

Abstract
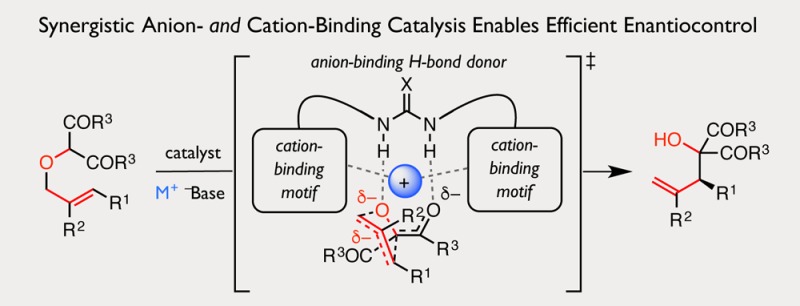
Sigmatropic rearrangements number among the most powerful complexity-building transformations in organic synthesis but have remained largely insensitive to enantioselective catalysis due to the diffuse nature of their transition structures. Here, we describe a synergistic ion-binding strategy for asymmetric catalysis of anionic sigmatropic rearrangements. This approach is demonstrated with the enantioselective [2,3]-Wittig rearrangement of α-allyloxy carbonyl compounds to afford highly enantioenriched homoallylic alcohol products. Chiral thiourea catalysts are shown to engage reactive anions and their countercations through a cooperative set of attractive, noncovalent interactions. Catalyst structure–reactivity–selectivity relationship studies and computational analyses provide insight into catalyst–substrate interactions responsible for enantioinduction and allude to the potential generality of this catalytic strategy.
Short abstract
Cooperative binding of an organic anion and its alkali metal counterion by a chiral, polyfunctional thiourea catalyst underlies stereocontrol in a classic sigmatropic rearrangement.
Introduction
As Doering and Roth wryly noted, sigmatropic rearrangements epitomize “no-mechanism” reactions.1 While these transformations have become staples of organic synthesis and have inspired landmark advances in diastereocontrol,2−6 the highly diffuse nature of their transition structures and the absence of discrete intermediates have rendered sigmatropic rearrangements frustratingly insensitive to catalysis, especially enantioselective variants thereof.7−9 In a step toward overcoming these limitations, we have previously demonstrated that chiral, polyfunctional hydrogen-bond donor catalysts accelerate enantioselective sigmatropic rearrangements of neutral substrates, such as the Claisen rearrangement10−12 and Cope-type hydroamination,13 by cooperatively engaging the charge-separated components of the dipolar transition structures (Figure 1A). In parallel, our group and others have demonstrated that similar chiral, small-molecule hydrogen-bond donors can bind the anions of tight ion-pair intermediates, thereby disposing the cations toward enantioselective nucleophilic trapping (Figure 1B).14−18 An analogous cation-binding catalysis strategy developed around chiral crown ethers has been employed for a limited number of transformations (Figure 1B).18−21
Figure 1.
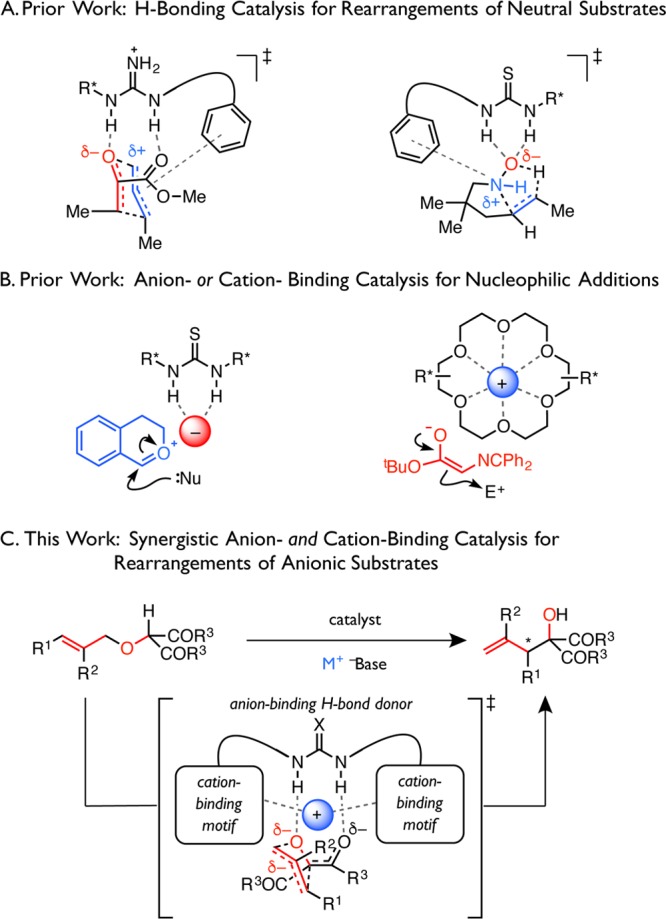
(A–C) Conceptual development of a synergistic ion-binding strategy for enantioselective catalysis of the [2,3]-Wittig rearrangement.
Because sigmatropic rearrangements of anionic species proceed through transition structures involving far greater charge delocalization than their corresponding substrates and products, we hypothesized that precise control of the relative positions of the reactive anions and their countercations could enable selective transition-state stabilization. We thus became interested in determining if the principles of anion- and cation-binding catalysis could be integrated to enable such sigmatropic rearrangements, which have been traditionally recalcitrant toward catalytic enantiocontrol.7,8,22 We envisioned that a polyfunctional hydrogen-bond donor catalyst could engage a prochiral, anionic substrate, while complementary cation-binding elements could orient the countercation precisely to stabilize one diastereomeric transition structure preferentially, thereby inducing stereoselective rearrangement (Figure 1C).
Herein, we report the realization of this strategy with a highly enantioselective thiourea and Brønsted base cocatalyzed [2,3]-Wittig rearrangement of α-allyloxy carbonyl compounds. Application of the [2,3]-Wittig rearrangement to the synthesis of hindered homoallylic alcohol fragments in a variety of natural products and pharmaceutical agents stands as a testament to its utility.4,5,23−27 Furthermore, this intramolecular O–C allyl transfer is representative of the broader class of anionic sigmatropic rearrangements.22 While significant advances have been made toward enantioselective catalysis of rearrangements with related ylides,5,6,28−32 development of a general, catalytic, enantioselective [2,3]-Wittig rearrangement has lagged.7,8 Accordingly, the transformation has been the subject of intense interest, and during the preparation of this manuscript, two methods for the enantioselective [2,3]-Wittig rearrangement of allyloxyoxindoles were disclosed.33,34 The work described here is complementary in scope to these methods, and we posit that the synergistic ion-binding approach it demonstrates can be extended to enable other anionic sigmatropic rearrangements and related transformations, which have hitherto resisted asymmetric catalysis.
Results and Discussion
Development of Reaction Methodology
While the Wittig rearrangement has been most widely explored using strong Brønsted bases,23−27 we sought to develop a system that would trigger rearrangement under mild conditions and would thereby be compatible with a wide variety of functional groups, including dual hydrogen-bond donor catalysts. Accordingly, we deemed that an appropriate substrate should be sufficiently acidic to undergo selective deprotonation in the presence of ureas and thioureas (pKa ≈ 9–20)35 and that the alkoxide produced in the rearrangement step should be sufficiently basic to enable regeneration of a catalytic amount of Brønsted base. With these factors in mind, the Wittig rearrangement of di-tert-butyl-2-cinnamyloxy malonate (1a) was selected as a model reaction, and representative chiral hydrogen-bond donor catalyst classes were evaluated for their ability to induce enantioselective rearrangement in cooperation with a variety of organic and inorganic Brønsted bases (see Supporting Information for details). Through this study, the combination of dialkylthiourea 3 and cesium di-tert-butyl malonate (CsCH(CO2tBu)2) generated in situ emerged as a particularly promising dual catalyst system for effective enantiocontrol (entry 2).
Initial optimization efforts (see Supporting Information for details) revealed that enantioselectivity is moderately sensitive to substituents on the 2-arylpyrrolidine (Ar1) and arylpyrrole (Ar2) components of the thiourea catalyst. However, in all cases, the 2-arylpyrrolidino amides exist as slowly interconverting mixtures of (E)- and (Z)-rotamers, which can be readily detected and quantified by NMR spectroscopic analysis. Given the likelihood that the two rotameric forms of the thiourea catalyst exert differing degrees of enantiocontrol, potentially even in opposite directions, we sought to limit the opportunity for reaction through multiple competing pathways. Accordingly, the 2-arylpyrrolidine was alkylated to constrain the amide in exclusively the (Z)-rotameric form, thereby positioning the aryl group proximal to the H-bond donor active site (see Supporting Information for NMR and crystallographic characterization).36 Catalysts bearing this constrained amide (4, 5, and 6) proved reactive and displayed improved enantioselectivity (entries 3–7). Complementary tuning of the arylpyrrole moiety revealed that catalysts 5b and 6b enable access to product 2a in high enantiomeric excess (entries 5 and 7), even with reduced catalyst loading (entry 8). While catalysts 5b and 6b may be used interchangeably, 5b is preferred for its greater synthetic accessibility.
The importance of each of the components of the thiourea was evinced through a series of structure–reactivity–enantioselectivity studies (Table 1). Truncation of the arylpyrrole moiety, as in cyclohyexyl thiourea 7, or replacement of the 2-arylpyrrolidino amide with an unsubstituted N-pyrrolidino amide, as in catalyst 8, impairs stereoinduction significantly (entries 9 and 10). Furthermore, C2-symmetric thiourea 9 affords low and reversed enantioselectivity relative to 8 (entry 11). These observations indicate that the arylpyrrole, the 2-arylpyrrolidine substituent, and the amide group all play crucial roles in enantiodifferentiation. The importance of the amide functional group has been noted in prior work with related catalysts and ascribed to the Lewis basic amide oxygen participating in stabilizing secondary interactions with reacting intermediates.37,38 While all of the dialkyl thioureas (3–9) examined afford good product yield, replacement of either the N-pyrrolidino amide or the arylpyrrole components with the privileged 3,5-bis(trifluoromethyl)phenyl motif 39 degrades both the reactivity and enantioselectivity of the system (entries 12 and 13). These results suggest that the acidity of the hydrogen-bond donor core must be carefully controlled and that weakly acidic dialkyl thioureas are well-suited to provide substrate activation while avoiding irreversible deprotonation under the basic reaction conditions.40
Table 1. Method Developmenta.
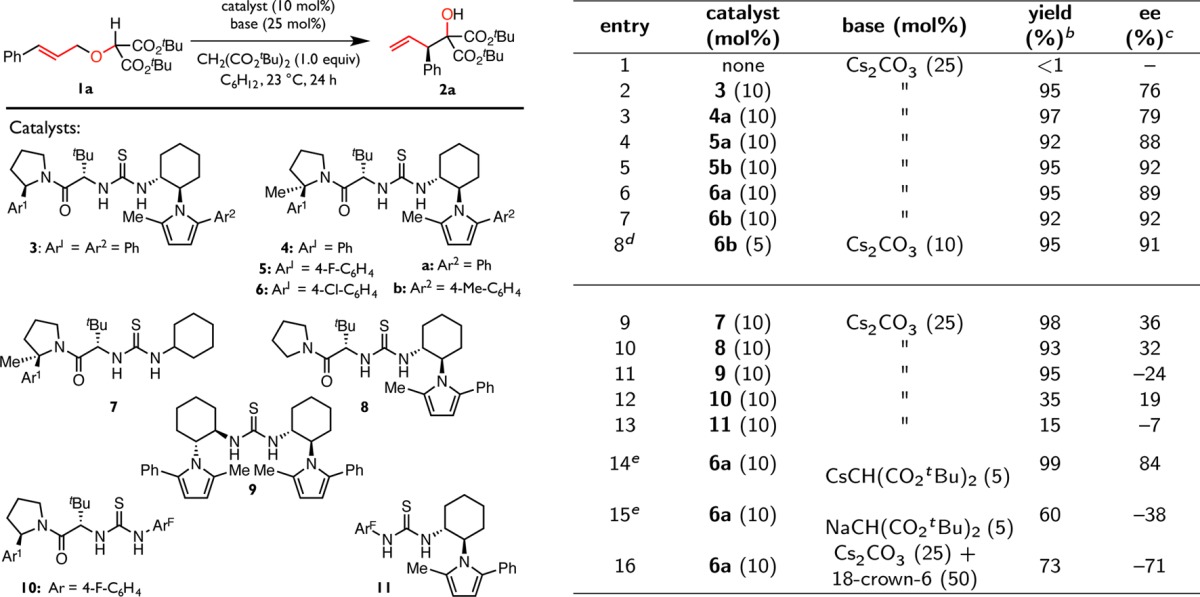
Reactions were performed in duplicate on a 0.1 mmol scale and were quenched with the addition of 1.0 M aqueous hydrochloric acid. Values reported are the averages of two trials. ArF = 3,5-bis(trifluoromethyl)phenyl.
Yields were determined by 1H NMR integration relative to a mesitylene internal standard.
Enantiomeric excesses were determined by CSP-HPLC analysis. Positive ee indicates an excess of the (R)-enantiomer, while negative ee indicates an excess of the (S)-enantiomer.
36 h.
4 h.
With optimized hydrogen-bond donor catalysts in hand, we carried out a systematic evaluation of inorganic Brønsted base cocatalysts. Substituting sodium or potassium carbonate for cesium carbonate leads to a complete suppression of reactivity, which may be ascribed to the superior solubility and increased degree of charge separation in Cs2CO3 and reactive CsX ion pair intermediates.41,42 Omission of di-tert-butyl malonate leads to highly variable yields and enantioselectivities. While the reaction exhibits little to no conversion for the first several hours under the conditions described in Table 1, premixing di-tert-butyl malonate and cesium carbonate overnight to form CsCH(CO2tBu)2 prior to the addition of substrate eliminates this induction period almost completely. Use of preformed CsCH(CO2tBu)2 accelerates the reaction dramatically but affords slightly reduced levels of enantioinduction due to competitive background reaction at elevated concentrations of soluble base (entry 14). Use of preformed NaCH(CO2tBu)2 reverses the observed sense of enantioinduction (entry 15). While reactivity differences between CsCH(CO2tBu)2 and NaCH(CO2tBu)2 are not unexpected, the reversal in the sense of enantioinduction is surprising and indicates that the size or charge density of the alkali metal cation plays a critical role in enantiodetermination. To probe the effect of cation sequestration, 18-crown-6, which is known to bind alkali metal cations including cesium effectively,43−47 was added to the reaction mixture. This change resulted in a dramatic reversal in the observed sense of enantioinduction (entry 16), thereby providing further definitive evidence that the cesium cation is intimately associated with the catalyst in the enantioselectivity-determining transition structure under the optimized reaction conditions.
A series of α-allyloxy carbonyl substrates was evaluated to establish the scope of the transformation and to provide additional insight into the reaction mechanism (Table 2). The identity of the ester substituent (R3) has a small effect on the reaction outcome (e.g., 2a–2c), with di-tert-butyl malonates generally affording the highest enantioselectivities. Prior computational studies have demonstrated that the allyl group accumulates significant partial negative charge in a typical rearrangement transition structure.48−50 Consistent with such charge buildup, substrates with aryl and electron-withdrawing substituents (R1) capable of stabilizing an allylic anion undergo facile rearrangement, while alkyl-substituted substrates (such as 1s) undergo reaction more sluggishly. Notably, substrates bearing trisubstituted alkenes (R2 ≠ H, as for 1d–f) or ortho-substituted arenes (1m,n) undergo rearrangement with modestly reduced rates, nonetheless affording products in good to excellent enantiomeric excess under the optimized conditions. Heteroaromatic substrates (1p–r) are also largely compatible with the optimized conditions, affording rearrangement products in good enantiomeric excess. The limitations of the methodology are illustrated with substrates 1u–w. The terminally disubstituted 1u preferentially undergoes [1,2]-rearrangement at 23 °C and only affords the [2,3]-rearrangement product with low conversion and stereoselectivity at reduced temperatures. Substrates possessing substitution at the allylic position (1v) or (Z)-alkenes (1w) also exhibit very poor reactivity (Table 2B). These observations indicate that the steric environment around each of the allylic termini is defined precisely in the enantioselectivity-determining step, consistent with the notion that the olefin substituent (R1) must adopt a pseudoequatorial orientation to avoid A1,3 strain in an envelope-like rearrangement transition structure (Figure 1C).5,23
Table 2. Substrate Scopea.
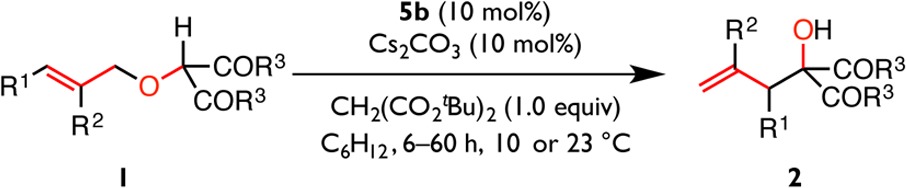
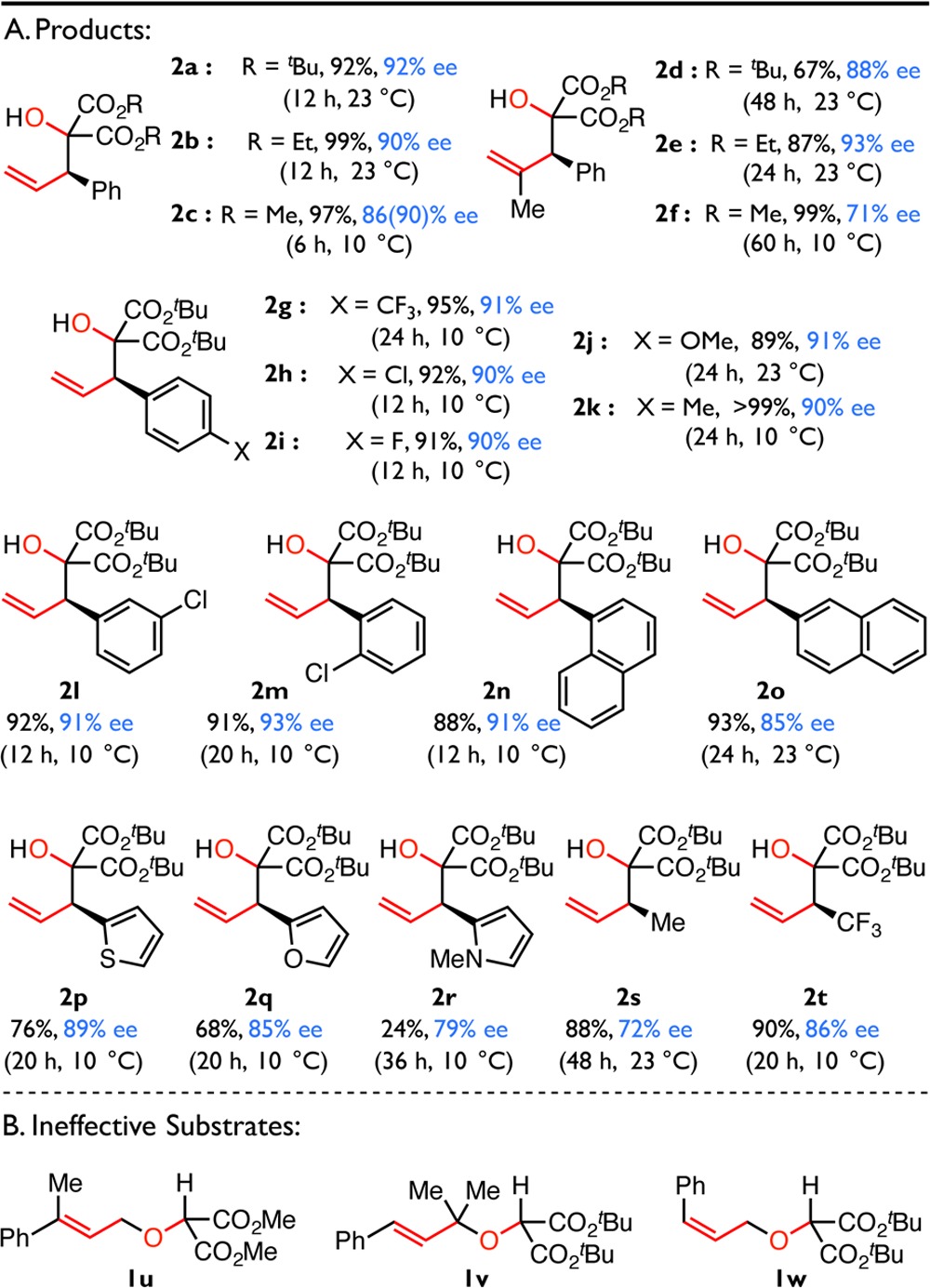
Reactions were performed in duplicate on a 0.1 mmol scale. Cs2CO3, di-tert-butyl malonate, and 5b were premixed for 12 h at 23 °C before the substrate was added at the indicated temperature. Reactions were quenched with the addition of 1.0 M aqueous hydrochloric acid. Values reported are the average of two trials. Enantiomeric excesses were determined by CSP-HPLC analysis, and values in parentheses were measured after a single recrystallization. The absolute configuration of 2c was determined as (R) by single crystal X-ray diffraction; all other absolute stereochemistry was assigned by analogy.
Identification of the Enantioselectivity-Determining Step
Given the potential for mechanistic insights to inform a general approach to asymmetric catalysis of anionic sigmatropic rearrangements, we undertook a series of experimental and computational studies to elucidate the basis for enantioselectivity in the thiourea-catalyzed [2,3]-Wittig rearrangement. Reaction progress kinetic analysis with substrate 1c revealed that the rate of the reaction is independent of catalyst concentration under synthetically relevant conditions.51 This observation suggests that mass transport or proton transfer is rate-determining and indicates that enantioinduction occurs after the rate-determining event. Accordingly, we were required to apply probes other than simple kinetic analysis to determine the identity of the enantioselectivity-determining step.
Stereoselective [2,3]-Wittig rearrangement reactions employing stoichiometric chiral ligands have been shown to proceed via rate- and enantioselectivity-determining deprotonation followed by stereospecific rearrangement.52−54 In the catalytic system described here, deprotonation, substrate-binding, or rearrangement could, in principle, be enantiodetermining. To distinguish between these possibilities, isotope effects were measured for substrates deuterated at the α (1a-d1,α) and allylic (1a-d2,allylic) positions (Scheme 1A,B). A significant inverse isotope effect (kH/kD = 0.53 at 50% conversion) was determined by absolute rate measurements. The fact that an inverse effect is observed is irreconcilable with a primary kinetic isotope effect (KIE) resulting from irreversible deprotonation, but it is indicative of an equilibrium isotope effect (EIE).55 Consistent with this assertion, hydrogen–deuterium scrambling at the α-positions of 1a and CsCH(CO2tBu)2 is observed by NMR spectroscopy at intermediate conversion. This indicates that only mechanisms involving reversible deprotonation of 1a-d1,α are plausible, thereby ruling out the possibility of enantiodetermining deprotonation. Furthermore, in an intermolecular competition experiment, substrate 1a-d2,allylic exhibits a normal secondary kinetic isotope effect (kH/kD = 1.19). Isotopic substitution at this site is unlikely to impact deprotonation or substrate binding, and KIEs of similar magnitude have been observed for analogously deuterated allyl vinyl ethers and 1,5-dienes undergoing Claisen and Cope rearrangements, respectively.56,57 Accordingly, this KIE is diagnostic of enantioselectivity-determining rearrangement.
Scheme 1. Evidence for Enantioselectivity-Determining Rearrangement by a Concerted Mechanism.
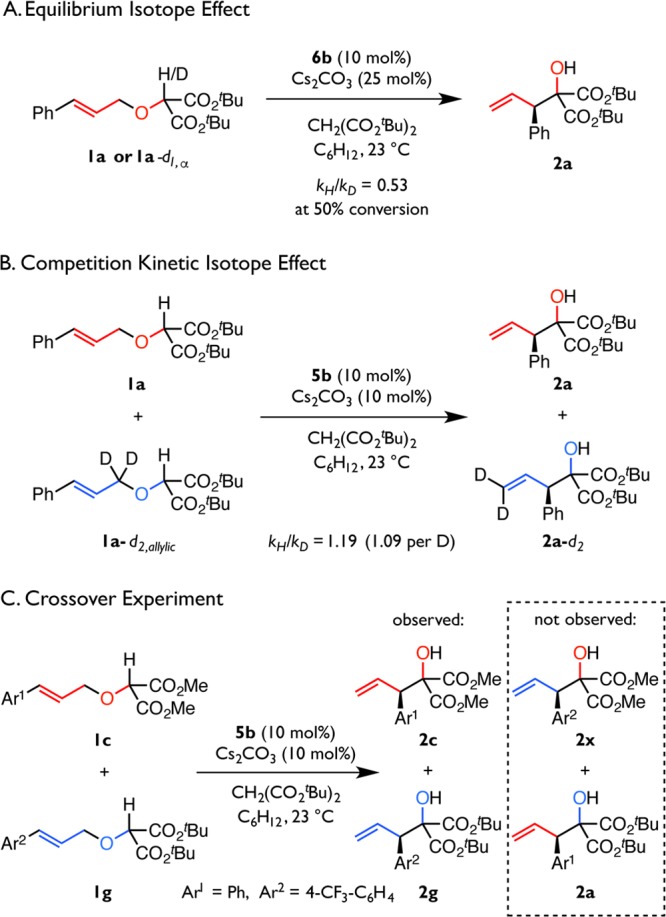
While [1,2]-Wittig rearrangements have been shown to proceed through radical cleavage/recombination, the [2,3]-Wittig rearrangement is generally understood to involve a concerted, pericyclic mechanism.5,25,48−50,58−60 The absence of competitive formation of [1,2]-rearrangement side products under the conditions described above is, therefore, suggestive that only the concerted mechanism is operative. Furthermore, a crossover experiment between substrates 1c and 1g affords 2c and 2g cleanly, while crossover products 2a and 2x are not detected (Scheme 1C). These results are consistent with concerted rearrangement; if homolytic or heterolytic cleavage does occur, recombination must be markedly faster than separation of the reactive partners. The combined weight of the extensive literature precedent, isotope effects, and absence of [1,2]-rearrangement or crossover side-products informed our decision to examine only the concerted reaction manifold in our subsequent analyses (vide infra).
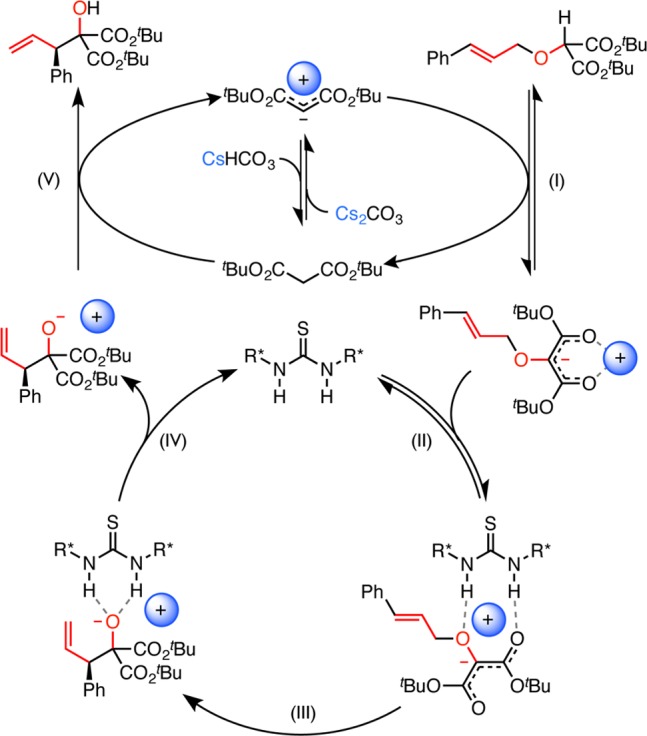
On the basis of these observations and the qualitative impact of substrate structural modification, we propose the following mechanism for the thiourea and Brønsted base cocatalyzed [2,3]-Wittig rearrangement of α-allyloxy malonates (Scheme 2). Substrate 1a undergoes reversible deprotonation (Step I) by CsCH(CO2tBu)2 to form an unreactive, cesium enolate tight ion pair. This equilibrium accounts for both the rate-dependence on CsCH(CO2tBu)2 and the inverse primary EIE. The absence of background rearrangement is consistent with literature reports that the [2,3]-Wittig rearrangement generally does not proceed in nonpolar solvents, even after quantitative deprotonation, unless a Lewis basic additive is employed to separate the tight ion pair.52,61−63 Catalyst 5b binds the ion pair (Step II), orienting the enolate and its cesium countercation in a reactive and charge-separated conformation. The enolate then undergoes irreversible, enantiodetermining rearrangement (Step III), accounting for the normal secondary KIE. Release of the product alkoxide (Step IV) and subsequent protonation by di-tert-butyl malonate (Step V) or another molecule of substrate (not shown) regenerates the active forms of both catalysts.
Scheme 2. Proposed Catalytic Cycle.
Elucidation of the Basis for Enantioinduction
The evidence described above unambiguously demonstrates that thiourea 5b engages the cesium enolate ion pair and the enantiodetermining rearrangement transition structure through specific attractive interactions. In particular, the strong dependence of enantioselectivity on the identities of the catalyst arenes indicates that these motifs play a direct role in the mechanism of stereoinduction. We next sought to determine whether their net effect on enantioselectivity is enthalpic or entropic in nature. An analysis of the temperature dependence of enantioselectivity revealed that the differential free energy of activation obtained with catalyst 4a is dominated by the enthalpic term (ΔΔH‡ = −1.45 kcal mol–1) with a small entropic compensation (−TΔΔS‡ = 0.43 kcal mol–1 at 295 K). Furthermore, changes to the arene substitution pattern (i.e., catalyst 5b vs 4a) substantially increase the differential enthalpy of activation (ΔΔH‡ = −3.52 kcal mol–1) with a smaller, countervailing impact on the differential entropy of activation (−TΔΔS‡ = 1.77 kcal mol–1 at 295 K). Given the remote position of these substituents relative to the thiourea active site, it seems unlikely that their impact on enantioinduction would be due to predominantly steric effects. In contrast, cation−π interactions involving the arylpyrrolidine groups of related hydrogen-bond donor catalysts have previously been implicated in anion-binding catalysis.64 The importance of the cesium countercation and the enthalpically driven influence of the catalyst arenes on enantioinduction thus suggest that such cation−π interactions may also be relevant here.
To examine these and other key interactions responsible enantioinduction, we performed a computational analysis of the stereodetermining rearrangement step using density functional theory (M06-2x/6-31G(d)/SDD(Cs)/PCM(cyclohexane)).65−68 Given the experimental evidence for enthalpy-controlled enantiodifferentiation, these calculations on the electronic energy surface are well founded.69,70 Consistent with the observations outlined above, all low-energy computed transition structures flank Cs+ with the π-faces of the pyrrole and arylpyrrolidine, positioning the cation for a complementary interaction with the Lewis basic amide oxygen (Figure 2).71 This arrangement is analogous to the “aromatic box” motif exhibited by many enzymes and protein receptors responsible for ammonium or metal cation-binding.72−75 While we initially speculated that the Lewis basicity of the sulfur might play an important role in enantioinduction, all attempts to locate energy-minimized ground-state or transition structures exhibiting a S···Cs+ interaction were unsuccessful. These energy minimizations instead returned the cesium cation to its aromatic box, and any further perturbation of the Cs-binding interactions resulted in much higher-energy structures. The lack of a specific role for the thiourea sulfur is consistent with the observation that the urea analogue of 4 is also a competent catalyst (see Supporting Information for details).
Figure 2.

Computed transition structures. (A) Energy-minimized lowest-energy transition structure for the [2,3]-Wittig rearrangement of 1a (leading to (R)-2a) in the active site of catalyst 5a, calculated at the M06-2x/6-31G(d)/SDD(Cs)/PCM(cyclohexane) level of density functional theory. (B) Analogous view of the transition structure leading to the minor enantiomer ((S)-2a). This transition structure places the allyl fragment exo to Cs+ and is disfavored relative to the structure in A by 1.4 kcal mol–1 to 2.1 kcal mol–1 using free energies or zero-point corrected electronic energies, respectively. Select bond distances are shown in angstroms. Carbon-bound hydrogen atoms are omitted for clarity. Black rods represent the π-faces of the arylpyrrole and arylpyrrolidine moieties forming an “aromatic box” around Cs+.
Taken together, these results illustrate that the cesium cation serves a central role in the structural organization of the intermediate ground state and transition structures, precisely orienting the substrate toward the hydrogen-bond donor portion of the catalyst. On the basis of the computational model, enantiodifferentiation thus may be ascribed to two key features distinguishing the major and minor transition structures. First, the N–H···Ocarbonyl hydrogen bond is 0.2 Å longer in the major transition structure leading to (R)-2a than in the minor transition structure leading to (S)-2a, while the N–H···Oether hydrogen bond is 0.1 Å shorter in the major transition structure than in the minor transition structure. Because negative charge must be redistributed from Ocarbonyl to Oether over the course of the rearrangement, these differences in hydrogen bond distances are consistent with greater stabilization of the transition state leading to the major enantiomer of product. Second, the migrating allyl group is positioned endo to Cs+ in the major transition structure leading to (R)-2a but exo to Cs+ en route to (S)-2a. Such an endo-preference is precedented for diastereoselective [2,3]-Wittig rearrangements. Consistent with the observations here, the preference is generally attributed to an attractive, electrostatic interaction between the negative charge accumulating on the migrating allyl fragment and the cationic character of the carbonyl carbon or its associated Lewis acid.48−50 Altogether, these subtle distinctions illustrate the advantages that a synergistic ion-binding strategy offers for achieving enantiocontrol.
In conclusion, the experimental and computational analyses presented for the enantioselective [2,3]-Wittig rearrangement provide strong evidence for a mechanistic model in which the chiral thiourea catalyst engages both the reactive anion and its countercation through a cooperative set of attractive, noncovalent interactions. Specifically, we advance that the polyfunctional catalyst stabilizes the enantiodetermining rearrangement transition state through the synergistic action of anion-binding and cation-binding motifs. These include the thiourea, which forms hydrogen bonds to the nascent alkoxide, along with the catalyst arenes and amide, which encapsulate the cesium cation through cooperative cation−π and Lewis base interactions. We envision that the features responsible for reactivity and selectivity may be extended to cooperative activation of other anionic, prochiral intermediates with polyfunctional hydrogen-bond donor catalysts. Accordingly, this activation mode has the potential to enable enantioselective approaches to important organic transformations, such as the oxy-Cope rearrangement or Ireland–Claisen rearrangement, that have hitherto resisted asymmetric catalysis. Our ongoing attention is directed toward the realization of these aims.
Methods
General Procedure for the Asymmetric [2,3]-Wittig Rearrangement Catalyzed by 5b
In an N2-atmosphere glovebox, a 10 mL round-bottom flask was charged with cesium carbonate (3.1 mg, 0.010 mmol, 10 mol %), catalyst 5b (6.0 mg, 0.010 mmol, 10 mol %), and a PTFE-coated magnetic stir bar. Di-tert-butyl malonate (22 μL, 0.10 mmol, 1.0 equiv) and anhydrous cyclohexane (2.0 mL) were then added. The flask was sealed, removed from the glovebox, and stirred (750 rpm) for 12 h at 23 °C over which time the reaction mixture became uniformly turbid. After 12 h, the reaction flask was cooled to 10 °C in a cryogenic cooling bath, and a stock solution of the desired substrate in anhydrous cyclohexane (2.0 mL, 50 mM, 0.10 mmol, 1.0 equiv) was injected directly into the precooled reaction mixture. The septum was quickly sealed with electrical tape and plastic paraffin film, and the reaction was maintained at 10 °C in the cooling bath with rapid stirring. After the indicated time, the reaction was quenched with the addition of 1 M aqueous hydrochloric acid (1.0 mL), diluted with ethyl ether and allowed to warm to ambient temperature. The layers were partitioned, and the aqueous layer was extracted with additional ethyl ether. The organic extracts were filtered through sodium sulfate and concentrated. The yield was determined from integration of the 1H NMR spectrum of the crude reaction mixture relative to an internal standard added after workup. The homoallylic alcohol product, 2, was purified by silica gel chromatography (on a pipet column), eluting with an appropriate ethyl ether/dichloromethane/pentane solvent mixture, and the enantiomeric excess was determined by chiral stationary phase–high performance liquid chromatographic (CSP-HPLC) analysis or chiral stationary phase–gas chromatographic (CSP-GC) analysis. The absolute configuration of 2c was determined by X-ray crystallographic analysis, and the configurations of all other products were assigned by analogy.
Acknowledgments
This work was supported by the NIH (GM43214) and through an NSF graduate research fellowship to C.R.K. (DGE1144152). The authors thank Dr. Dan Lehnherr for thoughtful conversation and synthetic assistance. The authors thank Dr. Shao-Liang Zheng for X-ray crystallographic assistance.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acscentsci.6b00125.
The authors declare no competing financial interest.
Supplementary Material
References
- von E. Doering W. E.; Roth W. The overlap of two allyl radicals or a four-centered transition state in the Cope rearrangement. Tetrahedron 1962, 18, 67–74. 10.1016/0040-4020(62)80025-8. [DOI] [Google Scholar]
- Nógrádi M.Stereoselective Synthesis: A Practical Approach, 2nd ed.; Wiley: Weinheim, 1995; pp 257–292. [Google Scholar]
- Marco-Contelles J.; Soriano E.. Rearrangements in natural product synthesis. In Modern Tools for the Synthesis of Complex Bioactive Molecules; Cossy J., Arseniyadis S., Eds.; Wiley: Hoboken, NJ, 2012; pp 243–269. [Google Scholar]
- Hiersemann M.; Jaschinski T.. Selected diastereoselective reactions: Diastereoface-differentiating Claisen Cope, and [2,3]-Wittig rearrangements in contemporary natural product synthesis. In Comprehensive Chirality; Carreira E. M., Yamamoto H., Eds.; Elsevier: Amsterdam, 2012; Vol. 2; pp 625–647. [Google Scholar]
- Seashore-Ludlow B.; Somfai P.. Sigmatropic rearrangements in stereoselective synthesis. In Stereoselective Synthesis of Drugs and Natural Products; Andrushko V., Andrushko N., Eds.; Wiley: Hoboken, NJ, 2013; Vol. 1; pp 475–499. [Google Scholar]
- Jones A. C.; May J. A.; Sarpong R.; Stoltz B. M. Toward a symphony of reactivity: Cascades involving catalysis and sigmatropic rearrangements. Angew. Chem., Int. Ed. 2014, 53, 2556–2591. 10.1002/anie.201302572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moyano A.; El-Hamdouni N.; Atlamsani A. Asymmetric organocatalytic rearrangement reactions. Chem. - Eur. J. 2010, 16, 5260–5273. 10.1002/chem.200903410. [DOI] [PubMed] [Google Scholar]
- West T. H.; Spoehrle S. S. M.; Kasten K.; Taylor J. E.; Smith A. D. Catalytic stereoselective [2,3]-rearrangement reactions. ACS Catal. 2015, 5, 7446–7479. 10.1021/acscatal.5b02070. [DOI] [Google Scholar]
- Tantillo D. J. Speeding Up Sigmatropic Shifts–To Halve or to Hold. Acc. Chem. Res. 2016, 49, 741–749. 10.1021/acs.accounts.6b00029. [DOI] [PubMed] [Google Scholar]
- Uyeda C.; Jacobsen E. N. Enantioselective Claisen rearrangements with a hydrogen-bond donor catalyst. J. Am. Chem. Soc. 2008, 130, 9228–9229. 10.1021/ja803370x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uyeda C.; Rötheli A. R.; Jacobsen E. N. Catalytic enantioselective Claisen rearrangements of O-allyl β-ketoesters. Angew. Chem., Int. Ed. 2010, 49, 9753–9756. 10.1002/anie.201005183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uyeda C.; Jacobsen E. N. Transition-state charge stabilization through multiple non-covalent interactions in the guanidinium-catalyzed enantioselective Claisen rearrangement. J. Am. Chem. Soc. 2011, 133, 5062–5075. 10.1021/ja110842s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown A. R.; Uyeda C.; Brotherton C. A.; Jacobsen E. N. Enantioselective thiourea-catalyzed intramolecular Cope-type hydroamination. J. Am. Chem. Soc. 2013, 135, 6747–6749. 10.1021/ja402893z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Z.; Schreiner P. R. (Thio)urea organocatalysis–What can be learnt from anion recognition?. Chem. Soc. Rev. 2009, 38, 1187–1198. 10.1039/b801793j. [DOI] [PubMed] [Google Scholar]
- Knowles R. R.; Jacobsen E. N. Attractive noncovalent interactions in asymmetric catalysis: Links between enzymes and small molecule catalysts. Proc. Natl. Acad. Sci. U. S. A. 2010, 107, 20678–20685. 10.1073/pnas.1006402107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brière J.-F.; Oudeyer S.; Dalla V.; Levacher V. Recent advances in cooperative ion pairing in asymmetric organocatalysis. Chem. Soc. Rev. 2012, 41, 1696–1707. 10.1039/C1CS15200A. [DOI] [PubMed] [Google Scholar]
- Phipps R. J.; Hamilton G. L.; Toste F. D. The progression of chiral anions from concepts to applications in asymmetric catalysis. Nat. Chem. 2012, 4, 603–614. 10.1038/nchem.1405. [DOI] [PubMed] [Google Scholar]
- Brak K.; Jacobsen E. N. Asymmetric ion-pairing catalysis. Angew. Chem., Int. Ed. 2013, 52, 534–561. 10.1002/anie.201205449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ooi T.; Maruoka K. Recent advances in asymmetric phase-transfer catalysis. Angew. Chem., Int. Ed. 2007, 46, 4222–4266. 10.1002/anie.200601737. [DOI] [PubMed] [Google Scholar]
- North M.Crown ethers, TADDOL, NOBIN and Metal(salen) complexes as chiral phase-transfer catalysts for asymmetric synthesis. In Asymmetric Phase Transfer Catalysis; Maruoka K., Ed.; Wiley: Weinheim, 2008; pp 161–187. [Google Scholar]
- Shirakawa S.; Maruoka K. Recent developments in asymmetric phase-transfer reactions. Angew. Chem., Int. Ed. 2013, 52, 4312–4348. 10.1002/anie.201206835. [DOI] [PubMed] [Google Scholar]
- Wilson S. R. Anion-assisted sigmatropic rearrangements. Org. React. 1993, 43, 93–250. 10.1002/0471264180.or043.02. [DOI] [Google Scholar]
- Nakai T.; Mikami K. [2,3]-Wittig sigmatropic rearrangements in organic synthesis. Chem. Rev. 1986, 86, 885–902. 10.1021/cr00075a011. [DOI] [Google Scholar]
- Marshall J. A.The Wittig rearrangement. In Comprehensive Organic Synthesis; Fleming I., Trost B. M., Eds.; Pergamon: Oxford, 1991; Vol. 3: Carbon-Carbon σ-Bond Formation; pp 975–1014. [Google Scholar]
- Nakai T.; Mikami K. The [2,3]-Wittig rearrangement. Org. React. 1994, 46, 105–209. 10.1002/0471264180.or046.02. [DOI] [Google Scholar]
- Nakai T.; Tomooka K. Asymmetric [2,3]-Wittig rearrangement as a general tool for asymmetric synthesis. Pure Appl. Chem. 1997, 69, 595–600. 10.1351/pac199769030595. [DOI] [Google Scholar]
- Isobe M.; Ploysuk C.. The [2,3]-Wittig Rearrangement. In Molecular Rearrangements in Organic Synthesis; Rojas C. M., Ed.; Wiley: Hoboken, NJ, 2015; pp 539–568. [Google Scholar]
- Hodgson D. M.; Pierard F. Y. T. M.; Stupple P. A. Catalytic enantioselective rearrangements and cycloadditions involving ylides from diazo compounds. Chem. Soc. Rev. 2001, 30, 50–61. 10.1039/b000708k. [DOI] [Google Scholar]
- Sweeney J. B. Sigmatropic rearrangements of ”onium” ylids. Chem. Soc. Rev. 2009, 38, 1027–1038. 10.1039/b604828p. [DOI] [PubMed] [Google Scholar]
- Bao H.; Qi X.; Tambar U. K. Stereoselective [2,3]-rearrangements of amine N-oxides. Synlett 2011, 2011, 1789–1792. 10.1055/s-0030-1260962. [DOI] [PubMed] [Google Scholar]
- Bao H.; Tambar U. K.. [2,3]-Rearrangements of ammonium zwitterions. In Molecular Rearrangements in Organic Synthesis; Rojas C. M., Ed.; Wiley: Hoboken, NJ, 2015; pp 459–496. [Google Scholar]
- Murphy G. K.; West F. G.. Oxonium ylide rearrangements in synthesis. In Molecular Rearrangements in Organic Synthesis; Rojas C. M., Ed.; Wiley: Hoboken, NJ, 2015; pp 497–538. [Google Scholar]
- Denmark and Cullen report modest enantioselectivies in the [2,3]-Wittig rearrangement of 2-allyloxy-1-tetralone and several allyloxyoxindoles. See; Denmark S. E.; Cullen L. R. Development of a phase-transfer-catalyzed [2,3]-Wittig rearrangement. J. Org. Chem. 2015, 80, 11818–11848. 10.1021/acs.joc.5b01759. [DOI] [PubMed] [Google Scholar]
- Oseka M.; Kimm M.; Kaabel S.; Järving I.; Rissanen K.; Kanger T. Asymmetric organocatalytic [2,3]-Wittig rearrangement of oxindoles. Org. Lett. 2016, 18, 1358–1361. 10.1021/acs.orglett.6b00291. [DOI] [PubMed] [Google Scholar]
- Jakab G.; Tancon C.; Zhang Z.; Lippert K. M.; Schreiner P. R. (Thio)urea organocatalyst equilibrium acidities in DMSO. Org. Lett. 2012, 14, 1724–1727. 10.1021/ol300307c. [DOI] [PubMed] [Google Scholar]
- Lehnherr D.; Ford D. D.; Bendelsmith A. J.; Kennedy C. R.; Jacobsen E. N.. Conformational control of chiral amido-thiourea catalysts enables improved activity and enantioselectivity. Org. Lett. 2016, http://dx.doi.org/10.1021/acs.orglett.6b01435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuend S. J.; Jacobsen E. N. Mechanism of amido-thiourea catalyzed enantioselective imine hydrocyanation: Transition state stabilization via multiple non-covalent interactions. J. Am. Chem. Soc. 2009, 131, 15358–15374. 10.1021/ja9058958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin S.; Jacobsen E. N. Thiourea-catalysed ring opening of episulfonium ions with indole derivatives by means of stabilizing non-covalent interactions. Nat. Chem. 2012, 4, 817–824. 10.1038/nchem.1450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lippert K. M.; Hof K.; Gerbig D.; Ley D.; Hausmann H.; Guenther S.; Schreiner P. R. Hydrogen-bonding thiourea organocatalysts: The privileged 3,5-bis(trifluoromethyl)phenyl group. Eur. J. Org. Chem. 2012, 2012, 5919–5927. 10.1002/ejoc.201200739. [DOI] [Google Scholar]
- Gilli P.; Pretto L.; Bertolasi V.; Gilli G. Predicting hydrogen-bond strengths from acid-base molecular properties. The pKa slide rule: Toward the solution of a long-lasting problem. Acc. Chem. Res. 2009, 42, 33–44. 10.1021/ar800001k. [DOI] [PubMed] [Google Scholar]
- Dijkstra G.; Kruizinga W. H.; Kellogg R. M. An assessment of the causes of the ”cesium effect. J. Org. Chem. 1987, 52, 4230–4234. 10.1021/jo00228a015. [DOI] [Google Scholar]
- Shukla R.; Kida T.; Smith B. D. Effect of competing alkali metal cations on neutral host’s anion binding ability. Org. Lett. 2000, 2, 3099–3102. 10.1021/ol0063104. [DOI] [PubMed] [Google Scholar]
- Mei E.; Dye J. L.; Popov A. I. Cesium-133 nuclear magnetic resonance study of the complexation of cesium tetraphenylborate by 18-crown-6 in pyridine solutions. J. Am. Chem. Soc. 1977, 99, 5308–5311. 10.1021/ja00458a014. [DOI] [Google Scholar]
- Bauer W. 133Cs, 1H two-dimensional heteronuclear Overhauser effect spectroscopy (HOESY): Structural analysis of organic caesium compounds by detection of short Cs,H contacts. Magn. Reson. Chem. 1991, 29, 494–499. 10.1002/mrc.1260290514. [DOI] [Google Scholar]
- Streeper R. T.; Khazaeli S. Complex formation of rubidium and caesium cations with crown ethers in acetonitrile: A 133Cs competitive NMR study. Polyhedron 1991, 10, 221–227. 10.1016/S0277-5387(00)81592-2. [DOI] [Google Scholar]
- More M. B.; Ray D.; Armentrout P. B. Intrinsic affinities of alkali cations for 15-crown-5 and 18-crown-6: Bond dissociation energies of gas-phase M+-crown ether complexes. J. Am. Chem. Soc. 1999, 121, 417–423. 10.1021/ja9823159. [DOI] [Google Scholar]
- Rodgers M. T.; Armentrout P. B. Cationic noncovalent interactions: Energetics and periodic trends. Chem. Rev. 2016, 116, 5642–5687. 10.1021/acs.chemrev.5b00688. [DOI] [PubMed] [Google Scholar]
- Wu Y. D.; Houk K.; Marshall J. A. Transition structure for the [2,3]-Wittig rearrangement and analysis of stereoselectivities. J. Org. Chem. 1990, 55, 1421–1423. 10.1021/jo00292a007. [DOI] [Google Scholar]
- Mikami K.; Uchida T.; Hirano T.; Wu Y.-d.; Houk K. N. Different transition structures for [2,3]-Wittig rearrangements of stabilized and unstabilized allyloxy methyl anions: Rationale for the dichotomous sense of stereoselection. Tetrahedron 1994, 50, 5917–5926. 10.1016/S0040-4020(01)90446-6. [DOI] [Google Scholar]
- Haeffner F.; Houk K. N.; Schulze S. M.; Lee J. K. Concerted rearrangement versus heterolytic cleavage in anionic [2,3]- and [3,3]-sigmatropic shifts. A DFT study of relationships among anion stabilities, mechanisms, and rates. J. Org. Chem. 2003, 68, 2310–2316. 10.1021/jo0268761. [DOI] [PubMed] [Google Scholar]
- Blackmond D. G. Reaction progress kinetic analysis: A powerful methodology for mechanistic studies of complex catalytic reactions. Angew. Chem., Int. Ed. 2005, 44, 4302–4320. 10.1002/anie.200462544. [DOI] [PubMed] [Google Scholar]
- Kawasaki T.; Kimachi T. Sparteine-mediated enantioselective [2,3]-Wittig rearrangement of allyl ortho-substituted benzyl ethers and ortho-substituted benzyl prenyl ethers. Tetrahedron 1999, 55, 6847–6862. 10.1016/S0040-4020(99)00338-5. [DOI] [Google Scholar]
- Tomooka K.; Komine N.; Nakai T. External chiral ligand-induced enantioselective versions of the [2,3]-Wittig sigmatropic rearrangement. Chirality 2000, 12, 505–509. . [DOI] [PubMed] [Google Scholar]
- Kitamura M.; Hirokawa Y.; Yoshioka Y.; Maezaki N. Bis(oxazoline)-ligand-mediated asymmetric [2,3]-Wittig rearrangement of benzyl ethers: Reaction mechanism based on the hydrogen/deuterium exchange effect. Tetrahedron 2012, 68, 4280–4285. 10.1016/j.tet.2012.03.059. [DOI] [Google Scholar]
- In general, equilibrium deuterium isotope effects (EIE) reflect the energetic preference for exchangeable deuteria to concentrate at the bond with the largest force constant. In the present case, the observed inverse EIE indicates that under the reaction conditions the pKa of CHD(CO2tBu)2 is higher than that of the substrate. The origin of this effect has not been established but could be attributable to the formation of an aggregated network of CsCH(CO2tBu)2 and CH2(CO2tBu)2 molecules, resulting in preferential partitioning of deuterium within that network.
- Gajewski J. J.; Conrad N. D. Variable transition state structure in 3,3-sigmatropic shifts from α-secondary deuterium isotope effects. J. Am. Chem. Soc. 1979, 101, 6693–6704. 10.1021/ja00516a035. [DOI] [Google Scholar]
- Gajewski J. J.; Conrad N. D. Aliphatic Claisen rearrangement transition state structure from secondary α-deuterium isotope effects. J. Am. Chem. Soc. 1979, 101, 2747–2748. 10.1021/ja00504a052. [DOI] [Google Scholar]
- Schöllkopf U. Recent results in carbanion chemistry. Angew. Chem., Int. Ed. Engl. 1970, 9, 763–773. 10.1002/anie.197007631. [DOI] [Google Scholar]
- Baldwin J. E.; Patrick J. E. Stereochemistry of [2,3]-sigmatropic reactions. Wittig rearrangement. J. Am. Chem. Soc. 1971, 93, 3556–3558. 10.1021/ja00743a060. [DOI] [Google Scholar]
- Hoffmann R. W. Stereochemistry of [2,3] sigmatropic rearrangements. Angew. Chem., Int. Ed. Engl. 1979, 18, 563–572. 10.1002/anie.197905633. [DOI] [Google Scholar]
- Kitamura M.; Hirokawa Y.; Maezaki N. Asymmetric [2,3]-Wittig rearrangement of oxygenated allyl benzyl ethers in the presence of a chiral di-tBu-bis(oxazoline) ligand: A novel synthetic approach to THF lignans. Chem. - Eur. J. 2009, 15, 9911–9917. 10.1002/chem.200901212. [DOI] [PubMed] [Google Scholar]
- Ikemoto H.; Sasaki M.; Takeda K. Solvent effects on the steric course of the [2,3]-Wittig rearrangement of (S,E)-[3-(allyloxy)prop-1-ene-1,3-diyl]dibenzene and derivatives. Eur. J. Org. Chem. 2010, 2010, 6643–6650. 10.1002/ejoc.201001061. [DOI] [Google Scholar]
- Kondoh A.; Terada M. Brønsted base catalyzed [2,3]-Wittig/phospha-Brook tandem rearrangement sequence. Org. Lett. 2013, 15, 4568–4571. 10.1021/ol402144q. [DOI] [PubMed] [Google Scholar]
- Kennedy C. R.; Lin S.; Jacobsen E. N. The cation−π interaction in small-molecule catalysis. Angew. Chem., Int. Ed. 2016, 10.1002/anie.201600547R1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frisch M. J.et al. Gaussian 09, Revision D.01; Gaussian Inc.: Wallingford, CT, 2009; see Supporting Information for full citation. [Google Scholar]
- Zhao Y.; Truhlar D. The M06 suite of density functionals for main group thermochemistry, thermochemical kinetics, noncovalent interactions, excited states, and transition elements: Two new functionals and systematic testing of four M06-class functionals and 12 other functionals. Theor. Chem. Acc. 2008, 120, 215–241. 10.1007/s00214-007-0310-x. [DOI] [Google Scholar]
- von Szentpály L.; Fuentealba P.; Preuss H.; Stoll H. Pseudopotential calculations on Rb2+, Cs2+, RbH+, CsH+ and the mixed alkali dimer ions. Chem. Phys. Lett. 1982, 93, 555–559. 10.1016/0009-2614(82)83728-7. [DOI] [Google Scholar]
- Scalmani G.; Frisch M. J. Continuous surface charge polarizable continuum models of solvation. I. General formalism. J. Chem. Phys. 2010, 132, 114110. 10.1063/1.3359469. [DOI] [PubMed] [Google Scholar]
- Ramadhar T. R.; Batey R. A. Accurate prediction of experimental free energy of activation barriers for the aliphatic-Claisen rearrangement through DFT calculations. Comput. Theor. Chem. 2011, 976, 167–182. 10.1016/j.comptc.2011.08.022. [DOI] [Google Scholar]
- Schenker S.; Schneider C.; Tsogoeva S. B.; Clark T. Assessment of popular DFT and semiempirical molecular orbital techniques for calculating relative transition state energies and kinetic product distributions in enantioselective organocatalytic reactions. J. Chem. Theory Comput. 2011, 7, 3586–3595. 10.1021/ct2002013. [DOI] [PubMed] [Google Scholar]
- In light of this model, the change in the sense of enantioinduction observed with NaCH(CO2tBu)2 vs CsCH(CO2tBu)2 might result directly from the differences in van der Waals radii of the two alkali metal ions. Like Cs+, Na+ can form strong cation−π interactions with the pyrrole and arylpyrrolidine moieties of catalyst 5. However, we hypothesize that these interactions cannot be sustained simultaneously with the smaller Na+. Reactions must therefore proceed through entirely different transition state geometries. For a representative computational study of Na+−π interactions; seeMecozzi S.; West A. P. Jr.; Dougherty D. A. Cation−π interactions in aromatics of biological and medicinal interest: Electrostatic potential surfaces as a useful qualitative guide. Proc. Natl. Acad. Sci. U. S. A. 1996, 93, 10566–10571. 10.1073/pnas.93.20.10566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salonen L. M.; Ellermann M.; Diederich F. Aromatic rings in chemical and biological recognition: Energetics and structures. Angew. Chem., Int. Ed. 2011, 50, 4808–4842. 10.1002/anie.201007560. [DOI] [PubMed] [Google Scholar]
- Cheng J.; Goldstein R.; Gershenson A.; Stec B.; Roberts M. F. The cation−π box is a specific phosphatidylcholine membrane targeting motif. J. Biol. Chem. 2013, 288, 14863–14873. 10.1074/jbc.M113.466532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagy G. N.; Marton L.; Contet A.; Ozohanics O.; Ardelean L.-M.; Révész Á; Vékey K.; Irimie F. D.; Vial H.; Cerdan R.; Vértessy B. G. Composite aromatic boxes for enzymatic transformations of quaternary ammonium substrates. Angew. Chem., Int. Ed. 2014, 53, 13471–13476. 10.1002/anie.201408246. [DOI] [PubMed] [Google Scholar]
- Davis M. R.; Dougherty D. A. Cation−π interactions: Computational analyses of the aromatic box motif and the fluorination strategy for experimental evaluation. Phys. Chem. Chem. Phys. 2015, 17, 29262–29270. 10.1039/C5CP04668H. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.