

Abstract
Rapid bioorthogonal reactivity can be induced by controllable, catalytic stimuli using air as the oxidant. Methylene blue (4 μM) irradiated with red light (660 nm) catalyzes the rapid oxidation of a dihydrotetrazine to a tetrazine thereby turning on reactivity toward trans-cyclooctene dienophiles. Alternately, the aerial oxidation of dihydrotetrazines can be efficiently catalyzed by nanomolar levels of horseradish peroxidase under peroxide-free conditions. Selection of dihydrotetrazine/tetrazine pairs of sufficient kinetic stability in aerobic aqueous solutions is key to the success of these approaches. In this work, polymer fibers carrying latent dihydrotetrazines were catalytically activated and covalently modified by trans-cyclooctene conjugates of small molecules, peptides and proteins. In addition to visualization with fluorophores, fibers conjugated to a cell adhesive peptide exhibited a dramatically increased ability to mediate contact guidance of cells.
Graphical abstract
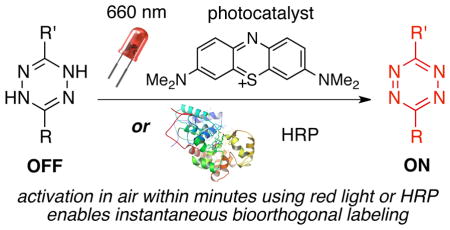
INTRODUCTION
Bioorthogonal chemistry has evolved into a field with broad-reaching applications in biology, medicine and materials science.1–4 Driving the field has been the vigorous development of unnatural transformations that proceed selectively in the presence of Nature’s functional groups.5–12 Recently, bioorthogonal chemistry has been utilized in payload release strategies, with the aim of triggering diverse events including drug delivery, gene expression, and modulating materials properties.13–15 There has also been a growing interest in using external stimuli to induce bioorthogonal reactivity. In particular, photoinducible reactions have emerged as a method for turning on bioorthogonal reactions with temporal and spatial control.9,16 Key advances include tetrazole10 and cyclopropenone11 based ligations, where photolysis produces reactive nitrile imines and cyclooctyne derivatives, respectively. Such ‘photoclick’ reactions generally utilize short-wavelength light to unleash more reactive species.10,11 The direct use of red or near IR light to induce bioorthogonal reactivity has not been described. Lin has recently described two-photon based photoinducible tetrazole reactions that utilize near IR light,8 and Popik has shown that cyclopropenones can be photodecarbonylated by a two photon process.17 While two-photon methods provide high spatial resolution,18 their very small focal volumes currently limit many practical applications. Recently, near-IR photodecaging strategies have been described based on cyanine19, BODIPY,20 and phthalocyanine21 dyes.22 A current challenge for red- and near-IR photodecaging strategies lies is the need to improve the kinetics of photorelease.
Enzymatic catalysis presents intriguing additional possibilities for turning on bioorthogonal reactivity. Proteins have been engineered with unnatural side chains capable of bioorthogonal coupling,2 Recently, bioorthogonal enzymatic decaging has been described in which engineered P450 proteins catalyze reactions to release alcohols.23 However, the use of an enzyme to create a bioorthogonal coupling partner had not been described. We considered that the enzyme horseradish peroxidase (HRP) might be an effective catalyst for the creation of compounds for bioorthogonal reactivity. HRP finds broad utility in biological assays by oxidizing phenols and other organic substrates.24 While H2O2 is typically required as the terminal oxidant, HRP can oxidize certain substrates (e.g. indole-3-acetic acid, NADH and hydroquinones) in the absence of peroxide.25–28
The inverse-electron demand Diels-Alder reaction of s-tetrazines with alkene or alkyne dienophiles, referred to as tetrazine ligation, has emerged as an important reaction in the bioorthogonal toolbox.29–31 A notable aspect of tetrazine ligations are the exceptionally high rates of reactivity that can be achieved.31 With strained trans-cyclooctenes, we have measured bimolecular rate constants as high as 3.3 × 106 M−1s−1 in reactions with tetrazines (Tz), representing the fastest bioorthogonal reactions reported to date.32,33 Tetrazines are typically synthesized through the chemical oxidation of dihydrotetrazine (DHTz) precursors,34 and the electroactivity of tetrazines is well established.35 Recently, Devaraj has demonstrated that electrochemistry can be used to control the redox state of pendant DHTz/Tz groups on the surface of microelectrodes, allowing selective bioconjugation to oxidized electrode surfaces.36 DHTz-containing metal organic frameworks have also been used as colorimetric materials for the detection of nitrous gases.37
Described herein are the first examples of catalytic turn-on of the tetrazine ligation, where rapid bioorthogonal reactivity can be induced by a controllable, catalytic stimulus (Figure 1A). Either visible light and a photosensitizer, or very low loadings of horseradish peroxidase can be used to catalyze the oxidation of a dihydrotetrazine to a tetrazine.
Figure 1.
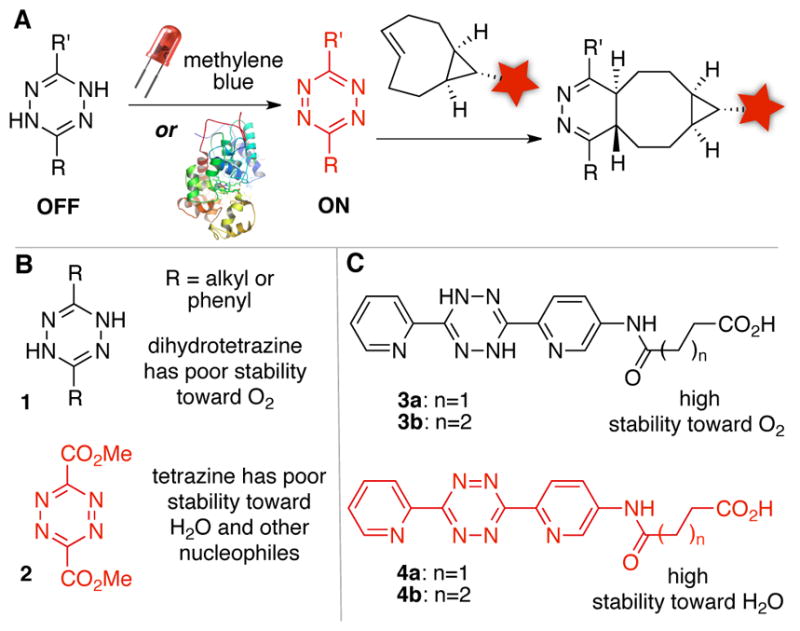
(A) Catalytic turn-on of tetrazine ligation. (B) Most dihydrotetrazine/tetrazine pairs have stability issues. (C) Identification of a dihydrotetrazine/tetrazine pair with high stability.
RESULTS AND DISCUSSION
A challenge to the development of catalytic methods for turning on the tetrazine ligation was the identification of a DHTz/Tz pair that would be stable in both oxidation states (Figure 1B). For most redox couples, either the DHTz is too readily oxidized in air (e.g. 1), or the Tz is too reactive toward water and other nucleophiles (e.g. 2). We show here that the dipyridyl DHTz/Tz pair38 (3/4) has good stability in both states (Figure 1C). Dihydrotetrazines 3 are highly resilient toward background oxidation in organic solvents, and a number of derivatives have been synthesized and shown stable even to silica gel chromatography. In ambient light, a 35 μM solution of 3a in MeOH was shown to retain 99 and 98% of the DHTz oxidation state after 1 and 2 hours, respectively (Figure S10c). Aqueous solutions of 3a were handled in glassware that had been first rinsed with 2.0 mM EDTA in PBS to remove adventitious metal impurities. After standing in the dark at 25 °C in PBS buffer, a solution of 3a was monitored by UV-vis and shown to retain 99 and 96% of the DHTz oxidation state over 30 min and 2.5 hours respectively (Figure S10a). In ambient light at 25 °C in PBS buffer, a solution of 3a was shown to retain 97 and 94% of the DHTz oxidation state after 1 and 2 hours, respectively (Figure S10d). In PBS containing 10% mouse serum, 90% of 3a was retained in the DHTz oxidation state after 1 h (Figure S10f). Analogs of tetrazines 4 have been described previously and used broadly for applications in nuclear medicine and cell imaging.31,39–43 In PBS buffer at 25 °C, tetrazine 4a (800 μM) shows 98% and 83% fidelity after 2 h and 24 h, respectively (Figure S11). The stability of a radiolabeled derivative of tetrazine 4b has been studied by Robillard at 37 °C in PBS, serum and blood, with 97%, 87% and 59% retention of the tetrazine observed after 2 hours.41
Compound 3a has a maximum in the UV-vis spectrum at 292 nm, and 4a has a maximum at 325 nm with a less intense peak at 525 nm (Figure 2). As a reference for our catalytic studies, we first monitored the electrochemical oxidation of 3a in phosphate buffer, for which the voltammogram displays a single peak centered at 0.02 V (Figure S13B). Under mildly oxidizing conditions (0.18 V relative to Ag/AgCl), a 1.1 mM solution of 3a turns pink (Figure S13A) and the oxidation to 4a proceeds cleanly with an isosbestic point at 303 nm (Figure S13C). This isosbestic point was also conserved in the photocatalytic (Figure S5) and enzymatic (Figure 2) oxidations of 3a described below, and the spectroscopic changes at 292 and 325 nm were routinely used for monitoring reaction progress.
Figure 2.
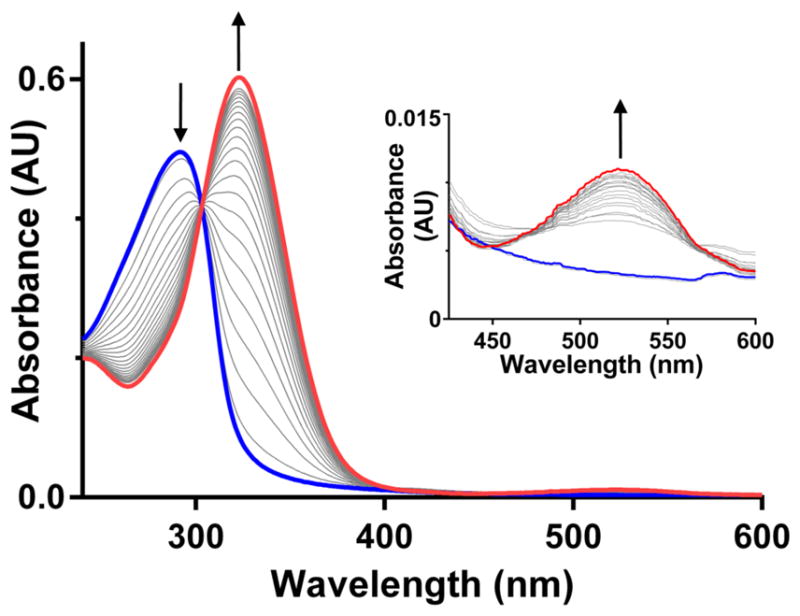
Compound 3a (17 μM in PBS, blue spectrum) displays a maximum in the UV-vis spectrum at 292 nm. Compound 4a (red spectrum) displays a maximum at 325 nm and a less intense peak at 525 nm (see inset). Upon addition of horseradish peroxidase (15 nM) the UV-vis spectrum was monitored every 10 seconds. With 50% conversion after 100 seconds, complete conversion of 3a to 4a is observed after 600 seconds, as evidenced by the decrease in the absorption at 292 nm and increase at 325 nm with an isosbestic point at 303 nm. Similar spectral changes are observed when 3a is electrochemically oxidized to 4a in aqueous solution, or when the oxidation of 3a to 4a is photocatalyzed by methylene blue.
A number of photosensitizers in the presence of long wavelength visible light were found to catalyze the oxidation of 3a to 4a in the presence of air.44 Methylene blue was considered a particularly attractive sensitizer due to its clinical relevance, low molecular weight, low toxicity, high solubility, and an absorption spectrum (lmax 665 nm) that extends to the near IR.45 Further, methylene blue has previously been explored for applications in photodynamic therapy based on oxidation of indole-3-acetic acid.46 Rose bengal (lmax 550 nm), used in a range of biomedical applications, was also identified as an excellent sensitizer. Experiments to study the catalytic photooxidation were conducted at 25 °C in a thermostatted cuvette with stirring capability and a single top-mounted LED. A custom 3D printed light fixture was used to mount the LED directly above the cuvette and block ambient light (Figure S4). As shown in Figure 3B, irradiation of 3a (21 μM) with a 660 nm LED (9.1 mW/cm2) in the presence of methylene blue (4 μM) in pH 7.4 PBS caused conversion to tetrazine 4a with quantitative yield within 200 seconds. Methylene blue (4 μM) also catalyzed the conversion of 3a to 4a in the presence of ambient light, with 47% conversion noted after 2 h (Figure S10e). The light dependence of the methylene blue catalyzed oxidation was demonstrated by turning the LED on and off (Figure 3C). Similar light dependent on/off behavior was exhibited with either rose bengal or carboxyfluorescein with irradiation centered at 528 nm (2.3 mW/cm2, Figure S8, S6). Both methylene blue and rose bengal are known 1O2 sensitizers, and we therefore queried the influence of a 1O2 quencher on the oxidation rate of 3a. Neither the methylene blue (Figure 3D) nor the rose bengal (Figure S9) catalyzed photooxidations are impeded by the addition of 60 mM NaN3. By contrast, the rate of the reaction between 2,5-diphenylisofuran with 1O2 was greatly reduced when 23 mM NaN3 was added (Figure S12). These experiments strongly imply that 1O2 is not the oxidant of 3a under photocatalytic conditions. The mechanism of photooxidation more likely involves electron transfer and is the subject of ongoing study.
Figure 3.

(A) Methylene blue catalyzed photooxidation of 3a to 4a was carried out with UV-vis monitoring and irradiation by a single LED centered at 660 nm. (B) After the onset of irradiation, reaction progress was monitored every 30 sec at 325 nm, which increased with formation of 4a, and 292 nm, which decreased upon consumption of 3a. (C) The reaction progress requires irradiation and stalls when the LED is turned off. (D) The addition of 60 mM NaN3, a singlet oxygen quencher, does not significantly slow the rate of conversion of 3a to 4a. In this experiment, the lamp intensity was reduced relative to experiments displayed in B and C.
As a complement to these photocatalyzed reactions, we observed that HRP can efficiently catalyze the oxidation of 3a in the dark at low enzyme concentration (15 nM) (Figure 4). While HRP typically requires H2O2 as the terminal oxidant, the addition of HRP to a peroxide-free solution of 3a (30 μM) in PBS led to the rapid formation of 4a (Figure 4B). The rate of formation of 4a was significantly slower in the presence of 2 mM H2O2, and was near baseline in the presence of H2O2 but absence of HRP. Neither cytochrome c nor hemoglobin were effective catalysts of DHTz oxidation, with only slow conversion of 3a to 4a even with heme concentrations that were nearly 3 orders of magnitude higher than that used with HRP (Figure S10b). As shown in Figure 4C, the addition of superoxide dismutase (SOD, 770 nM) does not suppress the rate of the oxidation of 3a by HRP, providing evidence that superoxide is not responsible for the oxidation. Finally, it was observed that the oxidation of 3a by HRP follows Michaelis-Menten kinetics, with Km = 1.0 × 10−4 M, kcat = 27 s−1, and kcat/Km = 2.7 × 105 M−1s−1 (Figure 4D).
Figure 4.

(A) Horseradish peroxidase catalyzed oxidation of 3a to 4a. Reactions were monitored by UV-vis every 30 sec at 325 nm. (B) Enzymatic oxidation is most efficient in absence of peroxide, and is suppressed upon addition of peroxide. Hydrogen peroxide without catalyst is not an effective oxidant of 3a. (C) The addition of superoxide dismutase does not suppress the rate of the oxidation of 3a by HRP, providing evidence that superoxide is not responsible for the oxidation. (D) The oxidation of 3a by HRP follows Michaelis-Menten kinetics. The reactions in (B, C) were carried out in PBS, and the reaction in (D) was carried out in PBS containing EDTA (2.0 mM).
The light and enzyme-catalyzed reactions developed here enable the functionalization of polymeric materials with potential biomedical applications. We previously demonstrated the production of peptide-containing polymer fibers through interfacial bioorthogonal polymerization based on tetrazine ligation using bis-tetrazine and bis-TCO monomers dissolved in immiscible solvents.47 These hydrogel-like polymer fibers, with diameters ranging from 6 to 11 μm when dry, are cytocompatible, biologically active and mechanically robust; they resemble many fibrous structures found in the human body and can be woven into complex, higher order assemblies for tissue engineering purposes. However, the use of interfacial polymerization to fabricate protein-containing polymer fibers is not trivial due to the possibility of protein denaturation by the required organic solvent.
We hypothesized that fibers could be synthesized with latent dihydrotetrazines, and subsequently be activated and functionalized through bioconjugation. Thus, an aqueous solution of a water soluble bis-tetrazine monomer was combined with an organic soluble bis-sTCO containing a tethered dihydrotetrazine (Figure 5A). Again, meter-long, mechanically robust polymer fibers were continuously pulled from the liquid-liquid interface without fiber breakage (Video 1), confirming that the molecular weight of the polymer exceeds that required for chain entanglement.47 Subsequent oxidation by long wavelength photocatalysis was used to generate reactive tetrazine functionality, and the fibers could then be functionalized by sTCO conjugates of proteins, fluorophores or peptides. Shown in Figure 5B are the monomers 5 and 6 that were used to create the DHT fibers. Notably, the DHT containing bis-sTCO 5 was readily purified, stored and handled without special precautions. The sTCO conjugates used to elaborate the fibers are displayed in Figure 5B.
Figure 5.

(A) Schematic representation of interfacial polymerization with a dihydrotetrazine-derived monomer. (B) Monomers 5 and 6 were used for interfacial polymerization. sTCO conjugates of the green fluorescent protein variant Clover, the dye Alexafluor 647, and an RGD peptide were used to modify the fibers.
As shown in Figure 6, the DHTz-fibers could be activated and then post-synthetically modified by treatment with Alexa-sTCO, Clover48-sTCO or RGD-sTCO. The fibers were immersed in 100 μM sensitizer in PBS and irradiated with a simple incandescent bulb for 5 min. Methylene blue was used to activate fibers toward conjugations of Clover-sTCO or RGD-sTCO, and rose bengal was used as the sensitizer in experiments with Alexa-sTCO due to the spectral overlap of the Alexa dye with methylene blue. After irradiation, the fibers were rinsed, allowed to react with an sTCO conjugate for 1 min, and rinsed again. Confocal microscopy images of activated fibers that were conjugated by Alexa-sTCO are displayed in Figure 6B and 6C. With this short incubation time, labeling was localized to the exterior of the fibers as clearly illustrated by the confocal z-stack images of the Clover-sTCO labeled fibers (Figure 6F, S19–20, Video 2) and Alexa-sTCO labeled fibers (Figures 6F, S14–15, Video 3). Control experiments illustrated that dye conjugation was not efficient if the sensitizer or light was excluded (Figure 6D, S16–18, S21–22). We note that HRP-catalyzed oxidation of dihydrotetrazines can also be used to activate fibers toward bioconjugation (Figure S23), however in this instance photocatalytic activation is faster and more efficient.
Figure 6.

(A) Schematic representation of fiber photoactivation and subsequent conjugation. DHTz-containing fibers were isolated in a silicone well on a glass substrate (for imaging) or in a polyHEMA coated Nunc chamber (for cell culture). The fibers were immersed in a PBS solution of sensitizer (100 μM), irradiated with visible light for 5 min, rinsed, allowed to react with an sTCO conjugate, and again rinsed. (B, C) Confocal images (10×, 40×) of activated fibers (rose bengal) that were treated with Alexa-sTCO (1 μM) for 1 min and rinsed. (D) Tagging by Alexa-sTCO was not observed in control experiments where (top) the rose bengal was excluded, and (bottom) where the sensitizer was included, but light was excluded. (E) Confocal (10×) image of activated fibers (methylene blue) that were treated with Clover-sTCO (5 μM) for 1 min and rinsed. (F) Confocal z-stack images of activated fibers that were conjugated with Alexa-sTCO (top) and Clover-sTCO (bottom). (G) Confocal images (10×) of activated fibers (methylene blue) that were treated with RGD-sTCO (10 μM). Cell culture with NIH 3T3 fibroblasts showed that the cells selectively adhered and spread on the fibers. Cell attachment and spreading on the fibers was not observed in control experiments where the sensitizer and/or the RGD-sTCO was excluded. The image above shows the control where both sensitizer and RGD-sTCO were excluded. Displayed in the supporting information are images from controls where only sensitizer was excluded, or where only RGD-sTCO was excluded.
The photocatalytic activation of tetrazines was also employed in the post-synthetic modification of the fibers with peptidic cues that promote cell adhesion and contact guidance. RGD-sTCO was conjugated to activated fibers through tetrazine ligation, and the resulting fibers were immobilized in silicone wells coated with poly(2-hydroxyethyl acrylate) to eliminate cellular adhesion to the culture wells. Here, fibroblasts selectively attached to RGD-tagged fibers and elongated along the long axis of the fibers, adopting a healthy fibroblastic morphology (Figure 6G). Cell attachment and spreading was not observed in control experiments where the sensitizer and/or the RGD-sTCO were excluded (Figures 6G, S26–S28). Instead, cells clustered to form multicellular spheroids, indicating the initiation of nemesis.49 These studies demonstrate the ability to functionalize biomimetic fibers with molecules that can enable visualization or promote cell adhesion.
As shown here, photocatalytic and enzymatic methods for turning on the tetrazine ligation provide a new tool for modulating the cell adhesive properties of a biomaterial. With future development we expect that the catalytically inducible tetrazine ligation will extend to a wide range of applications in materials, cellular and in vivo systems. We also expect that the electrochemical oxidation of dihydrotetrazines in solution will serve as an important stimulus for inducing bioorthogonal reactivity. We further anticipate that the new methods for inducing rapid bioorthogonal chemistry presented here should find a range of applications including cell imaging, pretargeted prodrug activation, and gene activation.
Supplementary Material
Acknowledgments
For funding this research, we are grateful to NSF DMR-1506613, NIH DC014461, NIH GM26643, P20GM104316 and the Osteoscience Foundation. K.T.D. is grateful for a Robert Gore Fellowship. Spectra were obtained with instrumentation supported by NIH grants P20GM104316, P30GM110758, S10RR026962, S10OD016267 and NSF grants CHE-0840401, CHE-1229234.
Footnotes
Supporting Information Synthetic procedures and compound characterization data; procedures for dihydrotetrazine (DHTz) and DHTz-containing microfiber oxidation/tagging experiments; confocal microscopic images of DHTz-containing microfiber tagging experiments and NIH 3T3 fibroblast cell attachment experiments.
References
- 1.Sletten EM, Bertozzi CR. Angew Chem Int Ed Engl. 2009;48:6974. doi: 10.1002/anie.200900942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lang K, Chin JW. Chem Rev. 2014;114:4764. doi: 10.1021/cr400355w. [DOI] [PubMed] [Google Scholar]
- 3.Azagarsamy MA, Anseth KS. ACS Macro Lett. 2013;2:5. doi: 10.1021/mz300585q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.McKay CS, Finn MG. Chem Biol. 2014;21:1075. doi: 10.1016/j.chembiol.2014.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Jewett JC, Bertozzi CR. Chem Soc Rev. 2010;39:1272. doi: 10.1039/b901970g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Shih HW, Kamber DN, Prescher JA. Curr Opin Chem Biol. 2014;21:103. doi: 10.1016/j.cbpa.2014.07.002. [DOI] [PubMed] [Google Scholar]
- 7.MacKenzie DA, Sherratt AR, Chigrinova M, Cheung LLW, Pezacki JP. Curr Opin Chem Biol. 2014;21:81. doi: 10.1016/j.cbpa.2014.05.023. [DOI] [PubMed] [Google Scholar]
- 8.Yu Z, Ohulchanskyy TY, An P, Prasad PN, Lin Q. J Am Chem Soc. 2013;135:16766. doi: 10.1021/ja407867a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tasdelen MA, Yagci Y. Angew Chem Int Ed Engl. 2013;52:5930. doi: 10.1002/anie.201208741. [DOI] [PubMed] [Google Scholar]
- 10.Ramil CP, Lin Q. Curr Opin Chem Biol. 2014;21:89. doi: 10.1016/j.cbpa.2014.05.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Arumugam S, Orski SV, Mbua NE, McNitt C, Boons GJ, Locklin J, Popik VV. Pure Appl Chem. 2013;85:1499. [Google Scholar]
- 12.Debets MF, van Berkel SS, Dommerholt J, Dirks ATJ, Rutjes FPJT, van Delft FL. Acc Chem Res. 2011;44:805. doi: 10.1021/ar200059z. [DOI] [PubMed] [Google Scholar]
- 13.Versteegen RM, Rossin R, ten Hoeve W, Janssen HM, Robillard MS. Angew Chem Int Ed Engl. 2013;52:14112. doi: 10.1002/anie.201305969. [DOI] [PubMed] [Google Scholar]
- 14.Matikonda SS, Orsi DL, Staudacher V, Jenkins IA, Fiedler F, Chen J, Gamble AB. Chem Sci. 2015;6:1212. doi: 10.1039/c4sc02574a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Li J, Jia S, Chen PR. Nat Chem Biol. 2014;10:1003. doi: 10.1038/nchembio.1656. [DOI] [PubMed] [Google Scholar]
- 16.Grim JC, Marozas IA, Anseth KS. J Control Release. 2015;219:95. doi: 10.1016/j.jconrel.2015.08.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Urdabayev NK, Poloukhtine A, Popik VV. Chem Commun. 2006:454. doi: 10.1039/b513248g. [DOI] [PubMed] [Google Scholar]
- 18.Dore TD, Wilson HC. Chromophores for the Delivery of Bioactive Molecules with Two-Photon Excitation. In: Chambers JJ, Kramer RH, editors. Neuromethods. Vol. 55. Humana Press; Totowa, NJ: 2011. pp. 57–92. [Google Scholar]
- 19.Gorka AP, Nani RR, Zhu J, Mackem S, Schnermann MJ. J Am Chem Soc. 2014;136:14153. doi: 10.1021/ja5065203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Palao E, Slanina T, Muchová L, Šolomek T, Vitek L, Klán P. J Am Chem Soc. 2016;138:126. doi: 10.1021/jacs.5b10800. [DOI] [PubMed] [Google Scholar]
- 21.Nkepang G, Bio M, Rajaputra P, Awuah SG, You Y. Bioconjug Chem. 2014;25:2175. doi: 10.1021/bc500376j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Olejniczak J, Carling CJ, Almutairi A. J Control Release. 2015;219:18. doi: 10.1016/j.jconrel.2015.09.030. [DOI] [PubMed] [Google Scholar]
- 23.Ritter C, Nett N, Acevedo-Rocha CG, Lonsdale R, Kräling K, Dempwolff F, Hoebenreich S, Graumann PL, Reetz MT, Meggers E. Angew Chem Int Ed Engl. 2015;54:13440. doi: 10.1002/anie.201506739. [DOI] [PubMed] [Google Scholar]
- 24.Azevedo AM, Martins VC, Prazeres DMF, Vojinović V, Cabral JMS, Fonseca LP. Biotechnol Annu Rev. 2003;9:199. doi: 10.1016/s1387-2656(03)09003-3. [DOI] [PubMed] [Google Scholar]
- 25.Wardman P. Curr Pharm Des. 2002;8:1363. doi: 10.2174/1381612023394610. [DOI] [PubMed] [Google Scholar]
- 26.Folkes LK, Wardman P. Biochem Pharmacol. 2001;61:129. doi: 10.1016/s0006-2952(00)00498-6. [DOI] [PubMed] [Google Scholar]
- 27.Klapper MH, Hackett DP. J Biol Chem. 1963;238:3736. [PubMed] [Google Scholar]
- 28.Yokota K, Yamazaki I. Biochemistry. 1977;16:1913. doi: 10.1021/bi00628a024. [DOI] [PubMed] [Google Scholar]
- 29.Wu H, Devaraj NK. Top Curr Chem. 2015;374:3. doi: 10.1007/s41061-016-0024-4. [DOI] [PubMed] [Google Scholar]
- 30.Devaraj NK, Weissleder R. Acc Chem Res. 2011;44:816. doi: 10.1021/ar200037t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Selvaraj R, Fox JM. Curr Opin Chem Biol. 2013;17:753. doi: 10.1016/j.cbpa.2013.07.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Darko A, Wallace S, Dmitrenko O, Machovina MM, Mehl RA, Chin JW, Fox JM. Chem Sci. 2014;5:3770. doi: 10.1039/C4SC01348D. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Taylor MT, Blackman ML, Dmitrenko O, Fox JM. J Am Chem Soc. 2011;133:9646. doi: 10.1021/ja201844c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Selvaraj R, Fox JM. Tetrahedron Lett. 2014;55:4795. doi: 10.1016/j.tetlet.2014.07.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Clavier G, Audebert P. Chem Rev. 2010;110:3299. doi: 10.1021/cr900357e. [DOI] [PubMed] [Google Scholar]
- 36.Ehret F, Wu H, Alexander SC, Devaraj NK. J Am Chem Soc. 2015;137:8876. doi: 10.1021/jacs.5b03371. [DOI] [PubMed] [Google Scholar]
- 37.Nickerl G, Senkovska I, Kaskel S. Chem Commun (Camb) 2015;51:2280. doi: 10.1039/c4cc08136f. [DOI] [PubMed] [Google Scholar]
- 38.Blackman ML, Royzen M, Fox JM. J Am Chem Soc. 2008;130:13518. doi: 10.1021/ja8053805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lang K, Davis L, Wallace S, Mahesh M, Cox DJ, Blackman ML, Fox JM, Chin JW. J Am Chem Soc. 2012;134:10317. doi: 10.1021/ja302832g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Rossin R, van den Bosch SM, Ten Hoeve W, Carvelli M, Versteegen RM, Lub J, Robillard MS. Bioconjug Chem. 2013;24:1210. doi: 10.1021/bc400153y. [DOI] [PubMed] [Google Scholar]
- 41.Rossin R, Renart Verkerk P, van den Bosch SM, Vulders RCM, Verel I, Lub J, Robillard MS. Angew Chem Int Ed. 2010;49:3375. doi: 10.1002/anie.200906294. [DOI] [PubMed] [Google Scholar]
- 42.Wu Z, Liu S, Hassink M, Nair I, Park R, Li L, Todorov I, Fox JM, Li Z, Shively JE, Conti PS, Kandeel F. J Nucl Med. 2013;54:244. doi: 10.2967/jnumed.112.109694. [DOI] [PubMed] [Google Scholar]
- 43.Rossin R, Robillard MS. Curr Opin Chem Biol. 2014;21:161. doi: 10.1016/j.cbpa.2014.07.023. [DOI] [PubMed] [Google Scholar]
- 44.Other successful sensitizers (irradiation wavelength) include acridine orange (528 nm), coomassie brilliant blue (528 nm), rhodamine B (590 nm), BODIPY (475 nm), safranin (528 nm), phenol red (528 nm) and carboxylfluorescein (528 nm).
- 45.Schirmer RH, Adler H, Pickhardt M, Mandelkow E. Neurobiol Aging. 2011;32:2325.e7. doi: 10.1016/j.neurobiolaging.2010.12.012. [DOI] [PubMed] [Google Scholar]
- 46.Folkes LK, Wardman P. Cancer Res. 2003;63:776. [PubMed] [Google Scholar]
- 47.Liu S, Zhang H, Remy RA, Deng F, Mackay ME, Fox JM, Jia X. Adv Mater. 2015;27:2783. doi: 10.1002/adma.201500360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lam AJ, St-Pierre F, Gong Y, Marshall JD, Cranfill PJ, Baird MA, McKeown MR, Wiedenmann J, Davidson MW, Schnitzer MJ, Tsien RY, Lin MZ. Nat Methods. 2012;9:1005. doi: 10.1038/nmeth.2171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Salmenperä P, Kankuri E, Bizik J, Sirén V, Virtanen I, Takahashi S, Leiss M, Fässler R, Vaheri A. Exp Cell Res. 2008;314:3444. doi: 10.1016/j.yexcr.2008.09.004. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.