

ABSTRACT
RBPjκ-dependent Notch signaling regulates multiple processes during cartilage development, including chondrogenesis, chondrocyte hypertrophy and cartilage matrix catabolism. Select members of the HES- and HEY-families of transcription factors are recognized Notch signaling targets that mediate specific aspects of Notch function during development. However, whether particular HES and HEY factors play any role(s) in the processes during cartilage development is unknown. Here, for the first time, we have developed unique in vivo genetic models and in vitro approaches demonstrating that the RBPjκ-dependent Notch targets HES1 and HES5 suppress chondrogenesis and promote the onset of chondrocyte hypertrophy. HES1 and HES5 might have some overlapping function in these processes, although only HES5 directly regulates Sox9 transcription to coordinate cartilage development. HEY1 and HEYL play no discernable role in regulating chondrogenesis or chondrocyte hypertrophy, whereas none of the HES or HEY factors appear to mediate Notch regulation of cartilage matrix catabolism. This work identifies important candidates that might function as downstream mediators of Notch signaling both during normal skeletal development and in Notch-related skeletal disorders.
KEY WORDS: HES1, HES5, Chondrogenesis, Chondrocyte hypertrophy, SOX9
Highlighted Article: HES1 and HES5 are new regulators of chondrogenesis and chondrocyte hypertrophy during cartilage development, with HES5 playing a previously unidentified role in modulating Sox9 transcription.
INTRODUCTION
The limb skeleton largely comprises endochondral bones, which initially form as cartilage templates and are ultimately replaced by bone. Cartilage formation of the limb skeleton begins with the migration of mesenchymal progenitor cells (MPCs) from the lateral plate mesoderm into the developing limb field. MPCs undergo rapid proliferation to expand the limb bud, followed by the formation of mesenchymal condensations that give rise to individual cartilage elements through chondrogenesis. This process, which generates mature chondrocytes or cartilage cells from MPCs through differentiation, is primarily driven by the expression and activity of the transcription factor Sry box 9 (SOX9). SOX9 induces and maintains the expression of numerous cartilage-related genes, including collagen type II (Col2a1) and aggrecan (Acan), and also drives growth of cartilage elements (Akiyama et al., 2002; Horton, 2003). As cartilage rudiments continue to develop, chondrocytes near the center of the elements undergo phenotypic and molecular changes known as pre-hypertrophy and hypertrophy, which are regulated and marked by the sequential activation of genes, including Indian hedgehog (Ihh), runt-related transcription factor 2 (Runx2), collagen type X (Col10a1) and matrix metalloproteinase 13 (Mmp13), and the concomitant downregulation of Sox9. The cartilage matrix is ultimately removed by the activity of terminally hypertrophic chondrocytes, which secrete MMP13 to catabolize or degrade the cartilage matrix, creating a scaffold for newly formed osteoblasts to lay down bone matrix (Zuscik et al., 2008).
The Notch signaling pathway is a known regulator of chondrogenesis, chondrocyte hypertrophy, cartilage matrix catabolism and osteoblastogenesis (Dong et al., 2010; Zanotti and Canalis, 2010; Kohn et al., 2012; Liu et al., 2015). Activation of the Notch pathway requires receptor–ligand interactions that initiate a cascade of cleavage events, leading to the release of the Notch intracellular domain (NICD) and translocation to the nucleus, where it forms a ternary transcriptional complex with recombination signal binding protein for immunoglobin κJ Region (RBPjκ; also known as RBPJ) and Mastermind-like (MAML) to activate downstream target genes (Bray, 2006). Recently, several groups have utilized various Notch pathway component loss-of-function (LOF) and gain-of-function (GOF) genetic approaches to study the roles of Notch signaling during cartilage and bone development. For example, utilization of the Prx1Cre transgene to remove RBPjκ floxed alleles (Notch LOF) within MPCs demonstrates an acceleration in chondrogenic and osteoblastic differentiation within the limb skeleton, whereas overexpression of NICD (Notch GOF) within MPCs potently inhibits chondrogenesis and osteogenesis while maintaining and expanding MPCs (Dong et al., 2010). Genetic removal of various Notch signaling components (presenilin1, presenilin2, Notch1, Notch2 and RBPjκ) within MPCs using Prx1Cre or within cartilage progenitor cells using a Col2Cre transgene delays the onset and progression of chondrocyte hypertrophy and cartilage matrix catabolism (Hilton et al., 2008; Kohn et al., 2012), whereas activation of NICD in committed chondrocytes both in vivo and in vitro promotes chondrocyte hypertrophy and cartilage matrix catabolism (Mead and Yutzey, 2009; Kohn et al., 2012). Recently, we have also demonstrated that several of the Notch-mediated effects on cartilage development occur in an RBPjκ-dependent manner (Dong et al., 2010) and are likely to be the consequence of an indirect transcriptional regulation of Sox9 (Kohn et al., 2015). Although the importance of Notch signaling in cartilage development has been well documented, the precise molecular mechanism(s) by which Notch regulates these distinct processes remain unclear or unknown.
Hairy and Enhancer of Split (HES) and Hairy and Enhancer of Split Related (HEY) proteins are bHLH transcription factors, of which several are molecular targets of RBPjκ-dependent Notch signaling and mediate aspects of Notch function within cells. The most well-documented RBPjκ-dependent Notch targets are Hes1, Hes3, Hes5, Hes7, Hey1 and HeyL. HES and HEY transcription factors are largely classified as transcriptional repressors that bind to unique N-box (CACNAG) and E-box (CANNAG) DNA sequences in the promoters of target genes (Kageyama et al., 2007). To determine whether specific HES and HEY factors function during cartilage development and mediate some aspects of Notch signaling in this process, we (1) analyzed HES and HEY gene expression using an in vitro model of chondrogenesis and chondrocyte hypertrophy, followed by further gene and protein expression analyses using in vivo models; (2) developed and analyzed individual and combined HES or HEY gene LOF and GOF mouse models for defects in cartilage development; and (3) tested the ability of specific HES factors to transcriptionally regulate Sox9 during chondrogenic differentiation in vitro and in vivo.
RESULTS
HES and HEY genes are expressed during chondrogenesis and chondrocyte hypertrophy
Previously, we have analyzed the expression and function of Hes1, Hey1 and HeyL during in vitro chondrogenesis using limb-bud micromass cultures and small hairpin (sh)RNA knockdown experiments. These data suggested that Hes1 is important in suppressing MPC differentiation and chondrogenesis, whereas Hey1 and HeyL are dispensable during chondrogenic differentiation (Dong et al., 2010). Here, we have further analyzed the expression of several RBPjκ-dependent Notch target genes of the HES and HEY families throughout both the processes of chondrogenesis and chondrocyte hypertrophy using a different in vitro system. ATDC5 cells were cultured in the presence of insulin–transferrin–selenium (ITS) supplements in order to induce chondrogenic differentiation and maturation. RNA was isolated for gene expression analysis at 7, 14, 21 and 28 days following chondrogenic induction. To monitor the progression of chondrogenesis and chondrocyte hypertrophy, we first examined the expression of the early chondrogenic markers Sox9, collagen type II (Col2a1) and Acan by performing quantitative PCR (qPCR). All early chondrogenic genes peaked in expression at day 21 and decreased by day 28, correlating with the ramping up of chondrogenic differentiation and the transition to hypertrophy (Fig. 1A). The hypertrophic chondrocyte markers Col10a1 and Mmp13 were most highly expressed at day 28, indicating that the majority of cells had reached hypertrophy (Fig. 1A). We next analyzed the expression of Hes1, Hes3, Hes5, Hes7 and Hey1. Hes1 reached peak expression at day 21 and then decreased at day 28 (Fig. 1A). Hes5 was highly expressed at day 7 followed by a reduction in expression at days 14 and 21, and then was elevated again at day 28 (Fig. 1A). When comparing the expression of Hes5 to that of Sox9, there appeared to be an inverse relationship between the two genes such that if one was highly expressed, the other decreased in expression. Hey1 expression was highest at day 28 and lower at earlier stages of chondrogenesis, suggesting a potential role in chondrocyte hypertrophy (Fig. 1A). Other HES genes were also analyzed but largely could not be detected during chondrogenesis and chondrocyte hypertrophy of ATDC5 cells in culture.
Fig. 1.

HES and HEY expression during chondrogenesis and chondrocyte hypertrophy. (A) qPCR gene expression analyses for Sox9, Col2a1, Acan, Col10a1, Mmp13, Hes1, Hes5 and Hey1 on RNA isolated from ATDC5 cells cultured for 7–28 days in ITS-supplemented medium. (B) qPCR analysis for Sox9, Hes1 and Hes5 on RNA isolated from WT limb buds at E10.5 and E11.5. The y-axis represents relative gene expression normalized to that of β-actin and then to expression in cultures at day 7. Bars represent means±s.d. *P<0.05 (one-way ANOVA followed by Bonferroni method and Student's t-test). Bars under asterisks identify relevant statistical comparisons. (C) Western blot analyses for HES5, HES1, SOX9 and β-actin using protein lysates from WT limb buds at E10.5 and E11.5. Numbers represent the fold change in protein abundance as compared to E10.5 samples for the representative blot.
To determine whether HES factors demonstrate similar expression profiles during chondrogenesis in vivo, we isolated RNA and protein from wild-type (WT) embryonic day 10.5 and 11.5 (E10.5 and E11.5) limb buds. Using qPCR, we observed a similar trend between Hes1 and Sox9, as well as between Hes5 and Sox9; that is, increased Hes1 expression was observed as Sox9 increased between E10.5 and E11.5, whereas Hes5 expression decreased as Sox9 expression increased between E10.5 and E11.5 (Fig. 1B). We then analyzed HES1 and HES5 protein expression in E10.5 and E11.5 limb buds using western blot analysis. Similar to the qPCR data, we again observed an increase of HES1 as SOX9 increased between E10.5 and E11.5, whereas HES5 levels decreased as SOX9 increased between E10.5 and E11.5 (Fig. 1C). Collectively, these data demonstrate that both Hes1 and Hes5 are expressed throughout chondrogenesis and chondrocyte hypertrophy, and suggest that HES5 is a negative regulator of Sox9, consistent with the transcriptional repressive role for most HES factors and the suppressive role of RBPjκ-dependent Notch signaling during chondrogenesis (Kageyama et al., 2005; Dong et al., 2010).
HES1 is dispensable for MPC differentiation, potentially owing to compensatory expression of HES5
As stated previously, HES1 is an RBPjκ-dependent Notch target gene that is capable of suppressing in vitro chondrogenesis in limb-bud micromass cultures (Dong et al., 2010). To determine whether the specific loss of Hes1 in MPCs can induce a similar acceleration of chondrogenesis in vivo, we generated and analyzed Prx1Cre;Hes1f/f (Hes1 LOF) embryos. This genetic targeting strategy allows for the specific removal of Hes1 floxed alleles within MPCs of the developing limbs. Using whole-mount in situ hybridization (WISH), we analyzed WT and Hes1 LOF mutant embryos at E12.5; however, we did not observe any change in Sox9 (Fig. S1Aa,Ab) or Col2a1 (Fig. S1Ac,Ad) expression. RNA was then isolated from whole limb buds of both WT and Hes1 LOF E12.5 embryos. Using qPCR analyses, we did not observe any change in expression of the chondrogenic markers Sox9, Col2a1 and Acan (Fig. S1B). This data was surprising because we had expected to observe an acceleration in chondrogenesis as previously observed in Hes1 LOF in vitro models of chondrogenesis and other Notch LOF in vivo models (Dong et al., 2010). To determine whether other HES or HEY genes were compensating for the loss of Hes1, we isolated RNA from WT and Hes1 LOF whole limb buds at E11.5. HES and HEY genes are prominently expressed in undifferentiated MPCs of the developing limb bud at E11.5 (Dong et al., 2010). Using qPCR analyses, we observed increased Hes5 gene expression in Hes1 LOF limb buds compared to that in WT controls (Fig. S1B). Additionally, we analyzed the expression of Hes3, Hes7, Hey1 and HeyL, but did not observe any change in expression in Hes1 LOF limb buds compared to WT controls, or the expression levels were too low to be reliably detected. These data suggest the potential for HES5 to compensate for the lack of HES1 during MPC differentiation in our Hes1 LOF embryos. This compensatory effect of HES factors has been well documented in other cells systems, such as neural progenitor cells (Hatakeyama et al., 2004).
Removal of multiple HES factors in MPCs accelerates differentiation and chondrogenesis
Owing to the compensatory increase in Hes5 expression following the conditional removal of Hes1 (Fig. S1B), we analyzed embryos in which both HES factors had been deleted from MPCs. We generated Prx1Cre;Hes1f/f;Hes5−/− (Hes1,5 LOF) and WT embryos to analyze cartilage development of the limb skeleton. We first confirmed genetic removal of Hes1 and Hes5 by analyzing their expression using qPCR on RNA isolated from E10.5 Hes1,5 LOF and WT limb buds (Fig. 2A). Using immunohistochemistry to detect SOX9 on E10.5 Hes1,5 LOF and WT control limb-bud sections, we observed a subsequent increase in the expression of SOX9 in Hes1,5 LOF limb buds as compared to that in WT mice (Fig. 2Ba,Bb). These data demonstrate an early acceleration in MPC differentiation and chondrogenesis. We next analyzed E11.5 Hes1,5 LOF and WT embryos. Similar to E10.5 limb-bud sections, we performed immunohistochemistry for SOX9 and observed increased SOX9 in the Hes1,5 LOF limb buds as compared to that in WT (Fig. 2Bc,Bd). We also performed qPCR on RNA that had been isolated from E11.5 Hes1,5 LOF and control limb buds. At E11.5, we observed a significant increase in both Sox9 and Col2a1 expression in Hes1,5 LOF limb buds (Fig. 2A), further suggesting an acceleration in MPC differentiation and chondrogenesis in the absence of Hes1 and Hes5. Lastly, we analyzed expression of COL2A1 and ACAN protein and RNA at E12.5 in WT and Hes1,5 LOF limb buds. Alcian Blue–Hematoxylin–Orange-G (ABH–OG) staining, which stains cartilage matrix blue, showed that Hes1,5 LOF mutant limbs displayed enhanced or accelerated cartilage formation in the radius and ulna, with more clearly defined joint formation in regions of the developing humerus and ulna when compared to WT sections (Fig. 2Ca,Cb). Protein analysis using immunohistochemistry demonstrated altered COL2A1 expression that was associated with advanced secondary chondrogenesis and joint formation (Fig. 2Cc–Cf) and an increase in ACAN expression (Fig. 2Cg–Cj) in E12.5 Hes1,5 LOF forelimbs as compared to WT controls. Further analysis at this time point revealed a change in cellular morphology. As chondrocytes mature prior to hypertrophy, they begin to flatten (Zuscik et al., 2008). In E12.5 Hes1,5 LOF limb buds, we observed more flattened cells compared to those in WT controls (Fig. 2Ci,Cj). This indicates that the cells in the Hes1,5 LOF limb bud are beginning to form the columnar zone of proliferating chondrocytes, which occurs just before hypertrophy. This was not observed as clearly in the WT control sections. Lastly, we analyzed RNA from E12.5 WT and Hes1,5 LOF mutant limb buds using qPCR. Gene expression analysis showed that the loss of Hes1 and Hes5 resulted in a significant increase in Col2a1 and Acan expression (Fig. 2A). We did not observe any change in Sox9 expression at this time point, which is probably owing to the fact that as chondrocytes begin to mature, Sox9 levels decrease (Fig. 2A). Collectively, these data demonstrate that Hes1 and Hes5 are necessary for appropriate MPC differentiation and chondrogenesis.
Fig. 2.
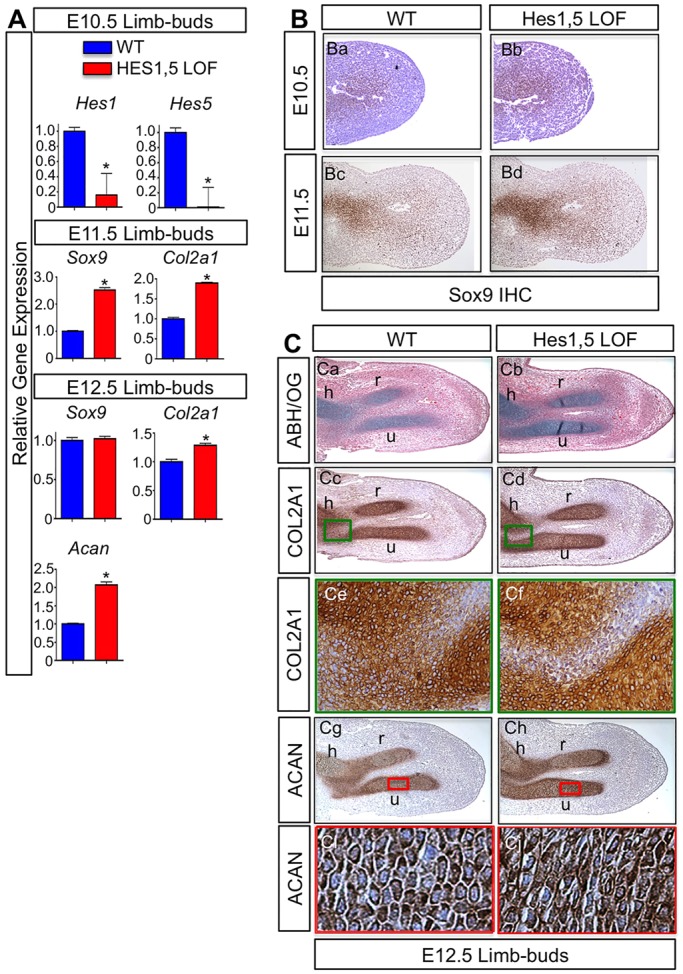
Loss of Hes1 and Hes5 in MPCs accelerates chondrogenesis. (A) qPCR gene expression analyses for Hes1, Hes5, Sox9, Col2a1 and Acan from RNA isolated from Prx1Cre;Hes1f/f;Hes5−/− (Hes1,5 LOF) and WT forelimbs at E10.5, E11.5 and E12.5. The y-axis represents relative gene expression normalized to that for β-actin then to the WT control. Bars represent means±s.d. *P<0.05 (two-tailed Student's t-test). (B) Immunohistochemistry (IHC) analyses of SOX9 expression in E10.5 (Ba,Bb) and E11.5 (Bc,Bd) WT and Hes1,5 LOF forelimb sections. (C) Histological and immunohistochemistry analyses on WT and Hes1,5 LOF forelimb sections at E12.5 using ABH–OG (ABH/OG) staining (Ca,Cb), COL2A1 immunohistochemistry (Cc–Cf) and ACAN immunohistochemistry (Cg–Cj). h, humerus; r, radius; u, ulna. Green box within the joint region (Cc,Cd) identifies accelerated secondary chondrogenesis and joint formation in Hes1,5 LOF forelimbs compared to WT (Ce,Cf). Red box within the ulna (Cg,Ch) annotates the region of interest for changes in cellular morphology in Hes1,5 LOF forelimbs compared to WT (Ci,Cj).
HES1 overexpression in MPCs delays chondrogenesis and induces MPC proliferation
To determine whether Hes1 is sufficient to suppress chondrogenesis, we analyzed Prx1Cre;Rosa-Hes1f/f [Hes1 gain-of-function (Rosa-Hes1)] embryos across multiple time points. These experiments were conducted using a mouse line in which Hes1 was targeted to the Rosa26 locus containing a transcriptional stop sequence flanked by loxP sites upstream of the Hes1 cassette. When crossed with the Prx1Cre mouse line, Hes1 is continuously overexpressed in MPCs of the developing limb buds (Kobayashi and Kageyama, 2010). We first analyzed Hes1 and chondrogenic gene expression from E12.5 Rosa-Hes1 and WT limb buds by using qPCR. We observed a significant overexpression of Hes1 and a significant decrease in both Col2a1 and Acan expression in Rosa-Hes1 limb buds as compared to WT (Fig. 3A). Interestingly, Sox9, Sox5 and Sox6 gene expression was largely unchanged in Rosa-Hes1 limb buds as compared to WT (Fig. 3A). Histological analyses of E12.5 Rosa-Hes1 and WT limb bud sections using ABH–OG staining demonstrated a reduction in proteoglycan content in Rosa-Hes1 developing hindlimbs as compared to WT sections (Fig. 3Ba,Bb). Similar to the decrease in ABH–OG staining, immunohistochemistry analyses showed decreased expression of COL2A1 (Fig. 3Bc,Bd) and ACAN (Fig. 3Be,Bf) in the Rosa-Hes1 hindlimbs compared to WT sections.
Fig. 3.

Overexpression of Hes1 increases MPC proliferation and delays chondrogenesis. (A) qPCR gene expression analyses for Hes1, Sox9, Sox5, Sox6, Col2a1 and Acan from RNA isolated from WT and Prx1Cre;Rosa-Hes1f/f [Hes1 gain-of-function (Rosa-Hes1)] limb buds at E12.5. The y-axis represents gene expression normalized to that for β-actin then to WT control. Bars represent means±s.d. *P<0.05 (two-tailed Student's t-test). (B) Histological and immunohistochemistry analyses on WT and Rosa-Hes1 hindlimb sections at E12.5 using ABH–OG (ABH/OG) staining (Ba,Bb), COL2A1 immunohistochemistry (Bc,Bd) and ACAN immunohistochemistry (Be,Bf). fe, femur; fi, fibula; t, tibia. Numbers indicate the relative change in expression intensity for COL2A1 and ACAN as compared to WT (normalized to 1.0). (C) BrdU immunohistochemistry analysis on sections from WT and Rosa-Hes1 forelimbs at E11.5 (Ca,Cb). Red boxes outline regions in which BrdU-positive and BrdU-negative cells were quantified. Statistical analysis shows a significant increase in BrdU-positive cells in Rosa-Hes1 limb buds compared to WT (Cc). The y-axis represents the percentage of BrdU-positive cells compared to the total number of cells counted. Bars represent means±s.d. *P<0.05 (by two-tailed Student's t-test). (D) Western blot analysis for CYCD1 and β-actin expression in Rosa-Hes1 limb buds compared to WT. Numbers represent the fold change in protein abundance as compared to WT samples for the representative blot.
To determine whether overexpression of Hes1 also affects MPC proliferation, we used BrdU immunohistochemistry to analyze Rosa-Hes1 and WT forelimbs. Similar to Notch gain-of-function in MPCs (Dong et al., 2010), we observed an increase in BrdU-positive MPCs in the Rosa-Hes1 forelimbs compared to WT (Fig. 3Ca,Cb). The red box outlines the area in which cells (both BrdU positive and BrdU negative) were counted. This region of the limb bud is just beyond the highly proliferative apical zone, where MPCs normally begin the differentiation process. Statistical analysis verified a significant increase in the percentage of BrdU-positive cells in the Rosa-Hes1 forelimbs compared to WT controls (Fig. 3Cc). We also isolated protein from E11.5 Rosa-Hes1 and WT limb buds to perform western blot analysis of the proliferative marker cyclinD1 (CYCD1). Rosa-Hes1 limb buds demonstrated increased CYCD1 protein as compared to WT controls, further validating the observed increase in proliferation (Fig. 3D). Collectively, these data demonstrate that Hes1 is sufficient to delay chondrogenesis by suppressing Col2a1 and Acan gene expression, and is also capable of expanding the MPC population during early limb development.
HES, but not HEY, factors regulate early chondrocyte hypertrophy, potentially through suppression of SOX9
We have shown previously that RBPjκ-dependent Notch signaling is an important regulator of the onset and progression of chondrocyte hypertrophy (Hilton et al., 2008; Kohn et al., 2012; Kohn et al., 2015). To determine whether HES1 regulates the onset and progression of chondrocyte hypertrophy, we first analyzed Hes1 LOF and WT embryos at E14.5. Hes1 LOF forelimbs were analyzed using histological staining and in situ hybridization for markers of chondrocyte maturation. ABH–OG staining of E14.5 Hes1 LOF mutant forelimbs revealed a mild and largely inconsistent decrease in the length of the hypertrophic zone (Fig. S1Ca,Cb), which was also revealed by reduced domains of Col10a1 (Fig. S1Cc,Cd) and Mmp13 (Fig. S1Ce,Cf) expression as compared to WT controls. Collectively, these data suggest that Hes1 plays a limited role in regulating the onset of chondrocyte hypertrophy at E14.5 or that compensation by other HES factors, such as HES5, blunt the effect of Hes1 LOF alone.
To determine whether Hes5 compensatory expression also affects chondrocyte hypertrophy in Hes1 LOF mutants, we generated and analyzed E13.5 Hes1,5 LOF mutant and WT embryos using histological approaches. Hematoxylin and eosin (H&E) staining of humerus sections demonstrated that Hes1,5 LOF mutants have a smaller hypertrophic zone compared to WT controls (Fig. 4Aa,Ab). In situ hybridization for the hypertrophic chondrocyte marker Col10a1 indicated a clear lack of expression in E13.5 Hes1,5 LOF mutant humerus sections compared to WT (Fig. 4Ac,Ad). To understand the potential mechanism underlying this delay in the onset of hypertrophy, we used immunohistochemistry to analyze the protein expression of SOX9. Analysis of E13.5 Hes1,5 LOF mutant and WT humerus sections demonstrated that Hes1,5 LOF mutants exhibited more continuous expression of SOX9 compared to WT controls within cells of the central regions of elements poised for hypertrophy (Fig. 4Ae,Af). These data suggest that the delay in the onset of chondrocyte hypertrophy is due to the maintenance of SOX9 expression.
Fig. 4.

Loss of Hes1 and Hes5 delays chondrocyte hypertrophy owing to prolonged SOX9 expression. (A) H&E staining (Aa,Ab), in situ hybridization for Col10a1 (Ac,Ad) and immunohistochemistry for SOX9 (Ae,Af) on WT and Prx1Cre;Hes1f/f;Hes5−/− (Hes1,5 LOF) humerus sections at E13.5. Black circles outline the hypertrophic zones. (B) H&E staining (Ba,Bb), in situ hybridization for Col10a1 (Bc,Bd) and Mmp13 (Be,Bf) and immunohistochemistry for SOX9 (Bg–Bj) on WT and Hes1,5 LOF humerus sections at E14.5. Red box denotes higher magnification images shown in Bi,Bj. (C) Statistical assessment of the hypertrophic zone lengths relative to the total lengths of the cartilage rudiments for WT and Hes1,5 LOF forelimbs. Bars represent means±s.d. *P<0.05 (two-tailed Student's t-test).
To determine whether the loss of Hes1 and Hes5 results in a continuous delay in chondrocyte hypertrophy, we analyzed E14.5 Hes1,5 LOF mutant and WT limb skeletons. H&E staining revealed a decrease in the length of the hypertrophic zone in E14.5 Hes1,5 LOF humerus sections as compared to WT (Fig. 4Ba,Bb). Furthermore, Hes1,5 LOF forelimbs exhibit a decrease in the Col10a1 (Fig. 4Bc,Bd) and Mmp13 (Fig. 4Be,Bf) expression domains at E14.5. To determine whether this continued delay in hypertrophy is due to altered SOX9 expression, we used immunohistochemistry analysis. Similar to the Hes1,5 LOF data at E13.5, we observed maintenance of SOX9 expression deeper into the hypertrophic zone of E14.5 mutant forelimbs (Fig. 4Bg–Bj). To determine whether this decrease in the size of the hypertrophic zone was statistically significant, we measured the length of the hypertrophic zones and normalized those values to the total lengths of the cartilage elements for WT and Hes1,5 LOF mutants. Quantitative analysis showed a significant decrease in the length of the hypertrophic zone in the Hes1,5 LOF mutant forelimbs compared to that in WT (Fig. 4C). To ensure this delay in hypertrophy was not due to changes in proliferation, we utilized BrdU immunohistochemistry analysis and did not observe any change in the percentage of BrdU-positive cells in Hes1,5 LOF humerus sections as compared to WT (Fig. S2A,B). Combined, these data demonstrate that HES1 and HES5 control the pace of chondrocyte hypertrophy, potentially through regulation of SOX9.
We next analyzed terminal chondrocyte hypertrophy and cartilage matrix catabolism using E18.5 Hes1,5 LOF and WT humerus sections. Interestingly, no delay in terminal chondrocyte hypertrophy or cartilage matrix turnover was observed between Hes1,5 LOF and WT cartilage elements, as indicated by similar zones of hypertrophy in ABH–OG-stained sections (Fig. S2Ca,Cb) and a lack of any change in MMP13 expression (Fig. S2Cc,Cd). This was consistent with data demonstrating that genetic removal of Hes1 alone within MPCs caused no obvious defects in terminal chondrocyte hypertrophy and cartilage matrix turnover, as indicated by similar zones of hypertrophy in E18.5 Hes1 LOF and WT H&E-stained sections (Fig. S2Da,Db), and similar expression domains of Ihh (Fig. S2Dc,Dd), Col10a1 (Fig. S2De,Df) and Mmp13 (Fig. S2Dg,Dh). Surprisingly, neither Hes1 LOF or Hes1,5 LOF mutants appeared to exhibit an expanded hypertrophic zone at E18.5, which has been observed previously in several Notch LOF mutant mice and is indicative of a continuous delay in terminal chondrocyte hypertrophy and cartilage matrix catabolism (Hilton et al., 2008; Mead and Yutzey, 2009; Kohn et al., 2012; Kohn et al., 2015). Based on these data, combined with the observation that HEY factor expression increases in maturing and hypertrophic chondrocytes (Fig. 1A), we obtained and analyzed E14.5 and E18.5 Hey1−/−; HeyL−/− double mutant (Hey1,HeyL LOF) and control embryos for defects in the onset and progression of chondrocyte hypertrophy and cartilage matrix catabolism. H&E staining (Fig. S3Aa,Ab,Ba,Bb) and in situ hybridization for Ihh (Fig. S3Ac,Ad,Bc,Bd), Col10a1 (Fig. S3Ae,Af,Be,Bf) and Mmp13 (Fig. S3Ag,Ah,Bg,Bh) on tibia sections at E14.5 and E18.5 demonstrate no obvious changes in the hypertrophic zones between Hey1,HeyL LOF and control cartilage elements. Collectively, these data demonstrate that HES factors (particularly HES5) primarily control the onset of chondrocyte hypertrophy during cartilage maturation, potentially through regulation of SOX9, whereas neither the HES nor HEY factors appear to control terminal chondrocyte hypertrophy or cartilage matrix catabolism.
HES1 overexpression in MPCs delays chondrocyte hypertrophy and inhibits skeletal growth
To determine whether Hes1 overexpression in MPCs affects chondrocyte proliferation and hypertrophy during cartilage development and maturation, we first examined Rosa-Hes1 and WT E14.5 skeletal preparations with Alcian-Blue staining. Rosa-Hes1 cartilage rudiments were shorter, and the limbs as a whole were smaller than those of WT controls (Fig. 5Aa–Ad). In the most severely affected Rosa-Hes1 forelimbs (Fig. 5Ab) and hindlimbs (Fig. 5Ad), we observed a hypoplastic or missing radius and/or fibula (black arrows). Distal cartilage rudiments appeared to be more severely affected as compared to proximal elements, and hindlimbs were more affected than forelimbs (Fig. 5Aa–Ad). Analysis of Alcian-Blue-stained hindlimbs showed the formation of a defined hypertrophic zone in WT cartilage rudiments (Fig. 5Ac) (red asterisks), although these were largely absent in severely affected Rosa-Hes1 mutants at this stage (Fig. 5Ad). ABH–OG staining of E14.5 humerus sections (the least affected proximal element) (Fig. 5Ba,Bb) demonstrated that Rosa-Hes1 mutants exhibited only a mild delay in chondrocyte hypertrophy as compared to WT controls, with only minor changes in Col10a1 (Fig. 5Bc,Bd) and Mmp13 (Fig. 5Be,Bf) expression within the hypertrophic zone.
Fig. 5.

Overexpression of Hes1 delays chondrocyte hypertrophy, and reduces chondrocyte proliferation and skeletal growth. (A) Alcian-Blue-stained skeletal analysis of Prx1Cre;Rosa-Hes1f/f (Rosa-Hes1) and WT forelimbs (Aa,Ab) and hindlimbs (Ac,Ad) at E14.5. Black arrows depict the missing radius in the forelimbs, and missing fibula in the hindlimbs of Rosa-Hes1 embryos. The red asterisks depict formation of the hypertrophic zone in the WT hindlimbs. (B) ABH–OG (ABH/OG) staining (Ba,Bb) and in situ hybridization for Col10a1 (Bc,Bd) and Mmp13 (Be,Bf) on WT and Rosa-Hes1 humerus sections at E14.5. (C) BrdU immunohistochemistry on E14.5 WT and Rosa-Hes1 humerus sections (Ca,Cb). Statistical analysis showing the percentage of BrdU-positive cells in WT and Rosa-Hes1 humerus sections (Cc) at E14.5. (D) Alcian-Blue and Alizarin-Red skeletal analyses of WT and Rosa-Hes1 proximal forelimb (FL) elements (Da,Db), distal forelimb elements (Dd,De), proximal hindlimb (HL) elements (Dg,Dh) and distal hindlimb elements (Dj,Dk) at E18.5. Statistical analysis of the lengths of WT and Rosa-Hes1 humeri (Dc), ulnae (Df), femurs (Di) and tibiae (Dl). Bars represent means±s.d. *P<0.05 (two-tailed Student's t-test).
To assess whether changes in chondrocyte proliferation could contribute to the Rosa-Hes1 skeletal phenotype, we performed BrdU staining on E14.5 Rosa-Hes1 and WT humerus sections (Fig. 5Ca,Cb). Consistent with the reduced size observed in most elements of Rosa-Hes1 mutants, we observed a decrease in the percentage of BrdU-positive chondrocytes (Fig. 5Cc). We next analyzed overall growth changes of skeletal elements at E18.5 using Rosa-Hes1 and WT forelimb and hindlimb skeletal preparations (Fig. 5Da,Db,Dd,De,Dg,Dh,Dj,Dk). Analyses indicated that the total lengths of these bones were significantly shorter in Rosa-Hes1 mutants as compared to those of WT controls, with the most prominent effects occurring on distal elements (Fig. 5Dc,Df,Di,Dl). Interestingly, Rosa-Hes1 mutant mice survive to adulthood and present with various skeletal anomalies, including alterations to skeletal patterning, bone ridge or tuberosity development, and digit number. These phenotypes are the likely result of the early and broad effects of the Prx1Cre transgene controlling Hes1 overexpression in skeletal progenitors. The precise cellular and molecular mechanisms underlying each of these peripheral phenotypes will be explored and described elsewhere. The data presented here suggest that Hes1 overexpression in MPCs can delay chondrocyte hypertrophy and reduce chondrocyte proliferation, although these effects might be secondary to the delay in chondrogenesis described above.
HES5 directly regulates Sox9 expression
Because chondrogenic differentiation from MPCs and early chondrocyte hypertrophy are coordinated by the expression and activity of SOX9, we next examined whether HES factors can transcriptionally regulate Sox9 expression. Above, we demonstrated that Hes1 overexpression in MPCs in vivo is sufficient to repress Col2a1 and Acan expression without affecting Sox9 expression (Fig. 3A). To determine whether HES5 is capable of regulating Sox9 expression, we first transfected ATDC5 chondrogenic cells with either Flag (control) or Hes5 overexpression constructs. After seven days in chondrogenic differentiation medium, RNA was isolated for qPCR analysis from each group. Hes5 overexpression resulted in a notable reduction of Sox9, Sox5 and Sox6, as well as chondrogenic genes such as Col2a1 and Acan, although to a lesser degree at this time point (Fig. 6A). These data suggest that HES5 is sufficient to downregulate or delay chondrogenesis in vitro and that it might directly regulate Sox9 expression.
Fig. 6.

HES5 inhibits Sox9 expression through direct transcriptional regulation and is sufficient to delay chondrogenesis. (A) qPCR gene expression analyses for Hes5, Sox9, Sox5, Sox6, Col2a1 and Acan from RNA isolated from ATDC5 cultures at day 7. The y-axis represents relative gene expression normalized to that for β-actin then to Flag controls. Bars represent means±s.d. *P<0.05 (two-tailed Student's t-test). (B) ChIP analysis for input genomic DNA (positive control), IgG (negative control) pull down, HES1 pull down and HES5 pull down (Ba). Lane 1 shows input genomic DNA amplification using primer set 1 (P1). Lane 2 shows input genomic DNA amplification using primer set 2 (P2). Lane 3 shows no amplification using P1 primers with an IgG pull down. Lane 4 shows no amplification using P2 primers with an IgG pull down. Lane 5 shows no amplification using P1 primers with a HES1 pull down. Lane 6 shows no amplification using P2 primers with HES1 pull down. Lane 7 shows no amplification using P1 primers with HES5 pull down. Lane 8 shows amplification using P2 primers with a HES5 pull down. qPCR on DNA from the HES5 pulldown ChIP assay using limb buds at E10.5 and E11.5 (Bb). The y-axis is the relative amplification normalized to the input control then to the expression at E10.5. Bars represent means±s.d. *P<0.05 (two-tailed Student's t-test). (C) C3H10T1/2 cells or ATDC5 cells co-transfected with (Ca) a 1-kb Sox9-promoter-driven luciferase construct, Renilla plasmid and over expression plasmids for Flag, NICD1 or HES5, or (Cb) with a 1-kb Sox9-promoter-driven luciferase construct containing an N-box mutation, Renilla plasmid and over expression plasmids for Flag, NICD1 or HES5. The y-axis is relative luciferase activity normalized to Renilla then to the Flag control. Bars represent means±s.d. *P<0.05 (two-tailed Student's t-test).
Previous studies have demonstrated RBPjκ-dependent Notch regulation of Sox9; however, the exact mechanism remains unknown or controversial (Mead and Yutzey, 2009; Dong et al., 2010; Kohn et al., 2012; Chen et al., 2013; Kohn et al., 2015). Recent data have suggested that the Notch-mediated transcriptional regulation of Sox9 occurs indirectly through secondary effectors (Kohn et al., 2015). Therefore, we first used a bioinformatics approach to search the Sox9 promoter for HES binding sites – N-box or E-box sequences (Fig. S4A). We identified two N-box and/or E-box sequences within the first kilobase of the Sox9 promoter that were 100% conserved between the mouse and human genomes (Fig. S4C). To determine whether HES5 directly binds to this region of the Sox9 promoter in MPCs and in chondrogenic cells in vivo, we utilized chromatin immunoprecipitation (ChIP) assays on DNA isolated from WT E10.5 and E11.5 limb buds. As previously indicated, Hes5 and Sox9 expression demonstrated an inverse relationship in E10.5 and E11.5 limb buds (Fig. 1Ba,Bb). Primers were designed to amplify the region of the Sox9 promoter containing the N-box and/or E-box (Fig. S4B, red arrows), and a negative control region approximately 19 kb upstream of the N-box and/or E-box sequences (Fig. S4B, green arrows). ChIP analysis at E11.5 using an antibody against HES5 revealed amplification of DNA when using primers flanking the N-box and/or E-box sequence (Primer 2; P2) (Lane 8), and no amplification when using primers targeting an upstream region of the Sox9 promoter (Primer 1; P1) (Lane 7) (Fig. 6Ba). Interestingly, no amplification of either primer set was observed when ChIP analyses were performed with an antibody against HES1 (Lanes 5 and 6) (Fig. 6Ba), indicating specificity of binding to the N-box and/or E-box site for HES5. We also observed amplification of the positive control – the sheared genomic DNA (Lanes 1 and 2) – when using P1 and P2 primers, whereas IgG pull downs showed no amplification (Lanes 3 and 4) (Fig. 6Ba). By performing qPCR on DNA pulled down during ChIP assays from E10.5 and E11.5 limb buds, we were able to determine that the occupancy of HES5 on the Sox9 promoter region was greater at E10.5 than at E11.5 (Fig. 6Bb). These data suggest that RBPjκ-dependent Notch regulation of Sox9 works, in part, through direct HES5 transcriptional activity.
Finally, we utilized a 1.0-kb Sox9-promoter-driven luciferase construct that included the N-box and/or E-box sequence to demonstrate the direct transcriptional regulation of Sox9 by HES5. When this construct was co-transfected with Flag, Notch 1 intracellular domain (NICD1) and HES5 expression vectors, we observed a significant and similar level of suppression of luciferase activity between NICD1- and HES5-transfected groups when compared to the Flag-transfected control (Fig. 6Ca). However, when we co-transfected a 1.0-kb Sox9-promoter-driven luciferase construct containing a mutated N-box sequence with Flag or HES5 expression vectors, we observed no change in luciferase activity (Fig. 6Cb). Collectively, these data demonstrate that the RBPjκ-dependent Notch target HES5 directly binds to the Sox9 promoter through the N-box sequence and is capable of downregulating Sox9 expression in MPCs and chondrogenic cells.
DISCUSSION
HES and HEY factors are well-known RBPjκ-dependent Notch target genes, which are capable of mediating several aspects of Notch function in various settings (Cau et al., 2000; Hirata et al., 2001; Zine et al., 2001; Kageyama et al., 2007). Previous in vitro studies have implicated HES1 as a potential suppressor of chondrogenesis, as well as a potential transcriptional regulator of the Col2a1 and Acan promoters (Grogan et al., 2008; Dong et al., 2010); however, the in vivo evidence for HES regulation of cartilage development has been lacking. Our results demonstrate that MPC-specific deletion of Hes1 alone is not sufficient to affect cartilage development, and that the additional removal of Hes5 is required to alter both chondrogenesis and the onset of chondrocyte hypertrophy, potentially owing to the compensatory expression of Hes5 in vivo. Interestingly, mutant mice in which Hes1had been deleted in more committed osteo-chondro progenitors and combined with conventional deletion of Hes5 (Col2Cre;Hes1f/f;Hes5−/−) failed to show any defects in cartilage development (Karlsson et al., 2010). Importantly, this study only analyzed embryos at E16.5 and later time points during endochondral bone development, thereby potentially missing the earlier defects we have described here. Alternatively, Col2Cre;Hes1f/f;Hes5−/− mutant mice might not have developed defects such as those observed in our study at E10.5–15.5 because the Col2Cre transgene targets a more committed osteo-chondro progenitor population. Prx1Cre;Hes1f/f ;Hes3−/−;Hes5−/− mutant mice have also previously been generated and shown to have increased postnatal bone mass that is consistent with other Notch LOF mutant mice (Hilton et al., 2008; Tu et al., 2012), although only limited late-stage embryonic skeletal analyses were performed that showed no obvious phenotype (Zanotti et al., 2011). Similarly, Hey1+/−; HeyL−/− mutant mice have been shown to have increased bone mass as compared to controls at late postnatal and adult time points (Tu et al., 2012). Therefore, although these studies have implicated HES and HEY factors in the regulation of postnatal bone development and homeostasis, they missed the important roles of specific HES factors during cartilage development of the limb skeleton.
Here, we report the first in vivo genetic evidence demonstrating that the RBPjκ-dependent Notch target genes Hes1 and Hes5 act as regulators of chondrogenesis and chondrocyte hypertrophy during cartilage development. We have demonstrated that HES1 is dispensable for normal MPC differentiation and chondrogenesis, probably owing to compensatory expression of Hes5. However, HES1 is sufficient to delay chondrogenesis by acting downstream of the SOX trio (SOX9, SOX5, SOX6), in addition to inducing MPC proliferation. Therefore, removal of both Hes1 and Hes5 in MPCs accelerates chondrogenesis and delays the onset of chondrocyte hypertrophy. Consistent with recent Notch LOF studies, the accelerated chondrogenesis and delay in the onset of chondrocyte maturation is likely to be the result of increased SOX9 expression (Kohn et al., 2015). We have identified HES5 as a direct transcriptional modifier of Sox9 gene expression, which in turn has direct influences on Sox5 and Sox6 gene regulation to coordinate chondrogenesis. However, HES1 is likely to mediate control of chondrogenesis and cartilage development through direct downstream regulation of other chondrogenic genes, such as Col2a1 and Acan (Grogan et al., 2008). Our work has also demonstrated that neither the removal of individual nor multiple HES or HEY genes alters terminal chondrocyte hypertrophy or cartilage matrix catabolism during normal development, thereby suggesting that the delayed terminal hypertrophy and cartilage matrix catabolism observed in other Notch LOF mutants (Hilton et al., 2008; Mead and Yutzey, 2009; Kohn et al., 2012, 2015) are the result of RBPjκ-dependent and HES- and HEY-independent signaling mechanisms. Interestingly, genetic removal of Hes1 within postnatal cartilages following joint injury in a murine model of osteoarthritis is capable of reducing Mmp13 expression as well as that of other cartilage-catabolizing enzymes, whereas prolonged overexpression of Hes1 has also been shown to induce some of these same catabolic genes in vitro (Sugita et al., 2015). Therefore, it is possible that HES, and potentially HEY, factor regulation of catabolic gene expression might only be evident in pathological or injury and/or inflammation contexts, and that during development, Notch signaling regulates terminal chondrocyte hypertrophy and cartilage matrix catabolism through alternative mechanisms. Alternatively, numerous HES and/or HEY factors might contribute to the regulation of this aspect of Notch function in cartilage, and therefore would require the elimination of nearly all HES and HEY genes simultaneously to uncover their requisite role in regulating cartilage catabolism during normal development.
Although the importance of Notch signaling in skeletal development, injury and disease has recently come to light, we are just beginning to learn about the underlying Notch-mediated molecular mechanisms that control these distinct events. Identifying the molecular players and their function would not only provide us with additional important molecules to consider when examining skeletal disorders but would also lead to the generation of additional drug targets for the treatment of these conditions or ailments. For example, Hajdu Cheney Syndrome (HCS) is a rare heritable multi-organ connective tissue disorder that presents with skeletal features such as skull deformities, short stature, joint laxity and a severe reduction in bone mass or osteoporosis, and is caused by heterozygous mutations in the NOTCH2 receptor (Majewski et al., 2011; Simpson et al., 2011). It has been recently discovered that these mutations lead to Notch GOF within connective tissue cells (Majewski et al., 2011; Simpson et al., 2011), but the precise downstream effectors that drive the pathology are unknown. Adams-Oliver Syndrome (AOS) is another rare heritable disorder characterized by skin and limb defects including hypoplastic or shortened digits, absence of bones in hands or the feet, as well as partial or complete absence of the lower legs (tibia, fibula and digits). AOS is an autosomal dominant disorder caused by mutations in RBPJ and/or NOTCH1 genes resulting in Notch LOF, although no additional molecular mechanisms underlying this disease are known (Hassed et al., 2012; Stittrich et al., 2014). Notch signaling defects, either GOF or LOF, have also been implicated in osteoarthritis (Mahjoub et al., 2012; Hosaka et al., 2013; Mirando et al., 2013; Sassi et al., 2014; Liu et al., 2015), rheumatoid arthritis (Nakazawa et al., 2001; Park et al., 2015) and osteoporosis (Engin et al., 2008; Hilton et al., 2008; Majewski et al., 2011; Simpson et al., 2011), and have been associated with a predisposition to pathologic fractures (Kung et al., 2010). Studies like the one presented here further our understanding of the molecular players and events that Notch signaling might control during normal skeletal development, as well as our understanding of how they contribute to the pathology of certain skeletal diseases and injury processes.
MATERIALS AND METHODS
Mouse strains
The Prx1Cre mouse line has been previously described (Logan et al., 2002). The Hes1f/f, Hes1f/f; Hes5−/− and Rosa-Hes f/f strains were a generous gift from Dr Ryoichiro Kageyama (Institute for Virus Research, Kyoto University) and have been described previously (Cau et al., 2000; Hirata et al., 2001; Imayoshi et al., 2008; Tateya et al., 2011; Kobayashi et al., 2009). Hey1−/−; HeyL−/− mutant and control embryos were provided by Dr Manfred Gessler (Biozentrum Universitat Wurzburg) and have been previously described (Fischer et al., 2007). All animal work was approved by both the Duke University and University of Rochester Institutional Animal Care and Use Committees (IACUC).
Real-time RT-PCR
Isolation of RNA from limb-bud tissues and ATDC5 cultures were performed as previously described (Kohn et al., 2015). Real-time reverse-transcriptase (RT)-PCR was used to analyze relative gene expression with the Bio-Rad CFX Connect Real-Time system. Gene expression was normalized to that of β-actin (Actb) before being normalized to control samples. Mouse specific primers for Sox9, Sox5, Sox6, Col2a1, Acan, Hes1, Hes3, Hes5, Hes7 and Hey1 were designed as described previously (Dong et al., 2010). Primer sequences are available upon request. Gene expression analyses from limb buds are from representative experiments of at least three biological replicates from pooled genotypes with statistical analyses performed on technical replicates of an individual experiment.
Tissue analysis
Embryos were harvested at E10.0–18.5 in cold 1× PBS, fixed in 10% neutral buffered formalin (NBF) and then processed. Embryos at >E12.5 were treated overnight with 14% EDTA. After processing, tissues were paraffin embedded. For embryos at E10.0–11.5, the whole embryo was embedded; limbs from embryos at E12.5–14.5 were dissected from a whole embryo before embedding. Tissue was then sectioned at 4 μm and 5 μm for limbs at E10.0–12.5 and E13.5–18.5, respectively. To analyze cartilage composition and general cellular morphology, standard histological staining using ABH–OG and H&E was performed. To analyze protein expression, immunohistochemistry was performed using the VectaStain ABC kits and developed with ImmPACT DAB (Vector Labs). Primary antibodies against the following proteins were used for immunohistochemistry analyses: ACAN (1:200; catalog AB1031, Chemicon), COL2A1 (1:100; catalog MS235-P, Thermo Scientific), SOX9 (1:100; catalog sc20095, Santa Cruz Biotechnology) and MMP13 (1:200; catalog MS-825P, Thermo Scientific). Standard heat-induced and sodium citrate antigen retrieval was performed for the previously listed antibodies, whereas no antigen retrieval was performed for SOX9 immunohistochemistry. Relative quantification of the intensities of some immunohistochemistry staining was calculated using the immunohistochemistry image analysis toolbox developed in ImageJ. Briefly, the software was first trained to build a statistical positive-stain detector using DAB-positive pixels. By selecting a region of interest in the image, pixels of the original image were displayed with original color values, while all other pixels were set to 255 as background and thus filtered out during the color detection process. Finally, only DAB-stained pixels were quantified and compared between groups. BrdU immunohistochemistry was performed as previously described (Dong et al., 2010). For in situ hybridization, embryos were prepared, fixed, processed and sectioned as described previously (Hilton et al., 2005, 2007, 2008; Dong et al., 2010). Dig-labeled whole-mount in situ hybridization was performed as described previously (Rutkowsky et al., 2014). Skeletal staining was performed using the protocol as previously described (Dong et al., 2010).
Chromatin immunoprecipitation assay
The ChIP assay was performed using the MAGnify Chromatin Immunoprecipitation system (Invitrogen) on limb buds at E10.5 and E11.5. Limb buds were homogenized in cold PBS using a 24 g syringe and immediately frozen using liquid nitrogen. Sonication was performed using a Covaris S2 sonicator according to manufacturer's instructions in order to shear chromatin to the lengths of 100–300 base pairs. The protocol was optimized for the use of six to ten limb buds. Antibodies against HES1 and HES5 (sc-25392 and sc-13859, respectively, Santa Cruz Biotechnology) were used at a concentration of 10 µg. Data analysis was performed using qPCR with primers specifically designed to amplify the region of interest within the Sox9 gene promoter.
ATDC5 cell analysis
ATDC5 cells (RIKEN BRC, Japan) were grown in a 12-well plate with Dulbecco's modified Eagle's medium (DMEM) with F12 1:1 (Invitrogen) supplemented with 5% fetal bovine serum (FBS) and 1% penicillin–streptomycin. Once cells were 70–80% confluent, they were treated with ITS media: standard DMEM/F12 medium supplemented with 1× ITS Premix [insulin (5 µg/ml), transferrin (5 µg/ml) and selenous acid (5 ng/ml)] (BD Biosciences). Treatment with ITS supplement has been previously reported to induce chondrocyte differentiation (Watanabe et al., 2001). Cells were incubated for 4 days with ITS medium before transfection with Flag, NICD1 and/or HES5 overexpression plasmids. Transfection was achieved using FuGENE HD (Promega) with 500 ng of each construct. Cells were cultured for 7 days, changing medium every 48 h. Luciferase assays were also performed using ATDC5 cells with a 1-kb Sox9 luciferase construct and an N-box mutant 1-kb Sox9 luciferase construct using the same protocol and reagents as described previously (Kohn et al., 2015). Western and luciferase analyses are representative experiments of at least three biological replicates with statistical analyses performed on technical replicates of an individual experiment. Real-time RT-PCR statistical analyses were performed on the means of three biological replicates.
Western blot
Total proteins were isolated from WT and mutant limb buds. Limb buds were dissociated in a standard lysis buffer and protease inhibitor solutions. Approximately, 10 µg of protein was separated using NuPAGE Novex 4–12% Bis-Tris pre-cast gels (Invitrogen), and the fractionated protein lysates were transferred onto a nitrocellulose membrane using the iBlot system (Invitrogen). Antibodies against HES1 (1:1000; catalog sc-25392, Santa Cruz Biotechnology), HES5 (1:1000; catalog ab194111 Abcam), SOX9 (1:1000; catalog sc20095, Santa Cruz Biotechnology), CYCD1 (1:1000; catalog 2926, Cell Signaling) and β-actin (1:2000; catalog 4970, Sigma-Aldrich) were used with the appropriate secondary antibody following the manufacturer's protocol. Quantification was performed on individual blots, and representative blots are shown. Western blot images were first converted to 8-bit images and then analyzed using ImageJ. Band intensity peak values were calculated and normalized to those of the loading control (β-actin) for comparison.
Statistical analysis
Statistical analyses were performed using two-tailed Student's t-test and one way ANOVA; a P-value <0.05 was considered significant.
Acknowledgements
We thank Drs Ryoichiro Kageyama and Manfred Gessler for providing important HES and HEY mouse strains. We would also like to gratefully acknowledge the technical expertise and assistance of Sarah Mack, Kathy Maltby and Ashish Thomas within the Histology, Biochemistry, and Molecular Imaging Core in the Center for Musculoskeletal Research at the University of Rochester Medical Center.
Footnotes
Competing interests
The authors declare no competing or financial interests.
Author contributions
M.J.H., T.P.R. and A.K. conceived and designed the study; T.P.R., A.K., D.S., A.J.M. and Y.R. conducted the study; T.P.R., A.K., D.S., A.J.M., Y.R. and M.J.H. analyzed and interpreted the data; T.R.P., A.K. and M.J.H. drafted and/or edited the manuscript; T.P.R., A.K., D.S., A.J.M., Y.R. and M.J.H. approved the final version of the manuscript.
Funding
This work was supported in part by the following United States National Institutes of Health grants (National Institute of Arthritis and Musculoskeletal and Skin Diseases): R01 grants [grant numbers AR057022 and AR063071 to M.J.H.]; R21 grant [grant number AR059733 to M.J.H.]; a P30 Core Center grant [grant number AR061307]; a T32 training grant that supported both T.P.R. and A.K. [grant number AR053459]. The work was also supported by departmental funds from the Department of Orthopaedic Surgery at Duke University School of Medicine. Deposited in PMC for release after 12 months.
Supplementary information
Supplementary information available online at http://jcs.biologists.org/lookup/doi/10.1242/jcs.181271.supplemental
References
- Akiyama H., Chaboissier M. C., Martin J. F., Schedl A. and de Crombrugghe B. (2002). The transcription factor Sox9 has essential roles in successive steps of the chondrocyte differentiation pathway and is required for expression of Sox5 and Sox6. Genes Dev. 16, 2813-2828. 10.1101/gad.1017802 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bray S. J. (2006). Notch signalling: a simple pathway becomes complex. Nat. Rev. Mol. Cell Biol. 7, 678-689. 10.1038/nrm2009 [DOI] [PubMed] [Google Scholar]
- Cau E., Gradwohl G., Casarosa S., Kageyama R. and Guillemot F. (2000). Hes genes regulate sequential stages of neurogenesis in the olfactory epithelium. Development 127, 2323-2332. [DOI] [PubMed] [Google Scholar]
- Chen S., Tao J., Bae Y., Jiang M. M., Bertin T., Chen Y., Yang T. and Lee B. (2013). Notch gain of function inhibits chondrocyte differentiation via Rbpj-dependent suppression of Sox9. J. Bone. Miner. Res. 28, 649-659. 10.1002/jbmr.1770 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong Y., Jesse A. M., Kohn A., Gunnell L. M., Honjo T., Zuscik M. J., O'Keefe R. J. and Hilton M. J. (2010). RBPjkappa-dependent Notch signaling regulates mesenchymal progenitor cell proliferation and differentiation during skeletal development. Development 137, 1461-1471. 10.1242/dev.042911 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engin F., Yao Z., Yang T., Zhou G., Bertin T., Jiang M. M., Chen Y., Wang L., Zheng H., Sutton R. E. et al. (2008). Dimorphic effects of Notch signaling in bone homeostasis. Nat. Med. 14, 299-305. 10.1038/nm1712 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischer A., Steidl C., Wagner T. U., Lang E., Jakob P. M., Friedl P., Knobeloch K.-P. and Gessler M. (2007). Combined loss of Hey1 and HeyL causes congenital heart defects because of impaired epithelial to mesenchymal transition. Circ. Res. 100, 856-863. 10.1161/01.RES.0000260913.95642.3b [DOI] [PubMed] [Google Scholar]
- Grogan S. P., Olee T., Hiraoka K. and Lotz M. K. (2008). Repression of chondrogenesis through binding of notch signaling proteins HES-1 and HEY-1 to N-box domains in the COL2A1 enhancer site. Arthritis Rheum. 58, 2754-2763. 10.1002/art.23730 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hassed S. J., Wiley G. B., Wang S., Lee J.-Y., Li S., Xu W., Zhao Z. J., Mulvihill J. J., Robertson J., Warner J. et al. (2012). RBPJ mutations identified in two families affected by Adams-Oliver syndrome. Am. J. Hum. Genet. 91, 391-395. 10.1016/j.ajhg.2012.07.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hatakeyama J., Bessho Y., Katoh K., Ookawara S., Fujioka M., Guillemot F. and Kageyama R. (2004). Hes genes regulate size, shape and histogenesis of the nervous system by control of the timing of neural stem cell differentiation. Development 131, 5539-5550. 10.1242/dev.01436 [DOI] [PubMed] [Google Scholar]
- Hilton M. J., Tu X., Cook J., Hu H. and Long F. (2005). Ihh controls cartilage development by antagonizing Gli3, but requires additional effectors to regulate osteoblast and vascular development. Development 132, 4339-4351. 10.1242/dev.02025 [DOI] [PubMed] [Google Scholar]
- Hilton M. J., Tu X. and Long F. (2007). Tamoxifen-inducible gene deletion reveals a distinct cell type associated with trabecular bone, and direct regulation of PTHrP expression and chondrocyte morphology by Ihh in growth region cartilage. Dev. Biol. 308, 93-105. 10.1016/j.ydbio.2007.05.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hilton M. J., Tu X., Wu X., Bai S., Zhao H., Kobayashi T., Kronenberg H. M., Teitelbaum S. L., Ross F. P., Kopan R. et al. (2008). Notch signaling maintains bone marrow mesenchymal progenitors by suppressing osteoblast differentiation. Nat. Med. 14, 306-314. 10.1038/nm1716 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirata H., Tomita K., Bessho Y. and Kageyama R. (2001). Hes1 and Hes3 regulate maintenance of the isthmic organizer and development of the mid/hindbrain. EMBO J. 20, 4454-4466. 10.1093/emboj/20.16.4454 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horton W. A. (2003). Skeletal development: insights from targeting the mouse genome. Lancet 362, 560-569. 10.1016/S0140-6736(03)14119-0 [DOI] [PubMed] [Google Scholar]
- Hosaka Y., Saito T., Sugita S., Hikata T., Kobayashi H., Fukai A., Taniguchi Y., Hirata M., Akiyama H., Chung U.-I. et al. (2013). Notch signaling in chondrocytes modulates endochondral ossification and osteoarthritis development. Proc. Natl. Acad. Sci. USA 110, 1875-1880. 10.1073/pnas.1207458110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imayoshi I., Shimogori T., Ohtsuka T. and Kageyama R. (2008). Hes genes and neurogenin regulate non-neural versus neural fate specification in the dorsal telencephalic midline. Development 135, 2531-2541. 10.1242/dev.021535 [DOI] [PubMed] [Google Scholar]
- Kageyama R., Ohtsuka T., Hatakeyama J. and Ohsawa R. (2005). Roles of bHLH genes in neural stem cell differentiation. Exp. Cell Res. 306, 343-348. 10.1016/j.yexcr.2005.03.015 [DOI] [PubMed] [Google Scholar]
- Kageyama R., Ohtsuka T. and Kobayashi T. (2007). The Hes gene family: repressors and oscillators that orchestrate embryogenesis. Development 134, 1243-1251. 10.1242/dev.000786 [DOI] [PubMed] [Google Scholar]
- Karlsson C., Brantsing C., Kageyama R. and Lindahl A. (2010). HES1 and HES5 are dispensable for cartilage and endochondral bone formation. Cells Tissues Organs 192, 17-27. 10.1159/000280416 [DOI] [PubMed] [Google Scholar]
- Kobayashi T. and Kageyama R. (2010). Hes1 regulates embryonic stem cell differentiation by suppressing Notch signaling. Genes cells 15, 689-698. 10.1111/j.1365-2443.2010.01413.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobayashi T., Mizuno H., Imayoshi I., Furusawa C., Shirahige K. and Kageyama R. (2009). The cyclic gene Hes1 contributes to diverse differentiation responses of embryonic stem cells. Genes Dev. 23, 1870-1875. 10.1101/gad.1823109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohn A., Dong Y., Mirando A. J., Jesse A. M., Honjo T., Zuscik M. J., O'Keefe R. J. and Hilton M. J. (2012). Cartilage-specific RBPjkappa-dependent and -independent Notch signals regulate cartilage and bone development. Development 139, 1198-1212. 10.1242/dev.070649 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohn A., Rutkowski T. P., Liu Z., Mirando A. J., Zuscik M. J., O'Keefe R. J. and Hilton M. J. (2015). Notch signaling controls chondrocyte hypertrophy via indirect regulation of Sox9. Bone Res. 3, 15021 10.1038/boneres.2015.21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kung A. W. C., Xiao S.-M., Cherny S., Li G. H., Gao Y., Tso G., Lau K. S., Luk K. D. K., Liu J.-M., Cui B. et al. (2010). Association of JAG1 with bone mineral density and osteoporotic fractures: a genome-wide association study and follow-up replication studies. Am. J. Hum. Genet. 86, 229-239. 10.1016/j.ajhg.2009.12.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Z., Chen J., Mirando A. J., Wang C., Zuscik M. J., O'Keefe R. J. and Hilton M. J. (2015). A dual role for NOTCH signaling in joint cartilage maintenance and osteoarthritis. Sci. Signal. 8, ra71 10.1126/scisignal.aaa3792 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Logan M., Martin J. F., Nagy A., Lobe C., Olson E. N. and Tabin C. J. (2002). Expression of Cre Recombinase in the developing mouse limb bud driven by a Prxl enhancer. Genesis 33, 77-80. 10.1002/gene.10092 [DOI] [PubMed] [Google Scholar]
- Mahjoub M., Sassi N., Driss M., Laadhar L., Allouche M., Hamdoun M., Romdhane K. B., Sellami S. and Makni S. (2012). Expression patterns of Notch receptors and their ligands in human osteoarthritic and healthy articular cartilage. Tissue Cell 44, 182-194. 10.1016/j.tice.2012.03.001 [DOI] [PubMed] [Google Scholar]
- Majewski J., Schwartzentruber J. A., Caqueret A., Patry L., Marcadier J., Fryns J.-P., Boycott K. M., Ste-Marie L.-G., McKiernan F. E., Marik I. et al. (2011). Mutations in NOTCH2 in families with Hajdu-Cheney syndrome. Hum. Mutat. 32, 1114-1117. 10.1002/humu.21546 [DOI] [PubMed] [Google Scholar]
- Mead T. J. and Yutzey K. E. (2009). Notch pathway regulation of chondrocyte differentiation and proliferation during appendicular and axial skeleton development. Proc. Natl. Acad. Sci. USA 106, 14420-14425. 10.1073/pnas.0902306106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mirando A. J., Liu Z., Moore T., Lang A., Kohn A., Osinski A. M., O'Keefe R. J., Mooney R. A., Zuscik M. J. and Hilton M. J. (2013). RBP-Jkappa-dependent Notch signaling is required for murine articular cartilage and joint maintenance. Arthritis Rheum. 65, 2623-2633. 10.1002/art.38076 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakazawa M., Ishii H., Aono H., Takai M., Honda T., Aratani S., Fukamizu A., Nakamura H., Yoshino S., Kobata T. et al. (2001). Role of Notch-1 intracellular domain in activation of rheumatoid synoviocytes. Arthritis Rheum. 44, 1545-1554. [DOI] [PubMed] [Google Scholar]
- Park J.-S., Kim S.-H., Kim K., Jin C.-H., Choi K. Y., Jang J., Choi Y., Gwon A.-R., Baik S.-H., Yun U. J. et al. (2015). Inhibition of notch signalling ameliorates experimental inflammatory arthritis. Ann. Rheum. Dis. 74, 267-274. 10.1136/annrheumdis-2013-203467 [DOI] [PubMed] [Google Scholar]
- Rutkowsky T., Sharma D. and Hilton M. J. (2014). Whole-mount in situ hybridization on murine skeletogenic tissues. Methods Mol. Biol. 1130, 193-201. 10.1007/978-1-62703-989-5_14 [DOI] [PubMed] [Google Scholar]
- Sassi N., Gadgadi N., Laadhar L., Allouche M., Mourali S., Zandieh-Doulabi B., Hamdoun M., Nulend J. K., Makni S. and Sellami S. (2014). Notch signaling is involved in human articular chondrocytes de-differentiation during osteoarthritis. J. Recept. Signal Transduct. Res. 34, 48-57. 10.3109/10799893.2013.856920 [DOI] [PubMed] [Google Scholar]
- Simpson M. A., Irving M. D., Asilmaz E., Gray M. J., Dafou D., Elmslie F. V., Mansour S., Holder S. E., Brain C. E., Burton B. K. et al. (2011). Mutations in NOTCH2 cause Hajdu-Cheney syndrome, a disorder of severe and progressive bone loss. Nat. Genet. 43, 303-305. 10.1038/ng.779 [DOI] [PubMed] [Google Scholar]
- Stittrich A.-B., Lehman A., Bodian D. L., Ashworth J., Zong Z., Li H., Lam P., Khromykh A., Iyer R. K., Vockley J. G. et al. (2014). Mutations in NOTCH1 cause Adams-Oliver syndrome. Am. J. Hum. Genet. 95, 275-284. 10.1016/j.ajhg.2014.07.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugita S., Hosaka Y., Okada K., Mori D., Yano F., Kobayashi H., Taniguchi Y., Mori Y., Okuma T., Chang S. H. et al. (2015). Transcription factor Hes1 modulates osteoarthritis development in cooperation with calcium/calmodulin-dependent protein kinase 2. Proc. Natl. Acad. Sci. USA 112, 3080-3085. 10.1073/pnas.1419699112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tateya T., Imayoshi I., Tateya I., Ito J. and Kageyama R. (2011). Cooperative functions of Hes/Hey genes in auditory hair cell and supporting cell development. Dev. Biol. 352, 329-340. 10.1016/j.ydbio.2011.01.038 [DOI] [PubMed] [Google Scholar]
- Tu X., Chen J., Lim J., Karner C. M., Lee S.-Y., Heisig J., Wiese C., Surendran K., Kopan R., Gessler M. et al. (2012). Physiological notch signaling maintains bone homeostasis via RBPjk and Hey upstream of NFATc1. PLoS Genet. 8, e1002577 10.1371/journal.pgen.1002577 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe H., de Caestecker M. P. and Yamada Y. (2001). Transcriptional cross-talk between Smad, ERK1/2, and p38 mitogen-activated protein kinase pathways regulates transforming growth factor-beta-induced aggrecan gene expression in chondrogenic ATDC5 cells. J. Biol. Chem. 276, 14466-14473. 10.1074/jbc.M005724200 [DOI] [PubMed] [Google Scholar]
- Zanotti S. and Canalis E. (2010). Notch and the skeleton. Mol. Cell. Biol. 30, 886-896. 10.1128/MCB.01285-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zanotti S., Smerdel-Ramoya A. and Canalis E. (2011). HES1 (hairy and enhancer of split 1) is a determinant of bone mass. J. Biol. Chem. 286, 2648-2657. 10.1074/jbc.M110.183038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zine A., Aubert A., Qiu J., Therianos S., Guillemot F., Kageyama R. and de Ribaupierre F. (2001). Hes1 and Hes5 activities are required for the normal development of the hair cells in the mammalian inner ear. J. Neurosci. 21, 4712-4720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuscik M. J., Hilton M. J., Zhang X., Chen D. and O'Keefe R. J. (2008). Regulation of chondrogenesis and chondrocyte differentiation by stress. J. Clin. Invest. 118, 429-438. 10.1172/JCI34174 [DOI] [PMC free article] [PubMed] [Google Scholar]