

Abstract
The direct catalytic conversion of methane to liquid oxygenated compounds, such as methanol or dimethyl ether, at low temperature using molecular oxygen is a grand challenge in C–H activation that has never been met with synthetic, heterogeneous catalysts. We report the first demonstration of direct, catalytic oxidation of methane into methanol with molecular oxygen over copper-exchanged zeolites at low reaction temperatures (483–498 K). Reaction kinetics studies show sustained catalytic activity and high selectivity for a variety of commercially available zeolite topologies under mild conditions (e.g., 483 K and atmospheric pressure). Transient and steady state measurements with isotopically labeled molecules confirm catalytic turnover. The catalytic rates and apparent activation energies are affected by the zeolite topology, with caged-based zeolites (e.g., Cu-SSZ-13) showing the highest rates. Although the reaction rates are low, the discovery of catalytic sites in copper-exchanged zeolites will accelerate the development of strategies to directly oxidize methane into methanol under mild conditions.
Short abstract
Copper-exchanged zeolites (e.g., MFI, MOR, and CHA) catalyze the direct oxidation of methane into methanol in the gas phase, at low temperature, and using only molecular oxygen and water.
To date, no man-made catalyst can convert methane (CH4) and oxygen (O2) directly into methanol (CH3OH) at low temperature. For more than 100 years, the selective oxidation of this simple alkane has remained unsolved. This transformation, however, is essential to exploit our highly abundant natural gas reserves, particularly those located in distributed fields or stranded wells that cannot be accessed with large, capital-intensive reforming facilities. Although oxidative C–H bond activation of CH4 is thermodynamically and kinetically accessible at low temperatures, the large bond dissociation energy (435 kJ mol–1) of this molecule hinders C–H cleavage reactions via homolytic or heterolytic pathways. Consequently, few catalysts are capable of preventing overoxidation to carbon dioxide (CO2). Several alternative strategies for activating methane have been reported, including multistep oxyfunctionalization with Periana catalysts,1,2 borylation,3 and electrophilic carbene insertion,4,5 but all have fallen short of producing methanol directly and do not use oxygen as the oxidizing agent. In contrast, methane monooxygenase enzymes are capable of oxidizing CH4 selectively into CH3OH using O2 at room temperature.6−8 While such biocatalysts are difficult to scale-up, the nature of their active sites provides inspiration for the development of synthetic oxidation catalysts. In this respect, iron-9,10 and copper-11−13 exchanged zeolites have emerged as a promising class of materials capable of selectively oxidizing CH4 into surface-bound methoxy species by hosting active sites akin to those found in CH4 monooxygenases. Despite their potential, these materials have only been shown to oxidize CH4 to CH3OH stepwise and stoichiometrically with molecular oxygen (O2)11,14,15 or catalytically with hydrogen peroxide,16 making the process prohibitively expensive. Here, we report the first instance of catalytic gas phase oxidation of CH4 into CH3OH with O2 under mild conditions. Copper-exchanged zeolite catalysts of various topologies maintain sustained activity and high CH3OH selectivity at 483 K, while transient kinetic experiments with isotopically labeled molecules confirm catalytic turnover. The discovery of catalytically active sites for CH3OH production in zeolites will be the foundation to develop new strategies for low temperature catalytic methane oxidation.
The stoichiometric oxidation of CH4 to CH3OH over copper-exchanged zeolites is believed to occur over mono-(μ-oxo) dicupric cores13,17 or [Cu3O3]2+ trimeric sites18,19 generated by flowing anhydrous O2 over the zeolite at temperatures above 723 K. These highly reactive electrophilic metal–oxygen species are adept at attacking the strong C–H bonds of CH4, generating surface-bound methoxy groups at temperatures below 473 K.12 Methoxy groups are typically extracted as CH3OH by reacting with H2O, which deactivates the copper sites and necessitates high temperature reactivation under anhydrous O2 to perform the next oxidation cycle.13,15 In the present study, stoichiometric and catalytic CH3OH production regimes are observed during the gas phase oxidation of CH4 over copper-exchanged zeolites with the MFI topology in the sodium (Cu-Na-ZSM-5) or proton (Cu-H-ZSM-5) forms at 483 K and atmospheric pressure (Figure 1). Similar to previous studies, the catalysts were activated under flowing O2 at 823 K, cooled to reaction temperature, purged with helium (He) for 1 h, and contacted with a pure CH4 stream for 0.5 h.11,13,14 However, unlike previous studies using only H2O for extraction, we hydrolyzed surface-bound methoxy species by flowing a gas mixture comprised of 3.2 kPa of H2O, 0.0025 kPa of O2 and balance CH4. Under these conditions, Cu-Na-ZSM-5 (Cu/Al = 0.37, Na/Al = 0.26) and Cu-H-ZSM-5 (Cu/Al = 0.31) evolved 37 μmol gcat–1 and 82 μmol gcat–1 of stoichiometric CH3OH, respectively. These values are more than two times higher than those obtained with Cu-Na-ZSM-5 using only He, O2, and H2O as the extracting gas (9.6 μmol gcat–1) or those reported by Lobo et al. (16 μmol gcat–1)20 using 3.2 kPa H2O in N2 at 473 K and Schoonheydt et al. (8 μmol gcat–1)11,14 using a 50% v/v acetonitrile aqueous solution for 1 h at 298 K to extract the oxidized products.
Figure 1.
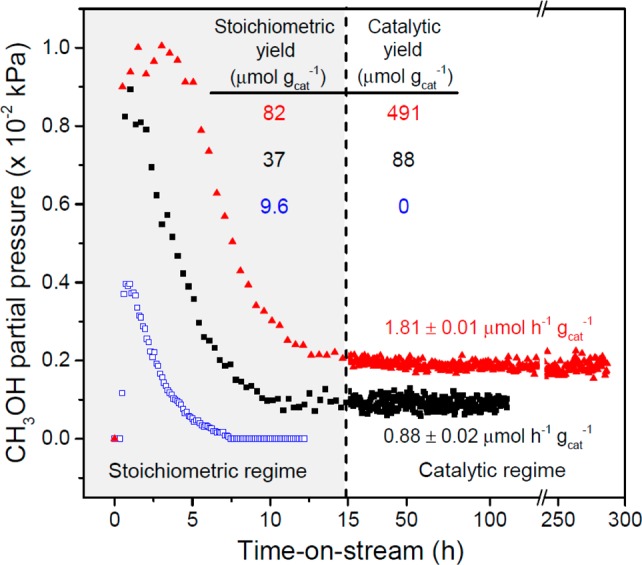
CH4 oxidation over Cu-ZSM-5. Catalyst pretreatment: 5 h at 823 K under flowing O2, cooled to 483 K under O2 flow and then purged under He for 0.5 h. Initial dry CH4 oxidation: 0.5 h under 2400 mL h–1 gcat–1 of CH4 at 483 K. CH3OH partial pressure (kPa) with He/H2O/O2 over (blue open squares) Cu-Na-ZSM-5 (Cu/Al = 0.37, Na/Al = 0.26): T = 483 K, WHSV = 2400 mL h–1 gcat–1, PHe = 98.1 kPa, PH2O = 3.2 kPa, PO2 = 0.0025 kPa (25 ppm). CH3OH partial pressure (kPa) with CH4, H2O, and O2 over (■) Cu-Na-ZSM-5 and (red solid triangles) Cu-H-ZSM-5 (Cu/Al = 0.31): T = 483 K, WHSV = 2400 mL h–1 gcat–1, PCH4 = 98.1 kPa, PH2O = 3.2 kPa, PO2 = 0.0025 kPa (25 ppm).
Remarkably, sustained CH4 oxidation activity was observed when continuing to feed the CH4, H2O, and O2 gas mixture after all stoichiometric CH3OH was extracted (Figure 1). Steady state CH3OH production rates of 0.88 ± 0.02 μmol h–1 gcat–1 and 1.81 ± 0.01 μmol h–1 gcat–1 were measured for Cu-Na-ZSM-5 and Cu-H-ZSM-5, respectively. Over Cu-Na-ZSM-5, CH3OH was the main product generated (as determined by 1H nuclear magnetic resonance, Figure S1) with a selectivity of 70.6 ± 0.4%, while CO2 was the only byproduct generated at a rate of 0.38 ± 0.02 μmol h–1 gcat–1 (Figure S2). CO2 selectivity did not increase when higher conversions were simulated by introducing CH3OH as a reagent at identical conditions (Supporting Information, Section S2, Figures S3–S4), although most of CH3OH was dehydrated into dimethyl ether in the presence of acid sites at these conditions (>60% yield). Notably, the steady state CH3OH production rates persisted without apparent deactivation, generating a total of 88 μmol gcat–1 over 108 h with Cu-Na-ZSM-5—a value roughly five times higher than that reported for the stoichiometric oxidation over Cu-Na-ZSM-5.20 Similarly, Cu-H-ZSM-5 generated a total of 491 μmol gcat–1 over 288 h—a value ca. 1.4 times larger than the total copper content of the zeolite (Figure S5). The excess CH3OH produced per copper atom in Cu-H-ZSM-5 coupled with the lack of sustained CH3OH production in the absence of CH4 in the extracting gas mixture over Cu-Na-ZSM-5 (Figure 1, open symbols) are strong evidence that CH4 is oxidized catalytically over H2O-tolerant copper sites.
Catalytic turnover was verified with transient experiments using isotopically labeled molecules coupled with online mass spectrometry (MS). 13C methoxy species were deposited on the zeolite by flowing 13CH4 (16.8 kPa 13CH4 [99 atom % 13C, Sigma-Aldrich] with balance He) over an activated Cu-Na-ZSM-5 sample for 0.5 h at 483 K and then flowing a regular 12CH4/H2O/O2 gas mixture to extract the methoxy species (detailed procedure found in Figure S6A). As shown in Figure 2, enriched 13CH3OH (m/z = 33) was detected in the stoichiometric regime, but unlabeled 12CH3OH was observed in the steady state regime, thus suggesting that new, unlabeled 12C methoxy species are formed after the initial 13C methoxy species are hydrolyzed. Next, the reaction was allowed to proceed at steady state at a measured rate of 0.88 μmol CH3OH h–1 gcat–1 for 21 h (equivalent to the production of 18.5 μmol gcat–1 of 12CH3OH). At this point, the weight hourly space velocity (WHSV) was reduced from 2400 mL h–1 gcat–1 to 300 mL h–1 gcat–1, and the gas mixture was switched to 13CH4/H2O/O2 for 0.5 h before resuming the flow of the regular, unlabeled gas mixture. This effective pulse of labeled 13CH4 resulted in the production of labeled 13CH3OH as evidenced by a detectable pulse of the m/z = 33 signal without significantly altering steady state production of CH3OH (Figure 2). Similar 13C enrichment profiles were observed for CO2 during both isotope switching experiments (Figure S7). Analogous behavior was observed for similar experiments carried out over Cu-H-ZSM-5 (Figure S8). Control experiments using 12CH4 to populate the activated catalyst with unlabeled 12C methoxy species did not generate a significant amount of 13CH3OH in the stoichiometric or steady state regimes (Figures S6B, S8B), thereby ruling out artifacts or potential effects arising from natural abundance 13CH4 in the unlabeled gas stream. Kinetic measurements on Cu-Na-ZSM-5 at 483 K show first, half, and zero order dependencies with respect to CH4, H2O, and O2, respectively (Supporting Information Section S7, Figure S9). Importantly, replacing CH4 with CD4 in the extracting gas during steady state operation at a WHSV of 240 mL h–1 gcat–1 decreased the CH3OH production rate from 0.090 to 0.055 μmol h–1 gcat–1 (Figure S10). This change corresponds to a kinetic isotope effect (KIE) of 1.6 ± 0.1, thus indicating that C–H abstraction is a kinetically relevant step in the catalytic cycle. Taken together, the data from the transient 13CH4 pulse experiments, the reaction rate order dependencies, and the kinetic isotope effect confirm catalytic turnover.
Figure 2.
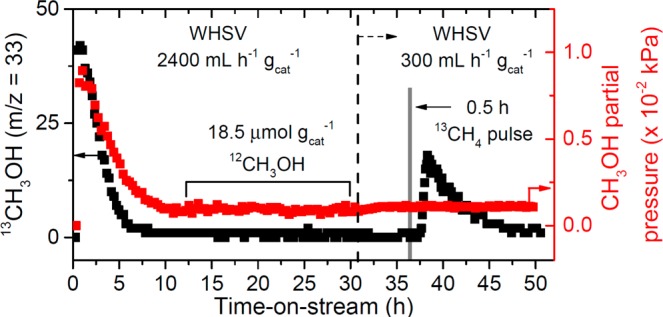
Transient, isotopically labeled kinetic experiments over Cu-Na-ZSM-5 (Cu/Al = 0.37, Na/Al = 0.26). Catalyst pretreatment: 5 h at 823 K under flowing O2, cooled to 483 K under O2 flow and purged under He for 0.5 h. Initial CH4 oxidation: 0.5 h under 2400 mL h–1 gcat–1 of 17% 13CH4/He at 483 K. Reaction conditions: T = 483 K, WHSV = 2400 mL h–1 gcat–1, P12CH4 = 98.1 kPa, PH2O = 3.2 kPa, PO2 = 0.0025 kPa (25 ppm). After 31 h on-stream, WHSV was reduced to 300 mL h–1 gcat–1 represented by the dashed line. Gray area denotes 0.5 h pulse of P13CH4 = 98 kPa, PH2O = 3.2 kPa, PO2 = 0.0025 kPa.
Gas pretreatments were varied to gain insight into the origin of catalytic and stoichiometric sites in Cu-Na-ZSM-5 (see Figures 3, S11–12). Specifically, Cu-Na-ZSM-5 samples were subjected to three pretreatments at 873 K prior to regular CH4 oxidation and extraction at 483 K: (i) calcination under O2 and then under He (20 ppm of O2); (ii) calcination under He (20 ppm of O2); or (iii) calcination under He without O2 (<0.1 ppm, Figure S11). In all cases, stoichiometric CH3OH production drastically diminished (98–100%), but catalytic CH3OH production was only moderately affected (0–60%) (Figure 3). Treating activated Cu-Na-ZSM-5 with He above 723 K has been shown to eliminate the mono-(μ-oxo) dicupric cores for stoichiometric CH4 oxidation.14 The absence of the mono-(μ-oxo) dicupric sites during steady state methane oxidation was confirmed with UV–visible spectroscopic and online gas chromatographic measurements over Cu-Na-ZSM-5 (Cu/Al = 0.37) (Supporting Information Section S9, Figures S13–S15). Interestingly, van Bokhoven et al. observed a small fraction of H2O-tolerant sites in copper-exchanged mordenite (Cu-MOR) that did not require high temperature reactivation for stoichiometric CH4 oxidation experiments after exposure to H2O.21,22 However, in the present study, exposing Cu-Na-ZSM-5 to H2O/O2/He at 483 K prior to contacting it with CH4 completely eliminated stoichiometric CH3OH production but did not affect catalytic CH3OH production. Note the onset of the catalytic regime was nearly identical for all samples (240 min on-stream) after exposure to CH4/H2O/O2 irrespective of the pretreatment used. The similarity of catalytic rates and onset of CH3OH production for all pretreated Cu-Na-ZSM-5 samples implies the catalytic sites are different from those responsible for stoichiometric CH4 oxidation, and the catalytic sites are either generated or activated when copper species are exposed to CH4, H2O, and O2 at reaction conditions rather than during the high temperature pretreatment.
Figure 3.

Thermal pretreatments and onset of steady state CH4 oxidation over Cu-Na-ZSM-5 (Cu/Al = 0.37). (A) Thermal pretreatments, reaction conditions for all pretreatments, and catalytic CH3OH production rates. The symbol ± denotes 95% confidence interval. (B) Evolution of catalytic CH3OH production for each pretreatment outlined in (A); t = 0 denotes when the CH4/H2O/O2 gas flow was started. Reaction conditions: T = 483 K, WHSV = 2400 mL h–1 gcat–1, PCH4 = 98.1 kPa, PH2O = 3.2 kPa, PO2 = 0.0025 kPa.
An induction period preceding the onset of catalytic CH4 oxidation suggests that copper speciation changes under reaction conditions. The CH3OH production profile for a regular CH4 oxidation and extraction experiment using Cu-Na-ZSM-5 pretreated at 823 K first with O2 and then with He (20 ppm of O2) shows that catalytic production began after 240 min on-stream despite H2O breaking through the catalyst bed after 25 min (Figure S12). A comparable induction process was observed when Cu-Na-ZSM-5 was pretreated without O2 and exposed to a CH4/H2O gas mixture at 483 K (Figure S11). In this case CH3OH was detected only ca. 300 min after O2 was introduced into the system. Hydrated copper species are known to weakly associate with the zeolite framework, becoming mobile and easily oxidized.23 In copper-exchanged zeolites with the chabazite topology (Cu-SSZ-13), hydrated Cu2+ ions have been shown to migrate under flowing wet O2/N2 (and trace NO and NH3) gas mixtures between 403–523 K during the selective catalytic reduction (SCR) of NOx24 to form transient dimeric active sites.25 The strong similarities between the reaction temperature and the gaseous atmosphere (H2O/O2)used during both the SCR of NOx and the oxidation of CH4 could imply that mobile, hydrated copper species also rearrange into active sites for catalytic CH4 oxidation as they do for the SCR of NOx.
Catalytic CH4 oxidation was investigated as a function of the copper content and Brønsted acidity of the zeolite (see Table S1). A control experiment with H-ZSM-5 (Si/Al = 11.5) not subjected to copper exchange did not generate CH3OH (Table 1), thereby confirming that trace transition metal impurities in the zeolite are not responsible for catalytic behavior. Higher Cu/Al ratios in Cu-Na-ZSM-5 and Cu-H-ZSM-5 increased the steady state specific activity (defined as μmolMeOH h–1 gcat–1), but decreased the site-time yield (STY, defined as molCH3OH molCu–1 h–1), suggesting that the number of active sites does not increase proportionally with the total amount of copper. The presence of Brønsted acid sites increased specific activity and STY for all samples with similar Cu/Al ratios. Indeed, different apparent activation energies (Eaapp) were observed between the sodium and proton forms of Cu-ZSM-5 (Table S1). Density functional theory calculations over Cu-ZSM-5 have shown that Brønsted acid sites change the energetics of the formation of mono-(μ-oxo) dicupric cores with NO.28 While mono-(μ-oxo) dicupric cores are likely not present under reaction conditions for catalytic CH4 oxidation, Brønsted acid sites could impart similar changes in the energetics of formation of the catalytic sites.
Table 1. Catalytic CH4 Oxidation Rates for Various Zeolite Topologiesa.
| material | framework | cage shape | cage size (Å)26 | channel size (Å)27 | Si/Alnomb | Si/Altotc | Cu/Altotd | specific activitye | STY (h–1 × 10–3)f |
|---|---|---|---|---|---|---|---|---|---|
| H-ZSM-5 | MFI | 5.3 × 5.6 | 11.5 | 13.2 | 0.31 | 1.79 ± 0.02 | 5.2 ± 0.05 | ||
| 5.1 × 5.5 | |||||||||
| H-Beta | BEA | 6.6 × 6.7 | 12.5 | 13.3 | 0.30 | 0.80 ± 0.01 | 2.4 ± 0.04 | ||
| 5.6 × 5.6 | |||||||||
| MCM-41 | MCM-41 | 30 | 12 | 16.1 | 0.74 | 0.36 ± 0.02 | 0.6 ± 0.03 | ||
| H-ZSM-5 | MFI | 5.3 × 5.6 | 11.5 | 13.9 | 0.13 | 0.84 ± 0.02 | 6.0 ± 0.17 | ||
| 5.1 × 5.5 | |||||||||
| H-mordenite | MOR | 6.5 × 7 | 10 | 11.1 | 0.14 | 0.84 ± 0.01 | 4.6 ± 0.08 | ||
| 2.6 × 5.7 | |||||||||
| H-ferrierite | FER | 4.2 × 5.4 | 10 | 10.6 | 0.12 | 0.44 ± 0.01 | 2.7 ± 0.04 | ||
| 3.5 × 4.8 | |||||||||
| Na-ZSM-5 | MFI | 5.3 × 5.6 | 11.5 | 13.6 | 0.37 | 0.88 ± 0.02 | 2.2 ± 0.04 | ||
| 5.1 × 5.5 | |||||||||
| Na-Y | FAU | spherical | 9.6 × 9.6 | 7.4 × 7.4 | 5.1 | 4.6 | 0.45 | 0.30 ± 0.01 | 0.3 ± 0.01 |
| Na-SAPO-34 | CHA | ellipsoidal | 9.4 × 9.4 × 12.7 | 3.8 × 3.8 | 0.3 | 0.6 | 0.02 | 0.84 ± 0.03 | 7.9 ± 0.29 |
| Na-SSZ-13 | CHA | ellipsoidal | 9.4 × 9.4 × 12.7 | 3.8 × 3.8 | 15 | 13.8 | 0.50 | 3.12 ± 0.01 | 6.1 ± 0.03 |
| CuOx-MFIg | MFI | 5.3 × 5.6 | ∞ | ∞ | ∞ | 0 | 0 | ||
| 5.1 × 5.5 | |||||||||
| CuOx-BEAg | BEA | 6.6 × 6.7 | ∞ | ∞ | ∞ | 0 | 0 | ||
| 5.6 × 5.6 | |||||||||
| H-ZSM-5g | MFI | 5.3 × 5.6 | 11.5 | 12.9 | 0 | 0 | 0 | ||
| 5.1 × 5.5 |
Catalyst pretreatment: 5 h at 823 K under flowing O2, cooled to 483 K under O2 flow and then purged under He for 0.5 h. Initial CH4 oxidation: 0.5 h under 2400 mL h–1 gcat–1 CH4 at 483 K. Reaction conditions: T = 483 K, WHSV = 2400 mL h–1 gcat–1, PCH4 = 98.1 kPa, PH2O = 3.2 kPa, PO2 = 0.0025 kPa (25 ppm).
Si/Alnom denotes the nominal silicon to aluminum ratio in the zeolite based on commercial figures or ratios of SiO2 to Al2O3 in synthesis procedures.
Si/Altot denotes the ratio of silicon to aluminum atoms ratio within the zeolite calculated using data from inductively coupled plasma mass spectrometry (ICP-MS) measurements.
Cu/Altot denotes the ratio of copper to aluminum atoms within the zeolite calculated using ICP-MS.
Specific activity = μmolCH3OH h–1 gcat–1.
Site time yield (STY) defined as mol CH3OH (mol Cu)−1 h–1.
T = 483 K, WHSV = 2400 mL h–1 gcat–1, PCH4 = 93.1 kPa, PH2O = 3.2 kPa, PO2 = 0.051 kPa. The ± symbol denotes 95% confidence intervals.
The catalytic rates and Eaapp values of CH4 oxidation are heavily influenced by zeolite topology (see Table 1, Table S1). Cu-MOR, featuring 12-membered ring (MR) pores intersected by sinusoidal 8-MR pores, exhibited either comparable or lower activity at 483 K than that of Cu-H-ZSM-5 (Table S1). However, a significantly higher Eaapp of 149 kJ/mol resulted in higher CH3OH rates at temperatures above 483 K when compared to those of Cu-H-ZSM-5 (Figure S16). These results suggest the site speciation and reaction environment within MOR may play a role in stabilizing kinetically relevant transition states. Other topologies, including ferrierite (FER), beta (BEA), Y (FAU), and caged-based SSZ-13 and SAPO-34 (CHA) also oxidize CH4 into CH3OH but with different rates than those of ZSM-5. Zeolites with large pores at high Cu/Al (0.30–0.50) including BEA (12-MR, 6.6 × 6.7 Å and 5.6 × 5.6 Å) and FAU (7.4 × 7.4 Å windows) showed 50% and 70% lower overall activity when compared to MFI. MCM-41, an amorphous aluminosilicate with large pores of 30 Å, had nearly an order of magnitude lower STY than ZSM-5, indicating that a crystalline, microporous structure with small pores is preferable for catalytic CH4 oxidation. At low Cu/Al (0.12–0.14), Cu-H-ZSM-5 has the highest specific activity and STY (Table 1), while FER (intersecting 10-MR [4.2 × 5.4 Å] and 8-MR [3.5 × 4.8 Å]) was half as active as ZSM-5. While Cu-ZSM-5 had the highest specific activity and STY compared to the small-pore zeolites tested, the cage-based aluminoslicate SSZ-13 and the silicoaluminophosphate SAPO-34 with the CHA topology (8-MR windows of 3.8 × 3.8 Å, ellipsoidal cages of 9.4 × 9.4 × 12.7 Å) featured higher STY of 6.1 × 10–3 h–1 and 7.9 × 10–3 h–1, respectively, than those in Cu-Na-ZSM-5 or Cu-H-ZSM-5 at similar Cu/Al ratios (Table 1). These studies indicate the catalytic sites or the kinetically relevant transition states are sensitive to the zeolite topology, with materials featuring small pores or cage-based structures showing enhanced performance when compared to those with large pores.
Taking advantage of the large Eaapp for Cu-Na-SSZ-13 (Cu/Al = 0.50, 100 ± 2.1 kJ mol–1), the reaction temperature was systematically increased to achieve higher catalytic rates. The site-time yield increased from 2.2 × 10–3 to 31.6 × 10–3 molCH3OH (mol Cu)−1 h–1 when increasing the temperature from 463 to 533 K before a decrease in rates was observed (Figure S17). The large Eaapp coupled with the stable CH3OH production over a wide range of temperature suggest that Cu-SSZ-13 zeolites could be further engineered to enhance further catalytic rates. In summary, copper-exchanged zeolites offer a broad and robust platform for the low temperature, catalytic oxidation of CH4 into CH3OH using O2.
The nature of the catalytic active sites for CH4 oxidation into CH3OH over copper-exchanged zeolites is currently unknown. A recent report by von Bokhoven et al. observed the formation of copper oxide clusters in Cu-MOR after the cyclic, isothermal, and stoichiometric oxidation of CH4 into CH3OH.22 It was hypothesized that these oxide clusters were active for isothermal cyclic CH4 oxidation. We did not observe steady state activity for large copper oxide nanoparticles supported on pure silica MFI between 483 and 543 K (average size 30 nm for CuOx, Table 1, Figures S18–20) or on pure silica BEA (average size 60 nm for Cu2O, 41 nm for CuO, Table 1, Figures S21–22). However, we cannot rule out that ultrasmall copper oxide clusters, or copper species stabilized by aluminum in the zeolite framework or in defect sites are responsible for catalytic activity. Diffuse reflectance UV–visible spectroscopic measurements show peaks forming at 20 800, 26 800, and 30 000 cm–1 during steady state CH3OH production (Figure S15C.1–2, D.1–2), possibly corresponding to copper oxide species29 generated during reaction. Future work will focus on identifying and characterizing the catalytic site(s) as well as devising strategies to maximize catalytic activity.
Acknowledgments
This work was supported by the Massachusetts Institute of Technology Energy Initiative’s Seed Fund (K.N. and Y.R.L.) and the Japanese Government via Postdoctoral Fellowships for Research Abroad from the Japan Society for the Promotion of Science (K.I.). 1H NMR spectra were collected at the Massachusetts Institute of Technology Department of Chemistry Instrumentation Facility (DCIF) supported by the National Science Foundation (Award Nos. CHE-9808061 and DBI-9729592).
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acscentsci.6b00139.
Experimental details, product identification for catalytic CH4 oxidation over Cu-Na-ZSM-5, simulation of high conversion CH4 oxidation over Cu-Na-ZSM-5, control experiments for isotopically labeled kinetic experiments, kinetic order of reactants over Cu-Na-ZSM-5, thermal and gas pretreatments on Cu-Na-ZSM-5, in-situ UV–visible spectra of Cu-Na-ZSM-5, kinetic data of CH4 oxidation over Cu-ZSM-5 and Cu-MOR, CH4 oxidation vs temperature over Cu-Na-SSZ-13, powder X-ray diffraction patterns and CH4 oxidation vs temperature over CuOx on silica MFI and BEA, powder X-ray diffraction patterns of ZSM-5 and MOR, elemental composition, pore volume, and surface area of ZSM-5, MOR, and SSZ-13 (PDF)
The authors declare the following competing financial interest(s): The authors and MIT have filed a patent on the results presented herein.
Supplementary Material
References
- Periana R. A.; Taube D. J.; Gamble S.; Taube H.; Satoh T.; Fujii H. Platinum catalysts for the high-yield oxidation of methane to a methanol derivative. Science 1998, 280, 560–564. 10.1126/science.280.5363.560. [DOI] [PubMed] [Google Scholar]
- Palkovits R.; Antonietti M.; Kuhn P.; Thomas A.; Schüth F. Solid Catalysts for the Selective Low-Temperature Oxidation of Methane to Methanol. Angew. Chem., Int. Ed. 2009, 48, 6909–6912. 10.1002/anie.200902009. [DOI] [PubMed] [Google Scholar]
- Smith K. T.; Berritt S.; González-Moreiras M.; Ahn S.; Smith M. R.; Baik M.-H.; Mindiola D. J. Catalytic borylation of methane. Science 2016, 351, 1424–1427. 10.1126/science.aad9730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caballero A.; Despagnet-Ayoub E.; Mar Díaz-Requejo M. M.; Díaz-Rodríguez A.; González-Núñez M. E.; Mello R.; Muñoz B. K.; Ojo W.-S.; Asensio G.; Etienne M.; Perez P. J. Silver-Catalyzed CC Bond Formation Between Methane and Ethyl Diazoacetate in Supercritical CO2. Science 2011, 332, 835–838. 10.1126/science.1204131. [DOI] [PubMed] [Google Scholar]
- Cavaliere V. N.; Mindiola D. J. Methane: a new frontier in organometallic chemistry. Chem. Sci. 2012, 3, 3356–3365. 10.1039/c2sc20530k. [DOI] [Google Scholar]
- Lieberman R. L.; Rosenzweig A. C. Crystal structure of a membrane-bound metalloenzyme that catalyses the biological oxidation of methane. Nature 2005, 434, 177–182. 10.1038/nature03311. [DOI] [PubMed] [Google Scholar]
- Balasubramanian R.; Smith S. M.; Rawat S.; Yatsunyk L. A.; Stemmler T. L.; Rosenzweig A. C. Oxidation of methane by a biological dicopper centre. Nature 2010, 465, 115–119. 10.1038/nature08992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shu L.; Nesheim J. C.; Kauffmann K.; Münck E.; Lipscomb J. D.; Que L. An Fe2IVO2 diamond core structure for the key intermediate Q of methane monooxygenase. Science 1997, 275, 515–518. 10.1126/science.275.5299.515. [DOI] [PubMed] [Google Scholar]
- Panov G.; Sobolev V.; Dubkov K.; Parmon V.; Ovanesyan N.; Shilov A.; Shteinman A. Iron complexes in zeolites as a new model of methane monooxygenase. React. Kinet. Catal. Lett. 1997, 61, 251–258. 10.1007/BF02478380. [DOI] [Google Scholar]
- Ovanesyan N.; Shteinman A.; Dubkov K.; Sobolev V.; Panov G. The state of iron in the Fe-ZSM-5-N2O system for selective oxidation of methane to methanol from data of Mössbauer spectroscopy. Kinet. Catal. 1998, 39, 792–797. [Google Scholar]
- Groothaert M. H.; Smeets P. J.; Sels B. F.; Jacobs P. A.; Schoonheydt R. A. Selective oxidation of methane by the bis (μ-oxo) dicopper core stabilized on ZSM-5 and mordenite zeolites. J. Am. Chem. Soc. 2005, 127, 1394–1395. 10.1021/ja047158u. [DOI] [PubMed] [Google Scholar]
- Narsimhan K.; Michaelis V. K.; Mathies G.; Gunther W. R.; Griffin R. G.; Román-Leshkov Y. Methane to acetic acid over Cu-exchanged zeolites: mechanistic insights from a site-specific carbonylation reaction. J. Am. Chem. Soc. 2015, 137, 1825–1832. 10.1021/ja5106927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woertink J. S.; Smeets P. J.; Groothaert M. H.; Vance M. A.; Sels B. F.; Schoonheydt R. A.; Solomon E. I. A [Cu2O] 2+ core in Cu-ZSM-5, the active site in the oxidation of methane to methanol. Proc. Natl. Acad. Sci. U. S. A. 2009, 106, 18908–18913. 10.1073/pnas.0910461106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smeets P. J.; Groothaert M. H.; Schoonheydt R. A. Cu based zeolites: A UV–vis study of the active site in the selective methane oxidation at low temperatures. Catal. Today 2005, 110, 303–309. 10.1016/j.cattod.2005.09.028. [DOI] [Google Scholar]
- Alayon E. M.; Nachtegaal M.; Ranocchiari M.; van Bokhoven J. A. Catalytic conversion of methane to methanol over Cu–mordenite. Chem. Commun. 2012, 48, 404–406. 10.1039/C1CC15840F. [DOI] [PubMed] [Google Scholar]
- Hammond C.; Forde M. M.; Ab Rahim M. H.; Thetford A.; He Q.; Jenkins R. L.; Dimitratos N.; Lopez-Sanchez J. A.; Dummer N. F.; Murphy D. M.; Carley A. F.; Taylor S. H.; Willock D. J.; Stangland E. E.; Kang J.; Hagen H.; Kiely C. J.; Hutchings G. J. Direct Catalytic Conversion of Methane to Methanol in an Aqueous Medium by using Copper-Promoted Fe-ZSM-5. Angew. Chem., Int. Ed. 2012, 51, 5129–5133. 10.1002/anie.201108706. [DOI] [PubMed] [Google Scholar]
- Vanelderen P.; Snyder B. E.; Tsai M.-L.; Hadt R. G.; Vancauwenbergh J.; Coussens O.; Schoonheydt R. A.; Sels B. F.; Solomon E. I. Spectroscopic definition of the copper active sites in Mordenite: selective methane oxidation. J. Am. Chem. Soc. 2015, 137, 6383–6392. 10.1021/jacs.5b02817. [DOI] [PubMed] [Google Scholar]
- Grundner S.; Markovits M. A.; Li G.; Tromp M.; Pidko E. A.; Hensen E. J.; Jentys A.; Sanchez-Sanchez M.; Lercher J. A. Single-site trinuclear copper oxygen clusters in mordenite for selective conversion of methane to methanol. Nat. Commun. 2015, 6, 7546. 10.1038/ncomms8546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grundner S.; Luo W.; Sanchez-Sanchez M.; Lercher J. A. Synthesis of single-site copper catalysts for methane partial oxidation. Chem. Commun. 2016, 52, 2553–2556. 10.1039/C5CC08371K. [DOI] [PubMed] [Google Scholar]
- Wulfers M. J.; Teketel S.; Ipek B.; Lobo R. Conversion of methane to methanol on copper-containing small-pore zeolites and zeotypes. Chem. Commun. 2015, 51, 4447–4450. 10.1039/C4CC09645B. [DOI] [PubMed] [Google Scholar]
- Alayon E. M. C.; Nachtegaal M.; Bodi A.; van Bokhoven J. A. Reaction Conditions of Methane-to-Methanol Conversion Affect the Structure of Active Copper Sites. ACS Catal. 2014, 4, 16–22. 10.1021/cs400713c. [DOI] [Google Scholar]
- Tomkins P.; Mansouri A.; Bozbag S. E.; Krumeich F.; Park M. B.; Alayon E. M. C.; Ranocchiari M.; van Bokhoven J. A. Isothermal Cyclic Conversion of Methane into Methanol over Copper-Exchanged Zeolite at Low Temperature. Angew. Chem. 2016, 128, 5557–5561. 10.1002/ange.201511065. [DOI] [PubMed] [Google Scholar]
- Palomino G. T.; Fisicaro P.; Bordiga S.; Zecchina A.; Giamello E.; Lamberti C. Oxidation states of copper ions in ZSM-5 zeolites. A multitechnique investigation. J. Phys. Chem. B 2000, 104, 4064–4073. 10.1021/jp993893u. [DOI] [PubMed] [Google Scholar]
- Gao F.; Kwak J. H.; Szanyi J.; Peden C. H. Current understanding of cu-exchanged Chabazite molecular sieves for use as commercial diesel engine DeNOx catalysts. Top. Catal. 2013, 56, 1441–1459. 10.1007/s11244-013-0145-8. [DOI] [Google Scholar]
- Gao F.; Walter E. D.; Kollar M.; Wang Y.; Szanyi J.; Peden C. H. F. Understanding ammonia selective catalytic reduction kinetics over Cu/SSZ-13 from motion of the Cu ions. J. Catal. 2014, 319, 1–14. 10.1016/j.jcat.2014.08.010. [DOI] [Google Scholar]
- Chen J.; Li J.; Wei Y.; Yuan C.; Li B.; Xu S.; Zhou Y.; Wang J.; Zhang M.; Liu Z. Spatial confinement effects of cage-type SAPO molecular sieves on product distribution and coke formation in methanol-to-olefin reaction. Catal. Commun. 2014, 46, 36–40. 10.1016/j.catcom.2013.11.016. [DOI] [Google Scholar]
- Baerlocher C.; McCusker L. B.. Database of Zeolite Structures, http://www.iza-structure.org/databases/.
- Sajith P. K.; Shiota Y.; Yoshizawa K. Role of Acidic Proton in the Decomposition of NO over Dimeric Cu(I) Active Sites in Cu-ZSM-5 Catalyst: A QM/MM Study. ACS Catal. 2014, 4, 2075–2085. 10.1021/cs500223z. [DOI] [Google Scholar]
- Vanelderen P.; Hadt R. G.; Smeets P. J.; Solomon E. I.; Schoonheydt R. A.; Sels B. F. Cu-ZSM-5: A biomimetic inorganic model for methane oxidation. J. Catal. 2011, 284, 157–164. 10.1016/j.jcat.2011.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.