChemPhysChem 2015, 16, 2069–2075. 10.1002/cphc.201500231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- In most cases, simulations of crystal nucleation do not embrace classical nucleation theory as a whole, taking into account instead only selected elements of the theory. Common examples (i) assuming the size of the largest crystalline cluster as the only relevant order parameter to describe the nucleation process, (ii) assuming the critical nuclei to be of spherical shape, and (iii) assuming a perfectly sharp interface between the crystalline and the liquid phase.
- Kelton K.; Greer A.. Crystallization in Glasses. In Nucleation in Condensed Matter: Applications in Materials and Biology; Kelton K., Greer A., Eds.; Pergamon Materials Series; Pergamon: Oxford, U.K., 2010; Vol. 15, Chapter 8, pp 279–329. [Google Scholar]
- Kalikmanov V.Nucleation Theory; Lecture Notes in Physics; Springer: Dordrecht, The Netherlands, 2013; Vol. 860. [Google Scholar]
- Vehkamäki H.Classical Nucleation Theory in Multicomponent Systems; Springer: Berlin, 2006. [Google Scholar]
- Farkas L. Nucleation Rates in Supersaturated Vapours. Z. Phys. Chem. 1927, 125, 236. [Google Scholar]
- Farkas, forced to flee Nazi Germany in 1933, was the first one to develop a real theory of nucleation, as acknowledged by Becker and Döring31 in 1935. Interestingly, Farkas wrote in ref (29) that his work was, in turn, inspired by an idea of Leo Szilard, who apparently never bothered to write anything on the topic.
- Becker R.; Döring W. Kinetische Behandlung Der Keimbildung in ÜbersÄttigten DÄmpfen. Ann. Phys. 1935, 416, 719–752. 10.1002/andp.19354160806. [DOI] [Google Scholar]
- Zeldovich J. B. On the Theory of New Phase Formation, Cavitation. Acta Physicochim. URSS 1943, 18, 1–22. [Google Scholar]
- Gibbs J. W.The Collected Works of J. Willard Gibbs; Longmans, Green and Co.: New York, 1928. [Google Scholar]
- In several cases, including, for instance, the aggregation of amyloid fibrils,471 nucleation effectively occurs in two dimensions.
- A number of additional assumptions have to be made to write down the steady-state nucleation rate. See, e.g., ref (472).
- Baidakov V. G.; Tipeev A. O. Crystal Nucleation and the Solid–Liquid Interfacial Free Energy. J. Chem. Phys. 2012, 136, 074510. 10.1063/1.3678214. [DOI] [PubMed] [Google Scholar]
- Hoffman J. D. Thermodynamic Driving Force in Nucleation and Growth Processes. J. Chem. Phys. 1958, 29, 1192–1193. 10.1063/1.1744688. [DOI] [Google Scholar]
- Thompson C. V.; Spaepen F. On the Approximation of the Free Energy Change on Crystallization. Acta Metall. 1979, 27, 1855–1859. 10.1016/0001-6160(79)90076-2. [DOI] [Google Scholar]
- Auer S.; Frenkel D. Prediction of Absolute Crystal-Nucleation Rate in Hard-Sphere Colloids. Nature 2001, 409, 1020–1023. 10.1038/35059035. [DOI] [PubMed] [Google Scholar]
- Schmelzer J. W. P. On. The Determination of the Kinetic Pre-Factor in Classical Nucleation Theory. J. Non-Cryst. Solids 2010, 356, 2901–2907. 10.1016/j.jnoncrysol.2010.02.026. [DOI] [Google Scholar]
- Vehkamäki H.; Määttänen A.; Lauri A.; Napari I.; Kulmala M. Technical Note: The Heterogeneous Zeldovich Factor. Atmos. Chem. Phys. 2007, 7, 309–313. 10.5194/acp-7-309-2007. [DOI] [Google Scholar]
- Raoux S.; Wuttig M.. Phase Change Materials: Science and Applications; Springer, New York, 2009. [Google Scholar]
- Fredriksson H.; Akerlind U.. Solidification and Crystallization Processing in Metals and Alloys; John Wiley & Sons, Ltd.: Chichester, U.K., 2012. [Google Scholar]
- Porter D.; Easterling K.; Sherif M.. Phase Transformations in Metals and Alloys; CRC Press: Boca Raton, FL, 2009. [Google Scholar]
- Kashchiev D.Nucleation: Basic Theory with Applications, 1st ed.; Butterworth-Heinemann: Oxford, U.K., 2000. [Google Scholar]
- Wette P.; Schöpe H. J. Nucleation Kinetics in Deionized Charged Colloidal Model Systems: A Quantitative Study by Means of Classical Nucleation Theory. Phys. Rev. E 2007, 75, 051405. 10.1103/PhysRevE.75.051405. [DOI] [PubMed] [Google Scholar]
- Angioletti-Uberti S.; Ceriotti M.; Lee P. D.; Finnis M. W. Solid–Liquid Interface Free Energy Through Metadynamics Simulations. Phys. Rev. B: Condens. Matter Mater. Phys. 2010, 81, 125416. 10.1103/PhysRevB.81.125416. [DOI] [Google Scholar]
- Davidchack R. L.; Handel R.; Anwar J.; Brukhno A. V. Ice Ih–Water Interfacial Free Energy of Simple Water Models with Full Electrostatic Interactions. J. Chem. Theory Comput. 2012, 8, 2383–2390. 10.1021/ct300193e. [DOI] [PubMed] [Google Scholar]
- Joswiak M. N.; Duff N.; Doherty M. F.; Peters B. Size-Dependent Surface Free Energy and Tolman-Corrected Droplet Nucleation of TIP4P/2005 Water. J. Phys. Chem. Lett. 2013, 4, 4267–4272. 10.1021/jz402226p. [DOI] [PubMed] [Google Scholar]
- Pereyra R. G.; Szleifer I.; Carignano M. A. Temperature Dependence of Ice Critical Nucleus Size. J. Chem. Phys. 2011, 135, 034508. 10.1063/1.3613672. [DOI] [PubMed] [Google Scholar]
- Sanz E.; Vega C.; Espinosa J. R.; Caballero-Bernal R.; Abascal J. L. F.; Valeriani C. Homogeneous Ice Nucleation at Moderate Supercooling from Molecular Simulation. J. Am. Chem. Soc. 2013, 135, 15008–15017. 10.1021/ja4028814. [DOI] [PubMed] [Google Scholar]
- Vekilov P. G. The Two-step Mechanism of Nucleation of Crystals in Solution. Nanoscale 2010, 2, 2346. 10.1039/c0nr00628a. [DOI] [PubMed] [Google Scholar]
- De Yoreo J. Crystal Nucleation: More than One Pathway. Nat. Mater. 2013, 12, 284–285. 10.1038/nmat3604. [DOI] [PubMed] [Google Scholar]
- Pan W.; Kolomeisky A. B.; Vekilov P. G. Nucleation of Ordered Solid Phases of Proteins via a Disordered High-density State: Phenomenological Approach. J. Chem. Phys. 2005, 122, 174905. 10.1063/1.1887168. [DOI] [PubMed] [Google Scholar]
- Vekilov P. G. Nucleation of Protein Condensed Phases. Rev. Chem. Eng. 2011, 27, 1–13. 10.1515/revce.2011.003. [DOI] [Google Scholar]
- Hu Q.; Nielsen M. H.; Freeman C. L.; Hamm L. M.; Tao J.; Lee J. R. I.; Han T. Y. J.; Becker U.; Harding J. H.; Dove P. M.; De Yoreo J. J. The Thermodynamics of Calcite Nucleation at Organic Interfaces: Classical vs. non-Classical Pathways. Faraday Discuss. 2012, 159, 509–523. 10.1039/c2fd20124k. [DOI] [Google Scholar]
- To be clear, it is perfectly legitimate to measure the contact angle of a water droplet of several millimeters on a macroscopically planar surface. In contrast, defining a contact angle for a critical crystalline nucleus containing just a few hundred molecules on a surface characterized by a roughness of the same extent is clearly much more difficult, if meaningful at all.
- Cardinaux F.; Gibaud T.; Stradner A.; Schurtenberger P. Interplay between Spinodal Decomposition and Glass Formation in Proteins Exhibiting Short-Range Attractions. Phys. Rev. Lett. 2007, 99, 118301. 10.1103/PhysRevLett.99.118301. [DOI] [PubMed] [Google Scholar]
- CNT is still expected to capture nucleation on very short timescales at strong supercooling when the free energy barrier for nucleation, although nonzero, is on the order of kBT.
- Binder K.; Fratzl P.. Phase Transformations in Materials; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2005; pp 409–480. [Google Scholar]
- Bartell L. S.; Wu D. T. Do Supercooled Liquids Freeze by Spinodal Decomposition?. J. Chem. Phys. 2007, 127, 174507. 10.1063/1.2779036. [DOI] [PubMed] [Google Scholar]
- Trudu F.; Donadio D.; Parrinello M. Freezing of a Lennard-Jones Fluid: From Nucleation to Spinodal Regime. Phys. Rev. Lett. 2006, 97, 105701. 10.1103/PhysRevLett.97.105701. [DOI] [PubMed] [Google Scholar]
- Peng L. J.; Morris J. R.; Lo Y. C. Temperature-Dependent Mechanisms of Homogeneous Crystal Nucleation in Quenched Lennard-Jones Liquids: Molecular Dynamics Simulations. Phys. Rev. B: Condens. Matter Mater. Phys. 2008, 78, 012201. 10.1103/PhysRevB.78.012201. [DOI] [Google Scholar]
- A complete list of the approximations contained in CNT would, itself, deserve a book chapter. The reader is referred to ref (472), for example.
- Schenter G. K.; Kathmann S. M.; Garrett B. C. Dynamical Nucleation Theory: A New Molecular Approach to Vapor–Liquid Nucleation. Phys. Rev. Lett. 1999, 82, 3484–3487. 10.1103/PhysRevLett.82.3484. [DOI] [Google Scholar]
- Kalikmanov V. I. Mean-Field Kinetic Nucleation Theory. J. Chem. Phys. 2006, 124, 124505. 10.1063/1.2178812. [DOI] [PubMed] [Google Scholar]
- Russell K. C. Linked Flux Analysis of Nucleation in Condensed Phases. Acta Metall. 1968, 16, 761–769. 10.1016/0001-6160(68)90148-X. [DOI] [Google Scholar]
- Peters B. On the Coupling between Slow Diffusion Transport and Barrier Crossing in Nucleation. J. Chem. Phys. 2011, 135, 044107. 10.1063/1.3613674. [DOI] [PubMed] [Google Scholar]
- Wei P. F.; Kelton K. F.; Falster R. Coupled-Flux Nucleation Modeling of Oxygen Precipitation in Silicon. J. Appl. Phys. 2000, 88, 5062–5070. 10.1063/1.1311309. [DOI] [Google Scholar]
- Kelton K. F. Time-Dependent Nucleation in Partitioning Transformations. Acta Mater. 2000, 48, 1967–1980. 10.1016/S1359-6454(99)00455-3. [DOI] [Google Scholar]
- Gránásy L. Diffuse Interface Theory of Nucleation. J. Non-Cryst. Solids 1993, 162, 301–303. 10.1016/0022-3093(93)91250-7. [DOI] [Google Scholar]
- Gránásy L.; Herlach D. M. Diffuse Interface Approach to Crystal Nucleation in Glasses. J. Non-Cryst. Solids 1995, 192–193, 470–473. 10.1016/0022-3093(95)00430-0. [DOI] [Google Scholar]
- Prestipino S.; Laio A.; Tosatti E. Systematic Improvement of Classical Nucleation Theory. Phys. Rev. Lett. 2012, 108, 225701. 10.1103/PhysRevLett.108.225701. [DOI] [PubMed] [Google Scholar]
- Kahl G.; Löwen H. Classical Density Functional Theory: An Ideal Tool to Study Heterogeneous Crystal Nucleation. J. Phys.: Condens. Matter 2009, 21, 464101. 10.1088/0953-8984/21/46/464101. [DOI] [PubMed] [Google Scholar]
- Löwen H.; Likos C. N.; Assoud L.; Blaak R.; Van Teefelen S. Critical Nuclei and Crystallization in Colloidal Suspensions. Philos. Mag. Lett. 2007, 87, 847–854. 10.1080/09500830701523033. [DOI] [Google Scholar]
- Neuhaus T.; Härtel A.; Marechal M.; Schmiedeberg M.; Löwen H. Density Functional Theory of Heterogeneous Crystallization. Eur. Phys. J.: Spec. Top. 2014, 223, 373–387. 10.1140/epjst/e2014-02097-x. [DOI] [Google Scholar]
- Lutsko J. F.Recent developments in classical density functional theory. In Advances in Chemical Physics; Rice S. A., Ed.; John Wiley & Sons, Inc., 2010; Vol. 144, pp 1–92. [Google Scholar]
- Martin R. M.Electronic Structure: Basic Theory and Practical Methods; Cambridge University Press: Cambridge, U.K., 2004; Vol. 1. [Google Scholar]
- Kelton K.; Greer A. L.. Nucleation in Condensed Matter: Applications in Materials and Biology; Elsevier: New York, 2010. [Google Scholar]
- By microscopic, we refer to insight into the smallest entities that make up the liquid phase.
- Pusey P. N.; van Megen W. Phase Behaviour of Concentrated Suspensions of Nearly Hard Colloidal Spheres. Nature 1986, 320, 340–342. 10.1038/320340a0. [DOI] [Google Scholar]
- Zhang T. H.; Liu X. Y. Experimental Modelling of Single-Particle Dynamic Processes in Crystallization by Controlled Colloidal Assembly. Chem. Soc. Rev. 2014, 43, 2324–2347. 10.1039/c3cs60398a. [DOI] [PubMed] [Google Scholar]
- Gasser U.; Weeks E. R.; Schofield A.; Pusey P. N.; Weitz D. A. Real-Space Imaging of Nucleation and Growth in Colloidal Crystallization. Science 2001, 292, 258–262. 10.1126/science.1058457. [DOI] [PubMed] [Google Scholar]
- Dinsmore A. D.; Weeks E. R.; Prasad V.; Levitt A. C.; Weitz D. A. Three-Dimensional Confocal Microscopy of Colloids. Appl. Opt. 2001, 40, 4152. 10.1364/AO.40.004152. [DOI] [PubMed] [Google Scholar]
- Sleutel M.; Lutsko J.; Van Driessche A. E. S.; Durán-Olivencia M. A.; Maes D. Observing Classical Nucleation Theory at Work by Monitoring Phase Transitions with Molecular Precision. Nat. Commun. 2014, 5, 5598. 10.1038/ncomms6598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pouget E. M.; Bomans P. H. H.; Goos J. A. C. M.; Frederik P. M.; de With G.; Sommerdijk N. A. J. M. The Initial Stages of Template-Controlled CaCO3 Formation Revealed by Cryo-TEM. Science 2009, 323, 1455–1458. 10.1126/science.1169434. [DOI] [PubMed] [Google Scholar]
- Nielsen M. H.; Aloni S.; De Yoreo J. J. In Situ TEM Imaging of CaCO3 Nucleation Reveals Coexistence of Direct and Indirect Pathways. Science 2014, 345, 1158–1162. 10.1126/science.1254051. [DOI] [PubMed] [Google Scholar]
- Chung S.-Y.; Kim Y.-M.; Kim J.-G.; Kim Y.-J. Multiphase Transformation and Ostwald’s Rule of Stages during Crystallization of a Metal Phosphate. Nat. Phys. 2009, 5, 68–73. 10.1038/nphys1148. [DOI] [Google Scholar]
- Baumgartner J.; Dey A.; Bomans P. H. H.; Le Coadou C.; Fratzl P.; Sommerdijk N. A. J. M.; Faivre D. Nucleation and Growth of Magnetite from Solution. Nat. Mater. 2013, 12, 310–314. 10.1038/nmat3558. [DOI] [PubMed] [Google Scholar]
- Sellberg J. A.; Huang C.; McQueen T. A.; Loh N. D.; Laksmono H.; Schlesinger D.; Sierra R. G.; Nordlund D.; Hampton C. Y.; Starodub D.; et al. Ultrafast X-Ray Probing of Water Structure Below the Homogeneous Ice Nucleation Temperature. Nature 2014, 510, 381–384. 10.1038/nature13266. [DOI] [PubMed] [Google Scholar]
- Laksmono H.; McQueen T. A.; Sellberg J. A.; Loh N. D.; Huang C.; Schlesinger D.; Sierra R. G.; Hampton C. Y.; Nordlund D.; Beye M.; et al. Anomalous Behavior of the Homogeneous Ice Nucleation Rate in “No-Man’s Land”. J. Phys. Chem. Lett. 2015, 6, 2826–2832. 10.1021/acs.jpclett.5b01164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell J. M.; Meldrum F. C.; Christenson H. K. Is Ice Nucleation from Supercooled Water Insensitive to Surface Roughness?. J. Phys. Chem. C 2015, 119, 1164–1169. 10.1021/jp5113729. [DOI] [Google Scholar]
- Li K.; Xu S.; Shi W.; He M.; Li H.; Li S.; Zhou X.; Wang J.; Song Y. Investigating the Effects of Solid Surfaces on Ice Nucleation. Langmuir 2012, 28, 10749–10754. 10.1021/la3014915. [DOI] [PubMed] [Google Scholar]
- Pusey P. N.; van Megen W.; Bartlett P.; Ackerson B. J.; Rarity J. G.; Underwood S. M. Structure of Crystals of Hard Colloidal Spheres. Phys. Rev. Lett. 1989, 63, 2753–2756. 10.1103/PhysRevLett.63.2753. [DOI] [PubMed] [Google Scholar]
- Zhu J.; Li M.; Rogers R.; Meyer W.; Ottewill R. H.; Russel W. B.; Chaikin P. M. Crystallization of Hard-Sphere Colloids in Microgravity. Nature 1997, 387, 883–885. 10.1038/43141. [DOI] [Google Scholar]
- Ehre D.; Lavert E.; Lahav M.; Lubomirsky I. Water Freezes Differently on Positively and Negatively Charged Surfaces of Pyroelectric Materials. Science 2010, 327, 672–675. 10.1126/science.1178085. [DOI] [PubMed] [Google Scholar]
- Ildefonso M.; Revalor E.; Punniam P.; Salmon J.; Candoni N.; Veesler S. Nucleation and Polymorphism Explored via an Easy-to-use Microfluidic Tool. J. Cryst. Growth 2012, 342, 9–12. 10.1016/j.jcrysgro.2010.11.098. [DOI] [Google Scholar]
- Kadam S. S.; Kulkarni S. A.; Coloma Ribera R.; Stankiewicz A. I.; ter Horst J. H.; Kramer H. J. A New View on the Metastable Zone Width during Cooling Crystallization. Chem. Eng. Sci. 2012, 72, 10–19. 10.1016/j.ces.2012.01.002. [DOI] [Google Scholar]
- Kubota N. A. New Interpretation of Metastable Zone Widths Measured for Unseeded Solutions. J. Cryst. Growth 2008, 310, 629–634. 10.1016/j.jcrysgro.2007.11.123. [DOI] [Google Scholar]
- Kashchiev D.; Borissova A.; Hammond R. B.; Roberts K. J. Effect of Cooling Rate on the Critical Undercooling for Crystallization. J. Cryst. Growth 2010, 312, 698–704. 10.1016/j.jcrysgro.2009.12.031. [DOI] [PubMed] [Google Scholar]
- Sangwal K. Recent Developments in Understanding of the Metastable Zone Width of Different Solute-Solvent Systems. J. Cryst. Growth 2011, 318, 103–109. 10.1016/j.jcrysgro.2010.11.078. [DOI] [Google Scholar]
- Peters B. Supersaturation Rates and Schedules: Nucleation Kinetics from Isothermal Metastable Zone Widths. J. Cryst. Growth 2011, 317, 79–83. 10.1016/j.jcrysgro.2011.01.017. [DOI] [Google Scholar]
- Sangwal K. Novel Approach to Analyze Metastable Zone Width Determined by the Polythermal Method: Physical Interpretation of Various Parameters. Cryst. Growth Des. 2009, 9, 942–950. 10.1021/cg800704y. [DOI] [Google Scholar]
- Kashchiev D.; Borissova A.; Hammond R. B.; Roberts K. J. Dependence of the Critical Undercooling for Crystallization on the Cooling Rate. J. Phys. Chem. B 2010, 114, 5441–5446. 10.1021/jp100202m. [DOI] [PubMed] [Google Scholar]
- Kashchiev D.; Verdoes D.; Van Rosmalen G. Induction Time and Metastability Limit in New Phase Formation. J. Cryst. Growth 1991, 110, 373–380. 10.1016/0022-0248(91)90273-8. [DOI] [Google Scholar]
- Lindenberg C.; Mazzotti M. Effect of Temperature on the Nucleation Kinetics of alpha l-glutamic Acid. J. Cryst. Growth 2009, 311, 1178–1184. 10.1016/j.jcrysgro.2008.12.010. [DOI] [Google Scholar]
- Roelands C. M.; Roestenberg R. R.; ter Horst J. H.; Kramer H. J.; Jansens P. J. Development of an Experimental Method to Measure Nucleation Rates in Reactive Precipitation. Cryst. Growth Des. 2004, 4, 921–928. 10.1021/cg034188k. [DOI] [Google Scholar]
- Roelands C. M.; ter Horst J. H.; Kramer H. J.; Jansens P. J. Analysis of Nucleation Rate Measurements in Precipitation Processes. Cryst. Growth Des. 2006, 6, 1380–1392. 10.1021/cg050678w. [DOI] [Google Scholar]
- Jiang S.; ter Horst J. H. Crystal Nucleation Rates from Probability Distributions of Induction Times. Cryst. Growth Des. 2011, 11, 256–261. 10.1021/cg101213q. [DOI] [Google Scholar]
- Teychené S.; Biscans B. Nucleation Kinetics of Polymorphs: Induction Period and Interfacial Energy Measurements. Cryst. Growth Des. 2008, 8, 1133–1139. 10.1021/cg0609320. [DOI] [Google Scholar]
- Kashchiev D.; Van Rosmalen G. Review: Nucleation in Solutions Revisited. Cryst. Res. Technol. 2003, 38, 555–574. 10.1002/crat.200310070. [DOI] [Google Scholar]
- Davey R. J.; Schroeder S. L.; ter Horst J. H. Nucleation of Organic Crystals - a Molecular Perspective. Angew. Chem., Int. Ed. 2013, 52, 2166–2179. 10.1002/anie.201204824. [DOI] [PubMed] [Google Scholar]
- Vetter T.; Iggland M.; Ochsenbein D. R.; Hänseler F. S.; Mazzotti M. Modeling Nucleation, Growth, and Ostwald Ripening in Crystallization Processes: a Comparison between Population Balance and Kinetic Rate Equation. Cryst. Growth Des. 2013, 13, 4890–4905. 10.1021/cg4010714. [DOI] [Google Scholar]
- Kulkarni S. A.; Kadam S. S.; Meekes H.; Stankiewicz A. I.; ter Horst J. H. Crystal Nucleation Kinetics from Induction Times and Metastable Zone Widths. Cryst. Growth Des. 2013, 13, 2435–2440. 10.1021/cg400139t. [DOI] [Google Scholar]
- Kubota N. Effect of Sample Volume on Metastable Zone Width and Induction Time. J. Cryst. Growth 2012, 345, 27–33. 10.1016/j.jcrysgro.2012.01.034. [DOI] [Google Scholar]
- Stan C. A.; Schneider G. F.; Shevkoplyas S. S.; Hashimoto M.; Ibanescu M.; Wiley B. J.; Whitesides G. M. A Microfluidic Apparatus for the Study of Ice Nucleation in Supercooled Water Drops. Lab Chip 2009, 9, 2293–2305. 10.1039/b906198c. [DOI] [PubMed] [Google Scholar]
- Bogoeva-Gaceva G.; Janevski A.; Mader E. Nucleation Activity of Glass Fibers towards IPP Evaluated by DSC and Polarizing Light Microscopy. Polymer 2001, 42, 4409–4416. 10.1016/S0032-3861(00)00659-5. [DOI] [Google Scholar]
- Davies S. R.; Hester K. C.; Lachance J. W.; Koh C. A.; Dendy Sloan E. Studies of Hydrate Nucleation with High Pressure Differential Scanning Calorimetry. Chem. Eng. Sci. 2009, 64, 370–375. 10.1016/j.ces.2008.10.017. [DOI] [Google Scholar]
- Charoenrein S.; Reid D. S. The Use of DSC to Study the Kinetics of Heterogeneous and Homogeneous Nucleation of Ice in Aqueous Systems. Thermochim. Acta 1989, 156, 373–381. 10.1016/0040-6031(89)87204-1. [DOI] [Google Scholar]
- Marcolli C.; Gedamke S.; Peter T.; Zobrist B. Efficiency of Immersion Mode Ice Nucleation on Surrogates of Mineral Dust. Atmos. Chem. Phys. 2007, 7, 5081–5091. 10.5194/acp-7-5081-2007. [DOI] [Google Scholar]
- Pinti V.; Marcolli C.; Zobrist B.; Hoyle C. R.; Peter T. Ice Nucleation Efficiency of Clay Minerals in the Immersion Mode. Atmos. Chem. Phys. 2012, 12, 5859–5878. 10.5194/acp-12-5859-2012. [DOI] [Google Scholar]
- Rasmussen D. H.; Loper C. R. Jr. DSC: A Rapid Method for Isothermal Nucleation Rate Measurement. Acta Metall. 1976, 24, 117–123. 10.1016/0001-6160(76)90014-6. [DOI] [Google Scholar]
- Ochshorn E.; Cantrell W. Towards Understanding Ice Nucleation by Long Chain Alcohols. J. Chem. Phys. 2006, 124, 054714. 10.1063/1.2166368. [DOI] [PubMed] [Google Scholar]
- Manka A.; Pathak H.; Tanimura S.; Wölk J.; Strey R.; Wyslouzil B. E. Freezing Water in No-Man’s Land. Phys. Chem. Chem. Phys. 2012, 14, 4505–4516. 10.1039/c2cp23116f. [DOI] [PubMed] [Google Scholar]
- Bhabhe A.; Pathak H.; Wyslouzil B. E. Freezing of Heavy Water (D2O) Nanodroplets. J. Phys. Chem. A 2013, 117, 5472–5482. 10.1021/jp400070v. [DOI] [PubMed] [Google Scholar]
- Yang X.; Lu J.; Wang X.-J.; Ching C.-B. Effect of Sodium Chloride on the Nucleation and Polymorphic Transformation of Glycine. J. Cryst. Growth 2008, 310, 604–611. 10.1016/j.jcrysgro.2007.11.072. [DOI] [Google Scholar]
- Fujiwara M.; Chow P. S.; Ma D. L.; Braatz R. D. Paracetamol Crystallization Using Laser Backscattering and ATR-FTIR Spectroscopy: Metastability, Agglomeration. Cryst. Growth Des. 2002, 2, 363–370. 10.1021/cg0200098. [DOI] [Google Scholar]
- Gebauer D.; Völkel A.; Cölfen H. Stable Prenucleation Calcium Carbonate Clusters. Science 2008, 322, 1819–1822. 10.1126/science.1164271. [DOI] [PubMed] [Google Scholar]
- Rogers D. C.; DeMott P. J.; Kreidenweis S. M.; Chen Y. A Continuous-Flow Diffusion Chamber for Airborne Measurements of Ice Nuclei. J. Atm. Oceanic Technol. 2001, 18, 725–741. 10.1175/1520-0426(2001)018<0725:ACFDCF>2.0.CO;2. [DOI] [Google Scholar]
- DeMott P. J.; Sassen K.; Poellot M. R.; Baumgardner D.; Rogers D. C.; Brooks S. D.; Prenni A. J.; Kreidenweis S. M. African Dust Aerosols as Atmospheric Ice Nuclei. Geophys. Res. Lett. 2003, 30, 1732. 10.1029/2003GL017410. [DOI] [Google Scholar]
- Tobo Y.; DeMott P. J.; Raddatz M.; Niedermeier D.; Hartmann S.; Kreidenweis S. M.; Stratmann F.; Wex H. Impacts of Chemical Reactivity on Ice Nucleation of Kaolinite Particles: A Case Study of Levoglucosan and Sulfuric Acid. Geophys. Res. Lett. 2012, 39, L19803. 10.1029/2012GL053007. [DOI] [Google Scholar]
- Lihavainen H.; Viisanen Y.; Kulmala M. Homogeneous Nucleation of N-Pentanol in a Laminar Flow Diffusion Chamber. J. Chem. Phys. 2001, 114, 10031–10038. 10.1063/1.1368131. [DOI] [PubMed] [Google Scholar]
- Konstantinov P.; Agopian T.; Tchokova I. Analysis on the ice-forming properties of the pyrotechnic composition of the AgI inside isothermal cloud chamber and drop freezing chamber. Bulg. J. Meteorol. Hydrol. 2000, 2000, 13–16. [Google Scholar]
- Finnegan W. G.; Chai S. K. A New Hypothesis for the Mechanism of Ice Nucleation on Wetted AgI and AgI·AgCl Particulate Aerosols. J. Atmos. Sci. 2003, 60, 1723–1731. 10.1175/1520-0469(2003)060<1723:ANHFTM>2.0.CO;2. [DOI] [Google Scholar]
- Hiranuma N.; Möhler O.; Yamashita K.; Tajiri T.; Saito A.; Kiselev A.; Hoffmann N.; Hoose C.; Jantsch E.; Koop T.; et al. Ice Nucleation by Cellulose and Its Potential Contribution to Ice Formation in Clouds. Nat. Geosci. 2015, 8, 273–277. 10.1038/ngeo2374. [DOI] [Google Scholar]
- Gard D. L. Organization, Nucleation, and Acetylation of Microtubules in Xenopus Laevis Oocytes: A Study by Confocal Immunofluorescence Microscopy. Dev. Biol. 1991, 143, 346–362. 10.1016/0012-1606(91)90085-H. [DOI] [PubMed] [Google Scholar]
- Harano K.; Homma T.; Niimi Y.; Koshino M.; Suenaga K.; Leibler L.; Nakamura E. Heterogeneous Nucleation of Organic Crystals Mediated by Single-Molecule Templates. Nat. Mater. 2012, 11, 877–881. 10.1038/nmat3408. [DOI] [PubMed] [Google Scholar]
- Nakamura E. Movies of Molecular Motions and Reactions: The Single-Molecule, Real-Time Transmission Electron Microscope Imaging Technique. Angew. Chem., Int. Ed. 2013, 52, 236–252. 10.1002/anie.201205693. [DOI] [PubMed] [Google Scholar]
- Regev O. Nucleation Events During the Synthesis of Mesoporous Materials Using Liquid Crystalline Templating. Langmuir 1996, 12, 4940–4944. 10.1021/la9602372. [DOI] [Google Scholar]
- Bauerecker S.; Ulbig P.; Buch V.; Vrbka L.; Jungwirth P. Monitoring Ice Nucleation in Pure and Salty Water via High-Speed Imaging and Computer Simulations. J. Phys. Chem. C 2008, 112, 7631–7636. 10.1021/jp711507f. [DOI] [Google Scholar]
- Nagy Z. K.; Fujiwara M.; Woo X. Y.; Braatz R. D. Determination of the Kinetic Parameters for the Crystallization of Paracetamol from Water using Metastable Zone Width Experiments. Ind. Eng. Chem. Res. 2008, 47, 1245–1252. 10.1021/ie060637c. [DOI] [Google Scholar]
- Bluhm H.; Ogletree D. F.; Fadley C. S.; Hussain Z.; Salmeron M. The Premelting of Ice Studied with Photoelectron Spectroscopy. J. Phys.: Condens. Matter 2002, 14, L227. 10.1088/0953-8984/14/8/108. [DOI] [Google Scholar]
- Ketteler G.; Yamamoto S.; Bluhm H.; Andersson K.; Starr D. E.; Ogletree D. F.; Ogasawara H.; Nilsson A.; Salmeron M. The Nature of Water Nucleation Sites on TiO2(110) Surfaces Revealed by Ambient Pressure X-Ray Photoelectron Spectroscopy. J. Phys. Chem. C 2007, 111, 8278–8282. 10.1021/jp068606i. [DOI] [Google Scholar]
- Barrere F.; Snel M. M. E.; van Blitterswijk C. A.; de Groot K.; Layrolle P. Nano-Scale Study of the Nucleation and Growth of Calcium Phosphate Coating on Titanium Implants. Biomaterials 2004, 25, 2901–2910. 10.1016/j.biomaterials.2003.09.063. [DOI] [PubMed] [Google Scholar]
- Zimmermann F.; Weinbruch S.; Schütz L.; Hofmann H.; Ebert M.; Kandler K.; Worringen A. Ice Nucleation Properties of the Most Abundant Mineral Dust Phases. J. Geophys. Res. 2008, 113, D23204. 10.1029/2008JD010655. [DOI] [Google Scholar]
- Auer S.; Frenkel D. Numerical Prediction of Absolute Crystallization Rates in Hard-Sphere Colloids. J. Chem. Phys. 2004, 120, 3015–3029. 10.1063/1.1638740. [DOI] [PubMed] [Google Scholar]
- Schilling T.; Dorosz S.; Schöpe H. J.; Opletal G. Crystallization in Suspensions of Hard Spheres: A Monte Carlo and Molecular Dynamics Simulation Study. J. Phys.: Condens. Matter 2011, 23, 194120. 10.1088/0953-8984/23/19/194120. [DOI] [PubMed] [Google Scholar]
- Punnathanam S.; Monson P. A. Crystal Nucleation in Binary Hard Sphere Mixtures: A Monte Carlo Simulation Study. J. Chem. Phys. 2006, 125, 024508. 10.1063/1.2208998. [DOI] [PubMed] [Google Scholar]
- For instance, in the case of sodium acetate crystal nucleation, it is used to craft hand warmers.473
- Zhang Z.; Walsh M. R.; Guo G.-J. Microcanonical Molecular Simulations of Methane Hydrate Nucleation and Growth: Evidence That Direct Nucleation to SI Hydrate Is Among the Multiple Nucleation Pathways. Phys. Chem. Chem. Phys. 2015, 17, 8870–8876. 10.1039/C5CP00098J. [DOI] [PubMed] [Google Scholar]
- Pérez A.; Rubio A. A Molecular Dynamics Study of Water Nucleation Using the TIP4P/2005 Model. J. Chem. Phys. 2011, 135, 244505. 10.1063/1.3672063. [DOI] [PubMed] [Google Scholar]
- Wedekind J.; Reguera D.; Strey R. Finite-Size Effects in Simulations of Nucleation. J. Chem. Phys. 2006, 125, 214505. 10.1063/1.2402167. [DOI] [PubMed] [Google Scholar]
- Bussi G.; Donadio D.; Parrinello M. Canonical Sampling Through Velocity Rescaling. J. Chem. Phys. 2007, 126, 014101. 10.1063/1.2408420. [DOI] [PubMed] [Google Scholar]
- Oxtoby D. W. Crystal Nucleation in Simple and Complex Fluids. Philos. Trans. R. Soc., A 2003, 361, 419–428. 10.1098/rsta.2002.1145. [DOI] [PubMed] [Google Scholar]
- Nielaba P.; Mareschal M.; Ciccotti G.. Bridging Time Scales: Molecular Simulations for the Next Decade; Springer: Berlin, 2002. [Google Scholar]
- Abrams C.; Bussi G. Enhanced Sampling in Molecular Dynamics Using Metadynamics, Replica-Exchange, and Temperature-Acceleration. Entropy 2014, 16, 163–199. 10.3390/e16010163. [DOI] [Google Scholar]
- Yasuoka K.; Matsumoto M. Molecular Dynamics of Homogeneous Nucleation in the Vapor Phase. I. Lennard-Jones Fluid. J. Chem. Phys. 1998, 109, 8451–8462. 10.1063/1.477509. [DOI] [PubMed] [Google Scholar]
- Skripov V. P.Metastable Liquids (translated from Russian by R. Kondor; translation edited by D. Slutzkin); Wiley: New York, 1974. [Google Scholar]
- ter Horst J. H.; Kashchiev D. Determination of the Nucleus Size from the Growth Probability of Clusters. J. Chem. Phys. 2003, 119, 2241–2246. 10.1063/1.1585020. [DOI] [Google Scholar]
- Yi P.; Locker C. R.; Rutledge G. C. Molecular Dynamics Simulation of Homogeneous Crystal Nucleation in Polyethylene. Macromolecules 2013, 46, 4723–4733. 10.1021/ma4004659. [DOI] [Google Scholar]
- Wedekind J.; Strey R.; Reguera D. New Method to Analyze Simulations of Activated Processes. J. Chem. Phys. 2007, 126, 134103. 10.1063/1.2713401. [DOI] [PubMed] [Google Scholar]
- Van Erp T. S.Dynamical Rare Event Simulation Techniques for Equilibrium and Nonequilibrium Systems. In Kinetics and Thermodynamics of Multistep Nucleation and Self-Assembly in Nanoscale Materials; Nicolis G., Maes D., Eds.; Advances in Chemical Physics Series; John Wiley & Sons, Inc.: New York, 2012; Vol. 151, Chapter 2, pp 27–60. [Google Scholar]
- Dellago C., Bolhuis P. G.. Transition Path Sampling and Other Advanced Simulation Techniques for Rare Events. In Advanced Computer Simulation Approaches for Soft Matter Sciences III; Holm P. C., Kremer P. K., Eds.; Advances in Polymer Science; Springer: Berlin, 2009; Vol. 221, Chapter 3, pp 167–233. [Google Scholar]
- Schlick T. Molecular Dynamics-Based Approaches for Enhanced Sampling of Long-Time, Large-Scale Conformational Changes in Biomolecules. F1000 Bio. Rep. 2009, 1, 51. 10.3410/B1-51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torrie G. M.; Valleau J. P. Monte Carlo Free Energy Estimates Using Non-Boltzmann Sampling: Application to the Sub-Critical Lennard-Jones Fluid. Chem. Phys. Lett. 1974, 28, 578–581. 10.1016/0009-2614(74)80109-0. [DOI] [Google Scholar]
- Torrie G. M.; Valleau J. P. Nonphysical Sampling Distributions in Monte Carlo Free-Energy Estimation: Umbrella Sampling. J. Comput. Phys. 1977, 23, 187–199. 10.1016/0021-9991(77)90121-8. [DOI] [Google Scholar]
- Kumar S.; Rosenberg J. M.; Bouzida D.; Swendsen R. H.; Kollman P. A. THE Weighted Histogram Analysis Method for Free-Energy Calculations on Biomolecules. I. The Method. J. Comput. Chem. 1992, 13, 1011–1021. 10.1002/jcc.540130812. [DOI] [Google Scholar]
- Laio A.; Parrinello M. Escaping Free-Energy Minima. Proc. Natl. Acad. Sci. U. S. A. 2002, 99, 12562–12566. 10.1073/pnas.202427399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laio A.; Gervasio F. L. Metadynamics: A Method to Simulate Rare Events and Reconstruct the Free Energy in Biophysics, Chemistry and Material Science. Rep. Prog. Phys. 2008, 71, 126601. 10.1088/0034-4885/71/12/126601. [DOI] [Google Scholar]
- Barducci A.; Bussi G.; Parrinello M. Well-Tempered Metadynamics: A Smoothly Converging and Tunable Free-Energy Method. Phys. Rev. Lett. 2008, 100, 020603. 10.1103/PhysRevLett.100.020603. [DOI] [PubMed] [Google Scholar]
- Eyring H. The Activated Complex in Chemical Reactions. J. Chem. Phys. 1935, 3, 107–115. 10.1063/1.1749604. [DOI] [Google Scholar]
- Wigner E. The Transition State Method. Trans. Faraday Soc. 1938, 34, 29–41. 10.1039/tf9383400029. [DOI] [Google Scholar]
- Anderson J. B. Statistical Theories of Chemical Reactions. Distributions in the Transition Region. J. Chem. Phys. 1973, 58, 4684–4692. 10.1063/1.1679032. [DOI] [Google Scholar]
- Hänggi P.; Talkner P.; Borkovec M. Reaction-Rate Theory: Fifty Years after Kramers. Rev. Mod. Phys. 1990, 62, 251. 10.1103/Revmodphys.62.251. [DOI] [Google Scholar]
- Chandler D. Statistical Mechanics of Isomerization Dynamics in Liquids and the Transition State Approximation. J. Chem. Phys. 1978, 68, 2959–2970. 10.1063/1.436049. [DOI] [Google Scholar]
- Bennett C. H.Molecular Dynamics and Transition State Theory: The Simulation of Infrequent Events. In Algorithms for Chemical Computations; Christoffersen R. E., Ed.; ACS Symposium Series; American Chemical Society: Washington, DC, 1977; Vol. 46, Chapter 4, pp 63–97. [Google Scholar]
- Van Erp T. S.; Moroni D.; Bolhuis P. G. A Novel Path Sampling Method for the Calculation of Rate Constants. J. Chem. Phys. 2003, 118, 7762–7774. 10.1063/1.1562614. [DOI] [Google Scholar]
- Moroni D.; van Erp T. S.; Bolhuis P. G. Investigating Rare Events by Transition Interface Sampling. Phys. A 2004, 340, 395–401. 10.1016/j.physa.2004.04.033. [DOI] [Google Scholar]
- Juraszek J.; Saladino G.; van Erp T. S.; Gervasio F. L. Efficient Numerical Reconstruction of Protein Folding Kinetics with Partial Path Sampling and Pathlike Variables. Phys. Rev. Lett. 2013, 110, 108106. 10.1103/PhysRevLett.110.108106. [DOI] [PubMed] [Google Scholar]
- Allen R. J.; Frenkel D.; ten Wolde P. R. Simulating Rare Events in Equilibrium or Nonequilibrium Stochastic Systems. J. Chem. Phys. 2006, 124, 024102. 10.1063/1.2140273. [DOI] [PubMed] [Google Scholar]
- Allen R. J.; Valeriani C.; Rein ten Wolde P. Forward Flux Sampling for Rare Event Simulations. J. Phys.: Condens. Matter 2009, 21, 463102. 10.1088/0953-8984/21/46/463102. [DOI] [PubMed] [Google Scholar]
- Dellago C.; Bolhuis P. G.; Chandler D. Efficient Transition Path Sampling: Application to Lennard-Jones Cluster Rearrangements. J. Chem. Phys. 1998, 108, 9236–9245. 10.1063/1.476378. [DOI] [Google Scholar]
- Bolhuis P. G.; Dellago C.; Chandler D. Sampling Ensembles of Deterministic Transition Pathways. Faraday Discuss. 1998, 110, 421–436. 10.1039/a801266k. [DOI] [Google Scholar]
- Geissler P. L.; Dellago C.; Chandler D. Kinetic Pathways of Ion Pair Dissociation in Water. J. Phys. Chem. B 1999, 103, 3706–3710. 10.1021/jp984837g. [DOI] [Google Scholar]
- Kirkwood J. G. Statistical Mechanics of Fluid Mixtures. J. Chem. Phys. 1935, 3, 300–313. 10.1063/1.1749657. [DOI] [Google Scholar]
- Gavezzotti A.; Filippini G.; Kroon J.; van Eijck B. P.; Klewinghaus P. The Crystal Polymorphism of Tetrolic Acid (CH3CCCOOH): A Molecular Dynamics Study of Precursors in Solution, and a Crystal Structure Generation. Chem. - Eur. J. 1997, 3, 893–899. 10.1002/chem.19970030610. [DOI] [Google Scholar]
- Gavezzotti A. Molecular Aggregation of Acetic Acid in a Carbon Tetrachloride Solution: A Molecular Dynamics Study with a View to Crystal Nucleation. Chem. - Eur. J. 1999, 5, 567–576. 10.1002/(SICI)1521-3765(19990201)5:2<567::AID-CHEM567>3.3.CO;2-Y. [DOI] [Google Scholar]
- Kawska A.; Brickmann J.; Kniep R.; Hochrein O.; Zahn D. An Atomistic Simulation Scheme for Modeling Crystal Formation from Solution. J. Chem. Phys. 2006, 124, 024513. 10.1063/1.2145677. [DOI] [PubMed] [Google Scholar]
- Kawska A.; Duchstein P.; Hochrein O.; Zahn D. Atomistic Mechanisms of ZnO Aggregation from Ethanolic Solution: Ion Association, Proton Transfer, and Self-Organization. Nano Lett. 2008, 8, 2336–2340. 10.1021/nl801169x. [DOI] [PubMed] [Google Scholar]
- Cacciuto A.; Auer S.; Frenkel D. Onset of Heterogeneous Crystal Nucleation in Colloidal Suspensions. Nature 2004, 428, 404–406. 10.1038/nature02397. [DOI] [PubMed] [Google Scholar]
- Browning A. R.; Doherty M. F.; Fredrickson G. H. Nucleation and Polymorph Selection in a Model Colloidal Fluid. Phys. Rev. E 2008, 77, 041604. 10.1103/PhysRevE.77.041604. [DOI] [PubMed] [Google Scholar]
- Kalikka J.; Akola J.; Larrucea J.; Jones R. O. Nucleus-Driven Crystallization of Amorphous Ge2Sb2Te5: A Density Functional Study. Phys. Rev. B: Condens. Matter Mater. Phys. 2012, 86, 144113. 10.1103/PhysRevB.86.144113. [DOI] [Google Scholar]
- Knott B. C.; Molinero V.; Doherty M. F.; Peters B. Homogeneous Nucleation of Methane Hydrates: Unrealistic under Realistic Conditions. J. Am. Chem. Soc. 2012, 134, 19544–19547. 10.1021/ja309117d. [DOI] [PubMed] [Google Scholar]
- Hoover W. G.; Ree F. H. Melting Transition and Communal Entropy for Hard Spheres. J. Chem. Phys. 1968, 49, 3609–3617. 10.1063/1.1670641. [DOI] [Google Scholar]
- Antl L.; Goodwin J. W.; Hill R. D.; Ottewill R. H.; Owens S. M.; Papworth S.; Waters J. A. The Preparation of Poly(Methyl Methacrylate) Latices in Non-Aqueous Media. Colloids Surf. 1986, 17, 67–78. 10.1016/0166-6622(86)80187-1. [DOI] [Google Scholar]
- Phan S.-E.; Russel W. B.; Cheng Z.; Zhu J.; Chaikin P. M.; Dunsmuir J. H.; Ottewill R. H. Phase Transition, Equation of State, and Limiting Shear Viscosities of Hard Sphere Dispersions. Phys. Rev. E: Stat. Phys., Plasmas, Fluids, Relat. Interdiscip. Top. 1996, 54, 6633–6645. 10.1103/PhysRevE.54.6633. [DOI] [PubMed] [Google Scholar]
- Palberg T. Colloidal Crystallization Dynamics. Curr. Opin. Colloid Interface Sci. 1997, 2, 607–614. 10.1016/S1359-0294(97)80053-2. [DOI] [Google Scholar]
- Palberg T. Crystallization Kinetics of Repulsive Colloidal Spheres. J. Phys.: Condens. Matter 1999, 11, R323. 10.1088/0953-8984/11/28/201. [DOI] [PubMed] [Google Scholar]
- Anderson V. J.; Lekkerkerker H. N. W. Insights into Phase Transition Kinetics from Colloid Science. Nature 2002, 416, 811–815. 10.1038/416811a. [DOI] [PubMed] [Google Scholar]
- Sear R. P. Nucleation: Theory and Applications to Protein Solutions and Colloidal Suspensions. J. Phys.: Condens. Matter 2007, 19, 033101. 10.1088/0953-8984/19/3/033101. [DOI] [Google Scholar]
- Gasser U. Crystallization in Three- and Two-Dimensional Colloidal Suspensions. J. Phys.: Condens. Matter 2009, 21, 203101. 10.1088/0953-8984/21/20/203101. [DOI] [PubMed] [Google Scholar]
- Palberg T. Crystallization Kinetics of Colloidal Model Suspensions: Recent Achievements and New Perspectives. J. Phys.: Condens. Matter 2014, 26, 333101. 10.1088/0953-8984/26/33/333101. [DOI] [PubMed] [Google Scholar]
- John B. S.; Stroock A.; Escobedo F. A. Cubatic Liquid-Crystalline Behavior in a System of Hard Cuboids. J. Chem. Phys. 2004, 120, 9383–9389. 10.1063/1.1711594. [DOI] [PubMed] [Google Scholar]
- Haji-Akbari A.; Engel M.; Keys A. S.; Zheng X.; Petschek R. G.; Palffy-Muhoray P.; Glotzer S. C. Disordered, Quasicrystalline and Crystalline Phases of Densely Packed Tetrahedra. Nature 2009, 462, 773–777. 10.1038/nature08641. [DOI] [PubMed] [Google Scholar]
- Damasceno P. F.; Engel M.; Glotzer S. C. Predictive Self-Assembly of Polyhedra into Complex Structures. Science 2012, 337, 453–457. 10.1126/science.1220869. [DOI] [PubMed] [Google Scholar]
- Agarwal U.; Escobedo F. A. Mesophase Behaviour of Polyhedral Particles. Nat. Mater. 2011, 10, 230–235. 10.1038/nmat2959. [DOI] [PubMed] [Google Scholar]
- Ni R.; Dijkstra M. Crystal Nucleation of Colloidal Hard Dumbbells. J. Chem. Phys. 2011, 134, 034501. 10.1063/1.3528222. [DOI] [PubMed] [Google Scholar]
- Thapar V.; Escobedo F. A. Localized Orientational Order Chaperones the Nucleation of Rotator Phases in Hard Polyhedral Particles. Phys. Rev. Lett. 2014, 112, 048301. 10.1103/PhysRevLett.112.048301. [DOI] [PubMed] [Google Scholar]
- Auer S.; Frenkel D. Crystallization of Weakly Charged Colloidal Spheres: A Numerical Study. J. Phys.: Condens. Matter 2002, 14, 7667. 10.1088/0953-8984/14/33/308. [DOI] [Google Scholar]
- Wette P.; Schöpe H. J.; Palberg T. Crystallization in Charged Two-component Suspensions. J. Chem. Phys. 2005, 122, 144901. 10.1063/1.1872752. [DOI] [PubMed] [Google Scholar]
- Punnathanam S.; Monson P. Crystal Nucleation in Binary Hard Sphere Mixtures: A Monte Carlo Simulation Study. J. Chem. Phys. 2006, 125, 024508. 10.1063/1.2208998. [DOI] [PubMed] [Google Scholar]
- Wette P.; Schöpe H. J. Nucleation Kinetics in Deionized Charged Colloidal Model Systems: A Quantitative Study by Means of Classical Nucleation Theory. Phys. Rev. E 2007, 75, 051405. 10.1103/PhysRevE.75.051405. [DOI] [PubMed] [Google Scholar]
- Williams S. R.; Royall C. P.; Bryant G. Crystallization of Dense Binary Hard-Sphere Mixtures with Marginal Size Ratio. Phys. Rev. Lett. 2008, 100, 225502. 10.1103/PhysRevLett.100.225502. [DOI] [PubMed] [Google Scholar]
- Peters B. Competing Nucleation Pathways in a Mixture of Oppositely Charged Colloids: Out-of-Equilibrium Nucleation Revisited. J. Chem. Phys. 2009, 131, 244103. 10.1063/1.3271024. [DOI] [PubMed] [Google Scholar]
- Schätzel K.; Ackerson B. J. Density Fluctuations During Crystallization of Colloids. Phys. Rev. E: Stat. Phys., Plasmas, Fluids, Relat. Interdiscip. Top. 1993, 48, 3766–3777. 10.1103/PhysRevE.48.3766. [DOI] [PubMed] [Google Scholar]
- Schöpe H. J.; Bryant G.; van Megen W. Two-Step Crystallization Kinetics in Colloidal Hard-Sphere Systems. Phys. Rev. Lett. 2006, 96, 175701. 10.1103/PhysRevLett.96.175701. [DOI] [PubMed] [Google Scholar]
- Schöpe H. J.; Bryant G.; van Megen W. Effect of Polydispersity on the Crystallization Kinetics of Suspensions of Colloidal Hard Spheres when Approaching the Glass Transition. J. Chem. Phys. 2007, 127, 084505. 10.1063/1.2760207. [DOI] [PubMed] [Google Scholar]
- Harland J. L.; Henderson S. I.; Underwood S. M.; van Megen W. Observation of Accelerated Nucleation in Dense Colloidal Fluids of Hard Sphere Particles. Phys. Rev. Lett. 1995, 75, 3572–3575. 10.1103/PhysRevLett.75.3572. [DOI] [PubMed] [Google Scholar]
- Cheng Z.; Zhu J.; Russel W. B.; Meyer W. V.; Chaikin P. M. Colloidal Hard-Sphere Crystallization Kinetics in Microgravity and Normal Gravity. Appl. Opt. 2001, 40, 4146–4151. 10.1364/AO.40.004146. [DOI] [PubMed] [Google Scholar]
- Francis P. S.; Martin S.; Bryant G.; van Megen W.; Wilksch P. A. A Bragg Scattering Spectrometer for Studying Crystallization of Colloidal Suspensions. Rev. Sci. Instrum. 2002, 73, 3878–3884. 10.1063/1.1512332. [DOI] [Google Scholar]
- Schilling T.; Schöpe H. J.; Oettel M.; Opletal G.; Snook I. Precursor-Mediated Crystallization Process in Suspensions of Hard Spheres. Phys. Rev. Lett. 2010, 105, 025701. 10.1103/PhysRevLett.105.025701. [DOI] [PubMed] [Google Scholar]
- Filion L.; Hermes M.; Ni R.; Dijkstra M. Crystal Nucleation of Hard Spheres Using Molecular Dynamics, Umbrella Sampling, and Forward Flux Sampling: A Comparison of Simulation Techniques. J. Chem. Phys. 2010, 133, 244115. 10.1063/1.3506838. [DOI] [PubMed] [Google Scholar]
- Filion L.; Ni R.; Frenkel D.; Dijkstra M. Simulation of Nucleation in Almost Hard-Sphere Colloids: The Discrepancy between Experiment and Simulation Persists. J. Chem. Phys. 2011, 134, 134901. 10.1063/1.3572059. [DOI] [PubMed] [Google Scholar]
- Kawasaki T.; Tanaka H. Formation of a Crystal Nucleus from Liquid. Proc. Natl. Acad. Sci. U. S. A. 2010, 107, 14036–14041. 10.1073/pnas.1001040107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brownian dynamics474 differs from molecular dynamics mainly with respect to the dynamics of the system at short times. This difference is thus not expected to play a role in the estimate of nucleation rates, which depend chiefly on the long-time dynamics of the system.
- Shiryayev A.; Gunton J. D. Crystal Nucleation for a Model of Globular Proteins. J. Chem. Phys. 2004, 120, 8318–8326. 10.1063/1.1695321. [DOI] [PubMed] [Google Scholar]
- Rosenbaum D. F.; Zukoski C. F. Protein Interactions and Crystallization. J. Cryst. Growth 1996, 169, 752–758. 10.1016/S0022-0248(96)00455-1. [DOI] [Google Scholar]
- Piazza R. Interactions and Phase Transitions in Protein Solutions. Curr. Opin. Colloid Interface Sci. 2000, 5, 38–43. 10.1016/S1359-0294(00)00034-0. [DOI] [Google Scholar]
- Lomakin A.; Asherie N.; Benedek G. B. Liquid–Solid Transition in Nuclei of Protein Crystals. Proc. Natl. Acad. Sci. U. S. A. 2003, 100, 10254–10257. 10.1073/pnas.1334069100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y.; Wang X.; Ching C. B. Toward Further Understanding of Lysozyme Crystallization: Phase Diagram, Protein-Protein Interaction, Nucleation Kinetics and Growth Kinetics. Cryst. Growth Des. 2010, 10, 548–558. 10.1021/cg900919w. [DOI] [Google Scholar]
- Liu H.; Kumar S. K.; Douglas J. F. Self-Assembly-Induced Protein Crystallization. Phys. Rev. Lett. 2009, 103, 018101. 10.1103/PhysRevLett.103.018101. [DOI] [PubMed] [Google Scholar]
- George A.; Wilson W. W. Predicting Protein Crystallization from a Dilute Solution Property. Acta Crystallogr., Sect. D: Biol. Crystallogr. 1994, 50, 361–365. 10.1107/S0907444994001216. [DOI] [PubMed] [Google Scholar]
- Doye J. P. K.; Louis A. A.; Lin I.-C.; Allen L. R.; Noya E. G.; Wilber A. W.; Kok H. C.; Lyus R. Controlling Crystallization and Its Absence: Proteins, Colloids and Patchy Models. Phys. Chem. Chem. Phys. 2007, 9, 2197. 10.1039/b614955c. [DOI] [PubMed] [Google Scholar]
- Dixit N. M.; Zukoski C. F. Crystal Nucleation Rates for Particles Experiencing Short-Range Attractions: Applications to Proteins. J. Colloid Interface Sci. 2000, 228, 359–371. 10.1006/jcis.2000.6944. [DOI] [PubMed] [Google Scholar]
- Dixit N. M.; Kulkarni A. M.; Zukoski C. F. Comparison of Experimental Estimates and Model Predictions of Protein Crystal Nucleation Rates. Colloids Surf., A 2001, 190, 47–60. 10.1016/S0927-7757(01)00664-1. [DOI] [Google Scholar]
- Chang J.; Lenhoff A. M.; Sandler S. I. Determination of Fluid–Solid Transitions in Model Protein Solutions Using the Histogram Reweighting Method and Expanded Ensemble Simulations. J. Chem. Phys. 2004, 120, 3003–3014. 10.1063/1.1638377. [DOI] [PubMed] [Google Scholar]
- Jones J. E. On. The Determination of Molecular Fields. II. From the Equation of State of a Gas. Proc. R. Soc. London, Ser. A 1924, 106, 463–477. 10.1098/rspa.1924.0082. [DOI] [Google Scholar]
- Baidakov V. G.; Tipeev A. O.; Bobrov K. S.; Ionov G. V. Crystal Nucleation Rate Isotherms in Lennard-Jones Liquids. J. Chem. Phys. 2010, 132, 234505. 10.1063/1.3439585. [DOI] [PubMed] [Google Scholar]
- van der Hoef M. A. Free Energy of the Lennard-Jones Solid. J. Chem. Phys. 2000, 113, 8142–8148. 10.1063/1.1314342. [DOI] [Google Scholar]
- de Wette F. W.; Allen R. E.; Hughes D. S.; Rahman A. Crystallization with a Lennard-Jones Potential: A Computer Experiment. Phys. Lett. A 1969, 29, 548–549. 10.1016/0375-9601(69)90430-7. [DOI] [Google Scholar]
- Khrapak S. A.; Morfill G. E. Accurate Freezing and Melting Equations for the Lennard-Jones System. J. Chem. Phys. 2011, 134, 094108. 10.1063/1.3561698. [DOI] [PubMed] [Google Scholar]
- Luo S.-N.; Strachan A.; Swift D. C. Nonequilibrium Melting and Crystallization of a Model Lennard-Jones System. J. Chem. Phys. 2004, 120, 11640–11649. 10.1063/1.1755655. [DOI] [PubMed] [Google Scholar]
- Morris J. R.; Song X. The Anisotropic Free Energy of the Lennard-Jones Crystal-Melt Interface. J. Chem. Phys. 2003, 119, 3920–3925. 10.1063/1.1591725. [DOI] [Google Scholar]
- Davidchack R. L.; Laird B. B. Direct Calculation of the Crystal-Melt Interfacial Free Energies for Continuous Potentials: Application to the Lennard-Jones System. J. Chem. Phys. 2003, 118, 7651–7657. 10.1063/1.1563248. [DOI] [Google Scholar]
- Broughton J. Q.; Gilmer G. H. Molecular Dynamics Investigation of the Crystal-Fluid Interface. VI. Excess Surface Free Energies of Crystal-Liquid Systems. J. Chem. Phys. 1986, 84, 5759–5768. 10.1063/1.449884. [DOI] [Google Scholar]
- Bolhuis P. G.; Frenkel D.; Mau S.-C.; Huse D. A. Entropy Difference between Crystal Phases. Nature 1997, 388, 235–236. 10.1038/40779.9230429 [DOI] [Google Scholar]
- Desgranges C.; Delhommelle J. Controlling Polymorphism During the Crystallization of an Atomic Fluid. Phys. Rev. Lett. 2007, 98, 235502. 10.1103/PhysRevLett.98.235502. [DOI] [PubMed] [Google Scholar]
- Rahman A. Correlations in the Motion of Atoms in Liquid Argon. Phys. Rev. 1964, 136, A405–A411. 10.1103/PhysRev.136.A405. [DOI] [Google Scholar]
- Verlet L. Computer “Experiments” on Classical Fluids. I. Thermodynamical Properties of Lennard-Jones Molecules. Phys. Rev. 1967, 159, 98–103. 10.1103/PhysRev.159.98. [DOI] [Google Scholar]
- McGinty D. J. Molecular Dynamics Studies of the Properties of Small Clusters of Argon Atoms. J. Chem. Phys. 1973, 58, 4733–4742. 10.1063/1.1679052. [DOI] [Google Scholar]
- Mandell M. J.; McTague J. P.; Rahman A. Crystal Nucleation in a Three Dimensional Lennard-Jones System: A Molecular Dynamics Study. J. Chem. Phys. 1976, 64, 3699–3702. 10.1063/1.432681. [DOI] [Google Scholar]
- Rein ten Wolde P.; Ruiz-Montero M. J.; Frenkel D. Numerical Calculation of the Rate of Crystal Nucleation in a Lennard-Jones System at Moderate Undercooling. J. Chem. Phys. 1996, 104, 9932–9947. 10.1063/1.471721. [DOI] [Google Scholar]
- ten Wolde P. R.; Ruiz-Montero M. J.; Frenkel D. Numerical Calculation of the Rate of Homogeneous Gas-Liquid Nucleation in a Lennard-Jones System. J. Chem. Phys. 1999, 110, 1591–1599. 10.1063/1.477799. [DOI] [Google Scholar]
- Lechner W.; Dellago C. Accurate Determination of Crystal Structures Based on Averaged Local Bond Order Parameters. J. Chem. Phys. 2008, 129, 114707. 10.1063/1.2977970. [DOI] [PubMed] [Google Scholar]
- Kalikmanov V. I.; Wölk J.; Kraska T. Argon Nucleation: Bringing Together Theory, Simulations, and Experiment. J. Chem. Phys. 2008, 128, 124506. 10.1063/1.2888995. [DOI] [PubMed] [Google Scholar]
- Bai X.-M.; Li M. Test of Classical Nucleation Theory via Molecular-Dynamics Simulation. J. Chem. Phys. 2005, 122, 224510. 10.1063/1.1931661. [DOI] [PubMed] [Google Scholar]
- Bai X.-M.; Li M. Calculation of Solid–Liquid Interfacial Free Energy: A Classical Nucleation Theory Based Approach. J. Chem. Phys. 2006, 124, 124707. 10.1063/1.2184315. [DOI] [PubMed] [Google Scholar]
- Wang H.; Gould H.; Klein W. Homogeneous and Heterogeneous Nucleation of Lennard-Jones Liquids. Phys. Rev. E 2007, 76, 031604. 10.1103/PhysRevE.76.031604. [DOI] [PubMed] [Google Scholar]
- Moroni D.; ten Wolde P. R.; Bolhuis P. G. Interplay between Structure and Size in a Critical Crystal Nucleus. Phys. Rev. Lett. 2005, 94, 235703. 10.1103/PhysRevLett.94.235703. [DOI] [PubMed] [Google Scholar]
- Huitema H.; van der Eerden J.; Janssen J.; Human H. Thermodynamics and Kinetics of Homogeneous Crystal Nucleation Studied by Computer Simulation. Phys. Rev. B: Condens. Matter Mater. Phys. 2000, 62, 14690–14702. 10.1103/PhysRevB.62.14690. [DOI] [Google Scholar]
- Peng L. J.; Morris J. R.; Aga R. S. A Parameter-Free Prediction of Simulated Crystal Nucleation Times in the Lennard-Jones System: From the Steady-State Nucleation to the Transient Time Regime. J. Chem. Phys. 2010, 133, 084505. 10.1063/1.3472301. [DOI] [PubMed] [Google Scholar]
- Klein W.; Leyvraz F. Crystalline Nucleation in Deeply Quenched Liquids. Phys. Rev. Lett. 1986, 57, 2845–2848. 10.1103/PhysRevLett.57.2845. [DOI] [PubMed] [Google Scholar]
- ten Wolde P.; Ruiz-Montero M.; Frenkel D. Numerical Evidence for bcc Ordering at the Surface of a Critical fcc Nucleus. Phys. Rev. Lett. 1995, 75, 2714–2717. 10.1103/PhysRevLett.75.2714. [DOI] [PubMed] [Google Scholar]
- Rein ten Wolde P.; Frenkel D. Homogeneous Nucleation and the Ostwald Step Rule. Phys. Chem. Chem. Phys. 1999, 1, 2191–2196. 10.1039/a809346f. [DOI] [Google Scholar]
- Desgranges C.; Delhommelle J. Molecular Mechanism for the Cross-Nucleation between Polymorphs. J. Am. Chem. Soc. 2006, 128, 10368–10369. 10.1021/ja063218f. [DOI] [PubMed] [Google Scholar]
- Wang X.; Mi J.; Zhong C. Density Functional Theory for Crystal-Liquid Interfaces of Lennard-Jones Fluid. J. Chem. Phys. 2013, 138, 164704. 10.1063/1.4802633. [DOI] [PubMed] [Google Scholar]
- As early as 1897, Ostwald asserted475 that the critical cluster nucleating from a supercooled liquid is not necessarily the thermodynamically most stable crystalline polymorph. In fact, it could very well be the polymorph closest in energy to the liquid phase. A nice review of the Ostwald rule can be found in ref (264).
- Delhommelle J. Crystal Nucleation and Growth from Supercooled Melts. Mol. Simul. 2011, 37, 613–620. 10.1080/08927022.2011.566611. [DOI] [Google Scholar]
- In this case, the equilibrium crystalline phase that forms upon homogeneous nucleation is an rhcp phase made of stacked hexagonal layers.
- Turnbull D.; Vonnegut B. Nucleation Catalysis. Ind. Eng. Chem. 1952, 44, 1292–1298. 10.1021/ie50510a031. [DOI] [Google Scholar]
- Mithen J. P.; Sear R. P. Computer Simulation of Epitaxial Nucleation of a Crystal on a Crystalline Surface. J. Chem. Phys. 2014, 140, 084504. 10.1063/1.4866035. [DOI] [PubMed] [Google Scholar]
- Jungblut S.; Dellago C. Heterogeneous Crystallization on Tiny Clusters. Europhys. Lett. 2011, 96, 56006. 10.1209/0295-5075/96/56006. [DOI] [Google Scholar]
- Page A. J.; Sear R. P. Crystallization Controlled by the Geometry of a Surface. J. Am. Chem. Soc. 2009, 131, 17550–17551. 10.1021/ja9085512. [DOI] [PubMed] [Google Scholar]
- Zhang H.; Peng S.; Long X.; Zhou X.; Liang J.; Wan C.; Zheng J.; Ju X. Wall-Induced Phase Transition Controlled by Layering Freezing. Phys. Rev. E 2014, 89, 032412. 10.1103/PhysRevE.89.032412. [DOI] [PubMed] [Google Scholar]
- Honeycutt J. D.; Andersen H. C. Small System Size Artifacts in the Molecular Dynamics Simulation of Homogeneous Crystal Nucleation in Supercooled Atomic Liquids. J. Phys. Chem. 1986, 90, 1585–1589. 10.1021/j100399a026. [DOI] [Google Scholar]
- Swope W.; Andersen H. 106-Particle Molecular-Dynamics Study of Homogeneous Nucleation of Crystals in a Supercooled Atomic Liquid. Phys. Rev. B: Condens. Matter Mater. Phys. 1990, 41, 7042–7054. 10.1103/PhysRevB.41.7042. [DOI] [PubMed] [Google Scholar]
- We are referring to the average population of critical nuclei at a given temperature and pressure; in other words, the number of critical nuclei expected per unit volume, which should not be confused with the density of the crystalline phase within the nuclei.
- Sutton A. P.; Chen J. Long-Range Finnis–Sinclair Potentials. Philos. Mag. Lett. 1990, 61, 139–146. 10.1080/09500839008206493. [DOI] [Google Scholar]
- Fumi F.; Tosi M. Ionic Sizes and Born Repulsive Parameters in the NaCl-Type Alkali Halides. J. Phys. Chem. Solids 1964, 25, 31–43. 10.1016/0022-3697(64)90159-3. [DOI] [Google Scholar]
- Stillinger F. H.; Weber T. A. Computer Simulation of Local Order in Condensed Phases of Silicon. Phys. Rev. B: Condens. Matter Mater. Phys. 1985, 31, 5262–5271. 10.1103/PhysRevB.31.5262. [DOI] [PubMed] [Google Scholar]
- Tersoff J. New Empirical Approach for the Structure and Energy of Covalent Systems. Phys. Rev. B: Condens. Matter Mater. Phys. 1988, 37, 6991–7000. 10.1103/PhysRevB.37.6991. [DOI] [PubMed] [Google Scholar]
- Tersoff J. Modeling Solid-State Chemistry: Interatomic Potentials for Multicomponent Systems. Phys. Rev. B: Condens. Matter Mater. Phys. 1989, 39, 5566–5568. 10.1103/PhysRevB.39.5566. [DOI] [PubMed] [Google Scholar]
- Brenner D. W.; Shenderova O. A.; Harrison J. A.; Stuart S. J.; Ni B.; Sinnott S. B. A second-generation reactive empirical bond order (REBO) potential energy expression for hydrocarbons. J. Phys.: Condens. Matter 2002, 14, 783. 10.1088/0953-8984/14/4/312. [DOI] [Google Scholar]
- Ackland G. J.; Thetford R. An Improved N-Body Semi-Empirical Model for Body-Centred Cubic Transition Metals. Philos. Mag. A 1987, 56, 15–30. 10.1080/01418618708204464. [DOI] [Google Scholar]
- Daw M. S.; Foiles S. M.; Baskes M. I. The Embedded-Atom Method: A Review of Theory and Applications. Mater. Sci. Rep. 1993, 9, 251–310. 10.1016/0920-2307(93)90001-U. [DOI] [Google Scholar]
- Ercolessi F.; Adams J. B. Interatomic Potentials from First-Principles Calculations: The Force-Matching Method. Europhys. Lett. 1994, 26, 583. 10.1209/0295-5075/26/8/005. [DOI] [Google Scholar]
- Ercolessi F.; Tosatti E.; Parrinello M. Au (100) Surface Reconstruction. Phys. Rev. Lett. 1986, 57, 719–722. 10.1103/PhysRevLett.57.719. [DOI] [PubMed] [Google Scholar]
- Hou Z.; Tian Z.; Liu R.; Dong K.; Yu A. Formation Mechanism of Bulk Nanocrystalline Aluminium with Multiply Twinned Grains by Liquid Quenching: A Molecular Dynamics Simulation Study. Comput. Mater. Sci. 2015, 99, 256–261. 10.1016/j.commatsci.2014.12.037. [DOI] [Google Scholar]
- Shibuta Y.; Oguchi K.; Takaki T.; Ohno M. Homogeneous Nucleation and Microstructure Evolution in Million-Atom Molecular Dynamics Simulation. Sci. Rep. 2015, 5, 13534. 10.1038/srep13534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shibuta Y.; Oguchi K.; Ohno M. Million-Atom Molecular Dynamics Simulation on Spontaneous Evolution of Anisotropy in Solid Nucleus During Solidification of Iron. Scr. Mater. 2014, 86, 20–23. 10.1016/j.scriptamat.2014.04.021. [DOI] [Google Scholar]
- Aguado A.; Jarrold M. F. Melting and Freezing of Metal Clusters. Annu. Rev. Phys. Chem. 2011, 62, 151–172. 10.1146/annurev-physchem-032210-103454. [DOI] [PubMed] [Google Scholar]
- Shibuta Y. A Molecular Dynamics Study of Effects of Size and Cooling Rate on the Structure of Molybdenum Nanoparticles. J. Therm. Sci. Technol. 2012, 7, 45–57. 10.1299/jtst.7.45. [DOI] [Google Scholar]
- Yakubovich A. V.; Sushko G.; Schramm S.; Solov’yov A. V. Kinetics of Liquid–Solid Phase Transition in Large Nickel Clusters. Phys. Rev. B: Condens. Matter Mater. Phys. 2013, 88, 035438. 10.1103/PhysRevB.88.035438. [DOI] [Google Scholar]
- Milek T.; Zahn D. Molecular Simulation of Ag Nanoparticle Nucleation from Solution: Redox-Reactions Direct the Evolution of Shape and Structure. Nano Lett. 2014, 14, 4913–4917. 10.1021/nl502503t. [DOI] [PubMed] [Google Scholar]
- Pan H.; Shou W. Single Crystal Formation in Micro/Nano-Confined Domains by Melt-Mediated Crystallization Without Seeds. J. Phys. D: Appl. Phys. 2015, 48, 225302. 10.1088/0022-3727/48/22/225302. [DOI] [Google Scholar]
- Lü Y.; Chen M. Surface Layering-Induced Crystallization of Ni-Si Alloy Drops. Acta Mater. 2012, 60, 4636–4645. 10.1016/j.actamat.2012.03.038. [DOI] [Google Scholar]
- Li T.; Donadio D.; Ghiringhelli L. M.; Galli G. Surface-Induced Crystallization in Supercooled Tetrahedral Liquids. Nat. Mater. 2009, 8, 726–730. 10.1038/nmat2508. [DOI] [PubMed] [Google Scholar]
- Molinero V.; Moore E. B. Water Modeled as an Intermediate Element between Carbon and Silicon. J. Phys. Chem. B 2009, 113, 4008–4016. 10.1021/jp805227c. [DOI] [PubMed] [Google Scholar]
- Haji-Akbari A.; DeFever R. S.; Sarupria S.; Debenedetti P. G. Suppression of Sub-Surface Freezing in Free-Standing Thin Films of a Coarse-Grained Model of Water. Phys. Chem. Chem. Phys. 2014, 16, 25916–25927. 10.1039/C4CP03948C. [DOI] [PubMed] [Google Scholar]
- Gianetti M. M.; Haji-Akbari A.; Paula Longinotti M.; Debenedetti P. G. Computational Investigation of Structure, Dynamics and Nucleation Kinetics of a Family of Modified Stillinger–Weber Model Fluids in Bulk and Free-standing Thin Films. Phys. Chem. Chem. Phys. 2016, 18, 4102–4111. 10.1039/C5CP06535F. [DOI] [PubMed] [Google Scholar]
- Li T.; Donadio D.; Galli G. Ice Nucleation at the Nanoscale Probes No Man’s Land of Water. Nat. Commun. 2013, 4, 1887. 10.1038/ncomms2918. [DOI] [PubMed] [Google Scholar]
- Li T.; Donadio D.; Galli G. Nucleation of Tetrahedral Solids: A Molecular Dynamics Study of Supercooled Liquid Silicon. J. Chem. Phys. 2009, 131, 224519. 10.1063/1.3268346. [DOI] [PubMed] [Google Scholar]
- Valeriani C.; Sanz E.; Frenkel D. Rate of Homogeneous Crystal Nucleation in Molten NaCl. J. Chem. Phys. 2005, 122, 194501. 10.1063/1.1896348. [DOI] [PubMed] [Google Scholar]
- Raoux S.; Welnich W.; Ielmini D. Phase Change Materials and Their Application to Nonvolatile Memories. Chem. Rev. 2010, 110, 240–267. 10.1021/cr900040x. [DOI] [PubMed] [Google Scholar]
- Lencer D.; Salinga M.; Wuttig M. Design Rules for Phase-Change Materials in Data Storage Applications. Adv. Mater. 2011, 23, 2030–2058. 10.1002/adma.201004255. [DOI] [PubMed] [Google Scholar]
- Wuttig M.; Yamada N. Phase-Change Materials for Rewriteable Data Storage. Nat. Mater. 2007, 6, 824–832. 10.1038/nmat2009. [DOI] [PubMed] [Google Scholar]
- Orava J.; Greer A. L.; Gholipour B.; Hewak D. W.; Smith C. E. Characterization of Supercooled Liquid Ge2Sb2Te5 and its Crystallization by Ultrafast-heating Calorimetry. Nat. Mater. 2012, 11, 279–283. 10.1038/nmat3275. [DOI] [PubMed] [Google Scholar]
- Zalden P.; von Hoegen A.; Landreman P.; Wuttig M.; Lindenberg A. M. How Supercooled Liquid Phase-Change Materials Crystallize: Snapshots after Femtosecond Optical Excitation. Chem. Mater. 2015, 27, 5641–5646. 10.1021/acs.chemmater.5b02011. [DOI] [Google Scholar]
- Hegedüs J.; Elliott S. R. Microscopic Origin of the Fast Crystallization Ability of Ge2Sb2Te5 Phase-Change Memory Materials. Nat. Mater. 2008, 7, 399–405. 10.1038/nmat2157. [DOI] [PubMed] [Google Scholar]
- Lee T. H.; Elliott S. R. Ab Initio Computer Simulation of the Early Stages of Crystallization: Application to Ge2Sb2Te5 Phase-Change Materials. Phys. Rev. Lett. 2011, 107, 145702. 10.1103/PhysRevLett.107.145702. [DOI] [PubMed] [Google Scholar]
- Sosso G. C.; Miceli G.; Caravati S.; Behler J.; Bernasconi M. Neural Network Interatomic Potential for the Phase Change Material GeTe. Phys. Rev. B: Condens. Matter Mater. Phys. 2012, 85, 174103. 10.1103/PhysRevB.85.174103. [DOI] [Google Scholar]
- Behler J.; Parrinello M. Generalized Neural-Network Representation of High-Dimensional Potential-Energy Surfaces. Phys. Rev. Lett. 2007, 98, 146401. 10.1103/PhysRevLett.98.146401. [DOI] [PubMed] [Google Scholar]
- Sosso G. C.; Miceli G.; Caravati S.; Giberti F.; Behler J.; Bernasconi M. Fast Crystallization of the Phase Change Compound GeTe by Large-Scale Molecular Dynamics Simulations. J. Phys. Chem. Lett. 2013, 4, 4241–4246. 10.1021/jz402268v. [DOI] [PubMed] [Google Scholar]
- Sosso G. C.; Salvalaglio M.; Behler J.; Bernasconi M.; Parrinello M. Heterogeneous Crystallization of the Phase Change Material GeTe via Atomistic Simulations. J. Phys. Chem. C 2015, 119, 6428–6434. 10.1021/acs.jpcc.5b00296. [DOI] [Google Scholar]
- Debenedetti P. G.; Stillinger F. H. Supercooled Liquids and the Glass Transition. Nature 2001, 410, 259–267. 10.1038/35065704. [DOI] [PubMed] [Google Scholar]
- Sosso G. C.; Behler J.; Bernasconi M. Breakdown of Stokes-Einstein Relation in the Supercooled Liquid State of Phase Change Materials. Phys. Status Solidi B 2012, 249, 1880–1885. 10.1002/pssb.201200355. [DOI] [Google Scholar]
- Potapczuk M. G. Aircraft Icing Research at NASA Glenn Research Center. J. Aerospace Eng. 2013, 26, 260–276. 10.1061/(ASCE)AS.1943-5525.0000322. [DOI] [Google Scholar]
- Ye Z.; Wu J.; Ferradi N. E.; Shi X. Anti-icing for key highway locations: fixed automated spray technology. Can. J. Civ. Eng. 2013, 40, 11–18. 10.1139/cjce-2012-0226. [DOI] [Google Scholar]
- Padayachee K.; Watt M.; Edwards N.; Mycock D. Cryopreservation as a tool for the conservation of Eucalyptus genetic variability: concepts and challenges. South. Forests: J. Forest. Sci. 2009, 71, 165–170. 10.2989/SF.2009.71.2.12.827. [DOI] [Google Scholar]
- Baker M. Cloud microphysics and climate. Science 1997, 276, 1072–1078. 10.1126/science.276.5315.1072. [DOI] [Google Scholar]
- Carslaw K. S.; Harrison R. G.; Kirkby J. Cosmic Rays, Clouds, and Climate. Science 2002, 298, 1732–1737. 10.1126/science.1076964. [DOI] [PubMed] [Google Scholar]
- Li T.; Donadio D.; Russo G.; Galli G. Homogeneous Ice Nucleation from Supercooled Water. Phys. Chem. Chem. Phys. 2011, 13, 19807–19813. 10.1039/c1cp22167a. [DOI] [PubMed] [Google Scholar]
- Moore E. B.; Molinero V. Structural Transformation in Supercooled Water Controls the Crystallization Rate of Ice. Nature 2011, 479, 506–508. 10.1038/nature10586. [DOI] [PubMed] [Google Scholar]
- Reinhardt A.; Doye J. P. K. Free Energy Landscapes for Homogeneous Nucleation of Ice for a Monatomic Water Model. J. Chem. Phys. 2012, 136, 054501. 10.1063/1.3677192. [DOI] [PubMed] [Google Scholar]
- Russo J.; Romano F.; Tanaka H. New Metastable Form of Ice and Its Role in the Homogeneous Crystallization of Water. Nat. Mater. 2014, 13, 733–739. 10.1038/nmat3977. [DOI] [PubMed] [Google Scholar]
- Espinosa J. R.; Sanz E.; Valeriani C.; Vega C. Homogeneous Ice Nucleation Evaluated for Several Water Models. J. Chem. Phys. 2014, 141, 18C529. 10.1063/1.4897524. [DOI] [PubMed] [Google Scholar]
- Haji-Akbari A.; Debenedetti P. G. Direct Calculation of Ice Homogeneous Nucleation Rate for a Molecular Model of Water. Proc. Natl. Acad. Sci. U. S. A. 2015, 112, 10582–10588. 10.1073/pnas.1509267112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hagen D. E.; Anderson R. J.; Kassner J. L. Jr Homogeneous condensation-freezing nucleation rate measurements for small water droplets in an expansion cloud chamber. J. Atmos. Sci. 1981, 38, 1236–1243. 10.1175/1520-0469(1981)038<1236:HCNRMF>2.0.CO;2. [DOI] [Google Scholar]
- Taborek P. Nucleation in emulsified supercooled water. Phys. Rev. B: Condens. Matter Mater. Phys. 1985, 32, 5902. 10.1103/PhysRevB.32.5902. [DOI] [PubMed] [Google Scholar]
- DeMott P. J.; Rogers D. C. Freezing nucleation rates of dilute solution droplets measured between-30 and-40 C in laboratory simulations of natural clouds. J. Atmos. Sci. 1990, 47, 1056–1064. 10.1175/1520-0469(1990)047<1056:FNRODS>2.0.CO;2. [DOI] [Google Scholar]
- Pruppacher H. A new look at homogeneous ice nucleation in supercooled water drops. J. Atmos. Sci. 1995, 52, 1924–1933. 10.1175/1520-0469(1995)052<1924:ANLAHI>2.0.CO;2. [DOI] [Google Scholar]
- Krämer B.; Hübner O.; Vortisch H.; Wöste L.; Leisner T.; Schwell M.; Rühl E.; Baumgärtel H. Homogeneous nucleation rates of supercooled water measured in single levitated microdroplets. J. Chem. Phys. 1999, 111, 6521–6527. 10.1063/1.479946. [DOI] [Google Scholar]
- Bartell L. S.; Chushak Y. G.. Nucleation of Ice in Large Water Clusters: Experiment and Simulation. In Water in Confining Geometries; Buch V., Devlin J. P., Eds.; Springer Series in Cluster Physics; Springer: Berlin, 2003; Chapter 17, pp 399–424. [Google Scholar]
- Stöckel P.; Weidinger I. M.; Baumgärtel H.; Leisner T. Rates of homogeneous ice nucleation in levitated H2O and D2O droplets. J. Phys. Chem. A 2005, 109, 2540–2546. 10.1021/jp047665y. [DOI] [PubMed] [Google Scholar]
- Stan C. A.; Schneider G. F.; Shevkoplyas S. S.; Hashimoto M.; Ibanescu M.; Wiley B. J.; Whitesides G. M. A microfluidic apparatus for the study of ice nucleation in supercooled water drops. Lab Chip 2009, 9, 2293–2305. 10.1039/b906198c. [DOI] [PubMed] [Google Scholar]
- Moore E. B.; Molinero V. Ice Crystallization in Water’s No-Man’s Land. J. Chem. Phys. 2010, 132, 244504. 10.1063/1.3451112. [DOI] [PubMed] [Google Scholar]
- Avrami M. Kinetics of Phase Change. I General Theory. J. Chem. Phys. 1939, 7, 1103–1112. 10.1063/1.1750380. [DOI] [Google Scholar]
- Avrami M. Kinetics of Phase Change. II Transformation Time Relations for Random Distribution of Nuclei. J. Chem. Phys. 1940, 8, 212–224. 10.1063/1.1750631. [DOI] [Google Scholar]
- Hage W.; Hallbrucker A.; Mayer E.; Johari G. P. Crystallization Kinetics of Water below 150 K. J. Chem. Phys. 1994, 100, 2743–2747. 10.1063/1.466468. [DOI] [Google Scholar]
- Hage W.; Hallbrucker A.; Mayer E.; Johari G. P. Kinetics of Crystallizing D2O Water near 150 K by Fourier Transform Infrared Spectroscopy and a Comparison with the Corresponding Calorimetric Studies on H2O Water. J. Chem. Phys. 1995, 103, 545–550. 10.1063/1.470140. [DOI] [Google Scholar]
- Gránásy L.; Pusztai T.; James P. F. Interfacial properties deduced from nucleation experiments: a Cahn-Hilliard analysis. J. Chem. Phys. 2002, 117, 6157–6168. 10.1063/1.1502652. [DOI] [Google Scholar]
- Malkin T. L.; Murray B. J.; Brukhno A. V.; Anwar J.; Salzmann C. G. Structure of Ice Crystallized from Supercooled Water. Proc. Natl. Acad. Sci. U. S. A. 2012, 109, 1041–1045. 10.1073/pnas.1113059109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Angell C.; Shuppert J.; Tucker J. Anomalous properties of supercooled water. Heat capacity, expansivity, and proton magnetic resonance chemical shift from 0 to-38%. J. Phys. Chem. 1973, 77, 3092–3099. 10.1021/j100644a014. [DOI] [Google Scholar]
- English N. J.; Tse J. S. Massively parallel molecular dynamics simulation of formation of ice-crystallite precursors in supercooled water: Incipient-nucleation behavior and role of system size. Phys. Rev. E 2015, 92, 032132. 10.1103/PhysRevE.92.032132. [DOI] [PubMed] [Google Scholar]
- Matsumoto M.; Saito S.; Ohmine I. Molecular dynamics simulation of the ice nucleation and growth process leading to water freezing. Nature 2002, 416, 409–413. 10.1038/416409a. [DOI] [PubMed] [Google Scholar]
- Vrbka L.; Jungwirth P. Homogeneous Freezing of Water Starts in the Subsurface. J. Phys. Chem. B 2006, 110, 18126–18129. 10.1021/jp064021c. [DOI] [PubMed] [Google Scholar]
- Moore E. B.; Molinero V. Is It Cubic? Ice Crystallization from Deeply Supercooled Water. Phys. Chem. Chem. Phys. 2011, 13, 20008. 10.1039/c1cp22022e. [DOI] [PubMed] [Google Scholar]
- Abascal J. L. F.; Sanz E.; Garciá Fernández R.; Vega C. A Potential Model for the Study of Ices and Amorphous Water: TIP4P/Ice. J. Chem. Phys. 2005, 122, 234511. 10.1063/1.1931662. [DOI] [PubMed] [Google Scholar]
- Palmer J. C.; Martelli F.; Liu Y.; Car R.; Panagiotopoulos A. Z.; Debenedetti P. G. Metastable Liquid–Liquid Transition in a Molecular Model of Water. Nature 2014, 510, 385–388. 10.1038/nature13405. [DOI] [PubMed] [Google Scholar]
- Chandler D.Illusions of phase coexistence: Comments on “Metastable liquid–liquid transition ...” by J. C. Palmer et al., Nature 510, 385 (2014). 2014, arXiv e-Print archive. arXiv:1407.6854. (accessed November 2015).
- Palmer J. C., Debenedetti P. G., Car R., Panagiotopoulos A. Z.. Response to Comment [arXiv: 1407.6854] on Palmer et al., Nature, 510, 385, 2014. 2014, arXiv e-Print archive. arXiv:1407.7884. (accessed November 2015).
- Herbert R. J.; Murray B. J.; Dobbie S. J.; Koop T. Sensitivity of Liquid Clouds to Homogenous Freezing Parameterizations. Geophys. Res. Lett. 2015, 42, 1599–1605. 10.1002/2014GL062729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carrasco J.; Hodgson A.; Michaelides A. A molecular perspective of water at metal interfaces. Nat. Mater. 2012, 11, 667–674. 10.1038/nmat3354. [DOI] [PubMed] [Google Scholar]
- Murray B. J.; O’Sullivan D.; Atkinson J. D.; Webb M. E. Ice nucleation by particles immersed in supercooled cloud droplets. Chem. Soc. Rev. 2012, 41, 6519–6554. 10.1039/c2cs35200a. [DOI] [PubMed] [Google Scholar]
- Nie S.; Feibelman P. J.; Bartelt N. C.; Thürmer K. Pentagons and heptagons in the first water layer on Pt(111). Phys. Rev. Lett. 2010, 105, 026102. 10.1103/PhysRevLett.105.026102. [DOI] [PubMed] [Google Scholar]
- Nutt D. R.; Stone A. J. Adsorption of Water on the BaF2 (111) Surface. J. Chem. Phys. 2002, 117, 800–807. 10.1063/1.1484377. [DOI] [Google Scholar]
- Nutt D. R.; Stone A. J. Ice nucleation on a model hexagonal surface. Langmuir 2004, 20, 8715–8720. 10.1021/la048958l. [DOI] [PubMed] [Google Scholar]
- Sadtchenko V.; Ewing G. E.; Nutt D. R.; Stone A. J. Instability of ice films. Langmuir 2002, 18, 4632–4636. 10.1021/la0255370. [DOI] [Google Scholar]
- Hu X. L.; Michaelides A. Ice formation on kaolinite: Lattice match or amphoterism?. Surf. Sci. 2007, 601, 5378–5381. 10.1016/j.susc.2007.09.012. [DOI] [Google Scholar]
- Hu X. L.; Michaelides A. Water on the hydroxylated (001) surface of kaolinite: From monomer adsorption to a flat 2D wetting layer. Surf. Sci. 2008, 602, 960–974. 10.1016/j.susc.2007.12.032. [DOI] [Google Scholar]
- Pruppacher H. R.; Klett J. D.. Microphysics of Clouds and Precipitation: Second Revised and Enlarged Edition with an Introduction to Cloud Chemistry and Cloud Electricity; Kluwer Academic Publishers: Dordrecht, The Netherlands, 1997. [Google Scholar]
- Croteau T.; Bertram A. K.; Patey G. N. Adsorption and structure of water on Kaolinite surfaces: Possible insight into ice nucleation from grand canonical Monte Carlo calculations. J. Phys. Chem. A 2008, 112, 10708–10712. 10.1021/jp805615q. [DOI] [PubMed] [Google Scholar]
- Croteau T.; Bertram A. K.; Patey G. N. Simulation of water adsorption on kaolinite under atmospheric conditions. J. Phys. Chem. A 2009, 113, 7826–7833. 10.1021/jp902453f. [DOI] [PubMed] [Google Scholar]
- Cygan R. T.; Liang J. J.; Kalinichev A. G. Molecular models of hydroxide, oxyhydroxide, and clay phases and the development of a general force field. J. Phys. Chem. B 2004, 108, 1255. 10.1021/jp0363287. [DOI] [Google Scholar]
- Berendsen H. J. C.; Grigera J. R.; Straatsma T. P. The Missing Term in Effective Pair Potentials. J. Phys. Chem. 1987, 91, 6269–6271. 10.1021/j100308a038. [DOI] [Google Scholar]
- Cox S. J.; Kathmann S. M.; Purton J. A.; Gillan M. J.; Michaelides A. Non-hexagonal ice at hexagonal surfaces: The role of lattice mismatch. Phys. Chem. Chem. Phys. 2012, 14, 7944–7949. 10.1039/c2cp23438f. [DOI] [PubMed] [Google Scholar]
- Jorgensen W. L.; Chandrasekhar J.; Madura J. D.; Impey R. W.; Klein M. L. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 1983, 79, 926. 10.1063/1.445869. [DOI] [Google Scholar]
- Yan J. Y.; Patey G. N. Heterogeneous Ice Nucleation Induced by Electric Fields. J. Phys. Chem. Lett. 2011, 2, 2555–2559. 10.1021/jz201113m. [DOI] [Google Scholar]
- Cox S. J.; Raza Z.; Kathmann S. M.; Slater B.; Michaelides A. The microscopic features of heterogeneous ice nucleation may affect the macroscopic morphology of atmospheric ice crystals. Faraday Discuss. 2014, 167, 389–403. 10.1039/c3fd00059a. [DOI] [PubMed] [Google Scholar]
- Abascal J. L. F.; Vega C. A general purpose model for the condensed phases of water: TIP4P/2005. J. Chem. Phys. 2005, 123, 234505. 10.1063/1.2121687. [DOI] [PubMed] [Google Scholar]
- Zielke S. A.; Bertram A. K.; Patey G. N. Simulations of Ice Nucleation by Kaolinite (001) with Rigid and Flexible Surfaces. J. Phys. Chem. B 2016, 120, 1726–1734. 10.1021/acs.jpcb.5b09052. [DOI] [PubMed] [Google Scholar]
- Lupi L.; Hudait A.; Molinero V. Heterogeneous Nucleation of Ice on Carbon Surfaces. J. Am. Chem. Soc. 2014, 136, 3156–3164. 10.1021/ja411507a. [DOI] [PubMed] [Google Scholar]
- Lupi L.; Molinero V. Does Hydrophilicity of Carbon Particles Improve Their Ice Nucleation Ability?. J. Phys. Chem. A 2014, 118, 7330–7337. 10.1021/jp4118375. [DOI] [PubMed] [Google Scholar]
- Cox S. J.; Kathmann S. M.; Slater B.; Michaelides A. Molecular simulations of heterogeneous ice nucleation. I. Controlling ice nucleation through surface hydrophilicity. J. Chem. Phys. 2015, 142, 184704. 10.1063/1.4919714. [DOI] [PubMed] [Google Scholar]
- Cox S. J.; Kathmann S. M.; Slater B.; Michaelides A. Molecular simulations of heterogeneous ice nucleation. II. Peeling back the layers. J. Chem. Phys. 2015, 142, 184705. 10.1063/1.4919715. [DOI] [PubMed] [Google Scholar]
- Whale T. F.; Rosillo-Lopez M.; Murray B. J.; Salzmann C. G. Ice Nucleation Properties of Oxidized Carbon Nanomaterials. J. Phys. Chem. Lett. 2015, 6, 3012–3016. 10.1021/acs.jpclett.5b01096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X.-X.; Chen M.; Fu M. Impact of Surface Nanostructure on Ice Nucleation. J. Chem. Phys. 2014, 141, 124709. 10.1063/1.4896149. [DOI] [PubMed] [Google Scholar]
- Fitzner M.; Sosso G. C.; Cox S. J.; Michaelides A. The Many Faces of Heterogeneous Ice Nucleation: Interplay between Surface Morphology and Hydrophobicity. J. Am. Chem. Soc. 2015, 137, 13658–13669. 10.1021/jacs.5b08748. [DOI] [PubMed] [Google Scholar]
- Bi Y.; Cabriolu R.; Li T. Heterogeneous Ice Nucleation Controlled by the Coupling of Surface Crystallinity and Surface Hydrophilicity. J. Phys. Chem. C 2016, 120, 1507–1514. 10.1021/acs.jpcc.5b09740. [DOI] [Google Scholar]
- Reinhardt A.; Doye J. P. K. Effects of surface interactions on heterogeneous ice nucleation for a monatomic water model. J. Chem. Phys. 2014, 141, 084501. 10.1063/1.4892804. [DOI] [PubMed] [Google Scholar]
- Cabriolu R.; Li T. Ice Nucleation on Carbon Surface Supports the Classical Theory for Heterogeneous Nucleation. Phys. Rev. E 2015, 91, 052402. 10.1103/PhysRevE.91.052402. [DOI] [PubMed] [Google Scholar]
- Aber J. E.; Arnold S.; Garetz B. A.; Myerson A. S. Strong dc Electric Field Applied to Supersaturated Aqueous Glycine Solution Induces Nucleation of the Gamma Polymorph. Phys. Rev. Lett. 2005, 94, 145503. 10.1103/PhysRevLett.94.145503. [DOI] [PubMed] [Google Scholar]
- Zielke S. A.; Bertram A. K.; Patey G. N. A Molecular Mechanism of Ice Nucleation on Model AgI Surfaces. J. Phys. Chem. B 2015, 119, 9049–9055. 10.1021/jp508601s. [DOI] [PubMed] [Google Scholar]
- Fraux G.; Doye J. P. K. Note: Heterogeneous ice nucleation on silver-iodide-like surfaces. J. Chem. Phys. 2014, 141, 216101. 10.1063/1.4902382. [DOI] [PubMed] [Google Scholar]
- Tasker P. W. The Stability of Ionic Crystal Surfaces. J. Phys. C: Solid State Phys. 1979, 12, 4977. 10.1088/0022-3719/12/22/036. [DOI] [Google Scholar]
- Anderson B. J.; Hallett J. Supersaturation and time dependence of ice nucleation from the vapor on single crystal substrates. J. Atmos. Sci. 1976, 33, 822–832. 10.1175/1520-0469(1976)033<0822:SATDOI>2.0.CO;2. [DOI] [Google Scholar]
- Price S. L. Predicting Crystal Structures of Organic Compounds. Chem. Soc. Rev. 2014, 43, 2098–2111. 10.1039/C3CS60279F. [DOI] [PubMed] [Google Scholar]
- Bauer J.; Spanton S.; Henry R.; Quick J.; Dziki W.; Porter W.; Morris J. Ritonavir: An Extraordinary Example of Conformational Polymorphism. Pharm. Res. 2001, 18, 859–866. 10.1023/A:1011052932607. [DOI] [PubMed] [Google Scholar]
- Datta S.; Grant D. J. W. Crystal Structures of Drugs: Advances in Determination Prediction and Engineering. Nat. Rev. Drug Discovery 2004, 3, 42–57. 10.1038/nrd1280. [DOI] [PubMed] [Google Scholar]
- Lee E. H. A. Practical Guide to Pharmaceutical Polymorph Screening & Selection. Asian J. Pharm. Sci. 2014, 9, 163–175. 10.1016/j.ajps.2014.05.002. [DOI] [Google Scholar]
- Davey R. J.; Schroeder S. L. M.; ter Horst J. H. Nucleation of Organic Crystals-A Molecular Perspective. Angew. Chem., Int. Ed. 2013, 52, 2166–2179. 10.1002/anie.201204824. [DOI] [PubMed] [Google Scholar]
- Agarwal V.; Peters B.. Solute precipitate nucleation: A review of theory and simulation advances. In Advances in Chemical Physics; Rice S. A., Dinner A. R., Eds.; John Wiley & Sons, Inc.: New York, 2014; Vol. 155, pp 97–160. [Google Scholar]
- Santiso E. E.; Trout B. L. A General Set of Order Parameters for Molecular Crystals. J. Chem. Phys. 2011, 134, 064109. 10.1063/1.3548889. [DOI] [PubMed] [Google Scholar]
- Yu T.-Q.; Chen P.-Y.; Chen M.; Samanta A.; Vanden-Eijnden E.; Tuckerman M. Order-Parameter-Aided Temperature-Accelerated Sampling for the Exploration of Crystal Polymorphism and Solid–Liquid Phase Transitions. J. Chem. Phys. 2014, 140, 214109. 10.1063/1.4878665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duff N.; Peters B. Polymorph Specific RMSD Local Order Parameters for Molecular Crystals and Nuclei: α-, β-, and γ-Glycine. J. Chem. Phys. 2011, 135, 134101. 10.1063/1.3638268. [DOI] [PubMed] [Google Scholar]
- Shetty R.; Escobedo F. A.; Choudhary D.; Clancy P. A Novel Algorithm for Characterization of Order in Materials. J. Chem. Phys. 2002, 117, 4000–4009. 10.1063/1.1494986. [DOI] [Google Scholar]
- Grossier R.; Veesler S. Reaching One Single and Stable Critical Cluster Through Finite-Sized Systems. Cryst. Growth Des. 2009, 9, 1917–1922. 10.1021/cg801165b. [DOI] [Google Scholar]
- Statistical mechanics tells us that macroscopic thermodynamic variables are well-behaved for a system with a number of molecules on the order of NA, where NA is Avogadro’s number.
- Zimmermann N. E. R.; Vorselaars B.; Quigley D.; Peters B. Nucleation of NaCl from Aqueous Solution: Critical Sizes, Ion-Attachment Kinetics, and Rates. J. Am. Chem. Soc. 2015, 137, 13352–13361. 10.1021/jacs.5b08098. [DOI] [PubMed] [Google Scholar]
- Duff N.; Peters B. Nucleation in a Potts Lattice Gas Model of Crystallization from Solution. J. Chem. Phys. 2009, 131, 184101. 10.1063/1.3250934. [DOI] [PubMed] [Google Scholar]
- Agarwal V.; Peters B. Nucleation Near the Eutectic Point in a Potts-Lattice Gas Model. J. Chem. Phys. 2014, 140, 084111. 10.1063/1.4865338. [DOI] [PubMed] [Google Scholar]
- Reguera D.; Bowles R. K.; Djikaev Y.; Reiss H. Phase Transitions in Systems Small Enough to be Clusters. J. Chem. Phys. 2003, 118, 340–353. 10.1063/1.1524192. [DOI] [Google Scholar]
- Salvalaglio M.; Perego C.; Giberti F.; Mazzotti M.; Parrinello M. Molecular-Dynamics Simulations of Urea Nucleation from Aqueous Solution. Proc. Natl. Acad. Sci. U. S. A. 2015, 112, E6–E14. 10.1073/pnas.1421192111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Agarwal A.; Wang H.; Schütte C.; Site L. D. Chemical Potential of Liquids and Mixtures via Adaptive Resolution Simulation. J. Chem. Phys. 2014, 141, 034102. 10.1063/1.4886807. [DOI] [PubMed] [Google Scholar]
- Perego C.; Salvalaglio M.; Parrinello M. Molecular Dynamics Simulations of Solutions at Constant Chemical Potential. J. Chem. Phys. 2015, 142, 144113. 10.1063/1.4917200. [DOI] [PubMed] [Google Scholar]
- Piana S.; Gale J. D. Understanding the Barriers to Crystal Growth: Dynamical Simulation of the Dissolution and Growth of Urea from Aqueous Solution. J. Am. Chem. Soc. 2005, 127, 1975–1982. 10.1021/ja043395l. [DOI] [PubMed] [Google Scholar]
- Piana S.; Reyhani M.; Gale J. D. Simulating Micrometre-Scale Crystal Growth from Solution. Nature 2005, 438, 70–73. 10.1038/nature04173. [DOI] [PubMed] [Google Scholar]
- Salvalaglio M.; Vetter T.; Giberti F.; Mazzotti M.; Parrinello M. Uncovering Molecular Details of Urea Crystal Growth in the Presence of Additives. J. Am. Chem. Soc. 2012, 134, 17221–17233. 10.1021/ja307408x. [DOI] [PubMed] [Google Scholar]
- Salvalaglio M.; Vetter T.; Mazzotti M.; Parrinello M. Controlling and Predicting Crystal Shapes: The Case of Urea. Angew. Chem., Int. Ed. 2013, 52, 13369–13372. 10.1002/anie.201304562. [DOI] [PubMed] [Google Scholar]
- Salvalaglio M.; Mazzotti M.; Parrinello M. Urea Homogeneous Nucleation Mechanism Is Solvent Dependent. Faraday Discuss. 2015, 179, 291–307. 10.1039/C4FD00235K. [DOI] [PubMed] [Google Scholar]
- Cornell W. D.; Cieplak P.; Bayly C. I.; Gould I. R.; Merz K. M.; Ferguson D. M.; Spellmeyer D. C.; Fox T.; Caldwell J. W.; Kollman P. A. A Second Generation Force Field for the Simulation of Proteins, Nucleic Acids, and Organic Molecules. J. Am. Chem. Soc. 1995, 117, 5179–5197. 10.1021/ja00124a002. [DOI] [Google Scholar]
- Wang J.; Wolf R. M.; Caldwell J. W.; Kollman P. A.; Case D. A. Development and Testing of a General Amber Force Field. J. Comput. Chem. 2004, 25, 1157–1174. 10.1002/jcc.20035. [DOI] [PubMed] [Google Scholar]
- Giberti F.; Salvalaglio M.; Mazzotti M.; Parrinello M. Insight into the Nucleation of Urea Crystals from the Melt. Chem. Eng. Sci. 2015, 121, 51–59. 10.1016/j.ces.2014.08.032. [DOI] [Google Scholar]
- Dey A.; Bomans P. H. H.; Müller F. A.; Will J.; Frederik P. M.; de With G.; Sommerdijk N. A. J. M. The Role of Prenucleation Clusters in Surface-Induced Calcium Phosphate Crystallization. Nat. Mater. 2010, 9, 1010–1014. 10.1038/nmat2900. [DOI] [PubMed] [Google Scholar]
- Yani Y.; Chow P. S.; Tan R. B. H. Glycine Open Dimers in Solution: New Insights into α-Glycine Nucleation and Growth. Cryst. Growth Des. 2012, 12, 4771–4778. 10.1021/cg300452n. [DOI] [Google Scholar]
- Ectors P.; Duchstein P.; Zahn D. From Oligomers towards a Racemic Crystal: Molecular Simulation of DL-Norleucine Crystal Nucleation from Solution. CrystEngComm 2015, 17, 6884–6889. 10.1039/C4CE02078B. [DOI] [Google Scholar]
- Ohtaki H.; Fukushima N. Nucleation Processes of NaCl and CsF Crystals from Aqueous Solutions Studied by Molecular Dynamics Simulations. Pure Appl. Chem. 1991, 63, 1743–1748. 10.1351/pac199163121743. [DOI] [Google Scholar]
- Zahn D. Atomistic Mechanism of NaCl Nucleation from an Aqueous Solution. Phys. Rev. Lett. 2004, 92, 040801. 10.1103/PhysRevLett.92.040801. [DOI] [PubMed] [Google Scholar]
- Nahtigal I. G.; Zasetsky A. Y.; Svishchev I. M. Nucleation of NaCl Nanoparticles in Supercritical Water: Molecular Dynamics Simulations. J. Phys. Chem. B 2008, 112, 7537–7543. 10.1021/jp709688g. [DOI] [PubMed] [Google Scholar]
- Chakraborty D.; Patey G. N. How Crystals Nucleate and Grow in Aqueous NaCl Solution. J. Phys. Chem. Lett. 2013, 4, 573–578. 10.1021/jz302065w. [DOI] [PubMed] [Google Scholar]
- Chakraborty D.; Patey G. N. Evidence That Crystal Nucleation in Aqueous NaCl Solution Occurs by the Two-Step Mechanism. Chem. Phys. Lett. 2013, 587, 25–29. 10.1016/j.cplett.2013.09.054. [DOI] [Google Scholar]
- Berendsen H. J. C.; Grigera J. R.; Straatsma T. P. The Missing Term in Effective Pair Potentials. J. Phys. Chem. 1987, 91, 6269–6271. 10.1021/j100308a038. [DOI] [Google Scholar]
- Chandrasekhar J.; Spellmeyer D. C.; Jorgensen W. L. Energy Component Analysis for Dilute Aqueous Solutions of Li+, Na+, F–, and Cl– Ions. J. Am. Chem. Soc. 1984, 106, 903–910. 10.1021/ja00316a012. [DOI] [Google Scholar]
- Äqvist J. Ion–Water Interaction Potentials Derived from Free Energy Perturbation Simulations. J. Phys. Chem. 1990, 94, 8021–8024. 10.1021/j100384a009. [DOI] [Google Scholar]
- Giberti F.; Tribello G. A.; Parrinello M. Transient Polymorphism in NaCl. J. Chem. Theory Comput. 2013, 9, 2526–2530. 10.1021/ct4002027. [DOI] [PubMed] [Google Scholar]
- Oostenbrink C.; Villa A.; Mark A. E.; Van Gunsteren W. F. A Biomolecular Force Field Based on the Free Enthalpy of Hydration and Solvation: The GROMOS Force-Field Parameter Sets 53A5 and 53A6. J. Comput. Chem. 2004, 25, 1656–1676. 10.1002/jcc.20090. [DOI] [PubMed] [Google Scholar]
- Alejandre J.; Hansen J.-P. Ions in Water: From Ion Clustering to Crystal Nucleation. Phys. Rev. E 2007, 76, 061505. 10.1103/PhysRevE.76.061505. [DOI] [PubMed] [Google Scholar]
- Joung I. S.; Cheatham T. E. Molecular Dynamics Simulations of the Dynamic and Energetic Properties of Alkali and Halide Ions Using Water-Model-Specific Ion Parameters. J. Phys. Chem. B 2009, 113, 13279–13290. 10.1021/jp902584c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aragones J. L.; Sanz E.; Vega C. Solubility of NaCl in Water by Molecular Simulation Revisited. J. Chem. Phys. 2012, 136, 244508. 10.1063/1.4728163. [DOI] [PubMed] [Google Scholar]
- Moucka F.; Lisal M.; Skvor J.; Jirsak J.; Nezbeda I.; Smith W. R. Molecular Simulation of Aqueous Electrolyte Solubility. 2. Osmotic Ensemble Monte Carlo Methodology for Free Energy and Solubility Calculations and Application to NaCl. J. Phys. Chem. B 2011, 115, 7849–7861. 10.1021/jp202054d. [DOI] [PubMed] [Google Scholar]
- Mester Z.; Panagiotopoulos A. Z. Temperature-Dependent Solubilities and Mean Ionic Activity Coefficients of Alkali Halides in Water from Molecular Dynamics Simulations. J. Chem. Phys. 2015, 143, 044505. 10.1063/1.4926840. [DOI] [PubMed] [Google Scholar]
- Anwar J.; Khan S.; Lindfors L. Secondary Crystal Nucleation: Nuclei Breeding Factory Uncovered. Angew. Chem., Int. Ed. 2015, 54, 14681–14684. 10.1002/anie.201501216. [DOI] [PubMed] [Google Scholar]
- Na H.-S.; Arnold S.; Myerson A. S. Cluster Formation in Highly Supersaturated Solution Droplets. J. Cryst. Growth 1994, 139, 104–112. 10.1016/0022-0248(94)90034-5. [DOI] [Google Scholar]
- Gao Y.; Yu L. E.; Chen S. B. Efflorescence Relative Humidity of Mixed Sodium Chloride and Sodium Sulfate Particles. J. Phys. Chem. A 2007, 111, 10660–10666. 10.1021/jp073186y. [DOI] [PubMed] [Google Scholar]
- Desarnaud J.; Derluyn H.; Carmeliet J.; Bonn D.; Shahidzadeh N. Metastability Limit for the Nucleation of NaCl Crystals in Confinement. J. Phys. Chem. Lett. 2014, 5, 890–895. 10.1021/jz500090x. [DOI] [PubMed] [Google Scholar]
- Sloan E. D.; Koh C. A.. Clathrate Hydrates of Natural Gases, 3rd ed.; CRC Press: Boca Raton, FL, 2008. [Google Scholar]
- Klauda J. B.; Sandler S. I. Global Distribution of Methane Hydrate in Ocean Sediment. Energy Fuels 2005, 19, 459–470. 10.1021/ef049798o. [DOI] [Google Scholar]
- Koh C. A.; Sloan E. D. Natural gas hydrates: Recent Advances and Challenges in Energy and Environmental Applications. AIChE J. 2007, 53, 1636–1643. 10.1002/aic.11219. [DOI] [Google Scholar]
- Muller-Bongartz B., Wildeman T., Sloan E. Jr.. A Hypothesis for Hydrate Nucleation Phenomena. Presented a the Second International Offshore and Polar Engineering Conference, San Francisco, CA, Jun 14–19, 1992.
- Sloan E. D.; Fleyfel F. A Molecular Mechanism for Gas Hydrate Nucleation from Ice. AIChE J. 1991, 37, 1281–1292. 10.1002/aic.690370902. [DOI] [Google Scholar]
- Radhakrishnan R.; Trout B. L. A new approach for studying nucleation phenomena using molecular simulations: Application to CO2 hydrate clathrates. J. Chem. Phys. 2002, 117, 1786–1796. 10.1063/1.1485962. [DOI] [Google Scholar]
- Hawtin R. W.; Quigley D.; Rodger P. M. Gas hydrate nucleation and cage formation at a water/methane interface. Phys. Chem. Chem. Phys. 2008, 10, 4853–4864. 10.1039/b807455k. [DOI] [PubMed] [Google Scholar]
- Moon C.; Hawtin R. W.; Rodger P. M. Nucleation and control of clathrate hydrates: insights from simulation. Faraday Discuss. 2007, 136, 367–382. 10.1039/b618194p. [DOI] [PubMed] [Google Scholar]
- Moon C.; Taylor P. C.; Rodger P. M. Molecular Dynamics Study of Gas Hydrate Formation. J. Am. Chem. Soc. 2003, 125, 4706–4707. 10.1021/ja028537v. [DOI] [PubMed] [Google Scholar]
- Walsh M. R.; Koh C. A.; Sloan E. D.; Sum A. K.; Wu D. T. Microsecond simulations of spontaneous methane hydrate nucleation and growth. Science 2009, 326, 1095–1098. 10.1126/science.1174010. [DOI] [PubMed] [Google Scholar]
- Vatamanu J.; Kusalik P. G. Unusual Crystalline and Polycrystalline Structures in Methane Hydrates. J. Am. Chem. Soc. 2006, 128, 15588–15589. 10.1021/ja066515t. [DOI] [PubMed] [Google Scholar]
- Jacobson L. C.; Hujo W.; Molinero V. Thermodynamic Stability and Growth of Guest-Free Clathrate Hydrates: A Low-Density Crystal Phase of Water. J. Phys. Chem. B 2009, 113, 10298–10307. 10.1021/jp903439a. [DOI] [PubMed] [Google Scholar]
- Jacobson L. C.; Hujo W.; Molinero V. Amorphous precursors in the nucleation of clathrate hydrates. J. Am. Chem. Soc. 2010, 132, 11806–11811. 10.1021/ja1051445. [DOI] [PubMed] [Google Scholar]
- Jacobson L. C.; Molinero V. A Methane-Water Model for Coarse-Grained Simulations of Solutions and Clathrate Hydrates. J. Phys. Chem. B 2010, 114, 7302–7311. 10.1021/jp1013576. [DOI] [PubMed] [Google Scholar]
- Jacobson L. C.; Molinero V. Can Amorphous Nuclei Grow Crystalline Clathrates? the Size and Crystallinity of Critical Clathrate Nuclei. J. Am. Chem. Soc. 2011, 133, 6458–6463. 10.1021/ja201403q. [DOI] [PubMed] [Google Scholar]
- Liang S.; Kusalik P. G. Exploring nucleation of H2S hydrates. Chem. Sci. 2011, 2, 1286–1292. 10.1039/c1sc00021g. [DOI] [Google Scholar]
- Sarupria S.; Debenedetti P. G. Homogeneous Nucleation of Methane Hydrate in Microsecond Molecular Dynamics Simulations. J. Phys. Chem. Lett. 2012, 3, 2942–2947. 10.1021/jz3012113. [DOI] [PubMed] [Google Scholar]
- Knott B. C.; Molinero V.; Doherty M. F.; Peters B. Homogeneous Nucleation of Methane Hydrates: Unrealistic under Realistic Conditions. J. Am. Chem. Soc. 2012, 134, 19544–19547. 10.1021/ja309117d. [DOI] [PubMed] [Google Scholar]
- Bai D.; Chen G.; Zhang X.; Wang W. Microsecond Molecular Dynamics Simulations of the Kinetic Pathways of Gas Hydrate Formation from Solid Surfaces. Langmuir 2011, 27, 5961–5967. 10.1021/la105088b. [DOI] [PubMed] [Google Scholar]
- Bai D.; Chen G.; Zhang X.; Wang W. Nucleation of the CO2 hydrate from three-phase contact lines. Langmuir 2012, 28, 7730–7736. 10.1021/la300647s. [DOI] [PubMed] [Google Scholar]
- Bai D.; Chen G.; Zhang X.; Sum A. K.; Wang W. How Properties of Solid Surfaces Modulate the Nucleation of Gas Hydrate. Sci. Rep. 2015, 5, 12747. 10.1038/srep12747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pirzadeh P.; Kusalik P. G. Molecular Insights into Clathrate Hydrate Nucleation at an Ice Solution Interface. J. Am. Chem. Soc. 2013, 135, 7278–7287. 10.1021/ja400521e. [DOI] [PubMed] [Google Scholar]
- Nguyen A. H.; Koc M. A.; Shepherd T. D.; Molinero V. Structure of the Ice-Clathrate Interface. J. Phys. Chem. C 2015, 119, 4104–4117. 10.1021/jp511749q. [DOI] [Google Scholar]
- Zhang Y.; Debenedetti P. G.; Prud’homme R. K.; Pethica B. A. Differential Scanning Calorimetry Studies of Clathrate Hydrate Formation. J. Phys. Chem. B 2004, 108, 16717–16722. 10.1021/jp047421d. [DOI] [Google Scholar]
- Poon G. G.; Peters B. A Stochastic Model for Nucleation in the Boundary Layer During Solvent Freeze-Concentration. Cryst. Growth Des. 2013, 13, 4642–4647. 10.1021/cg401172z. [DOI] [Google Scholar]
- Bi Y.; Li T. Probing Methane Hydrate Nucleation Through the Forward Flux Sampling Method. J. Phys. Chem. B 2014, 118, 13324–13332. 10.1021/jp503000u. [DOI] [PubMed] [Google Scholar]
- Walsh M. R.; Beckham G. T.; Koh C. A.; Sloan E. D.; Wu D. T.; Sum A. K. Methane hydrate nucleation rates from molecular dynamics simulations: effects of aqueous methane concentration, interfacial curvature, and system size. J. Phys. Chem. C 2011, 115, 21241–21248. 10.1021/jp206483q. [DOI] [Google Scholar]
- Handley C. M.; Behler J. Next Generation Interatomic Potentials for Condensed Systems. Eur. Phys. J. B 2014, 87, 1–16. 10.1140/epjb/e2014-50070-0. [DOI] [Google Scholar]
- Behler J. Representing Potential Energy Surfaces by High-Dimensional Neural Network Potentials. J. Phys.: Condens. Matter 2014, 26, 183001. 10.1088/0953-8984/26/18/183001. [DOI] [PubMed] [Google Scholar]
- Bartók A. P.; Payne M. C.; Kondor R.; Csányi G. Gaussian Approximation Potentials: The Accuracy of Quantum Mechanics, Without the Electrons. Phys. Rev. Lett. 2010, 104, 136403. 10.1103/PhysRevLett.104.136403. [DOI] [PubMed] [Google Scholar]
- Tribello G. A.; Bruneval F.; Liew C.; Parrinello M. A Molecular Dynamics Study of the Early Stages of Calcium Carbonate Growth. J. Phys. Chem. B 2009, 113, 11680–11687. 10.1021/jp902606x. [DOI] [PubMed] [Google Scholar]
- Zallen R.The Physics of Amorphous Solids; Wiley-VCH: New York, 1998. [Google Scholar]
- Cabriolu R.; Kashchiev D.; Auer S. Breakdown of nucleation theory for crystals with strongly anisotropic interactions between molecules. J. Chem. Phys. 2012, 137, 204903. 10.1063/1.4767531. [DOI] [PubMed] [Google Scholar]
- Sear R. P. The Non-Classical Nucleation of Crystals: Microscopic Mechanisms and Applications to Molecular Crystals, Ice and Calcium Carbonate. Int. Mater. Rev. 2012, 57, 328–356. 10.1179/1743280411Y.0000000015. [DOI] [Google Scholar]
- Menon N. A. Simple Demonstration of a Metastable State. Am. J. Phys. 1999, 67, 1109–1110. 10.1119/1.19092. [DOI] [Google Scholar]
- Schuss Z.Brownian Dynamics at Boundaries and Interfaces: In Physics, Chemistry, and Biology; Springer,New York, 2013; includes bibliographical references and index. [Google Scholar]
- Ostwald W. Studien über die Bildung und Umwandlung Fester Körper. 1. Abhandlung: Übersättigung und Überkaltung. Z. Phys. Chem. 1897, 22, 289–330. [Google Scholar]