

Abstract
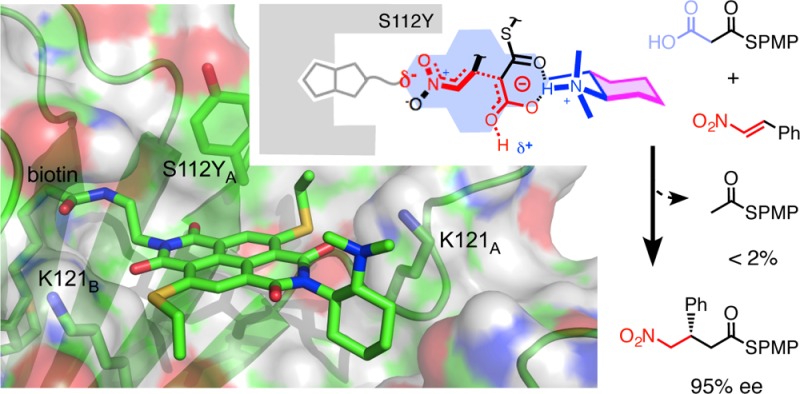
In this report, we introduce artificial enzymes that operate with anion-π interactions, an interaction that is essentially new to nature. The possibility to stabilize anionic intermediates and transition states on an π-acidic surface has been recently demonstrated, using the addition of malonate half thioesters to enolate acceptors as a biologically relevant example. The best chiral anion-π catalysts operate with an addition/decarboxylation ratio of 4:1, but without any stereoselectivity. To catalyze this important but intrinsically disfavored reaction stereoselectively, a series of anion-π catalysts was equipped with biotin and screened against a collection of streptavidin mutants. With the best hit, the S112Y mutant, the reaction occurred with 95% ee and complete suppression of the intrinsically favored side product from decarboxylation. This performance of anion-π enzymes rivals, if not exceeds, that of the best conventional organocatalysts. Inhibition of the S112Y mutant by nitrate but not by bulky anions supports that contributions from anion-π interactions exist and matter, also within proteins. In agreement with docking results, K121 is shown to be essential, presumably to lower the pKa of the tertiary amine catalyst to operate at the optimum pH around 3, that is below the pKa of the substrate. Most importantly, increasing enantioselectivity with different mutants always coincides with increasing rates and conversion, i.e., selective transition-state stabilization.
Short abstract
Proteins are interfaced with catalysts that operate with anion-π interactions. Mutant screening affords artificial enzymes that stabilize disfavored anionic transition states on aromatic surfaces with high enantioselectivity.
Introduction
The integration of unorthodox interactions into functional systems promises to advance the chemical sciences in the most fundamental way.1 Of particular interest is the creation of conceptually innovative catalysts that operate with anion-π interactions,2−5 ion pair-π interactions,6 halogen bonds,7−9 chalcogen bonds,10,11 and so on, i.e., the unorthodox counterparts of cation-π interactions, ion pairs, and hydrogen bonds. At the same time, explicit attention to more conventional interactions such as dispersion forces12,13 or to long-distance effects along α-helical macrodipoles14 contributes much to current progress with a conceptually innovative catalyst design. Among unorthodox interactions, anion-π interactions arguably are the youngest, most elusive, most debated, least used and, perhaps, the most promising.15−21 Complementary to the conventional cation-π interactions on π-basic surfaces, anion-π interactions occur on the π surfaces of electron-deficient aromatic rings with positive quadrupole moment. It is only very recently that the stabilization of anionic transition states on π-acidic aromatic surfaces has been realized.2−5 This remarkably reluctant integration of anion-π interactions15−21 in catalysis is intriguing also because the charge-inverted, conventional cation-π interactions occur more frequently,22−24 particularly in biosynthesis, including the spectacular cyclization of terpenes into steroids.1,22 Encouraged by the evidence of anion binding in transmembrane ion transport experiments,25 anion-π catalysis has been realized first with the Kemp elimination.2 Expansion into enamine catalysis quickly followed,3 but, because of its central importance in chemistry and biology, it was enolate chemistry with anion-π interactions that attracted the most attention.4,5
The addition of malonic acid half thioesters (MAHT) 1 to enolate acceptors is the beginning of all biosynthesis and reaches perfection during the synthesis of polyketide natural products (Figure 1). Without enzymes, however, this enolate chemistry does not really work. Under ambient conditions with a tertiary amine as base catalyst, the addition to acceptors such as nitroolefin 2 yields the addition product 3 only as a side product; decarboxylation to the simple thioester 4 dominates. With triethylamine, for example, an A/D ratio, i.e., the molar ratio between addition product 3 and decarboxylation product 4, of 0.6 has been reported.4
Figure 1.
The concept of anion-π enzymes (top), together with structures of substrates, products (A: addition, D: decarboxylation) and conceivable reactive intermediates (RI1) and transition states (TS1). Highlighted are π surface (blue), base catalyst (blue), fixed Leonard turns (magenta), and top-down approach of 2 (bold) to deprotonated 1 on the π surface (TS1). PMP = p-methoxyphenyl.
Recently, it has been suggested that, in the spirit of the precursive form of the Curtin–Hammett principle,26 the equilibrium between tautomers of the conjugate base of MAHT 1, i.e., the malonate half thioester (MHT) anion, could influence the selectivity between the two competing reactions.4 Deplanarized tautomers with a tetrahedral sp3 carbon between the two carbonyls and the negative charge localized on the carboxylate could favor decarboxylation. However, detailed mechanistic studies have shown that, at least in organocatalysis, decarboxylation occurs after enolate addition, and not before, as usually proposed in biosynthesis.27−32 With planarized tautomers with the negative charge delocalized beyond the sp2 carbon between the two carbonyls, enolate addition has to occur before decarboxylation (RI1, TS1, Figure 1).
To bias the equilibrium in favor of the planar and charge-delocalized tautomer, anion-π interactions on a π-acidic surface appeared well suited. To verify this hypothesis experimentally, tertiary amines were positioned next to the π-acidic surface of naphthalenediimides (NDIs). With increasing number and π acidity of the π surfaces next to the amine base, the selectivity of the reaction was inverted from A/D = 0.6 up to A/D = 1.9. Moreover, the existence of significant enolate-π stabilization was verified in covalent malonate macrodilactones,33 and Leonard turns have been introduced to best position a reaction on a π surface.5 With fully rigidified Leonard turns, selectivity inversions up to A/D = 4.4 could be observed under ambient conditions.5
With all small-molecule anion-π catalysts for MHT addition explored so far, product 3 was obtained as a racemic mixture.4,5 This contrasts sharply to high enantioselectivity obtained for this and related reactions with conventional organocatalysts, usually derivatives of cinchona alkaloids.27−32 To achieve the asymmetry, we considered to embed the catalytic moiety within the chiral environment of proteins. The prospect of artificial anion-π enzymes was particularly intriguing because, except for most unusual situations,34,35 enzymes do not operate with anion-π interactions due to the absence of canonical π-acidic amino acids.
Biotin–streptavidin technology was employed to interface anion-π catalysts with proteins. This reliable method has been used successfully to produce artificial metalloenzymes, which performed many different reactions with high stereoselectivity.36−39 Moreover, large mutant libraries are available for the screening. In the following, we describe how streptavidin mutant screening has led to the discovery of the first artificial anion-π enzyme.
Results and Discussion
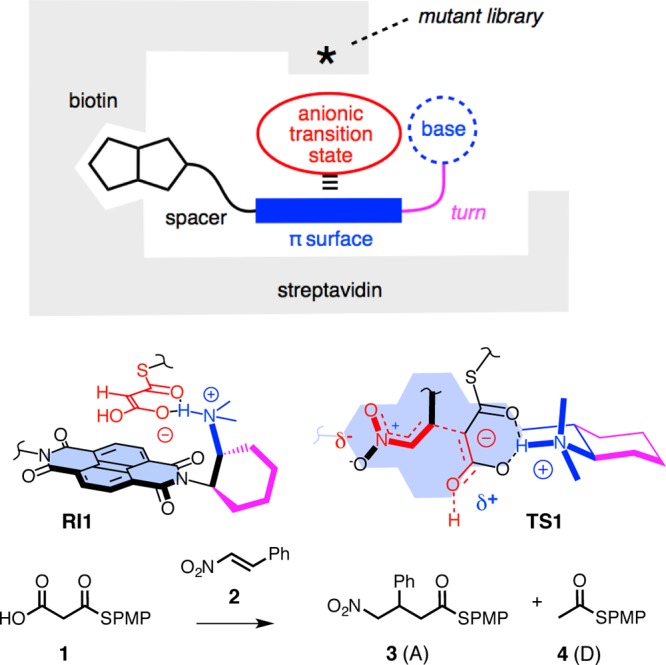
To interface anion-π catalysis with proteins, the small collection of triads 5–9 was designed and synthesized (Figure 2). Common to all are a π-acidic NDI surface, a base, and a biotin. In the biotin–NDI–base triad 5, a tertiary amine is attached to one imide at the right length to fold into a flexible Leonard turn.5 The other imide is equipped with an ethylenediamine to connect to a biotin. According to the electrochemical data, NDIs with two ethylsulfides as in 5 are already strong π acids with a Qzz ≥ 10 B.40 In 6, the sulfides in the core of 5 are oxidized to sulfoxides to further increase the π acidity of the NDI surface.5
Figure 2.
Structure of biotin–NDI–base triads. Note, 6 is a mixture of sulfoxide diastereoisomers.
In triad 7, the distance between biotin and NDI in 5 is shortened. In triad 8, the achiral and flexible Leonard turn in 5 is replaced by an enantiopure and rigidified one. Rigidified Leonard turns have been shown to increase the operational power of anion-π interactions, particularly of weaker ones.5 In 9, the distance between biotin and NDI in 8 is elongated. All conjugates were newly synthesized in a few steps from commercially available starting materials. Procedures and product characterization can be found in the Supporting Information.
Streptavidin is a homotetramer of eight-stranded β barrels that binds biotin with exceptional affinity.37 Spontaneous stoichiometric binding of the colored conjugates 5–9 to streptavidin tetramers could be observed by gel permeation chromatography (GPC), with the formed complexes absorbing at the maximum of the NDI (Figure 3A). Binding to the chiral protein tetramer caused the appearance of weak negative CD Cotton effects near the NDI absorptions at λmax = 527 nm and λmax = 370 nm (Figure 3B). The absence of strong bisignate Cotton effects, i.e., the occurrence of induced rather than exciton-coupled circular dichroism (CD), indicated that NDIs are not in close proximity, also when all four binding sites of streptavidin tetramer are occupied by NDIs.41
Figure 3.

Selected experimental data: (A) GPC chromatograms of S121Y with (solid) and without 8 (dashed, 2 equiv), detected at 220 nm (top) and 570 nm (bottom). (B) Absorption spectrum of 8 (top) and CD spectra of S112Y with increasing concentrations of 8 (0–4 equiv, bottom). (C) Dependence of the ee with WT + 8 on pH (red, pH < 3: Gly buffer, pKa = 2.4; pH > 3: DMG buffer (3,3-dimethylglutaric acid), pKa1 = 3.7, pKa2 = 6.3), compared to the pH dependent deprotonation of MAHT 1 (cyan). (D) Dependence of ee on the conversion into 3 after 24 h at pH 3.0 with 8 and different mutants (Table 1, entries 16-31). (E) Conversion into 3 with time for S112Y + 8 (●, ○) and WT + 8 (□) with (●) and without (□, ○) 200 mM NaNO3. (F) Dependence of the ee with WT + 8 on the concentration of NaNO3 (●) and glucose-6-phosphate (○).
Catalytic activity was assessed first with 5 mM substrate 1, 50 mM substrate 2, 10 mol % NDI catalyst, and 20 mol % protein in PBS buffer/CD3CN 1:1. Product formation was measured by 1H NMR spectroscopy after 24 h or, with slow reactions, after 96 h at ambient temperature. Enantioselectivities were determined by chiral HPLC analysis (Figures S2 and S3). With wild-type streptavidin (WT Sav) and catalyst 5, 4% addition product 3 was obtained after 1 day (Table 1, entry 1). The A/D = 2.0 was in the range of the selectivity observed without protein (under clearly different conditions).5 Within the WT Sav, increasing π acidity in triad 6 increased both the rate (28% in 24 h) and the selectivity of the reaction (A/D = 2.5, Table 1, entry 2). This finding was important because it supported1−5,19,22,25,27 that anion-π interactions contribute to both rate enhancement and chemoselectivity.
Table 1. Characteristics of Anion-π Enzymes.
| Cata | proteinb | conditionsc | η (%)d | A/Df | ee (%)g | |
|---|---|---|---|---|---|---|
| 1 | 5 | WT | PBS | 4 | 2.0 | 0 |
| 2 | 6 | WT | PBS | 28 | 2.5 | 0 |
| 3 | 6 | K121A | PBS | 16 | 2.6 | 0 |
| 4 | 6 | K121H | PBS | 18e | 3.8 | 0 |
| 5 | 6 | K121F | PBS | 7e | 0.4 | 0 |
| 6 | 6 | K121E | PBS | 12 | 0.6 | 0 |
| 7 | 6 | K121D | PBS | 14 | 2.5 | 0 |
| 8 | 6 | S112H | PBS | 23 | 3.3 | 0 |
| 9 | 6 | S112E | PBS | 0 | 0 | |
| 10 | 6 | S112A | PBS | 20 | 2.3 | 0 |
| 11 | 6 | L124V | PBS | 7 | 0.8 | 0 |
| 12 | 6 | L124G | PBS | 20 | 2.8 | 0 |
| 13 | 7 | WT | PBS | 54 | 5.8 | 0 |
| 14 | 8 | WT | PBS | 37 | 7.6 | 10 |
| 15 | 9 | WT | PBS | 3 | 1.0 | 0 |
| 16 | 8 | WT | Gly | 60 | >30 | 41 |
| 17 | 8 | S112A | Gly | 20 | >30 | 7 |
| 18 | 8 | S112Y | Gly | 90 | >30 | 95 |
| 19 | 8 | S112F | Gly | 90 | >30 | 91 |
| 20 | 8 | S112W | Gly | 50 | >30 | 53 |
| 21 | 8 | S112E | Gly | 71 | 18 | 63 |
| 22 | 8 | S112K | Gly | 52 | 7 | 38 |
| 23 | 8 | S112H | Gly | 70 | >30 | 64 |
| 24 | 8 | K121E | Gly | 0 | nd | nd |
| 25 | 8 | K121H | Gly | 0 | nd | nd |
| 26 | 8 | K121F | Gly | 15 | >30 | 0 |
| 27 | 8 | K121Y | Gly | 17 | 0.7 | 0 |
| 28 | 8 | K121W | Gly | 23 | 4.0 | 0 |
| 29 | 8 | K121A | Gly | 22 | 1.6 | 0 |
| 30 | 8 | L124F | Gly | 68 | >30 | 60 |
| 31 | 8 | L124Y | Gly | 30 | 1.6 | 0 |
| 32 | 5 | S112Y | Gly | 33 | 2.5 | 33 |
| 33 | 6 | S112Y | Gly | 0 | nd | nd |
| 34 | 7 | S112Y | Gly | 32 | 6.4 | 0 |
| 35 | 9 | S112Y | Gly | 41 | 10 | 0 |
| 36 | (±)-8 | S112Y | Gly | 83 | >30 | 90 |
| 37 | (±)-8 | WT | Gly | 38 | >30 | 20 |
| 38 | 8 | Gly | 0 | nd | nd | |
| 39 | WT | Gly | 0 | nd | nd | |
| 40 | S112Y | Gly | 0 | nd | nd |
Streptavidin, WT = wild type.
5 mM 1, 50 mM 2, 500 μM catalyst (10 mol %), 1 mM protein (20 mol %), rt; PBS: PBS buffer/CD3CN 1:1 (PBS = phosphate buffered saline, 274 mM NaCl, 5.4 mM KCl, 20 mM phosphate, pH 7.4). Gly: Gly/CD3CN 1:1 (Gly = 50 mM glycine buffer, pH 3.0).
Conversion into 3; after 24 h.
As d; but after 96 h.
Ratio of addition product A (3)/decarboxylation product D (4).
Enantiomeric excess. (S)-3 was always the preferred enantiomer.
A focused streptavidin mutant library was screened in combination with NDI 6 (Table 1, entries 3–12). However, none of the mutants gave significant improvements in activity or chemo-/enantioselectivities compared to the WT Sav. Nevertheless, the observed variation in activity demonstrated that indeed the reaction takes place within the protein pocket.
Next, the influence of the triads 7–9 on the catalytic performance was evaluated in the presence of WT Sav (Table 1, entries 13–15). Compared to triad 5, a shortened distance between biotin and NDI in triad 7 resulted in a significantly increased rate and a quite remarkable selectivity A/D = 5.8 (Table 1, entry 1 vs entry 13). Replacement of the flexible Leonard turn in 5 by a rigidified turn in 8 caused a further increase to A/D = 7.8 at preserved high rate, i.e., 37% product formation in 24 h (Table 1, entry 14). Elongation of the biotin–NDI distance in the presence of a rigidified Leonard turn in 9 removed most catalytic activity (Table 1, entry 15).
In the presence of wild-type streptavidin, biotin-NDI-base triad 8 with a chiral and rigidified Leonard turn yielded the addition product 3 with 10% ee (Table 1, entry 14). All other systems produced racemates under identical conditions. Considering possible interference of anion-π interactions with anions in the PBS buffer on the one hand and substrate protonation on the other, the system WT Sav + 8 was tested under different pH values between 7.4 and 2.0. Remarkably, the pH profile of the ee showed a bell-shaped behavior with a maximum around pH 3.0 (Figure 3C). This pH maximum is clearly below the pKa ≈ 4.5 of the MAHT substrate 1 (Figure 3C). Under optimized conditions, the MAHT substrate 1 will thus exist in protonated form in solution and liberate the proton only upon stabilization of the conjugate MHT base on the π-acidic surface. Recent systematic studies have shown that (a) anion-π interactions can increase the acidity of malonates by up to ΔpKa = 5.533 and (b) precise positioning by fixed Leonard turns significantly strengthens their impact.5 Facilitated protonation of nitronate anion3 might further contribute to the high activity at pH 3. According to CD spectroscopy and consistent with the literature,37 streptavidin remained intact at pH 3.0 (Figure S4).
At the optimum pH, the ee obtained with WT Sav + 8 was 41%, and the chemoselectivity reached an unexpected extent (>30, Table 1, entry 16). Further improvement could be achieved by screening of mutants (Table 1, entries 17–31). The best results were obtained with S112Y: The enolate addition product 3 was obtained in 95% ee and perfect chemoselectivity, without any detectable trace of decarboxylation product 4 (Table 1, entry 18). Similarly outstanding characteristics obtained for the S112F mutants suggested that aromatic interactions or simple sterics rather than hydrogen bonding or acid–base chemistry might account for this breakthrough (Table 1, entry 19). Mutations at position K121 resulted in much lower catalytic activities and loss of enantioselectivities, suggesting that the presence of the lysine residue at this position is important for catalysis and enantioselectivity (Table 1, entry 24–29, Figure 4). L124F, best among mutations below the NDI, was with 60% ee better than the WT (41% ee) but far from the performance of S112Y (95% ee, Table 1, entries 16, 18, 30). The top performance of S112Y was unique for triad 8 with a fixed and chiral Leonard turn: The original 5 with a flexible Leonard turn gave only 33% ee, and other catalysts produced racemates or were completely inactive (Table 1, entries 33–35). Interestingly, the excellent properties of S112Y + 8 did not suffer much by the “epimerization” of the catalyst to (±)-8, which was made from 1:1 mixtures of (R,R) and (S,S) cyclohexyldiamine and enantiopure biotin (Table 1, entry 37). With the WT, “epimerized” catalysts (±)-8 reduced conversion and enantioselectivity significantly (Table 1, entry 36). Finally, control experiments performed with catalyst 8 (Table 1, entry 38), WT streptavidin, or S112Y alone (Table 1, entries 39, 40) did not give any trace of product. These results demonstrated that the synergism between the streptavidin and the NDI catalyst at pH 3.0 is essential.
Figure 4.
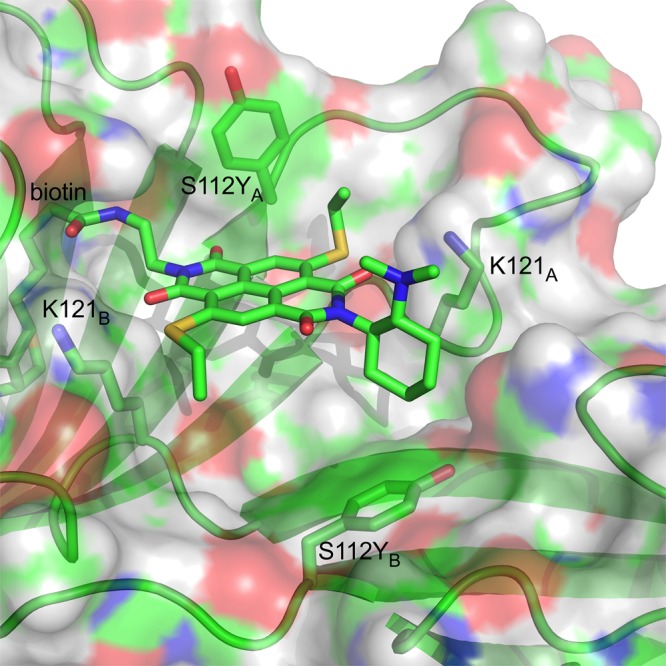
Docking simulations of S112Y dimers with 8, zoomed on the active site. Protein surfaces are rendered with their electrostatic potential (red: negative, blue: positive, green: aromatics), β sheets as faint green arrows. Exposed parts of 8 are in wireframe presentation, C green, N, blue, S yellow, O red.
In general, enantioselectivities increased almost linearly with the rate of the reaction (Figure 3D). This finding was important because it confirmed that fast reactions are selective; i.e., selectivity increases with transition-state stabilization. Reaction kinetics for WT Sav and the S112Y were in agreement with this important conclusion: Mutant S112Y (95% ee) was much (8.8 times) faster than WT Sav (41% ee, Figure 3E, ○ and □). With S112Y + 8, 1 mol % catalyst still achieved almost full conversion at still high enantioselectivity (η = 85%, 86% ee). With only 0.1 mol % catalyst, conversion and enantioselectivity decreased clearly (η = 66%, 20% ee). In the presence of increasing concentrations of sodium nitrate, both rate and enantioselectivity of S112Y with NDI 8 decreased significantly (Figure 3E, ● vs ○ and Figure 3F, ●). In contrast, inhibition by glucose-6-phosphate, a bulky anion, was much weaker (Figure 3F, ○). This inhibition of stereoselective product formation by nitrate was interesting because it provided corroborative experimental support that anion-π interactions contribute significantly to catalysis, particularly stereoselectivity, also in artificial anion-π enzymes. The found IC50 = 225 ± 24 mM was in agreement with this important conclusion. Nitrate-π interactions have been reported previously to be particularly favorable in several functional systems.25,42−44
Docking simulations revealed that NDI 8 fits very well in the biotin-binding vestibule on the surface of an S112Y dimer (consisting of streptavidin monomers A and B, Figures 4 and S7). On the left, the biotin anchor is buried deep inside the β barrel of the monomer A. The biotin binding vestibule being widely exposed to the solvent, flexibility was observed in the docking results, especially concerning the orientation of the diaminocyclohexane. Importantly, the orientation of both tyrosines S112YA and S112YB was consistent among the results. The key S112YA of the 8-containing monomer points its hydroxyl group toward the solvent, hence exposing the aromatic surface toward the NDI, consistent with preserved activity of S112F. The ethylsulfide next to this tyrosine is twisted out of the usual coplanarity with the NDI core, stabilized by an intramolecular S–O chalcogen bond.1,40 In contrast, the S112YB (of the opposite monomer B) points toward the protein, shielding the opposite biotin binding site B. This residue however is too far from the exposed NDI π surface to play a significant role in catalysis. Both K121A and K121B pinch the NDI into place. The K121A ammonium is only 3.8 Å away from the catalytic NMe2 moiety. This essential ammonium cation is hypothesized to lower the pKa of the tertiary amine in the fixed Leonard turn to keep it in the functional neutral form even at pH 3.0, at least partially. Similar proximity effects are abundant in biological enzymes, not only with amines (e.g., class I aldolases)45,46 but also carboxylates (e.g., glycosidases,47 proteases48) and operate also for transport across bilayer membranes.49 Consistent with this interpretation is the observation that at pH 3.0, mutation of K121 reliably annihilates most catalytic activity, most chemoselectivity (except K121F) and all stereoselectivity (Table 1, entries 24–29), whereas at pH 7.4, conversion and chemoselectivity can be preserved (Table 1, entries 3–7). The importance of K121 for the enantioselectivity also suggests its involvement in the stereodetermining step by probably acting as a proton donor to the nitronate group.3,28
Docking results also provided convincing explanations for why triad 8 performed best. In 7, for example, the linker with biotin is too short to comfortably accommodate the NDI in the vestibule on the protein surface (Table 1, entry 34; Figures 4 and S7). With S112Y, 6 was inactive despite increased π acidity, presumably because the oxygen atoms added to the sulfur atoms do not fit into the tight NDI binding site (Table 1, entry 33, Figures 4 and S7).
Conclusions
The objective of this study was to create artificial enzymes that operate with anion-π interactions, an interaction essentially new to nature, and display emergent properties that are otherwise beyond reach. Our results, based on a chemogenetic optimization relying on screening a collection of catalysts with protein mutants, fully live up to these expectations. The best anion-π enzymes catalyze the intrinsically disfavored but chemically and biologically most relevant addition of MHTs to enolate acceptors with perfect chemoselectivity. Moreover, a stereoselectivity of 95% ee with η = 90% conversion clearly exceeds the performance of conventional organocatalysts (88% ee with η = 57% or 63% ee with η = 94%).27 The existence of operational anion-π interactions are supported by increasing activity with (a) increasing π acidity, (b) precise positioning of the reaction on the π surface with rigidified Leonard turns, and most importantly, (c) inhibition of the artificial anion-π enzyme with nitrate. The found best performance at pH 3 suggests that the enolate forms only due to the stabilization by π-acidic surfaces. The tertiary amine catalysts are kept operational under these acidic conditions by repulsive proximity effects with two essential lysines (K121).
In summary, asymmetric catalysis with the first artificial anion-π enzyme outperforms conventional organocatalysts, i.e., derivatives of cinchona alkaloids.27 Attractive perspectives include anion-π enzymes that operate in living cells, the interfacing of anion-π catalysts with other complex systems, and the expansion of the successful approach to reactions that otherwise do not work, and to other unorthodox interactions.
Acknowledgments
We thank the NMR and the Sciences Mass Spectrometry (SMS) platforms for services, and the National Centre of Competence in Research (NCCR) Molecular Systems Engineering, the University of Geneva, the University of Basel, the European Research Council (ERC Advanced Investigator), the NCCR Chemical Biology and the Swiss NSF for financial support.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acscentsci.6b00097.
Detailed experimental procedures (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Zhao Y.; Cotelle Y.; Sakai N.; Matile S. Unorthodox interactions at work. J. Am. Chem. Soc. 2016, 138, 4270–4277. 10.1021/jacs.5b13006. [DOI] [PubMed] [Google Scholar]
- Zhao Y.; Domoto Y.; Orentas E.; Beuchat C.; Emery D.; Mareda J.; Sakai N.; Matile S. Catalysis with anion-π interactions. Angew. Chem., Int. Ed. 2013, 52, 9940–9943. 10.1002/anie.201305356. [DOI] [PubMed] [Google Scholar]
- Zhao Y.; Cotelle Y.; Avestro A.-J.; Sakai N.; Matile S. Asymmetric anion-π catalysis: enamine addition to nitroolefins on π-acidic surfaces. J. Am. Chem. Soc. 2015, 137, 11582–11585. 10.1021/jacs.5b07382. [DOI] [PubMed] [Google Scholar]
- Zhao Y.; Benz S.; Sakai N.; Matile S. Selective acceleration of disfavored enolate addition reactions by anion-π interactions. Chem. Sci. 2015, 6, 6219–6223. 10.1039/C5SC02563J. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cotelle Y.; Benz S.; Avestro A.-J.; Ward T. R.; Sakai N.; Matile S. Anion-π catalysis of enolate chemistry: rigidified Leonard turns as a general motif to run reactions on aromatic surfaces. Angew. Chem., Int. Ed. 2016, 55, 4275–4279. 10.1002/anie.201600831. [DOI] [PubMed] [Google Scholar]
- Fujisawa K.; Humbert-Droz M.; Letrun R.; Vauthey E.; Wesolowski T. A.; Sakai N.; Matile S. Ion pair-π interactions. J. Am. Chem. Soc. 2015, 137, 11047–11056. 10.1021/jacs.5b05593. [DOI] [PubMed] [Google Scholar]
- Jungbauer S. H.; Huber S. M. Cationic multidentate halogen-bond donors in halide abstraction organocatalysis: catalyst optimization by preorganization. J. Am. Chem. Soc. 2015, 137, 12110–12120. 10.1021/jacs.5b07863. [DOI] [PubMed] [Google Scholar]
- Zong L.; Ban X.; Kee C. W.; Tan C.-H. Catalytic enantioselective alkylation of sulfenate anions to chiral heterocyclic sulfoxides using halogenated pentanidium salts. Angew. Chem., Int. Ed. 2014, 53, 11849–11853. 10.1002/anie.201407512. [DOI] [PubMed] [Google Scholar]
- Cavallo G.; Metrangolo P.; Milani R.; Pilati T.; Priimagi A.; Resnati G.; Terraneo G. The halogen bond. Chem. Rev. 2016, 116, 2478–2601. 10.1021/acs.chemrev.5b00484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Birman V. B.; Li X. Benzotetramisole: a remarkably enantioselective acyl transfer catalyst. Org. Lett. 2006, 8, 1351–1354. 10.1021/ol060065s. [DOI] [PubMed] [Google Scholar]
- Beno B. R.; Yeung K.-S.; Bartberger M. D.; Pennington L. D.; Meanwell N. A. A Survey of the role of noncovalent sulfur interactions in drug design. J. Med. Chem. 2015, 58, 4383–4438. 10.1021/jm501853m. [DOI] [PubMed] [Google Scholar]
- Wagner J. P.; Schreiner P. R. London dispersion in molecular chemistry - reconsidering steric effects. Angew. Chem., Int. Ed. 2015, 54, 12274–12296. 10.1002/anie.201503476. [DOI] [PubMed] [Google Scholar]
- Wende R. C.; Seitz A.; Niedek D.; Schuler S. M. M.; Hofmann C.; Becker J.; Schreiner P. R. The enantioselective Dakin-West reaction. Angew. Chem., Int. Ed. 2016, 55, 2719–2723. 10.1002/anie.201509863. [DOI] [PubMed] [Google Scholar]
- Le Bailly B. A. F.; Byrne L.; Clayden J. Refoldable foldamers: Global conformational switching by deletion or insertion of a single hydrogen bond. Angew. Chem., Int. Ed. 2016, 55, 2132–2136. 10.1002/anie.201510605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frontera A.; Gamez P.; Mascal M.; Mooibroek T. J.; Reedijk J. Putting anion-π interactions into perspective. Angew. Chem., Int. Ed. 2011, 50, 9564–9583. 10.1002/anie.201100208. [DOI] [PubMed] [Google Scholar]
- Giese M.; Albrecht M.; Rissanen K. Experimental investigation of anion-π interactions - applications and biochemical relevance. Chem. Commun. 2016, 52, 1778–1795. 10.1039/C5CC09072E. [DOI] [PubMed] [Google Scholar]
- Chifotides H. T.; Dunbar K. R. Anion-π interactions in supramolecular architectures. Acc. Chem. Res. 2013, 46, 894–906. 10.1021/ar300251k. [DOI] [PubMed] [Google Scholar]
- Wang D.-X.; Wang M.-X. Anion recognition by charge neutral electron-deficient arene receptors. Chimia 2011, 65, 939–943. 10.2533/chimia.2011.939. [DOI] [PubMed] [Google Scholar]
- Ballester P. Experimental quantification of anion-π interactions in solution using neutral host-guest model systems. Acc. Chem. Res. 2013, 46, 874–884. 10.1021/ar300080f. [DOI] [PubMed] [Google Scholar]
- Schneebeli S. T.; Frasconi M.; Liu Z.; Wu Y.; Gardner D. M.; Strutt N. L.; Cheng C.; Carmieli R.; Wasielewski M. R.; Stoddart J. F. Electron sharing and anion-π recognition in molecular triangular prisms. Angew. Chem., Int. Ed. 2013, 52, 13100–13104. 10.1002/anie.201307984. [DOI] [PubMed] [Google Scholar]
- Bauzà A.; Mooibroek T. J.; Frontera A. The bright future of unconventional σ/π-hole interactions. ChemPhysChem 2015, 16, 2496–2517. 10.1002/cphc.201500314. [DOI] [PubMed] [Google Scholar]
- Knowles R. R.; Lin S.; Jacobsen E. N. Enantioselective thiourea-catalyzed cationic polycyclizations. J. Am. Chem. Soc. 2010, 132, 5030–5032. 10.1021/ja101256v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Q.; Tiefenbacher K. Terpene cyclization catalysed inside a self-assembled cavity. Nat. Chem. 2015, 7, 197–202. 10.1038/nchem.2181. [DOI] [PubMed] [Google Scholar]
- Holland M. C.; Paul S.; Schweizer W. B.; Bergander K.; Mück-Lichtenfeld C.; Lakhdar S.; Mayr H.; Gilmour R. Noncovalent interactions in organocatalysis: Modulating conformational diversity and reactivity in the MacMillan catalyst. Angew. Chem., Int. Ed. 2013, 52, 7967–7971. 10.1002/anie.201301864. [DOI] [PubMed] [Google Scholar]
- Vargas Jentzsch A.; Hennig A.; Mareda J.; Matile S. Synthetic ion transporters that work with anion-π interactions, halogen bonds and anion-macrodipole interactions. Acc. Chem. Res. 2013, 46, 2791–2800. 10.1021/ar400014r. [DOI] [PubMed] [Google Scholar]
- Seeman J. I. Effect of conformational change on reactivity in organic chemistry. Evaluations, applications, and extensions of Curtin-Hammett Winstein-Holness kinetics. Chem. Rev. 1983, 83, 83–134. 10.1021/cr00054a001. [DOI] [Google Scholar]
- Lubkoll J.; Wennemers H. Mimicry of polyketide synthases - Enantioselective 1,4-addition reactions of malonic acid half-thioesters to nitroolefins. Angew. Chem., Int. Ed. 2007, 46, 6841–6844. 10.1002/anie.200702187. [DOI] [PubMed] [Google Scholar]
- Pan Y.; Kee C. W.; Jiang Z.; Ma T.; Zhao Y.; Yang Y.; Xue H.; Tan C.-H. Expanding the utility of Brønsted base catalysis: Biomimetic enantioselective decarboxylative reactions. Chem. - Eur. J. 2011, 17, 8363–8370. 10.1002/chem.201100687. [DOI] [PubMed] [Google Scholar]
- Saadi J.; Wennemers H. Enantioselective aldol reactions with masked fluoroacetates. Nat. Chem. 2016, 8, 276–280. 10.1038/nchem.2437. [DOI] [PubMed] [Google Scholar]
- Wang Z.-L. Recent advances in catalytic asymmetric decarboxylative addition reactions. Adv. Synth. Catal. 2013, 355, 2745–2755. 10.1002/adsc.201300375. [DOI] [Google Scholar]
- Nakamura S. Catalytic enantioselective decarboxylative reactions using organocatalysts. Org. Biomol. Chem. 2014, 12, 394–405. 10.1039/C3OB42161A. [DOI] [PubMed] [Google Scholar]
- Blaquiere N.; Shore D. G.; Rousseaux S.; Fagnou K. Decarboxylative ketone aldol reactions: Development and mechanistic evaluation under metal-free conditions. J. Org. Chem. 2009, 74, 6190–6198. 10.1021/jo901022j. [DOI] [PubMed] [Google Scholar]
- Miros F. N.; Zhao Y.; Sargsyan G.; Pupier M.; Besnard C.; Beuchat C.; Mareda J.; Sakai N.; Matile S. Enolate stabilization by anion-π interactions: Deuterium exchange in malonate dilactones on π-acidic surfaces. Chem. - Eur. J. 2016, 22, 2648–2657. 10.1002/chem.201504008. [DOI] [PubMed] [Google Scholar]
- Bauza A.; Quinonero D.; Deya P. M.; Frontera A. Long-range effects in anion-π interactions: their crucial role in the inhibition mechanism of mycobacterium tuberculosis malate synthase. Chem. - Eur. J. 2014, 20, 6985–6990. 10.1002/chem.201304995. [DOI] [PubMed] [Google Scholar]
- Lucas X.; Bauzá A.; Frontera A.; Quiñonero D. A thorough anion−π interaction study in biomolecules: on the importance of cooperativity effects. Chem. Sci. 2016, 7, 1038–1050. 10.1039/C5SC01386K. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson M. E.; Whitesides G. M. Conversion of a protein to a homogeneous asymmetric hydrogenation catalyst by site-specific modification with a diphosphinerhodium(I) moiety. J. Am. Chem. Soc. 1978, 100, 306–307. 10.1021/ja00469a064. [DOI] [Google Scholar]
- Ward T. R. Artificial metalloenzymes based on the biotin-avidin technology: enantioselective catalysis and beyond. Acc. Chem. Res. 2011, 44, 47–57. 10.1021/ar100099u. [DOI] [PubMed] [Google Scholar]
- Reetz M. T.; Peyralans J. J.-P.; Maichele A.; Fu Y.; Maywald M. Directed evolution of hybrid enzymes: evolving enantioselectivity of an achiral Rh-complex anchored to a protein. Chem. Commun. 2006, 41, 4318–4320. 10.1039/b610461d. [DOI] [PubMed] [Google Scholar]
- Hyster T. K.; Knörr L.; Ward T. R.; Rovis T. Biotinylated Rh (III) complexes in engineered streptavidin for accelerated asymmetric C–H activation. Science 2012, 338, 500–503. 10.1126/science.1226132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miros F. N.; Matile S.. Core-substituted naphthalenediimides: LUMO levels revisited, in comparison to perylenediimides with sulfur redox switches in the core. ChemistryOpen, 2016, DOI: 10.1002/open.201500222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pescitelli G.; Di Bari L.; Berova N. Application of electronic circular dichroism in the study of supramolecular systems. Chem. Soc. Rev. 2014, 43, 5211–5233. 10.1039/C4CS00104D. [DOI] [PubMed] [Google Scholar]
- Wang D.-X.; Wang M.-X. Anion-π interactions: Generality, binding strength, and structure. J. Am. Chem. Soc. 2013, 135, 892–897. 10.1021/ja310834w. [DOI] [PubMed] [Google Scholar]
- Giese M.; Albrecht M.; Ivanova G.; Valkonen A.; Rissanen K. Geometrically diverse anions in anion−π interactions. Supramol. Chem. 2012, 24, 48–55. 10.1080/10610278.2011.622384. [DOI] [Google Scholar]
- Adriaenssens L.; Estarellas C.; Vargas Jentzsch A.; Martinez Belmonte M.; Matile S.; Ballester P. Quantification of nitrate-π interactions and selective transport of nitrate using calix[4]pyrroles with two aromatic walls. J. Am. Chem. Soc. 2013, 135, 8324–8330. 10.1021/ja4021793. [DOI] [PubMed] [Google Scholar]
- Machajewski T. D.; Wong C. H. The catalytic asymmetric aldol reaction. Angew. Chem., Int. Ed. 2000, 39, 1352–1375. . [DOI] [PubMed] [Google Scholar]
- Muller M. M.; Windsor M. A.; Pomerantz W. C.; Gellman S. H.; Hilvert D. A rationally designed aldolase foldamer. Angew. Chem., Int. Ed. 2009, 48, 922–925. 10.1002/anie.200804996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heightman T. D.; Vasella A. T. Recent insights into inhibition, structure, and mechanism of configuration-retaining glycosidases. Angew. Chem., Int. Ed. 1999, 38, 750–770. . [DOI] [PubMed] [Google Scholar]
- Schramm V. L. Transition states, analogues, and drug development. ACS Chem. Biol. 2013, 8, 71–81. 10.1021/cb300631k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gasparini G.; Bang E.-K.; Montenegro J.; Matile S. Cellular uptake: lessons from supramolecular organic chemistry. Chem. Commun. 2015, 51, 10389–10402. 10.1039/C5CC03472H. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.