

Abstract
Analysis of the 3D structure of DNA in tumour cells reveals how mutations in the IDH1 gene, and associated changes in methyl groups attached to DNA, elevate the expression of cancer-promoting genes.
The discovery in the late 2000s that mutations in the gene that encodes the enzyme isocitrate dehydrogenase 1 (IDH1) are often associated with glioma, the most common form of brain cancer, was unexpected and tantalizing1,2. The IDH1 protein is involved in the citric-acid cycle — a metabolic process that is used by nearly all cells to generate energy, and that in 2008 had only recently been connected to cancer3,4. The discovery therefore supported the longstanding theory that altered metabolism could transform normal cells into cancerous ones. On page 110 of this issue, Flavahan et al.5 report that an abnormal metabolite generated by mutant IDH1 may drive cancer primarily by altering the 3D conformation of DNA.
Mutant IDH1 converts the citric-acid-cycle molecule isocitrate into an abnormal metabolite that inhibits TET enzymes6, which remove methyl groups from DNA. The presence of methyl groups can alter gene expression by preventing some proteins from binding DNA, and an excess of methyl groups in promoter sequences (which drive gene expression) can silence tumour-suppressor genes, leading to cancer. It has been suggested7 that inhibition of TET enzymes leads to such hypermethylation in IDH1-mutant tumours. However, promoter hypermethylation in these tumours is not generally correlated with changes in gene expression8, suggesting that cancer-associated changes in methylation may occur at other DNA sequences.
In addition to promoter regions, gene expression can be regulated by the 3D structure of chromatin (the complex in which DNA is wound around histone proteins for packaging in the cell). Chromatin structure is exceptionally intricate, and is defined in part by evolutionarily conserved loops called topologically associated domains (TADs). Interactions between DNA sequences — for instance, those that bring promoters into contact with distant enhancer elements to activate gene expression — are more common within than between TADs, and there is evidence9 that gene expression is coordinated in these loops.
TADs are insulated from one another by DNA-binding proteins such as the CCCTC-binding factor (CTCF). Deletion of the DNA sequence encompassing one CTCF binding site has been shown to cause changes in TAD structure and gene expression that lead to limb malformations10, highlighting the importance of maintaining these boundaries. Notably, CTCF binding is sensitive to changes in DNA methylation11,12.
Flavahan et al. demonstrated that a subset of CTCF binding sites is methylated in IDH1-mutant gliomas, and that CTCF binding at these sites is subsequently reduced. Leveraging gene-expression data from hundreds of gliomas and normal brain specimens, and using 3D chromosome-conformation data from various cell lines, the authors found previously unknown gene-expression correlations between TADs in IDH1-mutant gliomas, suggesting that TAD borders are disrupted.
Hundreds of the pairs of genes that are correlated in the mutant cells straddle a disrupted TAD border. Of these, PDGFRA and FIP1L1 are among the most highly expressed. PDGFRA is an appealing candidate for further study, because it is a well-documented oncogene (it promotes cancer when mutationally activated or overexpressed) and is amplified genetically in some 20% of advanced (high-grade) gliomas13. The authors find that, in IDH1-mutant gliomas, which are low grade, the CTCF site at the TAD boundary between PDGFRA and FIP1L1 is methylated and CTCF binding is reduced. Thus, an increase in PDGFRA expression, although arising through different mechanisms in low- and high-grade tumours, may be a common theme in glioma.
Flavahan and colleagues showed that, in glioma cells in which IDH1 is not mutated, the PDGFRA promoter strongly interacts with its own enhancer. The interaction patterns are markedly different in IDH1-mutant tumours. Here, there is a strong interaction between the PDGFRA promoter and the unrelated enhancer of FIP1L1, despite the fact that these two genetic elements are separated by almost 900,000 base pairs. This aberrant interaction is approximately five times stronger than that between the PDGFRA promoter and its own enhancer. Together, these results suggest that disruption of a boundary element by hypermethylation allows a potent FIP1L1 enhancer to interact with the PDGFRA promoter, increasing gene expression (Fig. 1).
Figure 1. Breaking down boundaries to cancer.
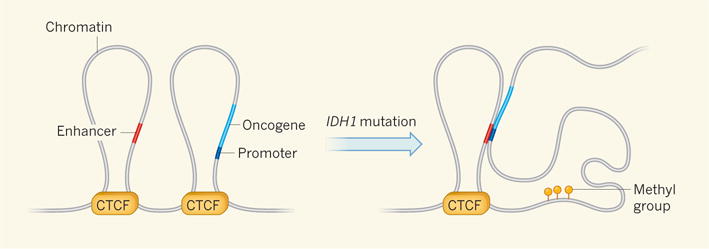
Structural boundaries between regions of chromatin (the complex of DNA and proteins in which DNA is packaged in the nucleus) define loops called topologically associated domains (TADs), within which gene activity is coordinated. DNA binding by the insulator protein CTCF separates these domains. Flavahan et al.5 provide evidence that CTCF insulation prevents the activation of oncogenes (genes whose hyperactivity promotes cancer) by distant enhancer elements from different TADs. The authors find that mutations in the gene IDH1 increase the number of methyl groups that are attached to CTCF binding sites, reducing CTCF–DNA binding. This breaks down the TAD border structure, allowing aberrant association between enhancers and the promoter regions of oncogenes. Oncogene expression is subsequently amplified, leading to cancer.
To confirm that DNA hypermethylation is responsible for the elevated PDGFRA expression that they observed, the authors treated IDH1-mutant cells with a drug that reduces DNA methylation. In agreement with their hypothesis, the treatment reduced methylation of the relevant CTCF binding site, increasing CTCF binding and reducing PDGFRA expression. Conversely, experimental disruption of the CTCF binding site in cells that lacked the IDH1 mutation led to increased PDGFRA expression. The altered expression presumably occurs because of changes in enhancer–promoter interactions, but this was not tested directly. Elevated PDGFRA expression doubled cell growth compared with untreated cells. This suggests that the increased PDGFRα protein in IDH1-mutant glioma cells provides a selective growth advantage over cells lacking the mutation.
Flavahan and colleagues’ study focuses on one CTCF site out of hundreds, so other oncogenes might also be activated by newly formed enhancer–promoter interactions in IDH1-mutant tumours. Many newly activated genes may also be ‘passenger’ events, which have no functional consequences. The methylation states of CTCF sites and the activity of enhancers vary widely across cell types, suggesting that 3D chromosome-conformation analysis of high-grade gliomas, colorectal cancers, lymphomas, leukaemias and other IDH1-mutant cancers could reveal different targets of genomic hypermethylation. These targets may also include those normally bound by methylation-sensitive factors other than CTCF.
Consistent with the fact that DNA methylation is highly stable, aberrant hypermethylation persists in IDH1-mutant tumours after treatment with an inhibitor of mutant IDH1 (ref. 14). Assuming that hypermethylation is involved in the transition to cancer, as is strongly suggested by the current study, such stability could pose a challenge for the success of IDH1-inhibitor treatments in patients. Unravelling the effects of DNA hypermethylation on gene dysregulation will lead to a more complete survey of the forces downstream of TET and other enzymes that drive the evolution of IDH1-mutant cancer cells15. Flavahan and colleagues’ study provides a fresh perspective on which to base such future analyses. ■
Footnotes
J.F.C. declares competing financial interests. See online article for details.
Contributor Information
MATTHEW R. GRIMMER, Email: joseph.costello@ucsf.edu.
JOSEPH F. COSTELLO, Email: matthew.grimmer@ucsf.edu.
References
- 1.Parsons DW, et al. Science. 2008;321:1807–1812. doi: 10.1126/science.1164382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Yan H, et al. N Engl J Med. 2009;360:765–773. doi: 10.1056/NEJMoa0808710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Selak MA, et al. Cancer Cell. 2005;7:77–85. doi: 10.1016/j.ccr.2004.11.022. [DOI] [PubMed] [Google Scholar]
- 4.The Multiple Leiomyoma Consortium. Nature Genet. 2002;30:406–410. [Google Scholar]
- 5.Flavahan WA, et al. Nature. 2015;529:110–114. doi: 10.1038/nature16490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Xu W, et al. Cancer Cell. 2011;19:17–30. doi: 10.1016/j.ccr.2010.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Turcan S, et al. Nature. 2012;483:479–483. doi: 10.1038/nature10866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Noushmehr H, et al. Cancer Cell. 2010;17:510–522. doi: 10.1016/j.ccr.2010.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Nora EP, et al. Nature. 2012;485:381–385. doi: 10.1038/nature11049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lupiáñez DG, et al. Cell. 2015;161:1012–1025. doi: 10.1016/j.cell.2015.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bell AC, Felsenfeld G. Nature. 2000;405:482–485. doi: 10.1038/35013100. [DOI] [PubMed] [Google Scholar]
- 12.Hark AT, et al. Nature. 2000;405:486–489. doi: 10.1038/35013106. [DOI] [PubMed] [Google Scholar]
- 13.Phillips JJ, et al. Brain Pathol. 2013;23:565–573. doi: 10.1111/bpa.12043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rohle D, et al. Science. 2013;340:626–630. doi: 10.1126/science.1236062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Koivunen P, et al. Nature. 2012;483:484–488. doi: 10.1038/nature10898. [DOI] [PMC free article] [PubMed] [Google Scholar]