

Abstract
We developed VariantBam, a C ++ read filtering and profiling tool for use with BAM, CRAM and SAM sequencing files. VariantBam provides a flexible framework for extracting sequencing reads or read-pairs that satisfy combinations of rules, defined by any number of genomic intervals or variant sites. We have implemented filters based on alignment data, sequence motifs, regional coverage and base quality. For example, VariantBam achieved a median size reduction ratio of 3.1:1 when applied to 10 lung cancer whole genome BAMs by removing large tags and selecting for only high-quality variant-supporting reads and reads matching a large dictionary of sequence motifs. Thus VariantBam enables efficient storage of sequencing data while preserving the most relevant information for downstream analysis.
Availability and implementation: VariantBam and full documentation are available at github.com/jwalabroad/VariantBam.
Contact: rameen@broadinstitute.org
Supplementary information: Supplementary data are available at Bioinformatics online.
1 Introduction
As the cost of genome sequencing decreases, the storage and computational burden of handling large sequencing datasets is an increasing concern. Whole genome sequencing of a human genome to 30× coverage can result in approximately 1 billion reads, requiring more than 100 GB of disk space even in the compressed BAM format (Li et al., 2009). To mitigate this, the CRAM format was established to provide reference-based compression of BAM files, resulting in a 2–3× reduction in file size (Hsi-Yang Fritz et al., 2011). However, the CRAM format is not currently supported by many analysis tools, and can still leave whole-genome files at 30× coverage at > 30 GB.
An alternative is to create trimmed BAM files that contain only the sequence information relevant for a particular set of scientific questions. For instance, one group may be interested in only mutations in exons, while another may be interested in only reads that support structural variants. Furthermore, depending on the quality of the sequencing library, a large fraction of the low-quality reads can be removed with little effect on downstream analyses. Even further reductions in size can be achieved by removing alignment tags or subsampling reads in areas of high coverage. Read filtering that meets all of these needs could reduce the data footprint while preserving the most relevant information in a compact file. The original BAMs could then be moved to low-cost archival storage, or retained on an external server (e.g. dbGaP) and filtered during retrieval.
Read filtering can also be used directly in analysis pipelines that operate only on a subset of reads. Research questions in different fields (e.g. epigenomics, metagenomics, etc.) will tend to focus on different subsets of the data. Efficient and accurate retrieval of those sequences would facilitate development of such analyses.
To address these issues, we developed VariantBam, a scriptable BAM/CRAM/SAM filtering tool designed to provide a robust and flexible method for retrieving custom sets of reads. VariantBam uses a hierarchical system of filtering rules to determine if a given read should be included in the output. Unique combinations of filtering rules can be applied to different genomic intervals, allowing for the needs of various analyses (e.g. SNPs, exon variants) to be met with a single pass through the sequencing file. VariantBam provides advanced filtering options not available through other filtering tools, including motif matching to a dictionary of sequences, CIGAR parsing, considerations for base-quality scores, and the ability to subsample to a maximum sequence coverage.
VariantBam is written in C ++ and utilizes the rapid C htslib (github.com/samtools/htslib) to perform I/O with high efficiency. Output to BAM, CRAM, SAM and standard output is supported.
2 Methods
To be included in the final output, a read must satisfy one of a set of user-defined rules constructed from the filters listed in Table 1. VariantBam allows the construction of logic statements by combining these filters with logical AND, OR and NOT operations.
Table 1.
VariantBam filtering options
| Filter | Command |
|---|---|
| Insert size | isize[min,max] |
| Mapping quality | mapq[min,max] |
| Phred-trimmed length | phred[min];length[min,max] |
| Alignment flags | e.g.! qcfail |
| Alignment tags | e.g. nm[min,max], xp[min,max] |
| Motif matching | motif[dictionary file] |
| CIGAR parsing | ins[min,max]/del[min,max]/clip[min,max] |
| Number of N bases | nbases[min,max] |
| Subsampling | subsample[percentage] |
| Read-pair orientation | e.g. FR |
| Retrieve all reads | all |
VariantBam can extract reads containing an arbitrarily large dictionary of specified sequence motifs. It uses an implementation of the Aho-Corasick string-matching algorithm to find exact sub-strings in the sequences (sourceforge.net/projects/multifast). This enables efficient queries for dictionaries containing even hundreds of thousands of motifs (see Section 3).
VariantBam can apply these rules for read profiling, keeping track of the number of reads satisfying a given rule. Read-group specific profiles can be plotted with the included R script (Supplementary Fig. S1).
2.1 Regions and mate-linking
VariantBam allows users to specify unique sets of rules on genomic intervals, input as a coordinate string, a VCF or a BED file. A read that belongs to a region and satisfies any single rule will be included in the output. Intervals can be padded to expand to the surrounding region. Regions can also be inverted, such that a read satisfying a rule in an inverted region will be excluded from the final output (e.g. remove reads in centromeres).
To extract reads whose pair-mate (but not the read itself) intersects a region, we introduce the concept of ‘mate-linked’ regions. A read is considered a member of a mate-linked region if either itself or its pair-mate intersects the region. Coordinate queries against mate-linked regions are accomplished efficiently with an interval tree. An example of using mate-linking to extract all read-pairs intersecting SNV sites is provided in Figure 1A and Supplementary Figure S2.
Fig. 1.
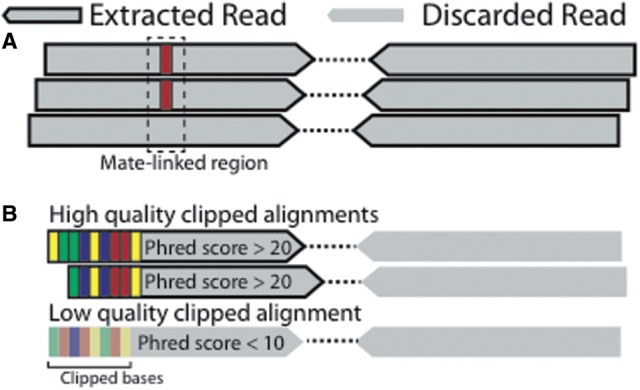
Example applications of two VariantBam rules. (A) A mate-linked region cov ering a SNP site. All reads and their pair-mates that intersect this region are extracted. (B) Genome-wide rule to extract high-quality clipped reads. Bases with low-quality Phred scores do not count as clipped bases
2.2 Quality handling
Poor optical quality (Phred scores) can lead to incorrect base-pair calls in reads, which in turn will decrease the accuracy of the read alignment (Ewing et al., 1998). VariantBam can apply minimum Phred score thresholds before applying rules involving read length, number of clipped bases and existence of a subsequence. For instance, using the Phred filter, a read with n soft-clipped bases containing k low-quality bases will be considered as having only n–k clipped bases (Fig. 1B). This is useful when enriching for high-quality variant supporting reads, where reads with mismatches to the reference due to their low sequence quality should be excluded.
2.3 Subsampling
Sequence alignment artifacts can result in massive pileups of thousands of reads at certain low-complexity regions of the genome (Ross et al., 2013). For most genome analysis applications, these low-information reads can be ignored. To remove them, VariantBam can enforce a maximum or minimum coverage for its output, randomly sub-sampling reads aligned to regions with higher coverage. To ensure that read-pairs are not separated by this subsampling, they are subsampled using a random number generated by hashing the query name. VariantBam also supports subsampling of reads at region-specific rates (e.g. subsample non-SNP sites).
3 Results and discussion
We filtered reads for 10 BAMs, each containing reads representing a lung cancer whole genome at 60× coverage. After completing a first pass analysis with MuTect (Cibulskis et al., 2013), we used VariantBam to keep smaller BAM and CRAM files containing all read-pairs intersecting candidate variant sites, all reads with high quality soft-clips, and all reads containing any one of 700 000 twenty base-pair motifs. We also removed the large original (before recalibration) base-score tag (OQ) for further size reduction. The CPU time to apply these filters was 286 ± 62 (s.d.) min per BAM. The mean size reduction ratio was 3.1:1 (Supplementary Fig. S3). We re-genotyped the full BAMs with FreeBayes v0.9.21 (Garrison and Marth, 2012) in 2531 ± 1175 min, and the filtered BAMs in 873 ± 422 min. We further compared the performance of VariantBam to BamTools filter (Barnett et al., 2011) on the 10 lung cancer tumor BAMs, using a simple filter to extract reads with mapping qualities greater than 30 and with 4 or more mismatches. VariantBam completed in 184 ± 19 min and BamTools completed in 477 ± 61 min.
We have developed an efficient, flexible and robust filtering tool for next-generation sequencing files. The wide-array of filtering options we have described make VariantBam appropriate for filtering and profiling data across a range of bioinformatics needs. VariantBam is easy to compile and run, and is extensively documented with a number of use cases and examples.
Supplementary Material
Funding
JW receives support from the National Institutes of Health (T32 HG002295/HG/NHGRI, U54CA143798) and the DFCI-Novartis Drug Discovery Program. JW and RB receive support from the Pediatric Low-Grade Astrocytoma Foundation. RB is also supported by the Sontag Foundation.
Conflict of Interest: none declared.
References
- Barnett D.W. et al. (2011) BamTools: a C ++ API and toolkit for analyzing and managing BAM files. Bioinformatics, 27, 1691–1692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cibulskis K. et al. (2013) Sensitive detection of somatic point mutations in impure and heterogeneous cancer samples. Nat. Biotechnol., 31, 213–219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ewing B. et al. (1998) Base-calling of automated sequencer traces using phred. I. Accuracy assessment. Genome Res., 8, 175–185. [DOI] [PubMed] [Google Scholar]
- Garrison E, Marth G. (2012) Haplotype-based variant detection from short-read sequencing. arXiv preprint arXiv: 1207.3907 [q-bio.GN]
- Hsi-Yang Fritz M. et al. (2011) Efficient storage of high throughput DNA sequencing data using reference-based compression. Genome Res., 21, 734–740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H. et al. (2009) The Sequence Alignment/Map format and SAMtools. Bioinformatics, 25, 2078–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ross M. et al. (2013) Characterizing and measuring bias in sequence data. Genome Biol., 14, R51. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.