

Abstract
We describe a general method to synthesize the iminium tetrahydrothiophene embedded in the dimeric Nuphar alkaloids. In contrast to prior studies, the sulfur atom of the thiaspirane pharmacophore is shown to be electrophilic. This α-thioether reacts with thiophenol or glutathione at ambient temperature to cleave the C–S bond and form a disulfide. Rates of conversion are proportional to the corresponding ammonium ion pKa and exhibit half-lives less than 5 h at a 5 mM concentration of thiol. A simple thiophane analogue of the Nuphar dimers causes apoptosis at single-digit micromolar concentration and labels reactive cysteines at similar levels as the unsaturated iminium “warhead”. Our experiments combined with prior observations suggest the sulfur of the Nuphar dimers can react as an electrophile in cellular environments and that sulfur-triggered retrodimerization can occur in the cell.
Short abstract
We describe a new method for the synthesis of the Nuphar iminium thiophane pharmacophore and disclose its previously unidentified sulfur electrophilicity.
Background
The Nuphar dimers (1–4, Figure 1) are a small family of bioactive terpene alkaloids defined by the presence of a nonsymmetric spiro-tetrahydrothiophene (thiaspirane) linker.1,2 Members of the class exhibit weak antimicrobial activity,3−5 but strong immunosuppressive activity.6 Some members also promote apoptosis7−10 and inhibit metastasis,11,12 both in vitro and in a murine model. The most potent member, 6-hydroxythiobinupharidine (1b), exhibits antimetastatic activity with an in vitro IC50 of 29 nM and inhibits mouse lung tumor formation at 90% versus control over 10 days.11 Semipurified mixtures of the dimers downregulate NFκB at low concentrations, although the active constituents in this mixture were not definitively proven and therefore potency values could not be defined.13
Figure 1.

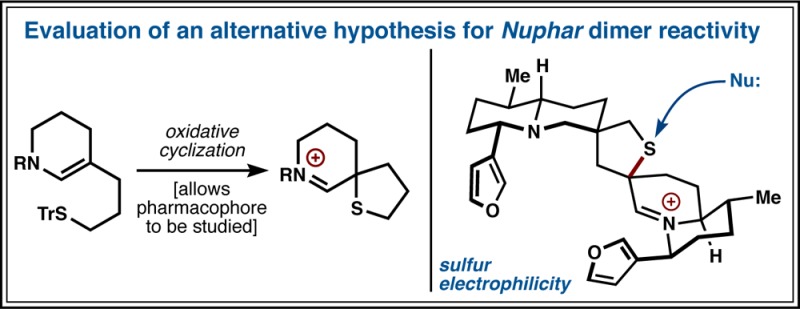
(a) The Nuphar family of thiaspirane diastereomers and congeners (a: X, Y = OH; b: X = H, Y = OH; c: X = OH, Y = H; d: X, Y = H). (b) Activity was proposed to involve an electrophilic carbon. (c) Here we propose an electrophilic sulfur as a basis for bioactivity.
The structural basis for much of this bioactivity is not well explained. Cytotoxicity assays indicate that a hemiaminal adjacent to the tert-alkyl thioether (C6) is necessary for induction of apoptosis, whereas a thioether adjacent to a quaternary center (C6′) displays low potency.7,8 For example, 6-OH-TNL (2b, Figure 1a) is strongly apoptotic below 1 μM, whereas 6′-OH-TNL (2c) is completely inactive at 10 μM.8 Simplified analogues that replace the thioether with a hydroxyl are also reported to lose all activity in fungicidal experiments.14 One explanation put forth for these structural requirements is the enhanced stability of the iminium adjacent to a thioether in 6-hydroxydimers (Figure 1b),15−17 which may allow this otherwise strong carbon electrophile to penetrate the cell and reach a specific target.2 In contrast, the electrophilic promiscuity of the unstabilized iminium/hemiaminal of 6′-hydroxy dimers is thought to prevent access to a specific target.2 Dihydroxy-thiaspirane dimers would then be expected to not show activity or exhibit the low activity of 6′-hydroxydimers since they also give rise to an unstabilized iminium. Yet 6,6′-dihydroxy-1 (1a) and 6-hydroxy-1 (1b) inhibit invasion of collagen-matrix by B-16 melanoma cells11 and promote apoptosis7,8 at nearly identical levels. Therefore, the promiscuity of an unstabilized iminium and the preservation of a stabilized iminium cannot be the basis for the different activity of C6-hydroxy versus C6′-hydroxy dimers. A recent report by MacMillan, Eastman, and Wu demonstrates that all diastereomers and enantiomers of dihydroxydimers 1a, 2a, 3a, 4a have similar apoptotic activity,8 suggesting a common mechanism of action between the stereoisomers, at least for the dihydroxy-dimers.
We hypothesize that a dormant electrophilic sulfur atom embedded in the Nuphar dimers might better account for these unexplained observations. Such reactivity also explains the stereochemical equilibration observed in 6-hydroxy dimers (see Figure 2 below). Here we report an oxidative cyclization to synthesize the thiaspirane-iminium pharmacophore and demonstrate proof-of-principle selective thiol capture at the electrophilic sulfur. Rates of thiol capture can be tuned with the electronics of the imine and correlated to ammonium pKa. We show analogous reactivity under cell-relevant conditions in a synthetic dimer by observing retrodimerization to highly electrophilic monomers. This simple dimeric thiaspirane analogue exhibits sub-micromolar cytotoxicity and causes rapid cell death. The thiol-triggered activation exhibited by this pharmacophore and its Trojan horse-capacity suggest a future role as a component of biological tools or therapeutics.18
Figure 2.
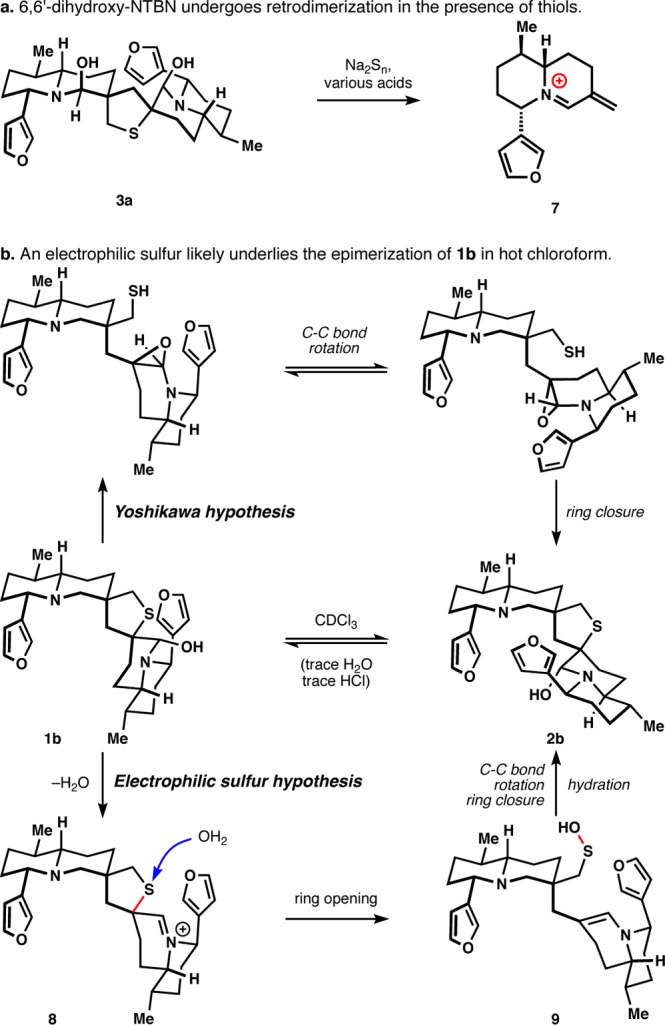
(a) Retrodimerization of 3a could be effected by nucleophilic addition to sulfur. (b) An electrophilic sulfur probably also underlies the epimerization of 1b to 2b.
Initial Observations of S-Electrophilicity
We recently reported the first chemical synthesis of a Nuphar dimer, (−)-neothiobinupharidine (3d), via the intermediacy of its probable biosynthetic precursor 6,6′-dihydroxyneothiobinupharidine (3a).19 This latter compound proved difficult to isolate because the sulfurous reagents used for its synthesis caused retrodimerization to 7 upon acidification, concentration, and addition to silica (Figure 2a). These observations indicated that the final ring closure of the tetrahydrothiophene could be easily reversed via cleavage of the C–S bond, which surprised us since retrodimerization had not been previously reported in the extensive isolation literature.5,20−37 However, this same tert-alkyl center undergoes epimerization upon heating in chloroform, which was rationalized by a stereoretentive epoxide formation and reopening by a pendant thiol (Figure 2, top pathway).20 More likely, the presence of trace hydrochloric acid at high temperature induces iminium ion formation (8), whereby water or chloride anion attacks the thioether to form a sulfenic acid/enamine pair (9) that collapses to 2b after σ-bond rotation (bottom pathway).38 A sulfur nucleophile would not require the same activation energy to cleave the C–S bond as water or chloride,39 but it is unlikely that dihydroxydimers would have been subjected to acids and thiols simultaneously during the isolation process.
This invocation of an electrophilic thioether during retrodimerization or epimerization relies on circumstantial evidence rather than direct observation of an electrophile-nucleophile adduct.40 Therefore, we sought a simple monomeric system that would allow us to fully characterize a thiol adduct and study its formation in more detail.41−43 So, we devised a new method to easily generate a monomeric Nuphar thiaspirane, which would allow us to study its reactivity.
Oxidative Spirocyclization
During our work on the syntheses of 3a–d, we observed that triphenylmethanethiol (TrSH) could mediate formation of acyclic dimers from monomeric iminium ions. Addition of an oxidant completed the tetrahydrothiophene—a stepwise variant of the dimerization we disclosed (i.e., 7 to 3a).19 Using this information, we designed a simple oxidative cyclization method to forge the thiaspirane.
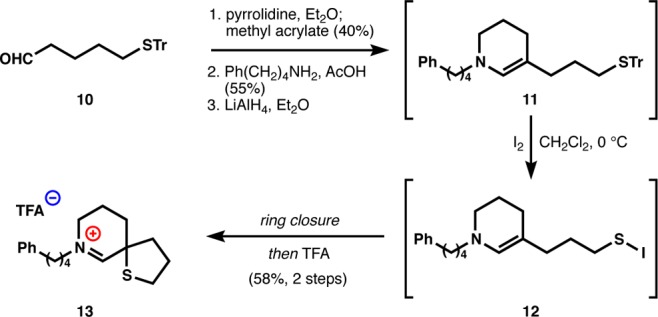
As shown in Scheme 1, tetrahydropyridine 11 is readily available in three steps.44 Michael reaction of 5-(tritylthio)pentanal (10) with methyl acrylate is effected through the intermediacy of a pyrrolidine enamine. Subsequent amidation with 4-phenyl-butylamine occurred smoothly, as did lactam reduction with lithium aluminum hydride. While enamines similar to 11 were isolated in prior work,25 we used crude solutions due to its sensitivity to water and oxygen. After some experimentation, we found that treatment of 11 with I2 in a solution of dichloromethane at 0 °C effected tandem trityl removal45,46 and oxidative cyclization to directly form iminium tetrahydrothiophene 13. Trifluoroacetic acid was added prior to purification to exchange the iodide counteranion, which was sensitive to autoxidation. This simple protocol could yield significant quantities of the isolated pharmacophore and could be adapted to many analogues (see Scheme 3).
Scheme 1. General Strategy for Synthesis of Monomeric Iminium Thiaspiranes.
Scheme 3. Synthesis of a Small Library of Spirocyclic Iminiums.
Yields from enamide (two steps) based on 1H NMR internal standard, unless noted.
Yield after HPLC.
Observation of Disulfide Formation
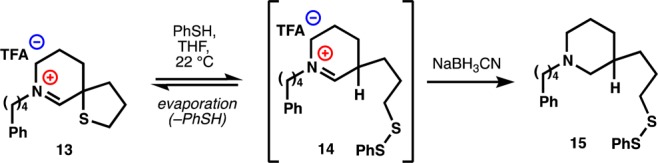
The ability to form the pharmacophore in isolation from other functional groups and not embedded in a dimeric structure allowed us to study its reactivity with nucleophiles. Obviously, the tetrahydrothiophene is stable to alcoholic nucleophiles1,8—it can be formed in methanol and can be characterized by NMR in methanol-d4. No hemiaminal is observed in equilibrium even at −50 °C (see Supporting Information), reflecting the stability of the Nuphar thiaspirane noted previously (see below).15−17 However, addition of thiophenol to a solution of 13 (Scheme 2) resulted in gradual formation of a single thiophenol adduct stable enough to be observed by liquid chromatography–mass spectrometry (LCMS). Complete removal of the volatile reaction components by evaporation over several hours delivered very pure starting material (13), indicating that if formation of disulfide 14 had occurred, it could also be rapidly reversed. Nevertheless, none of this information proved whether bond formation had occurred at the iminium carbon or the thiophane sulfur. Unfortunately, reduction of the reaction mixture with sodium borohydride did not deliver a thiophenol adduct, but a fully reduced piperidine-thiol. Eventually, we discovered that addition of sodium cyanoborohydride to the reaction cleanly delivered disulfide 15, demonstrating for the first time that the Nuphar thiaspirane pharmacophore can function as a sulfur electrophile and that it is selective for thiol nucleophiles over water or alcohols. The disulfide in 15 must be lower in electrophilicity than thiophane 13; otherwise, disulfide exchange would occur in the presence of excess thiophenol to yield diphenyldisulfide. However, only a trace amount of the free thiol derived from 15 was observed in any given experiment.
Scheme 2. Demonstration of S-Electrophilicity of Thiaspiranes.
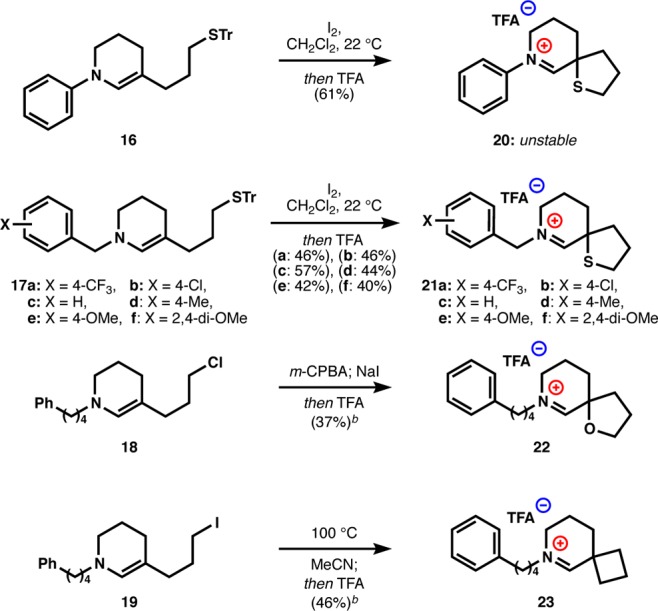
Using the general strategy of Scheme 1, we were then able to assemble a small library of spirocyclic iminium ions (20–23) to study their stability and reactivity. Initially, we explored aniline enamines (e.g., 16), which might have formed the basis of a straightforward Hammett analysis.47 Although substrates of type 16 did undergo oxidative cyclization, the iminium-thiaspiranes 20 proved too unstable to study extensively. Instead we turned to benzyl substituents, since para-substitution on benzylamines modulate their pKa by about 1 unit—an observable but not destabilizing electronic effect.48−50 These benzyl substituted enamines (17a–f) cyclized cleanly and could be purified by high-performance liquid chromatography (HPLC) as stable iminium salts 21a–f. As benchmark iminiums for these and future studies, we were also able to adapt this route to the synthesis of a spirocyclic tetrahydrofuran iminium (18 → 22) and a cyclobutane iminium (19 → 23) as shown in Scheme 3. In the first case, peracid-mediated epoxidation/ring-opening of 18 delivers an alpha-hydroxy iminium, which undergoes SN2 displacement to tetrahydrofuran 22. In the second case, direct 4-exo-tet cyclization of the enamine onto a pendant organoiodide furnishes cyclobutane 23.
The solvolytic stabilities of the iminiums 21–23 were probed by measuring their NMR spectra in methanol-d4 (see Supporting Information). Like N-4-phenylbutyl-iminium 13, N-benzyl-thiophanes 21a–f exist exclusively as the iminium ions on the NMR time scale at temperatures as low as −60 °C even in the case of electron-deficient 4-trifluoromethylbenzyl-iminium 21f. Interestingly, spiro-tetrahydrofuran 22 exists mostly as hemiaminal diastereomers in MeOH-d4, either reflecting decreased donation of the ether n-electrons into the C=N antibonding orbital compared to the thiophane1 or increased inductive withdraw from the oxygen. Supporting this latter idea, spiro-cyclobutane 23 is observed as the iminium ion in methanol, not the hemiaminal (see Supporting Information).
Rates of Thiol Addition to Sulfur
Access to 21a–f allowed us to study the effect of N-substitutent electronics on the electrophilicity at sulfur. In order to reach rates that would allow us to easily study relative reactivity, we increased thiophenol concentration to 5 M. Not surprisingly, the tetrahydrofuran 22 did not react under these conditions, but neither did the strained cyclobutane 23. The large amounts of thiophenol frustrated our attempts to follow the reactions in situ using standard techniques (NMR, HPLC) due to high background signals and peak broadening. Therefore, we turned to transmission IR spectroscopy to effectively screen out the signal from the solvent and thiophenol. An isolated IR signal from the iminium ion was observed at 1778 cm–1, and by following the changes in absorbance at this wavenumber, we could compare initial rates of reaction for each substrate.
We had expected to observe a positive Hammett or negative pKa correlation reflecting increased reactivity of a C–S bond adjacent to an electron-deficient iminium due to loss of positive charge in the transition state.51 In fact, we observed the opposite: more electron-rich rings reacted more quickly. We hypothesized that this increased rate of electron-rich iminiums reflects either a decreased susceptibility to nucleophilic addition at carbon, i.e., a higher concentration of iminium reactant,52 or a more rapid protonation of electron-rich intermediate enamines. To probe these possibilities, we added thiophenol in low concentration to electron-deficient substrate 21a and measured its 1H NMR. At ambient temperature, peak broadening prevented interpretation, but at −40 °C, we observed a decrease of imium peaks and an appearance of a small diagnostic signal at 4.5 ppm, which increased at higher concentrations of thiophenol. On the basis of the NMR data of isolated thiophenol hemiaminals,53,54 this peak appears attributable to the thioaminal methine (25a). This adduct is unstable and cannot be observed by LCMS, unlike a disulfide adduct (for additional spectra, see Supporting Information). Electron-rich substrate 21f, on the other hand, does not display these new peaks at ambient or low temperature even in the presence of 10 equiv of PhSH. Therefore, correlation of initial rates of reaction with pKa of the ammonium/iminium ion appears to reflect varying concentrations of iminium ion in solution (Figure 4). Addition of Brønsted acids did not affect rates, ruling out enamine protonation as the rate-determining step.
Figure 4.
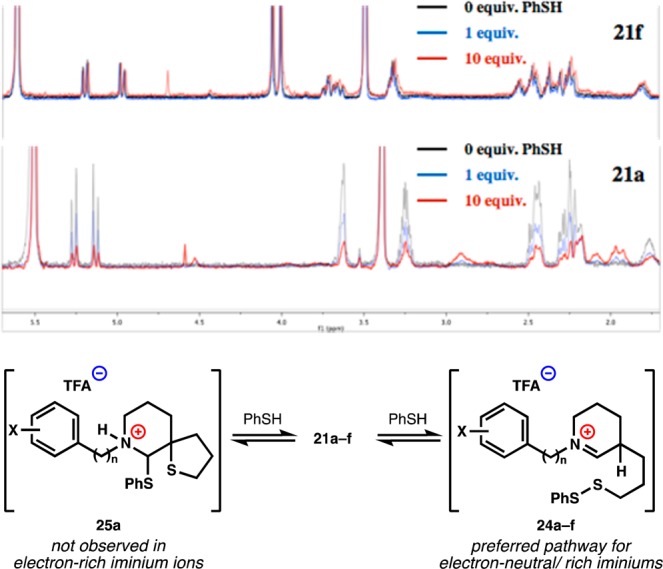
Titration of PhSH to electron-rich or -deficient iminiums. Measured by 1H NMR in methanol-d4 at −40 °C.
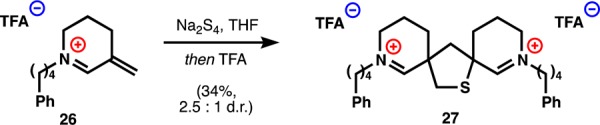
Although these small electronic changes held a profound influence on the reactivity of the iminium, neither thiohemiaminal 25a nor disulfides 24a–f formed at concentrations of nucleophile relevant to a cellular environment (1–5 M). Whereas monomeric thiaspiranes were useful for characterizing nucleophile–electrophile adducts, we hypothesized that because 21a–f can undergo intramolecular resulfenylation of the nascent disulfide,55 these compounds show appreciable reaction with thiols only at high concentrations. So, we generated a simple analogue of the dimers (analogous to 1–4a), in which a retro-Mannich reaction would compete with resulfenylation, and tested its reactivity with thiols. Dimer 27 can be synthesized from 26 in analogy to our biomimetic dimerization of Nuphar monomers and the similar reactions of chalcones42 reported by Reinecke (Scheme 4).
Scheme 4. Dimerization of Simple Iminium 26 to an Unnatural Dimer 27.
In contrast to the monomers, dimer 27 retrodimerizes to 28a with a half-life of less than 5 h at a PhSH concentration of only 5 mM (Figure 5)—a relevant concentration to cellular environments since the integrated, intracellular concentration of glutathione lies between 1 and 10 mM.56 This retrodimerization reaction also occurs with 5 mM aqueous glutathione but more slowly because the dimers are poorly water-soluble. The resulting unsaturated iminium 26 captures thiols at the β-position to form a stable adduct (28a) in solution. The disulfide itself apparently funnels to 28a (as determined by 1H NMR), probably due to the lower pKa of persulfides relative to thiols and the instability of persulfides.57 Release of H2S,58 a common degradation product of persulfides, was identified by odor and confirmed by reaction with AgNO3 to yield Ag2S. However, β-thioether 28 itself acts as a latent Michael acceptor and will exchange with other thiols, like 4-methoxythiophenol, to give mixtures of the two adducts (1H NMR). The identities of adducts 28a–c were confirmed by independent synthesis from iminium ion 26, which rapidly captures thiols at the β-carbon exclusively (not the iminium carbon).
Figure 5.
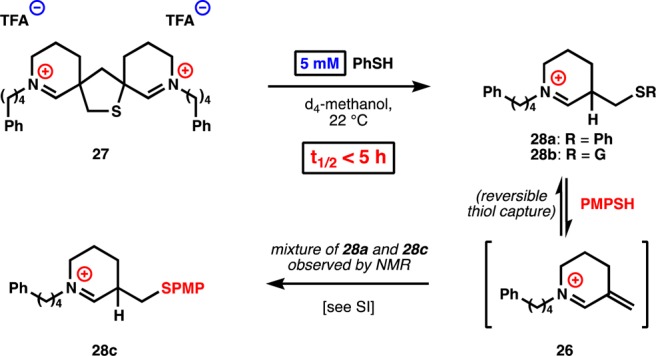
Dimeric thiaspiranes retrodimerize quickly at 5 mM thiol.
Using the tetrasulfide mediated dimerization shown in Scheme 4, we prepared a small series of analogues (29–31) that differed in the electronics of the nitrogen substituent (Figure 6). We measured the rate of retrodimerization at 2.5 mM substrate, a lower concentration than Figure 5 to induce measurable rate differences. Substituent electronics changed the half-lives of retrodimerization of the analogues, which could be roughly correlated to the pKa of the corresponding amine.59,60 This trend matches the same correlation made in the monomeric iminium tetrahydrothiophene series (Figure 3). Whether this reactivity in organic solvent under acidic conditions reflected behavior of these compounds in cellular environments still remained an open question.
Figure 6.
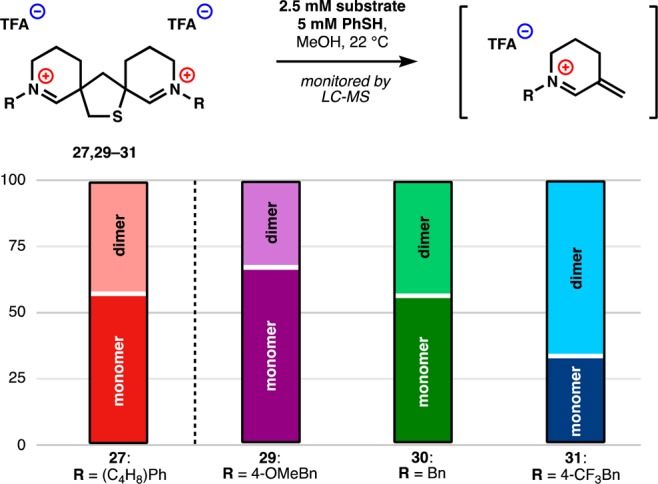
Extent of retrodimerization after 5 h at 2.5 mM dimer and 5 mM thiophenol depends on iminium electronics.
Figure 3.
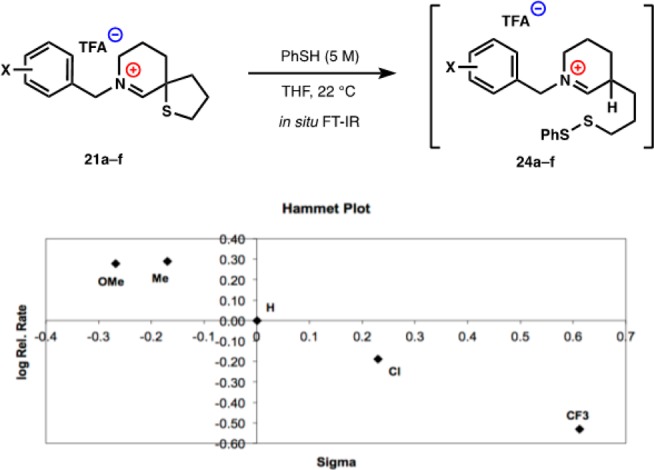
Relative rates of benzyl analogues plotted against sigma values.
Rates of Cell Death
In order to evaluate the cellular relevance of this sulfur electrophilicity and its release of an unsaturated iminium “warhead,” we measured the potency and rate of killing of 3a, 27, and 29–31. Our chosen method for analyzing cell death promotion by Nuphar analogues was the CellTiter-Glo luminescence assay that measures depletion of cellular ATP.61,62 The dynamic range and normalization of samples for this luminescence-based approach supersedes traditional Western blot-based detection of protein cleavage events that occur during cell death. All cell-based luminescence assays are pragmatically normalized to the number of cells per well, whereas Western blotting normalization is dependent on densitometry analysis of antibody detection of general cellular proteins (i.e., GAPDH and actin). The latter technique can be compromised by sample overloading issues where the densitometry values of antibody binding extends beyond the limited linear range of detection and thus impedes the ability to correlate samples. Importantly, only certain general cellular proteins can be employed as loading controls during apoptosis experiments, as many candidate proteins are subject to degradation during cell death, including actin.63 A recent publication on Nuphar analogues employed Western blot-based detection of the apoptosis marker poly(ADP-ribose) polymerase (PARP) and actin as a loading control.8,9 Use of actin as a loading control for this publication’s end point assays would likely not be compromised, as the authors assessed PARP cleavage at early stages of cell death (i.e., 2 and 6 h).
We found that simple dimeric analogues 27 and 29–31 kill Jurkat and HT29 cells rapidly at 5 and 10 μM, especially butylphenyl analogue 27, which exhibits rates of killing close to the metabolite 3a. Correlation to bulk-solvent retrodimerization rates is imperfect, which is not surprising given potential differences in cell permeability between the dimers and the different pH encountered by the dimers in the cell versus bulk solvent. However, the simplicity of these dimers make them valuable tools and potential replacements for the complex and difficult to access metabolites 1–4. In contrast, the monomeric iminiums 22–23 did not approach the rates of killing of the dimers, even at 25 μM. The monomeric thiophane iminium 13 did kill rapidly, but only at this much higher concentration. Therefore, it is not merely the iminium, nor merely the electrophilic sulfur, that is required to effect strong and rapid apoptosis.
Rather, this data taken along with observations by Wu et al. that both enantiomers of 1–4 are all actively apoptotic, make it likely that the observed high potency of dihydroxy-dimers derives from retrodimerization to electrophilic monomers (27).8 However, this correlation is imperfect. Recent work from Wu demonstrates cyctotoxicity and rapid apoptosis in monomeric α-thioether quinolizidine iminium ions.8 And to our surprise, the proposed warhead 26 demonstrated low potency after 48 h incubation (Figure 7). Given its reactivity and polarity, we suspected this low activity of 26 might be due to poor cellular stability or low membrane permeability. Together, the foregoing data do not indicate whether retrodimerization occurs in vivo.
Figure 7.
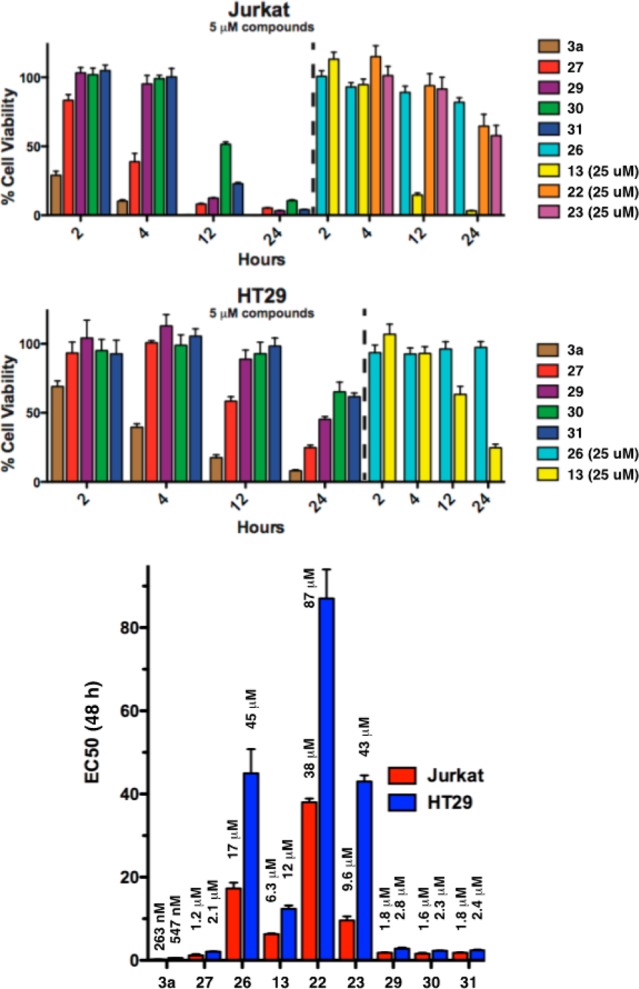
Dimers promote rapid apoptosis and kill at low concentrations, whereas monomeric iminiums do not.
S-Electrophilicity in Cell Lysate
Therefore, in order to determine if retrodimerization to the unsaturated iminium warhead occurs in a native biological environment, we performed isotopic tandem orthogonal proteolysis-activity-based protein profiling (isoTOP-ABPP).64−66 In this method, cells or cell lysate are treated with an electrophile of interest, then with the general electrophilic probe iodoacetamide alkyne (IA-alkyne). Proteinaceous Cys residues that react with the electrophile will not react with IA-alkyne; this difference can be read out stoichometrically, and at single amino acid resolution, using mass spectrometry. A primary advantage of the isoTOP-ABPP technology is that the electrophile of interest does not need to be chemically modified for target identification, which is particularly useful when studying low molecular weight electrophiles.
As shown in Figure 8, monomers 13, 22, and 23 react with proteinaceous Cys residues of Jurkat cell lysate only modestly, whereas dimer 27 and warhead 26 display stronger reactivity. These high extents of cysteine modification are consistent with cytotoxicity—there is clear correlation between the cytotoxicity and cysteine adduction. Although these data stop short of definitively proving a detailed mechanism of Nuphar dimer activity in cells, the similar isoTOP-ABPP profiles of dimer 27 and warhead 26, and their deviation from the monomers, support a shared degree and mechanism of reactivity.
Figure 8.
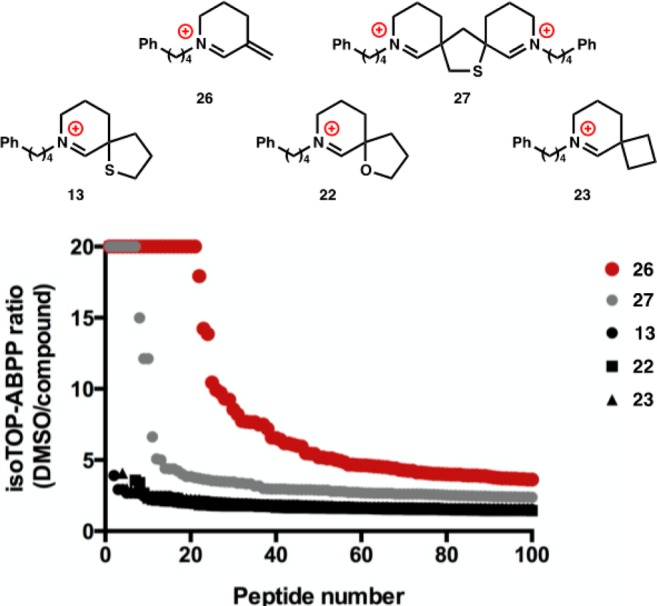
isoTOP-ABPP of Jurkat cell lysate treated with dimer, warhead, or unsaturated monomers. Jurkat cell lysate was treated with dimer (25 μM), warhead (50 μM), or unsaturated monomers (50 μM) for 1 h at ambient temperature. isoTOP-ABPP was performed as previously described.41,43 Shown on x-axis are top-100 changing Cys-peptides for each tested compound, where change is defined as a high isoTOP-ABPP ratio (DMSO/compound).
It is important to note that not all biological activity exhibited by Nuphar dimers can be ascribed to retrodimerizations and production of an iminium warhead such as 7 or 26. Even though both enantiomers of 1b are apoptotic,8 these 6-monohydroxy dimers cannot retrodimerize. But it also seems unlikely that both enantiomers of the topologically complex dimers are subject to lock-and-key or induced fit. Whether an electrophilic carbon or an electrophilic sulfur underlies the mechanism of 1b or the related monomeric species reported by Wu9 is unknown, but this study opens up the latter possibility to experimentation.
Conclusion
We have demonstrated that the sulfur atom of the Nuphar thiaspirane iminium pharmacophore reacts as an electrophile with nucleophilic thiols at ambient temperature. The rate of disulfide formation is proportional to the pKa of the embedded iminium and may reflect susceptibility to rapidly reversible thiohemiaminal formation in electron deficient substrates. In dihydroxy-dimers (metabolites or their analogues), this reactivity causes rapid retrodimerization to electrophilic, unsaturated iminium ions. In monomeric models of the pharmacophore, the initial disulfide adduct can be isolated and characterized. On the basis of this observed reactivity and rapid induction of apoptosis by 3a and synthetic dimer 27, we propose that some of the observed activity of the dihydroxy Nuphar dimers can be ascribed to retrodimerization; i.e., the dihydroxydimers are prodrugs for a highly reactive covalent binder. Clearly, retrodimerization is not an option for monohydroxydimers, so the mechanism of action for these species is still an open and potentially valuable question; capture of nucleophiles at the thioether is one possible answer. We anticipate that these observations of sulfur electrophilicity in the iminium thiaspirane could lead to the design of new therapeutics based on the Nuphar pharmacophore and new tools for selective capture of free thiols in a cellular setting. The ability to tune the structure (steric environment) and electronics of the iminium α-thioether could lead to an arsenal of probes with varying activity to profile or inhibit biologically important thiols. In particular, caged iminium thioethers could represent an important class of probes for further exploration.67
Acknowledgments
We thank Dr. Milan Gembicky, Dr. Curtis Moore, and Professor Arnold L. Rheingold for X-ray crystallographic analysis, and Professors Jacob Gopas and Avi Golan for helpful discussions.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acscentsci.6b00113.
Author Present Address
† National Center for Advancing Translational Sciences, National Institutes of Health, 9800 Medical Center Drive, Rockville, Maryland 20850, USA.
Author Contributions
‡ N.T. and D.J.J. contributed equally.
Financial support for this work was provided by the NIH (GM105766 and CA132630). Additional support was provided by Eli Lilly, Novartis, Bristol-Myers Squibb, Amgen, Boehringer-Ingelheim, the Sloan Foundation, and the Baxter Foundation.
The authors declare the following competing financial interest(s): A provisional patent was filed in collaboration with Ben Gurion University (J. Gopas) on the use of the compounds in this manuscript for treatment of inflammation.
Supplementary Material
References
- For reviews, see:LaLonde R. T.; Wong C. Properties of sulfur containing Nuphar alkaloids. Pure Appl. Chem. 1977, 49, 169–181. 10.1016/B978-0-08-021204-3.50007-4. [DOI] [Google Scholar]
- LaLonde R. T. Intramolecular iminium ion-sulfide charge-transfer association: a recurring theme in the study of thiaspirane alkaloids. Acc. Chem. Res. 1980, 13, 39–44. 10.1021/ar50146a002. [DOI] [Google Scholar]
- Vichkanova S. A.; Adgina V. V.; Fedorchenko T. S.; Rubinchik M. A. Study of antibacterial and antifungal activity of lutenurine in experiments in vitro. Farmakol. Toksikol. 1969, 32, 620–623. [PubMed] [Google Scholar]
- Okamura S.; Nishiyama E.; Yamazaki T.; Otsuka N.; Taniguchi S.; Ogawa W.; Hatano T.; Tsuchiya T.; Kuroda T. Action mechanism of 6, 6′-dihydroxythiobinupharidine from Nuphar japonicum, which showed anti-MRSA and anti-VRE activities. Biochim. Biophys. Acta, Gen. Subj. 2015, 1850, 1245–1252. 10.1016/j.bbagen.2015.02.012. [DOI] [PubMed] [Google Scholar]
- Cullen W. P.; LaLonde R. T.; Wang C. J.; Wong C. F. Isolation and in-vitro antifungal activity of 6,6′-dihydroxythiobinupharidine. J. Pharm. Sci. 1973, 62, 826–827. 10.1002/jps.2600620530. [DOI] [PubMed] [Google Scholar]
- Matsuda H.; Shimoda H.; Yoshikawa M. Dimeric sesquiterpene thioalkaloids with potent immunosuppressive activity from the rhizome of Nuphar pumilum: structural requirements of Nuphar alkaloids for immunosuppressive activity. Bioorg. Med. Chem. 2001, 9, 1031–1035. 10.1016/S0968-0896(00)00327-8. [DOI] [PubMed] [Google Scholar]
- Matsuda H.; Yoshida K.; Miyagawa K.; Nemoto Y.; Asao Y.; Yoshikawa M. Nuphar alkaloids with immediately apoptosis-inducing activity from Nuphar pumilum and their structural requirements for the activity. Bioorg. Med. Chem. Lett. 2006, 16, 1567–1573. 10.1016/j.bmcl.2005.12.032. [DOI] [PubMed] [Google Scholar]
- Korotkov A.; Li H.; Chapman C. W.; Xue H.; MacMillan J. B.; Eastman A.; Wu J. Total syntheses and biological evaluation of both enantiomers of several hydroxylated dimeric Nuphar alkaloids. Angew. Chem., Int. Ed. 2015, 54, 10604–10607. 10.1002/anie.201503934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H.; Korotkov A.; Chapman C. W.; Eastman A.; Wu J. Enantioselective Formal Syntheses of 11 Nuphar Alkaloids and Discovery of Potent Apoptotic Monomeric Analogues. Angew. Chem., Int. Ed. 2016, 55, 3509–3513. 10.1002/anie.201600106. [DOI] [PubMed] [Google Scholar]
- Dul’tsina S. M.; Zhizhina G. P.; Kruglyak S. A.; Vermel E. M.; Emanuel N. M. Kinetics of the destruction of tumor cells under the influence of chemotherapeutic preparations in vitro. Vopr. Onkol. 1966, 12, 60–66. [PubMed] [Google Scholar]
- Matsuda H.; Morikawa T.; Oda M.; Asao Y.; Yoshikawa M. Potent anti-metastatic activity of dimeric sesquiterpene thioalkaloids from the rhizome of Nuphar pumilum. Bioorg. Med. Chem. Lett. 2003, 13, 4445–4449. 10.1016/j.bmcl.2003.09.019. [DOI] [PubMed] [Google Scholar]
- Kawahara Y.; Shimoda H.; Yoshikawa M.. (Morishita Jintan Co. Ltd. Japan) Hair growth stimulants containing sesquiterpene dimer thioalkaloids. JP 2000–233085, August 1, 2000.
- Ozer J.; Eisner N.; Ostrozhenkova E.; Bacher A.; Eisenreich W.; Benharroch D.; Golan-Goldhirsh A.; Gopas J. Nuphar lutea thioalkaloids inhibit the nuclear factor κB pathway, potentiate apoptosis and are synergistic with cisplatin and etoposide. Cancer Biol. Ther. 2009, 8, 1860–1868. 10.4161/cbt.8.19.9567. [DOI] [PubMed] [Google Scholar]
- LaLonde R. T.; Tsai A. I.-M.; Wang C. J.; Wong C.; Lee G. Preparation and in vitro antifungal activities of some quinolizidine derived hemiaminals and β-tert-amino sulfides. J. Med. Chem. 1976, 19, 214–219. 10.1021/jm00224a004. [DOI] [PubMed] [Google Scholar]
- LaLonde R. T.; Wong C. F.; Das K. C. Thiospiran, C30, Nuphar alkaloids. Structure and evidence for intramolecular sulfur-immonium ion interactions. J. Am. Chem. Soc. 1973, 95, 6342–6349. 10.1021/ja00800a030. [DOI] [PubMed] [Google Scholar]
- LaLonde R. T.; Wong C. F.; Das K. C. The stereospecificty of α-thiohemiaminal reduction. A 300 MHz proton magnetic resonance investigation of deuteride reduction products from 6,6′-dihydroxythiobinupharidine and 6-hydroxythiobinupharidine. Can. J. Chem. 1974, 52, 2714–2716. 10.1139/v74-395. [DOI] [Google Scholar]
- LaLonde R. T.; Wong C. F. Hemiaminal derivatives of neothiobinupharidine. J. Org. Chem. 1976, 41, 291–294. 10.1021/jo00864a022. [DOI] [Google Scholar]
- Cf. the neocarzinostatin chromophore:Myers A. G. Proposed structure of the neocarzinostatin chromophore-methyl thioglycolate adduct; a mechanism for the nucleophilic activation of neocarzinostatin. Tetrahedron Lett. 1987, 28, 4493–4496. 10.1016/S0040-4039(00)96545-6. [DOI] [Google Scholar]
- Jansen D. J.; Shenvi R. A. Synthesis of (−)-neothiobinupharidine. J. Am. Chem. Soc. 2013, 135, 1209–1212. 10.1021/ja310778t. [DOI] [PubMed] [Google Scholar]
- Achmatowicz O.; Bellen Z. Alkaloids of Nuphar luteum (L) Sm. Isolation of alkaloids containing sulphur. Tetrahedron Lett. 1962, 3, 1121–1124. 10.1016/S0040-4039(00)70971-3. [DOI] [Google Scholar]
- Achmatowicz O.; Bellen Z. Alkaloids of Nuphar luteum (L) Sm. II. Isolation of alkaloids containing sulfur. Roczniki Chim. 1962, 36, 1815–1825. [Google Scholar]
- Achmatowicz O.; Wrobel J. T. Alkaloids from Nuphar luteum. III. A new alkaloid, neothiobinupharidine. Spectroscopic studies on the structure of thiobinupharidine and neothiobinupharidine. Tetrahedron Lett. 1964, 5, 129–136. 10.1016/S0040-4039(00)90341-1. [DOI] [Google Scholar]
- Achmatowicz O.; Banaszek H.; Spiteller G.; Wrobel J. T. Alkaloids from Nuphar luteum. IV. Mass spectroscopy of thiobinupharidine, neothiobinupharidine, and their desulfuration products. Tetrahedron Lett. 1964, 5, 927–934. 10.1016/S0040-4039(00)90409-X. [DOI] [Google Scholar]
- Birnbaum G. I. Structure of neothiobinupharidine. Tetrahedron Lett. 1965, 6, 4149–4152. 10.1016/S0040-4039(01)99580-2. [DOI] [Google Scholar]
- Il’inskaya T. N.; Kuzovkov A. D.; Monakhova T. G. Alkaloids of Nuphar luteum. Khim. Priro. Soed. 1967, 3, 178–182. [Google Scholar]
- Birnbaum G. Crystal and molecular structure of neothiobinupharidine dihydrobromide tetrahydrate. Acta Crystallogr. 1967, 23, 526–535. 10.1107/S0365110X67003147. [DOI] [Google Scholar]
- Wrobel J. T. Thionupharoline a new alkaloid from yellow water-lily. Roczniki Chim. 1970, 44, 457–458. [Google Scholar]
- Wrobel J. T.; Galuszko K. Furan chemistry. IV. IR spectra of monosubstituted derivatives. Roczniki Chim. 1970, 44, 769–776. [Google Scholar]
- LaLonde R. T.; Wong C. F.; Cullen W. P. Two new Nuphar alkaloids, 6,6′-dihydroxythionuphlutine-A and – B. Tetrahedron Lett. 1970, 11, 4477–4480. 10.1016/S0040-4039(01)83954-X. [DOI] [Google Scholar]
- Wrobel J. T.; Iwanow A.; Szychowski J.; Poplawski J.; Yu C. K.; Martin T. I.; MacLean D. B. Neothiobinupharidine sulfoxide, a new alkaloid of Nuphar luteum. Can. J. Chem. 1972, 50, 1968–1971. 10.1139/v72-314. [DOI] [Google Scholar]
- LaLonde R. T.; Wong C. F. Sulfur containing alkaloids from Nuphar luteum. Phytochemistry 1972, 11, 3305–3306. 10.1016/S0031-9422(00)86394-3. [DOI] [Google Scholar]
- Wong C. F.; LaLonde R. T. 6- and 6′-hydroxyneothiobinupharidine. Monohemiaminals from Nuphar luteum and their possible role in thiaspirane Nuphar alkaloid biogenesis. Experientia 1975, 31, 15–16. 10.1007/BF01924651. [DOI] [PubMed] [Google Scholar]
- LaLonde R. T.; Wong C. Thiobinupharidines epimeric at C-1 and C-1′Can. Can. J. Chem. 1975, 53, 3545–3550. 10.1139/v75-510. [DOI] [Google Scholar]
- Wrobel J. T.; Iwanow A.; Wojtasiewicz K. New hemiaminals from Nuphar luteum. 6,6′-Dihydroxyneothiobinupharidine and monohydroxyhemiaminals. Bull. Acad. Polym. Sci. 1975, 23, 735–737. [Google Scholar]
- Wrobel J.; Iwanow A.; Wojtasiewicz K. New sulfoxides from Nuphar luteum. Thiobinupharidine and thionuphlutine sulfoxides. Bull. Acad. Polym. Sci. 1976, 24, 99–100. [Google Scholar]
- Iwanow A.; Wojtasiewicz K.; Wrobel J. T. Sulfoxides of thiobinupharidine thiohemiaminals from Nuphar lutea. Phytochemistry 1986, 25, 2227–2231. 10.1016/0031-9422(86)80097-8. [DOI] [Google Scholar]
- Yoshikawa M.; Murakami T.; Ishikado A.; Wakao S.; Murakami N.; Yamahara J.; Matsuda H. Crude drugs from aquatic plants. VI. On the alkaloid constituents of Chinese Nupharis Rhizoma, the dried rhizoma of Nuphar pumilum (TIMM.) DC. (Nymphaceae): structures and rearrangement reaction of thiohemiaminal type Nuphar alkaloids. Heterocycles 1997, 45, 1815–1824. 10.3987/COM-97-7878. [DOI] [Google Scholar]
- Lalonde observed sulfenylation of an intermediate sulfenic acid formed by thermolysis of neothiobinupharidine sulfoxide:LaLonde R. T.; Wong C. F. Nuphar thiaspirane sulfoxides. 13C nuclear magnetic resonance determined configuration and utilization in the interconversion of thiaspirane stereochemical types. Can. J. Chem. 1978, 56, 56–61. 10.1139/v78-010. [DOI] [Google Scholar]
- Singh R.; Whitesides G. M. In Sulfur-Containing Functional Groups; Supplement S; Patai S., Ed.; J. Wiley and Sons, Ltd: New York, 1993. [Google Scholar]
- As in the reduction of vinblastine analogs for example:Magnus P.; Ladlow M.; Elliott J. Models for a Hypothetical Mechanism of Action of the Anticancer Agent Vinblastine. J. Am. Chem. Soc. 1987, 109, 7929. 10.1021/ja00259a078. [DOI] [Google Scholar]
- For other strategies to synthesize the nuphar pharmacophore, see ref (41−43), ref (8), and ref (19).LaLonde R. T.; Florence R. A.; Horenstein B. A.; Fritz R. C.; Silveira L.; Clardy J.; Krishnan B. S. Variable reaction pathways for the action of polysulfide on Michael acceptors. J. Org. Chem. 1985, 50, 85–91. 10.1021/jo00201a017. [DOI] [Google Scholar]
- Del Mazza D.; Reinecke M. G. Reactions of α,β-unsaturated ketones with hydrogen sulfide. γ-Keto sulfides or tetrahydrothiopyranols?. J. Org. Chem. 1981, 46, 128–134. 10.1021/jo00314a028. [DOI] [Google Scholar]
- Lu P.; Herrmann A. T.; Zakarian A. Toward the synthesis of Nuphar sesquiterpene thioalkaloids: stereodivergent rhodium-catalyzed synthesis of the thiolane subunit. J. Org. Chem. 2015, 80, 7581–7589. 10.1021/acs.joc.5b01177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Norman M. H.; Heathcock C. H. Improved synthesis of N-benzyl-5-ethyl-1,2,3,4-tetrahydropyridine. J. Org. Chem. 1988, 53, 3370–3371. 10.1021/jo00249a050. [DOI] [Google Scholar]
- Kamber B.; Rittel W. A new, simple method for the synthesis of cysteine peptides. Helv. Chim. Acta 1968, 51, 2061–2064. 10.1002/hlca.19680510826. [DOI] [PubMed] [Google Scholar]
- Moyer M. P.; Feldman P. L.; Rapoport H. Intramolecular N-H, O-H and S-H insertion reactions. Synthesis of heterocycles from α-diazo β-keto esters. J. Org. Chem. 1985, 50, 5223. 10.1021/jo00225a047. [DOI] [Google Scholar]
- Zuend S. J.; Jacobsen E. J. Mechanism of Amido-Thiourea Catalyzed Enantioselective Imine Hydrocyanation: Transition State Stabilization via Multiple Non-Covalent Interactions. J. Am. Chem. Soc. 2009, 131, 15358–15374. 10.1021/ja9058958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bunting J. W.; Stefanidis D. A systematic entropy relationship for the general-base catalysis of the deprotonation of a carbon acid. A quantitative probe of transition-state solvation. J. Am. Chem. Soc. 1990, 112, 779–786. 10.1021/ja00158a043. [DOI] [Google Scholar]
- Hall H. K. Jr. Correlation of the Base Strengths of Amines. J. Am. Chem. Soc. 1957, 79, 5441–5444. 10.1021/ja01577a030. [DOI] [Google Scholar]
- Brown H. C.et al. In Determination of Organic Structures by Physical Methods; Braude E. A., Nachod F. C., Ed.; Academic Press: New York, 1955. [Google Scholar]
- Anslyn E. V.; Dougherty D. A.. Modern Physical Chemistry; University Science Books: Sausalito, 2006; p 445. [Google Scholar]
- Eldin S.; Jencks W. P. Lifetimes of Iminium Ions in Aqueous Solution. J. Am. Chem. Soc. 1995, 117, 4851–4857. 10.1021/ja00122a015. [DOI] [Google Scholar]
- Kessar S. V.; Singh P.; Singh K. N.; Singh S. K. Facile α-deprotonation–electrophilic substitution of quinuclidine and DABCO. Chem. Commun. 1999, 1927–1928. 10.1039/a905359j. [DOI] [Google Scholar]
- Jarvis C. L.; Richers M. T.; Breugst M.; Houk K. N.; Seidel D. Redox-Neutral α-Sulfenylation of Secondary Amines: Ring-Fused N,S-Acetals. Org. Lett. 2014, 16, 3556–3559. 10.1021/ol501509b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SeeJarboe S. G.; Terrazas M. S.; Beak P. The Endocyclic Restriction Test: The Geometries of Nucleophilic Substitutions at Sulfur(VI) and Sulfur(II). J. Org. Chem. 2008, 73, 9627–9632. and references therein for considerations of geometry in sulfur(II) transfer 10.1021/jo8016428. [DOI] [PubMed] [Google Scholar]
- Meister A.; Anderson M. E. Glutathione. Annu. Rev. Biochem. 1983, 52, 711–760. 10.1146/annurev.bi.52.070183.003431. [DOI] [PubMed] [Google Scholar]
- Park C.-M.; Weerasinghe L.; Day J. J.; Fukuto J. M.; Xian M. Persulfides: current knowledge and challenges in chemistry and chemical biology. Mol. BioSyst. 2015, 11, 1775–1785. 10.1039/C5MB00216H. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benavides G. A.; Squadrito G. L.; Mills R. W.; Patel H. D.; Isbell T. S.; Patel R. P.; Darley-Usmar V. M.; Doeller J. E.; Kraus D. W. Hydrogen sulfide mediates the vasoactivity of garlic. Proc. Natl. Acad. Sci. U. S. A. 2007, 104, 17977–17982. 10.1073/pnas.0705710104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Because of peak broadening and instability on the column, the rate of retrodimerization of 3a could not be reliably measured. It appeared comparable to 27.
- On the basis of integration of LCMS peak areas calibrated to standardized references.
- Crouch S. P.; Kozlowski R.; Slater K. J.; Fletcher J. The use of ATP bioluminescence as a measure of cell proliferation and cytotoxicity. J. Immunol. Methods 1993, 160, 81–88. 10.1016/0022-1759(93)90011-U. [DOI] [PubMed] [Google Scholar]
- Bradbury D. A.; Simmons T. D.; Slater K. J.; Crouch S. P. Measurement of the ADP:ATP ratio in human leukaemic cell lines can be used as an indicator of cell viability, necrosis and apoptosis. J. Immunol. Methods 2000, 240, 79–92. 10.1016/S0022-1759(00)00178-2. [DOI] [PubMed] [Google Scholar]
- Brown S. B.; Bailey K.; Savill J. Actin is cleaved during constitutive apoptosis. Biochem. J. 1997, 323, 233–237. 10.1042/bj3230233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weerapana E.; Speers A. E.; Cravatt B. F. Tandem orthogonal proteolysis-activity-based protein profiling (TOP-ABPP)—a general method for mapping sites of probe modification in proteomes. Nat. Protoc. 2007, 2, 1414–1425. 10.1038/nprot.2007.194. [DOI] [PubMed] [Google Scholar]
- Weerapana E.; Wang C.; Simon G. M.; Richter F.; Khare S.; Dillon M. B.; Cravatt B. F.; Bachovchin D. A.; Mowen K.; Baker D. Quantitative reactivity profiling predicts functional cysteines in proteomes. Nature 2010, 468, 790–795. 10.1038/nature09472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang C.; Weerapana E.; Blewett M. M.; Cravatt B. F. A chemoproteomic platform to quantitatively map targets of lipid-derived electrophiles. Nat. Methods 2014, 11, 79–85. 10.1038/nmeth.2759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abo M.; Weerapana E. A Caged Electrophilic Probe for Global Analysis of Cysteine Reactivity in Living Cells. J. Am. Chem. Soc. 2015, 137, 7087–7090. 10.1021/jacs.5b04350. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.