

ABSTRACT
Kinesin-12 (also named Kif15) participates in important events during neuronal development, such as cell division of neuronal precursors, migration of young neurons and establishment of axons and dendritic arbors, by regulating microtubule organization. Little is known about the molecular mechanisms behind the functions of kinesin-12, and even less is known about its roles in other cell types of the nervous system. Here, we show that kinesin-12 depletion from cultured rat cortical astrocytes decreases cell proliferation but increases migration. Co-immunoprecipitation, GST pulldown and small interfering RNA (siRNA) experiments indicated that kinesin-12 directly interacts with myosin-IIB through their tail domains. Immunofluorescence analyses indicated that kinesin-12 and myosin-IIB colocalize in the lamellar region of astrocytes, and fluorescence resonance energy transfer analyses revealed an interaction between the two. The phosphorylation at Thr1142 of kinesin-12 was vital for their interaction. Loss of their interaction through expression of a phosphorylation mutant of kinesin-12 promoted astrocyte migration. We suggest that kinesin-12 and myosin-IIB can form a hetero-oligomer that generates force to integrate microtubules and actin filaments in certain regions of cells, and in the case of astrocytes, that this interaction can modulate their migration.
KEY WORDS: Kinesin-12, Kif15, Microtubule, Myosin-IIB, Protein interaction, Rat astrocytes, Migration
Summary: Kinesin-12 and myosin-IIB can form a hetero-oligomer that generates force to integrate microtubules and actin filaments in certain regions of cells. In astrocytes, this interaction can modulate their migration.
INTRODUCTION
Kinesin-12 (also called Kif15, KSNL7 and HKLP2) is a microtubule-based plus-end-directed motor protein originally studied in the context of its role in mitosis. Recent studies have resolved key aspects of the structure and functionality of kinesin-12 (Drechsler et al., 2014; Klejnot et al., 2014), with the idea that interfering with kinesin-12 function could be exploited for cancer therapy. In previous studies, kinesin-12 was found to promote bipolar spindle formation by cooperating with kinesin-5 (also called Eg5 or Kif11), a tetrameric motor protein important for cell division (Kashina et al., 1996; Scholey, 2009; Ferenz et al., 2010). These studies revealed that kinesin-12 is not required for spindle bipolarity in cells with full Eg5 activity but becomes essential when Eg5 is partially inhibited (Tanenbaum et al., 2009). In support of this, kinesin-12 expression starts at embryonic day (E)3.5 of mouse embryo development, which coincides with the disappearance of kinesin-5 dependency for the spindle maintenance (Courtois et al., 2012). A persistent question has been how kinesin-12 can duplicate the function of kinesin-5 when the latter has been considered unique in its capacity to form tetramers with four motor domains projected outward. A recent study indicates that kinesin-12 can also form tetramers, but with some unique track-switching properties not shared by kinesin-5 (Drechsler et al., 2014).
In terms of re-purposing kinesin-12 for non-mitotic functions, there is a report of its involvement in plasma membrane trafficking in neurons (Eskova et al., 2014), but most studies have suggested that the functions of kinesin-12 in neurons are mainly to regulate microtubule movements and organization during important events such as growth cone mobility and navigation, axonal growth and branching, dendrite differentiation and neuronal migration (Buster et al., 2003; Lin et al., 2012; Eskova et al., 2014). It would appear that some of the neuronal functions of kinesin-12 involve its capacity to regulate microtubule–microtubule interactions, and this would presumably be explained by the recently described capacity of kinesin-12 to form tetramers. Other functions, however, seem to involve the capacity of kinesin-12 to regulate interactions between microtubules and actin filaments, for example in axonal growth cones (Buster et al., 2003; Liu et al., 2010). Such actin interactions have not been revealed in studies of kinesin-12 in mitosis, and might be specific to the non-mitotic functions of kinesin-12. Whether or not the interaction of kinesin-12 with actin filaments is direct or indirect is not known. Another unknown issue is whether kinesin-12 has important non-mitotic roles in non-neuronal cells of the nervous system, such as glia. Here, we set forth to ascertain whether kinesin-12 has a role in the migration of rat cortical astrocytes, and to shed light on the mechanistic basis for its capacity to integrate microtubules with the actin cytoskeleton.
RESULTS
Treatment of astrocytes with kinesin-12 siRNA decreases cell proliferation
Because mitotic kinesins are known for their importance to cell proliferation, we first investigated the effect of kinesin-12 knockdown on astrocyte proliferation. We used cultures of astrocytes prepared by a procedure that results in greater than 90% astrocytes in the culture, as confirmed by immunostaining for cell-type-specific markers (see Materials and Methods). The EdU incorporation DNA assay was then applied to cultured astrocytes depleted of kinesin-12 with small interfering RNA (siRNA) (Fig. 1). We also included in our studies siRNA-based depletion of kinesin-5, as this other motor is known to be essential for mitosis across many vertebrate cell types. By contrast, kinesin-12 has been reported in some cell types to be dispensable as long as kinesin-5 is present (see Introduction). With kinesin-12 depletion, the astrocytes showed less-frequent cell division, with a 24.7% decrease compared to control siRNA (P<0.05). There was a 32.9% decrease with kinesin-5 siRNA treatment (P<0.05), and a 46.9% decrease with siRNA to both motor proteins combined (P<0.05). These results demonstrate that kinesin-12 depletion (to the degree that we are able to knock it down with siRNA) is about half as effective as kinesin-5 knockdown in curtailing cell division, and that knocking down the two motors together has an additive effect.
Fig. 1.

Depletion of kinesin-12 from cultured astrocytes of adult rat spinal cords decreases proliferation but increases migration. (A) The efficiency of kinesin-12 (K-12) siRNA treatment for the indicated number of days in cultured astrocytes was detected by qRT-PCR (a) and western blotting (b). (c) A representative image of western blotting. (B) The efficiency of kinesin-5 (K-5) siRNA treatment for the indicated number of days in cultured astrocytes was detected by qRT-PCR (a) and western blotting (b). (c) A representative image of western blotting. (C) Representative images of EdU staining of astrocyte proliferation after siRNA treatment for 3 days with (a) control (Ctrl) siRNA, (b) kinesin-12 (K-12) siRNA, (c) kinesin-5 (K-5) siRNA and (d) K-12 and K-5 siRNA. (D) Graph showing astrocyte proliferation after siRNA treatment for 3 days. The data are mean±s.d., n=5 (ctrl siRNA, 18.6±1.7%; K-12 siRNA, 13.9±2.9%; K-5 siRNA, 12.4±2.5%; K12 and K-5 siRNAs, 9.8±1.4%). *P<0.05, **P<0.01 (two-tailed t test).
Depletion of kinesin-12 increases astrocytes migration
Given that previous studies have shown that kinesin-5 and kinesin-12 are enriched in migratory neurons, and that depletion of either promoted neuronal migration (Buster et al., 2003; Falnikar et al., 2011; Klejnot et al., 2014), we were curious to learn whether depletion of kinesin-12 or kinesin-5 affects cell migration in astrocytes. For these studies, we performed a Transwell assay, in which identical medium conditions were used in an upper chamber and a lower chamber, divided by a porous membrane, such that migration of cells in all directions from the upper chamber resulted in a portion of the migrating cells appearing in the lower chamber (Fig. 2). We found that depletion of kinesin-12 promoted cell migration, with a 79.3% increase compared to the control (P<0.01). We also applied kinesin-5 siRNA treatment in parallel, and the data showed no significant promotion (22.9%) of astrocyte migration (P>0.05). When astrocytes were treated with kinesin-12 and kinesin-5 siRNA together, the transferred cells showed a 63.2% increase compared to the control (P<0.01). Therefore, it seems that kinein-12 possesses a property that kinesin-5 does not, to affect astrocyte migration. It might be useful in a future study to determine whether depleting kinesin-5 or kinesin-12 affects astrocyte migration in response to a stimulant (for example, 1–2% FBS in the upper chamber and 10% FBS in the lower chamber).
Fig. 2.

Depletion of kinesin-12 increases astrocyte migration. (A) Representative Transwell images of Crystal Violet staining of transferred cultured astrocytes after siRNA treatment for 3 days with (a) control (ctrl) siRNA, (b) kinesin-12 (K-12) siRNA, (c) kinesin-5 (K-5) siRNA, and (d) K-12 and K-5 siRNA. (B) Graph showing the number of transferred cells number after siRNA treatment for 3 days. The data are mean±s.d., n=5 (ctrl siRNA, 87±32; K-12 siRNA, 156±37; K-5 siRNA, 107±25; K12 and K-5 siRNAs, 142±16). **P<0.01 (two-tailed t test).
Kinesin-12 interacts with myosin-IIB tail domain
In our previous study comparing kinesin-5 and kinesin-12 in cultured primary neurons, we concluded that the effects of knocking them down were similar with regard to events attributable to microtubule–microtubule interactions, but different with regard to events attributable to microtubule–actin interactions (Liu et al., 2010). Neuronal migration and astrocyte migration are quite different, and hence this might provide a good explanation for why only kinesin-12 depletion affects astrocyte migration but depletion of either of the two motors affects neuronal migration. It is known that kinesin-12 has a myosin-like domain (Buster et al., 2003) that is not present on kinesin-5, and we showed in our earlier work by co-immunoprecipitation assay that kinesin-12, but not kinesin-5, can interact with actin filaments (Liu et al., 2010). However, we did not determine whether kinesin-12 directly or indirectly interacts with actin filaments. Previously, we sequenced, by performing mass spectrometry, the co-immunoprecipitation complex that was pulled down by anti-kinesin-12 antibody from a total protein of newborn rat brain tissue, and the result showed two non-muscle myosin-II heavy chains, namely myosin-IIA (MYH9, or NMIIA) and myosin-IIB (MYH10, or NMIIB) existing in the complex. This is functionally important because myosin-II is the type of myosin relevant to force generation between microfilaments that would be expected to provide the traction needed for astrocyte migration (as well as growth cone motility of neurons). In the present study, we further investigated whether kinesin-12 might interact directly with myosin-II. As shown in Fig. 3A, our results indicate that myosin-IIB, but not myosin-IIA, could be pulled down by the anti-kinesin-12 antibody from total protein of cultures astrocytes.
Fig. 3.

Kinesin-12 interacts with the myosin-IIB tail domain. (A) Results of the co-immunoprecipitation (IP) showing that the non-muscle myosin heavy chain myosin-IIB exists in the complex pulled down by anti-kinesin-12 antibody, but that myosin-IIA does not exist in that complex (complete blots are provided in Fig. S1). IB, immunoblot. (B) Results of GST pulldown assay. (a) Schematic diagram of the truncated GST-tagged rat kinesin-12 constructs (GST–K-12, motor domain removal) containing amino acids 743–1333 of the 1385-amino-acid full-length protein, and three truncated His-tagged myosin-IIB constructs, His–myosin-IIB-1 (842–1349), His–myosin-IIB-2 (1345–1976) and His–myosin-IIB-3 (947–1648), of the 1976-amino-acid full-length protein. (b) Representative image of GST pulldown assay showing that kinesin-12 strongly interacts with myosin-IIB through the 1345–1976 amino acid domain. (C) Result of siRNA and co-immunoprecipitation assay. (a) Results of co-immunoprecipitation showing that kinesin-12 protein exists in the complex pulled down by anti-myosin-IIB antibody. Complete blots are provided in Fig. S1. (b) Top panel, results of siRNA and co-immunoprecipitation showing that anti-kinesin-12 antibody pulls down myosin-IIB-bound actin, and after myosin-IIB depletion, actin is notably reduced in the co-immunoprecipitation complex pulled down by the anti-kinesin-12 antibody. The two left lanes (Input) show that the levels of myosin-IIB protein decreased after siRNA treatment (the results of effects of myosin IIB siRNA treatment in astrocytes for 1 day, 3 days and 5 days were detected by western blotting as shown in Fig. S2), and the two right lanes (Co-IP) show that when myosin-IIB is decreased, then the amount of actin is notably reduced in the co-immunoprecipitation complex pulled down by the anti-kinesin-12 antibody. Bottom panel, graph showing a 60% diminution of actin protein in the pulldown after myosin-IIB knockdown (mean±s.d., n=3). **P<0.01 (two-tailed t test).
Online software analysis for conserved domains of proteins indicated that kinesin-12 has one myosin tail 1 domain whereas myosin-IIB has two of these. In the case of conventional myosin, the myosin tail region forms a long coiled-coil that facilitates dimerization and oligomerization (Syamaladevi et al., 2012). We considered that kinesin-12 and myosin-IIB might form hetero-oligomers through their myosin tail 1 domains. To explore this possibility, we used a GST pulldown assay to confirm a direct interaction between kinesin-12 and myosin-IIB, and to further narrow down the interacting domain. As shown in the schematic diagram in Fig. 3B, we generated a truncated GST-tagged rat kinesin-12 construct (GST–K-12, motor domain removal) containing amino acids 743–1333 of the 1385-amino-acid full-length protein, and three truncated His-tagged myosin-IIB constructs, His–myosin-IIB-1(842–1349), His–myosin-IIB-2 (1345–1976), His–myosin-IIB-3 (947–1648) containing the indicated fragments of the 1976-amino-acid full-length protein. The results showed (Fig. 3B) that kinesin-12 could strongly interact with myosin-IIB in the region corresponding to amino acids 1345–1976, but not in the regions corresponding to amino acids 842–1349. There was a weak interaction in the region corresponding to amino acids 947–1648.
To further confirm the interaction between kinesin-12 and myosin-IIB, we performed a reverse co-immunoprecipitation assay, with the results showing kinesin-12 protein existing in the complex pulled down by the anti-myosin-IIB antibody (Fig. 3Ca). We were then curious to know whether myosin IIB depletion would affect the pulldown of actin by the kinesin-12 antibody. For this, we set up an siRNA and co-immunoprecipitation assay. Briefly, siRNA was introduced to knock down myosin-IIB from cultured astrocytes, after which we performed co-immunoprecipitation using the anti-kinesin-12 antibody, followed by western blotting of the resulting immunoprecipitant with anti-myosin-IIB and anti-actin antibodies, as in our previously published study (Liu et al., 2010). As shown in the top panel of Fig. 3Cb, we found that, in the Input lanes, myosin-IIB levels decreased after siRNA treatment, and in co-immunoprecipitation lanes, after myosin-IIB depletion, actin was notably reduced in the co-immunoprecipitation complex pulled down by the kinesin-12 antibody. The statistical analyses shown in the bottom panel of Fig. 3Cb revealed a 60% diminution of actin protein in the pulldown after myosin-IIB knockdown. We suspect that the incomplete diminution of actin is due to the fact that actin is a sticky protein, and in fact, this is why conducting the experiment with an actin antibody is difficult. In addition, the capacity of myosin-IIB to interact with actin in the immunoprecipitation preparation is almost certainly diminished by the fact that actin is not polymeric in the preparation. On the basis of the present results, we tentatively propose that kinesin-12 interacts with actin filaments through the myosin IIB protein, and when myosin-IIB protein is depleted, kinesin-12 cannot bind to actin filaments.
Colocalization of kinesin-12 and myosin-IIB as well as interaction by FRET in lamellar region of astrocytes
We would anticipate that kinesin-12 and myosin-IIB should show at least partially overlapping distributions if they are interacting in astrocytes, and especially if they are forming a hetero-oligomer. Using immunocytochemistry, we found this to be the case in the lamellar region of astrocytes (Fig. 4A). (We use the term colocalization as it is the typical vernacular, but we acknowledge that the immunocytochemistry alone indicates that the proteins are in close proximity but cannot reveal whether they perfectly colocalize.) Furthermore, we used the fluorescence resonance energy transfer (FRET) method to investigate a potential interaction of the two proteins of interest by co-transfecting them with fluorescent tags into living cells (Day and Davidson, 2012; Suzuki et al., 2013). We co-transfected kinesin-12–GFP or myosin-IIA–GFP with myosin-IIB–mCherry constructs into cultured astrocytes, and then performed a FRET assay. At 48 h after transfection, the cells were fixed with 4% paraformaldehyde (PFA), and were then subject to the acceptor photobleaching program of FRET provided by the confocal microscope (for details, see Materials and Methods). In this assay, GFP served as a donor, and when mCherry was photobleached, it served as the acceptor. The co-transfection of myosin-IIA–GFP and myosin-IIB–mCherry served as a positive control in this study, whereas co-transfection of GFP and myosin-IIB–mCherry served as a negative control. We found that in astrocytes expressing kinesin-12–GFP and myosin-IIB–mCherry, after mCherry was photobleached, the fluorescence of GFP was raised, as shown in Fig. 4B, indicating interaction of the two proteins.
Fig. 4.

Kinesin-12 colocalizes with myosin-IIB in the lamellar region of astrocytes. (A) Representative images of immunocytochemistry show colocalization of kinesin-12 (red) and myosin-IIB (green) in the astrocyte lamellar region (yellow). (B) Result of FRET assay. (a) A representative image of FRET seen after using the photobleach program in the confocal microscope software. Kinesin-12–GFP (green) and myosin-IIB–mCherry (red) constructs were co-transfected into cultured astrocytes, with the GFP fluorophore serving as donor and the mCherry fluorophore serving as the acceptor. The panels show spectral imaging of cells producing the GFP–kinesin-12 fusion protein and mCherry–myosin IIB protein. preBleach, the cell was illuminated at the donor excitation wavelength of 488 nm and spectral measurements were acquired from the region of interest (ROI) indicated by the circles (top-right panel). The linked mCherry fluorophore was then completely photobleached using the 561-nm laser line in the ROI. The lower left panel shows a spectral image after photobleaching, with the ROI bleached. postBleach, the spectral measurements were then reacquired under identical conditions and from the same ROI, and changes in the donor signal were measured. The FRET efficiency data were acquired directly from the software. (b) Graph showing FRET efficiency in cells co-expressing of GFP and myosin-IIB–mCherry constructs, myosin-IIA–GFP and myosin-IIB–mCherry constructs, and kinesin-12–GFP and myosin-IIB–mCherry constructs. Data are mean±s.d., n=5 (control, 0%; myosin-IIA–GFP and myosin-IIB–mCherry, 2.62±2.42%; kinesin-12–GFP and myosin-IIB–mCherry, 2.07±2.0%). *P<0.05 (two-tailed t test).
Phosphorylation of rat kinesin-12 at T1142 modulates rat cortical astrocytes migration
Functions of kinesin and myosin family members are often closely related to their phosphorylation status. We hypothesized that the interaction of kinesin-12 and myosin-IIB depends on the phosphorylation status of kinesin-12, and that the ability of the two proteins to participate together in cell migration depends on the phosphorylation of kinesin-12 at one or more functionally important sites on the molecules. We regarded Thr1142 of rat kinesin-12 as a likely site for this, on the basis of the online tool PhosphoSitePlus (http://www.phosphosite.org) and the corresponding site Thr1144 of human or mouse kinesin-12 sequence (Olsen et al., 2010). We performed site-directed mutagenesis and checked effects on cell migration, and detected the colocalization and interaction of the two motor proteins by expressing wild-type GFP–kinesin-12, GFP–kinesin-12-T1142A (phospho-mutant), or GFP–kinesin-12-T1142D (phospho-mimic) (the constructs were confirmed by DNA sequencing; data not shown) in cultured astrocytes. All constructs were transfected and expressed for the same time, the cells were dissociated for cell sorting, depending on the GFP fluorescence channel, using flow cytometery. As shown by anti-GFP antibody immunostaining, all obtained cells were GFP positive. The cells that were selected for analyses had similar levels of GFP fluorescence (moderate levels as opposed to extremely bright or extremely dim, as assessed qualitatively). As shown in Fig. 5Aa–e, in Transwell assays, we found that, compared to the wild-type GFP–kinesin-12 construct, the T1142D mutant expression reduced cell migration, with a 34.1% decrease (P<0.05), and similarly, T1142A mutant expression promoted cell migration, with a 30.2% increase (P<0.05). The cells transfected with the different constructs also showed subtle differences in morphology, which would be expected on the basis of differences in the functionality of kinesin-12. As shown in Fig. 5Ba–e, by double immunostaining of GFP (green) and myosin-IIB (red), we defined as positive cells with colocalized kinesin-12 and myosin-IIB that displayed a distinct colocalization of green and red (labeled with an asterisk in Fig. 5), and calculated the ratio of these positive cells to total cells (number of blue nuclei). We found that compared to the wild-type GFP–kinesin-12 construct, the GFP T1142D mutant protein displayed a 124% more colocalization with myosin-IIB protein, and the T1142A mutant showed a 54% less colocalization with myosin-IIB motor protein. Next, we used anti-GFP antibody to immunoprecipitate GFP-fused wild-type kinesin-12 protein, and the T1142D (phospho-mimic) or T1142A (phospho-mutant) proteins together with their interacting protein complex. Then, as determined by western blotting with anti-myosin-IIB or kinesin-12 antibodies (Fig. 5C), we found that T1142D mutant kinesin-12 protein immunoprecipitated with notably increased levels of myosin-IIB protein compared to the wild-type kinesin-12 protein, and T1142A mutant kinesin-12 protein only immunoprecipitated with a small amount of myosin-IIB protein. These data are consistent with kinesin-12-T1142 phosphorylation being necessary for the interaction of kinesin-12 and myosin-IIB, and the interaction of the two motors affecting cell migration.
Fig. 5.

Phosphorylation of rat kinesin-12 at T1142 is necessary for the interaction of kinesin-12 and myosin-IIB. (A) Representative Transwell images of Crystal Violet staining of transferred cultured astrocytes after expression of constructs encoding wild-type (wt) GFP–kinesin-12-T1142 (a), phospho-mimic GFP–kinesin-12-T1142D (b), GFP–kinesin-12-T1142A phospho-mutant (c) and GFP empty vector (d) for 3 days. K-12, kinesin-12. (e) Graph showing the number of cells that had migrated after expression for 3 days. The data are mean±s.d., n=5; the relative cell number of migration for GFP empty vector construct expression was set as 1 [GFP–K-12, 0.95±0.146; T1142D, 0.62±0.05; T1142A, 1.24±0.10], *P<0.05 (two-tailed t test). (B) Representative confocal images of immunocytochemistry showing the colocalization of GFP-fused kinesin-12 (green) and myosin-IIB (red). Nuclei are blue. Colocalization is indicated by yellow in the merged images. Cells were transfected with constructs encoding GFP–kinesin-12-T1142 wild-type (a), GFP–kinesin-12-T1142D (b), GFP–kinesin-12-T1142A (c) and GFP empty vector (d). We directly defined cells with colocalized kinesin-12 and myosin-IIB that displayed a distinct colocalization of green and red as positive cells (*), and calculated the ratio of positive cells to total cells (number of blue nuclei). (e) Graph showing the ratio of positive cells with colocalized kinesin-12 and myosin-IIB after GFP-fused protein expression for 3 days. The data are represented as mean±s.d. ratio of cells with colocalization for myosin-IIB the indicated GFP construct [GFP vector, 0.12±0.039 (n=33); GFP–K-12 wt, 0.35±0.135 (n=39); T1142D, 0.78±0.153 (n=58); T1142A, 0.16±0.05 (n=54)] n=cell number. *P<0.05 (two-tailed t test). (C) Results of co-immunoprecipitation showing that the amount of myosin-IIB protein combined with GFP-fused kinesin-12 protein. GFP antibody was used to immunoprecipitate GFP-fused kinesin-12 wild-type protein, T1142D or T1142A mutant proteins, together with their interacting protein complex. Western blots (IB) were probed with anti-myosin-IIB or anti-kinesin-12 antibody. Scale bars: 100 μm (A); 75 μm (B).
DISCUSSION
In previous studies on fibroblasts, kinesin-12 alternately colocalized with either microtubules or actin filaments, depending on the phase of the cell cycle (Buster et al., 2003). During mitosis, kinesin-12 decorated microtubules but not actin filaments, whereas in interphase, kinesin-12 decorated actin filaments but not microtubules. In terminally post-mitotic neurons, kinesin-12 could interact with both filament systems. Although the major localization of kinesin-12 is microtubule-associated, knocking down kinesin-12 from cultured neurons reveals actin-related effects. Depleting kinesin-12 decreases axonal branching and growth cone size, whereas inhibiting kinesin-5 increases these parameters. Depleting kinesin-12 diminishes the appearance of growth-cone-like waves along the length of the axon, an effect not observed with depletion of kinesin-5 (Liu et al., 2010). Finally, and most interestingly, depletion of kinesin-12 abolishes the ‘waggling’ behavior of microtubules that occurs as they assemble along actin bundles within filopodia, whereas inhibition of kinesin-5 does not. Depletion of either kinesin-12 or kinesin-5 has a similar effect to speed axonal growth and increase the frequency of transport of short microtubules in the axon (Myers and Baas, 2007; Liu et al., 2010). Depletion of either motor speeds the movement of migrating neurons in culture (Falnikar et al., 2011; Klejnot et al., 2014). These observations are consistent with kinesin-12 acting in two ways in neurons – one that mimics the ability of kinesin-5 tetramers to regulate microtubule–microtubule interactions, and another that is distinct from kinesin-5 and regulates microtubule–actin interactions.
Astrocytes are a major cell type in the central nervous system (CNS) that proliferate and migrate, and also have broad lamellar regions that are ideal for colocalization and FRET analyses. They are also important cells of the nervous system, and are the most abundant of any cell type in the brain. In addition to serving a number of structural and functional roles, astrocytes are important in the response of the CNS to injury. Astrocytes migrate into lesions of the injured spinal cord, for example, to participate in reactive astrogliosis forming the glial scar. Although the glial scar is inhibitory to axon regeneration, some studies have indicated that immature astrocytes introduced at an injury site can be conducive to axonal regeneration, as they can form bridges along which regenerating axons can grow (Filous et al., 2010). For these various reasons, we chose to pursue the next phase of our kinesin-12 studies on cultures of astrocytes. We found that knocking down kinesin-12 suppressed astrocyte proliferation, although not as much as knocking down kinesin-5. Knocking the two down together had an additive affect, suggesting their mechanisms of action are not entirely overlapping. Consistent with this, flow cytometry showed that kinesin-12 depletion induced G1 phase arrest in cultured astrocytes whereas kinesin-5 depletion causes G2/M phase arrest (Feng et al., 2015). As for astrocyte migration, we found that depletion of kinesin-12 promoted cell migration significantly whereas depletion of kinesin-5 had no effect. Given that astrocytes migrate by an actin-based crawling mechanism, the dependence of their migration on kinesin-12 but not kinesin-5 conforms to our view that functions of kinesin-12 not shared by kinesin-5 are generally actin-based in nature.
The remaining studies reported here used the astrocyte cultures to further elucidate the interaction between kinesin-12 and actin that we previously reported in neurons and using brain homogenates (Buster et al., 2003; Liu et al., 2010; Klejnot et al., 2014). Our data indicate that kinesin-12 interacts directly with a member of non-muscle myosin-II (NMII), and hence that kinesin-12 probably does not interact directly with actin filaments. NMII plays core roles in many developmental and cellular processes, such as adhesion, migration, morphogenesis and cytokinesis, by pulling on actin filaments to generate contractile forces in cells. Recent studies have shown that individual NMII isoforms can perform both isoform-specific and isoform-redundant functions by assembling with other NMII isoforms (Beach et al., 2014), and have demonstrated the existence of NMII monomers, co-polymerization of NMIIA and NMIIB molecules, and contribution of both isoforms to early stages of contractile system assembly in cells (Shutova et al., 2014). These data show the potential of different NMII isoforms to assemble in different cellular localizations, related to different cytoskeleton functions.
Our data, using various techniques (Co-IP, GST pulldown, FRET and immunocytochemistry), indicate that kinesin-12 interacts directly with myosin-IIB (also named Myh10) but not myosin-IIA (also named Myh9). This makes sense because in neurons, myosin-IIB predominates in growth cones (Rochlin et al., 1995). In astrocytes, it has already been reported that inhibition of myosin-II increases migration (Peng et al., 2013) (which we found also in our experiments, data not shown), which supports the idea of an interaction between myosin-IIB and kinesin-12, the depletion of either of which increases migration. Moreover, we found in our present studies that it is within amino acids 743–1333 of kinesin-12 and amino acids 1345–1976 of myosin-IIB that the interaction occurs. Additional data showed that kinesin-12-T1142 phosphorylation is necessary for the interaction of kinesin-12 and myosin-IIB, and for the two motors to participate together during cell migration. Our recent studies of kinesin-12, myosin-IIA and myosin-IIB expression during zebrafish embryonic development have demonstrated that kinesin-12 and myosin-IIB are concentrated in the CNS and share similar expression patterns, whereas myosin-IIA has a different expression pattern, and is not concentrated in CNS but instead in the epidermis enveloping layer (Huang et al., 2013; Xu et al., 2014).
Based on these various findings, we propose a model wherein kinesin-12 can either form homo-tetramers that regulate microtubule–microtubule interactions or hetero-oligomers with myosin-IIB that regulate microtubule–actin interactions (shown in Fig. 6). This idea is supported by our previous observations that microtubules ‘waggle’ as they assemble along actin bundles into filopodia, and this waggling is eliminated when kinesin-12 is depleted. We envisage the hypothetical hetero-oligomer of kinesin-12 and myosin-IIB interacting between the microtubule and the actin bundle in a manner that functionally links the two, and assists the microtubule in assembling relative to actin filaments undergoing retrograde transport as they generate tension for growth cone advance. We suggest that this also occurs during astrocyte migration. At a broader level, we propose that kinesin-12 might function across many cell types in these two capacities, either behaving as homo-tetramers to regulate microtubule–microtubule interactions, or as hetero-oligomers with myosin-IIB to regulate microtubule–actin interactions. We suggest that it is not the levels of kinesin-12 but rather the regulation of kinesin-12 by its phosphorylation that permits migration to occur in a kinesin-12-regulated manner.
Fig. 6.
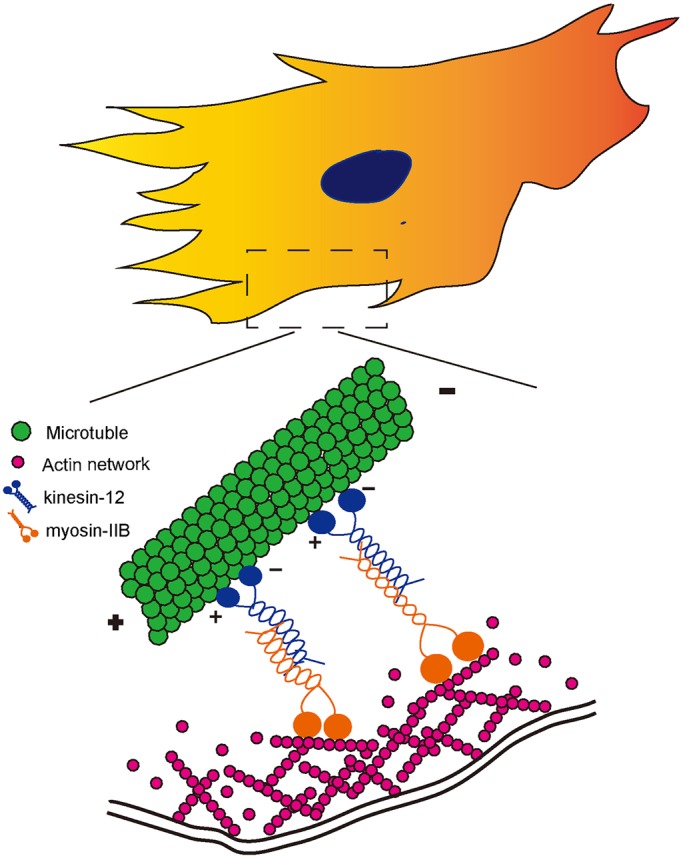
Schematic diagram of a model for interaction between kinesin-12 and myosin-IIB during astrocyte migration. Kinesin-12 is a microtubule-based plus-end-directed motor, and myosin IIB, a non-muscle myosin-II heavy chain, is an actin-dependent molecular motor. In the lamellar region of astrocytes, kinesin-12 directly interacts with myosin-IIB through their tail domains. We posit that the two motors form a hetero-oligomer that generates force to integrate microtubules and actin filaments during the migration of astrocytes.
MATERIALS AND METHODS
Cell culture and reagents
Newborn rat pups (postnatal day 1, termed P1) were obtained from the Laboratory Animal Centre of Nantong University (Nantong, Jiangsu Province, China). All animal surgeries were conducted under the Institutional Animal Care guidelines as well as according to NIH (National Institutes of Health, USA) guidelines, and the procedures were approved by the Administration Committee of Experimental Animals, Jiangsu Province, China. Primary astrocytes from cerebral cortex were prepared as previously described (Sarc et al., 2011; de Vellis and Cole, 2012), cultured in Dulbecco's modified Eagle's medium (DMEM; Invitrogen, CA) medium supplemented with 10% fetal bovine serum (FBS; Invitrogen), 0.5 mM glutamine (Invitrogen), 1% penicillin–streptomycin (Invitrogen), and incubated in a humidified atmosphere of 95% air and 5% CO2 at 37°C. Briefly, cerebral hemispheres from P1 rat pups were isolated aseptically and meninges were removed. The cortices were dissected out and then gently dropped through a sterile 75-μm Nitex mesh. The cell suspension was plated into tissue-culture dishes. In order to purify the cultures, when cells became confluent, the cultures were shaken at 150 rpm for 14–16 h to remove microglial cells. This procedure was repeated a second time. The purification of the sub-cultured astrocytes was confirmed by GFAP [Signalway Antibody LLC (SAB), MD] immunocytochemical staining, and astrocyte cultures were considered appropriate for use when they were 95% positive for GFAP.
siRNA treatment and western blotting
All siRNAs used in this study were designed and synthesized by Biomics Biotechnologies Inc. (Nantong, China). The sequence of the universal control (Ctrl) siRNA and gene-specific kinesin-12 (K-12) siRNA and kinesin-5 (K-5) siRNAs are listed in Table S1. For studies involving siRNA in cultured cells, the cultured astrocytes were transfected using Lipofectamine 2000 (Invitrogen), according to the manufacturer's instructions. After transfection for 24 h, the cells were re-plated on 35-mm culture dishes that had been coated with 0.1 mg/ml poly-L-Lysine (Gibco), after which the cultures were expanded for indicated numbers of days. The efficiency of kinesin-5 siRNA, kinesin-12 siRNA or myosin IIB siRNA transfection was over 80% as indicated by control siRNA-tagged FAM fluorescence, and the knockdown levels in the entire population were confirmed by quantitative real-time PCR (qRT-PCR) and western blotting. After 2–3 days in culture, to permit protein depletion, cells for morphological and functional assays were re-plated on poly-L-Lysine-coated glass coverslips or Transwell dishes.
For western blotting, protein samples (that had been BCA-method quantified) were separated using SDS-PAGE. After transfer to a PVDF membrane (Millipore), the membrane was blocked with 5% non-fat dried milk in Tris-buffered saline (TBS, pH 7.4) and was incubated overnight with primary antibody [anti-kinesin-12 (1:400; custom antibody produced, project no. 11313-1hz, rabbit, polyclonal, Abmart Co., Shanghai, China), anti-kinesin-5 (1:400; catalog no. 27083, mouse, monoclonal, SAB LLC, MD), anti-myosin-IIA (1:1000; catalog no. 3403, rabbit, monoclonal, Cell Signaling Technology, Boston, MA), or anti-myosin-IIB (1:1000; catalog no. ab684, mouse, monoclonal, Abcam, Cambridge, UK)] at 4°C. After washing with TBST (TBS with 0.1% Tween 20), IRDye-800-conjugated affinity-purified goat anti-mouse-IgG (1:5000; Rockland, Philadelphia, PA) or goat anti-rabbit-IgG (1:5000; Rockland) antibodies were applied at room temperature for 2 h. The images were scanned with an Odyssey infrared imaging system (LI-COR), and the data were analyzed with PDQuest 7.2.0 software (Bio-Rad). For loading normalization and relative quantitative analysis of target proteins expression, GAPDH was used as an internal control protein.
Analyses of cell proliferation and migration
We used the EdU and Transwell methods to evaluate astrocyte proliferation and migration as described previously (Yu et al., 2012). Briefly, the astrocytes were dissociated, counted and re-suspended in fresh pre-warmed culture medium, and plated at a density of 1×105 cells/ml in 96-well plates. At the indicated time after cell transfection, 50 µM 5-ethynyl-2′-deoxyuridine (EdU) was applied to the cells. After incubation for an additional 24 h, the cells were fixed with 4% formaldehyde in PBS for 30 min. The cells were then assayed using a Cell-Light™ EdU DNA Cell Proliferation Kit (Ribobio, Guangzhou, China), following the manufacturer's protocol. Astrocyte proliferation (ratio of EdU-positive cells to all cells) was analyzed by using images of randomly selected fields obtained on a DMR fluorescence microscope (Leica Microsystems, Bensheim, Germany). Astrocyte migration was examined using 6.5-mm Transwell chambers with 8-μm pores (Costar, Cambridge, MA). 200 μl of medium containing dissociated astrocytes of 3×104 was transferred to the top chambers of each transwell and 600 μl of complete medium was added into the lower cell-free chambers. We simply monitored the migration of cells from the upper to the lower chamber as a measure of their migration in all directions, without any chemotactic factors. After allowing the cells to migrate for the indicated time, the non-migrated cells on the upper surface of each membrane were cleaned with a cotton swab. Cells adhering to the bottom surface of each membrane were stained with 0.1% Crystal Violet, imaged and counted using the DMR inverted microscope. Assays were performed three times using triplicate wells.
Co-immunoprecipitation
By methods described previously (Liu et al., 2010), a commercially available co-immunoprecipitation Kit (Pierce, Rockford, IL) was used to investigate a potential interaction between kinesin-12 and myosin-IIA or myosin-IIB. Briefly, total protein from cultured astrocytes was extracted using NP-40 Cell Lysis Buffer (containing Protease Inhibitor Cocktail, Pierce). In different assays, anti-kinesin-12 antibody (Abmart Co., Shanghai, China), anti-myosin-IIB (Cell Signaling Technology), or anti-GFP antibody (Santa Cruz Biotechnology) were immobilized in antibody-coupling resin. The control agarose resin supplied in the kit, or normal serum IgG was used as negative control. The total input of co-IP, elution of the immunoprecipitated complex and preparation of samples for SDS-PAGE were performed as per the instructions in the kit. Western blotting was used to determine the presence of myosin-IIA or -IIB and kinesin-12 (Fig. 3A), or actin in the co-immunoprecipitated complex (Fig. 5C).
GST pulldown assay
GST-tagged kinesin-12 and His-tagged myosin-IIB (NM_031520.1) truncated constructs were generated from rat fetal brain cDNA and identified by DNA sequencing. The primers used are listed in Table S1. IPTG (0.4 mM) was added to induce target protein expression in the E. coli BL21 system (Tiangen Biotech, Beijing, China). The proteins were harvested and lysed with a ultrasonic homogenizer (JY92-2D, Ningbo Scientz Biotechnology Co. Ltd, Zhejiang, China). GST-tagged proteins, GST-4T1 control and GST–kinesin-12, were immobilized on a 40-μl glutathione gel (7Sea Pharmatech Co. Ltd, Shanghai, China) for 3 h for each sample at 4°C. After washing with PBS three times, each time for 30 min, beads were incubated with His fusion protein (pET30a-myosin-IIB-1, pET30a-myosin-IIB-2, and pET30a-myosin-IIB-3 transformation vectors) overnight at 4°C. Then the beads were washed with PBS three times, each time for 30 min. The beads were suspended in 40 μl of 2×SDS sample buffer, boiled and centrifuged briefly. 10-μl volumes of each sample were then subjected to SDS-PAGE (10%) and western blotting detection using anti-His6 tag antibody (1:1000; catalog no. ab18184, mouse, monoclonal, Abcam) and anti-GST antibody (1:500; catalog no. ab9058, rabbit, polyclonal, Abcam).
Fluorescence resonance energy transfer
FRET was used for detecting molecular interactions between kinesin-12 and myosin-IIB following the procedure as previously described (Day and Davidson, 2012; Suzuki et al., 2013). The constructs expressing the following fusion proteins were used: pEGFP-N1, kinesin-12–GFP, myosin-IIA–GFP, and myosin-IIB–mCherry. The tagged constructs were made in our laboratory and confirmed by DNA sequencing. The full-length cDNA sequences of rat kinesin-12 (GenBank no. NM_181635.2), rat myosin-IIA (NM_013194), myosin-IIB (NM_031520) were obtained with High Fidelity PCR (KOD, Toyobo, Japan) using rat fetal brain cDNA which was reverse transcribed (Thermo, Rockford, USA) from total RNA extracted using Trizol reagent (Invitrogen). Following treatment with indicated restriction enzymes (Takara Biotechnology, Dalian, China), PCR products were cloned into pEGFP-N1 (Takara-Clontech) and confirmed by DNA sequencing (BGI, Shanghai, China). The primers used are listed in Table S1. Three co-transfection constructs: (1) pEGFP-N1 and myosin-IIB–mCherry (negative control), (2) kinesin-12–GFP and myosin-IIB–mCherry (assay), and (3) myosin-IIA–GFP and myosin-IIB–mCherry (positive control) were set up. For the GFP and mCherry fluorescent proteins, GFP served as the energy donor and mCherry served as acceptor. Astrocytes (0.5×105 cells) were plated into one well of a 24-well plate and transfected using HP X-tremeGENE DNA Transfection Reagent (Roche, Basel, Switzerland) as recommended by the manufacturer. After 48 h, the cells were fixed with 4% PFA. Then, as an approach for measuring FRET, acceptor photobleaching was conducted using the included program FRET AB wizard of a confocal laser scanning fluorescence microscope (Leica TCS SP5, Wetzlar, Germany). Briefly, the cell was illuminated at the donor excitation wavelength of 488 nm and spectral measurements were acquired from the region of interest (ROI) indicated by the circle. The linked mCherry fluorophore was then completely photobleached using the 561-nm laser line in the ROI. The spectral measurements were then reacquired under identical conditions and from the same ROI, and changes in the donor signal were measured. The FRET efficiency data were taken directly from the software. Any increase in the donor fluorescence signal was measured, which occurs when FRET is disrupted by photobleaching of the acceptor fluorophore if the two proteins interact in cells.
Immunocytochemistry
For investigating potential colocalization of kinesin-12 and myosin-IIB, cultures were incubated with rabbit polyclonal anti-rat-kinesin-12 antibody (1:200; custom antibody produced, project no. 11313-1hz, rabbit, polyclonal, ABmart Co. Shanghai, China) or GFP antibody (1:100, catalog no. AG279, rabbit, monoclonal, BiYunTian Co., Nantong, China) for detecting exogenous GFP-tagged kinesin-12 expression, together with mouse monoclonal anti-myosin-IIB antibody (1:500; catalog no. SC-376954, mouse, monoclonal, Santa Cruz Biotechnology, CA) at 4°C for 14–16 h and then incubated in the corresponding secondary antibodies (Cy3-conjugated goat anti-mouse-IgG at 1:800, Cy3-conjugated goat anti-rabbit-IgG at 1:800, Alexa-Fluor-488-conjugated goat anti-mouse IgG at 1:200, Alexa-Fluor-488-conjugated goat anti-rabbit IgG at 1:200, Jackson ImmunoResearch) for 2 h at room temperature. Fluorescence images were obtained on an Olympus microscope using identical camera, microscope, and imaging criteria such as gain, brightness and contrast, and exposure time.
Site-directed mutagenesis and gene overexpression of kinesin-12 mutants
The QuikChange® Lightning Site-Directed Mutagenesis Kit (Stratagene) was used to generate the mutant kinesin-12-T1142A (phospho-mutant) and kinesin-12-T1142D (phospho-mimic) constructs. The mutagenesis primers (listed in Table S1) design and performance procedures were according to the manufacturer's instructions, and all the constructs were confirmed by DNA sequencing (BGI, Shanghai). The GFP empty vector, GFP–kinesin-12 wild-type, GFP–kinesin-12-T1142D or GFP–kinesin-12-T1142A constructs were transfected into cultures astrocytes using X-tremeGENE HP DNA Transfection Reagent following the manufacturer's instructions. After expression for the scheduled time, the cells were dissociated for cell sorting, depending on the GFP fluorescence channel, using Flow cytometery (FACS-Aria, BD). GFP-positive cells were identified using a 488-nm laser. The obtained cells were used for the Transwell migration assay, for colocalization analysis by immunostaining, or in the co-immunoprecipitation assay as above.
Statistics
Comparisons between different experimental groups were analyzed by using repeated measures ANOVA with the GraphPad Prism 5.0 software (GraphPad Software Inc., La Jolla, CA). Data are expressed as the means±s.d. of three to five separate experiments for each assay, and subjected to a one-way analysis of variance (ANOVA) using SPSS software package (SPSS Software Inc., Chicago, IL). If there was a significant overall difference among groups, pair-wise comparisons were legitimately conducted by Fisher's least significant difference test. Values of P<0.05 were considered statistically significant.
Acknowledgements
We thank Professor Tianyi Zhang of the Cytological analysis laboratory for helping us to set up the FRET assay. We thank the two anonymous reviewers for their great help in improving our work.
Footnotes
Competing interests
The authors declare no competing or financial interests.
Author contributions
M.L., Y.L. and P.W.B. conceived and designed the experiments; J.F., Z.H., H.C. and J.H. performed the experiments; J.F., M.L., Y.L. and L.Q. analyzed the data; R.W. and Z.D. contributed reagents, materials and analysis tools; and M.L. and P.W.B. wrote the article.
Funding
This study was supported by grants from the National Natural Science Foundation of China [grant numbers 31171007, 31371078]; Natural Science Foundation of Jiangsu Province [grant number BK20150404 to Z.D.]; Jiangsu Provincial Department of Education [grant number 11KJA180004 and Priority Academic Program Development (PAPD)], and Scientific innovation project [grant number KYZZ14_0354 to Z.H., CXLX13_868 to J.F.] for postgraduate students in Jiangsu Province; as well as a grant from the National Institutes of Health [grant number R01 NS28785 to P.W.B.]. Deposited in PMC for release after 12 months.
Supplementary information
Supplementary information available online at http://jcs.biologists.org/lookup/doi/10.1242/jcs.181867.supplemental
References
- Beach J. R., Shao L., Remmert K., Li D., Betzig E. and Hammer J. A. III (2014). Nonmuscle myosin II isoforms coassemble in living cells. Curr. Biol. 24, 1160-1166. 10.1016/j.cub.2014.03.071 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buster D. W., Baird D. H., Yu W., Solowska J. M., Chauvière M., Mazurek A., Kress M. and Baas P. W. (2003). Expression of the mitotic kinesin Kif15 in postmitotic neurons: implications for neuronal migration and development. J. Neurocytol. 32, 79-96. 10.1023/A:1027332432740 [DOI] [PubMed] [Google Scholar]
- Courtois A., Schuh M., Ellenberg J. and Hiiragi T. (2012). The transition from meiotic to mitotic spindle assembly is gradual during early mammalian development. J. Cell Biol. 198, 357-370. 10.1083/jcb.201202135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Day R. N. and Davidson M. W. (2012). Fluorescent proteins for FRET microscopy: monitoring protein interactions in living cells. Bioessays 34, 341-350. 10.1002/bies.201100098 [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Vellis J. and Cole R. (2012). Preparation of mixed glial cultures from postnatal rat brain. Methods Mol. Biol. 814, 49-59. 10.1007/978-1-61779-452-0_4 [DOI] [PubMed] [Google Scholar]
- Drechsler H., McHugh T., Singleton M. R., Carter N. J. and McAinsh A. D. (2014). The Kinesin-12 Kif15 is a processive track-switching tetramer. eLife 3, e01724 10.7554/eLife.01724 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eskova A., Knapp B., Matelska D., Reusing S., Arjonen A., Lisauskas T., Pepperkok R., Russell R., Eils R., Ivaska J. et al. (2014). An RNAi screen identifies KIF15 as a novel regulator of the endocytic trafficking of integrin. J. Cell Sci. 127, 2433-2447. 10.1242/jcs.137281 [DOI] [PubMed] [Google Scholar]
- Falnikar A., Tole S. and Baas P. W. (2011). Kinesin-5, a mitotic microtubule-associated motor protein, modulates neuronal migration. Mol. Biol. Cell 22, 1561-1574. 10.1091/mbc.E10-11-0905 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng J., Hua J., Hu Z., Yan Y., Wu R. and Liu M. (2015). Effect of Kinesin-12 knockdown on cell cycle arresting and apoptosis in rat astrocyte, a comparison study with Kinesin-5. J. Nantong Univ. 35, 173-177. [Google Scholar]
- Ferenz N. P., Gable A. and Wadsworth P. (2010). Mitotic functions of kinesin-5. Semin. Cell Dev. Biol. 21, 255-259. 10.1016/j.semcdb.2010.01.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Filous A. R., Miller J. H., Coulson-Thomas Y. M., Horn K. P., Alilain W. J. and Silver J. (2010). Immature astrocytes promote CNS axonal regeneration when combined with chondroitinase ABC. Dev. Neurobiol. 70, 826-841. 10.1002/dneu.20820 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang Y., Wang X., Wang X., Xu M., Liu M. and Liu D. (2013). Nonmuscle myosin II-B (myh10) expression analysis during zebrafish embryonic development. Gene Expr. Patterns 13, 265-270. 10.1016/j.gep.2013.04.005 [DOI] [PubMed] [Google Scholar]
- Kashina A. S., Baskin R. J., Cole D. G., Wedaman K. P., Saxton W. M. and Scholey J. M. (1996). A bipolar kinesin. Nature 379, 270-272. 10.1038/379270a0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klejnot M., Falnikar A., Ulaganathan V., Cross R. A., Baas P. W. and Kozielski F. (2014). The crystal structure and biochemical characterization of Kif15: a bifunctional molecular motor involved in bipolar spindle formation and neuronal development. Acta Crystallogr. D Biol. Crystallogr. 70, 123-133. 10.1107/S1399004713028721 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin S., Liu M., Mozgova O. I., Yu W. and Baas P. W. (2012). Mitotic motors coregulate microtubule patterns in axons and dendrites. J. Neurosci. 32, 14033-14049. 10.1523/JNEUROSCI.3070-12.2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu M., Nadar V. C., Kozielski F., Kozlowska M., Yu W. and Baas P. W. (2010). Kinesin-12, a mitotic microtubule-associated motor protein, impacts axonal growth, navigation, and branching. J. Neurosci. 30, 14896-14906. 10.1523/JNEUROSCI.3739-10.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Myers K. A. and Baas P. W. (2007). Kinesin-5 regulates the growth of the axon by acting as a brake on its microtubule array. J. Cell Biol. 178, 1081-1091. 10.1083/jcb.200702074 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olsen J. V., Vermeulen M., Santamaria A., Kumar C., Miller M. L., Jensen L. J., Gnad F., Cox J., Jensen T. S., Nigg E. A. et al. (2010). Quantitative phosphoproteomics reveals widespread full phosphorylation site occupancy during mitosis. Sci. Signal. 3, ra3 10.1126/scisignal.2000475 [DOI] [PubMed] [Google Scholar]
- Peng H., Ong Y. M., Shah W. A., Holland P. C. and Carbonetto S. (2013). Integrins regulate centrosome integrity and astrocyte polarization following a wound. Dev. Neurobiol. 73, 333-353. 10.1002/dneu.22055 [DOI] [PubMed] [Google Scholar]
- Rochlin M. W., Itoh K., Adelstein R. S. and Bridgman P. C. (1995). Localization of myosin II A and B isoforms in cultured neurons. J. Cell Sci. 108, 3661-3670. [DOI] [PubMed] [Google Scholar]
- Sarc L., Wraber B. and Lipnik-Stangelj M. (2011). Ethanol and acetaldehyde disturb TNF-alpha and IL-6 production in cultured astrocytes. Hum. Exp. Toxicol. 30, 1256-1265. 10.1177/0960327110388533 [DOI] [PubMed] [Google Scholar]
- Scholey J. M. (2009). Kinesin-5 in Drosophila embryo mitosis: sliding filament or spindle matrix mechanism? Cell Motil. Cytoskeleton 66, 500-508. 10.1002/cm.20349 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shutova M. S., Spessott W. A., Giraudo C. G. and Svitkina T. (2014). Endogenous species of mammalian nonmuscle myosin IIA and IIB include activated monomers and heteropolymers. Curr. Biol. 24, 1958-1968. 10.1016/j.cub.2014.07.070 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki Y., Yamamura H., Ohya S. and Imaizumi Y. (2013). Direct molecular interaction of caveolin-3 with KCa1.1 channel in living HEK293 cell expression system. Biochem. Biophys. Res. Commun. 430, 1169-1174. 10.1016/j.bbrc.2012.12.015 [DOI] [PubMed] [Google Scholar]
- Syamaladevi D. P., Spudich J. A. and Sowdhamini R. (2012). Structural and functional insights on the Myosin superfamily. Bioinform. Biol. Insights 6, 11-21. 10.4137/BBI.S8451 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanenbaum M. E., Macůrek L., Janssen A., Geers E. F., Alvarez-Fernández M. and Medema R. H. (2009). Kif15 cooperates with eg5 to promote bipolar spindle assembly. Curr. Biol. 19, 1703-1711. 10.1016/j.cub.2009.08.027 [DOI] [PubMed] [Google Scholar]
- Xu M., Liu D., Dong Z., Wang X., Wang X., Liu Y., Baas P. W. and Liu M. (2014). Kinesin-12 influences axonal growth during zebrafish neural development. Cytoskeleton 71, 555-563. 10.1002/cm.21193 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu B., Qian T., Wang Y., Zhou S., Ding G., Ding F. and Gu X. (2012). miR-182 inhibits Schwann cell proliferation and migration by targeting FGF9 and NTM, respectively at an early stage following sciatic nerve injury. Nucleic Acids Res. 40, 10356-10365. 10.1093/nar/gks750 [DOI] [PMC free article] [PubMed] [Google Scholar]