

Abstract
Most molecular phylogenetic studies place all placental mammals into four superordinal groups, Laurasiatheria (e.g. dogs, bats, whales), Euarchontoglires (e.g. humans, rodents, colugos), Xenarthra (e.g. armadillos, anteaters) and Afrotheria (e.g. elephants, sea cows, tenrecs), and estimate that these clades last shared a common ancestor 90–110 million years ago. This phylogeny has provided a framework for numerous functional and comparative studies. Despite the high level of congruence among most molecular studies, questions still remain regarding the position and divergence time of the root of placental mammals, and certain ‘hard nodes’ such as the Laurasiatheria polytomy and Paenungulata that seem impossible to resolve. Here, we explore recent consensus and conflict among mammalian phylogenetic studies and explore the reasons for the remaining conflicts. The question of whether the mammal tree of life is or can be ever resolved is also addressed.
This article is part of the themed issue ‘Dating species divergences using rocks and clocks’.
Keywords: phylogenomics, time-tree, supermatrix, coalesence, fossils, molecules
1. Introduction
Of all classes of animals, humans are most concerned with and fascinated by class Mammalia, of which we are members. Class Mammalia contains warm-blooded animals that have hair or fur, produce milk and typically give birth to live young [1]. As of 2005, there are approximately 5400 living mammalian species that inhabit every biome on earth, range in size from the 2 g bumblebee bat to the 170 tonne blue whale, and exhibit unprecedented phenomic diversity and ecological adaptations [1]. Class Mammalia is divided into two subclasses: Prototheria, which contains the egg-laying monotremes (platypus and echidna), and Theria, which contains the placental and marsupial clades [2]. The mammalian fossil record extends deep into the Triassic (approx. 220 Ma) and records the evolution of mammalian lineages through extreme changes in flora, environments and landmasses during the Cretaceous Terrestrial Revolution (KTR) and the Cretaceous–Palaeogene (KPg) mass extinction events [3–6].
Despite the keen interest in mammals, the evolutionary history of this clade has been and remains at the centre of heated scientific debates [4,5,7–12]. In part, these controversies stem from the widespread occurrence of convergent morphological characters in mammals, which makes it difficult to tease apart homology and homoplasy in phylogenetic analyses that are solely based on these characters [4,9,13,14]. Molecules have proven more successful in recovering relationships among extant taxa in the mammalian tree [4,5,15,16], but molecular data cannot be obtained for most extinct taxa. Notable exceptions include South American ungulates and glyptodonts, which have been positioned in the mammalian tree based on protein sequences of type I collagen [17] and complete mitogenomic DNA sequences [18], respectively. Even for molecular data, different data types require different phylogenetic models [14], each of which has its own limitations [10]. Whole genome analyses have promised to revolutionize our understanding of animal evolutionary history, but some claims for the robust resolution of difficult nodes with phylogenomic data are underpinned by problematic analyses and/or poor data [10,19,20]. Despite these difficulties, there is consensus over the main topology [4–6] even though a few local polytomies are still unresolved (e.g. placental root; branching pattern within Paenungulata; position of Scandentia within Euarchontoglires; branching pattern within Laurasiatheria [4,7,21,22]). Consensus on the timing and biogeographic history of the placental mammal radiation has proved more elusive [4–6,8,9] owing to morphological convergence and uncertain phylogenetic relationships of fossil to living taxa [9,13]; rapid cladogenesis at difficult to resolve nodes [23]; disagreement over appropriate calibration strategies for molecular dating analyses; and higher levels of incompleteness in the Cretaceous and Early Cenozoic fossil record of the Southern Hemisphere than Northern Hemisphere. Indeed, the history of debate on these issues, in conjunction with the rich database of mammalian fossils and genomes, provides an unprecedented opportunity to explore the pros and cons of modern methods for species tree estimation and timetree construction.
Here, we showcase some of the past and present debates pertaining to the phylogenetic and evolutionary history of placental mammals. We discuss the next generation of phylogenetic methods and data that have been employed to resolve the placental mammal tree of life. We analyse a new molecular dataset comprised of sequences for 286 mammals and four outgroups, and date the divergence of these species to generate a novel timetree. We provide a roadmap for future analyses, detailing the pros and cons of current methods, and highlight the ways forward to finally resolve the mammal tree of life.
2. Shaking the morphological tree
In 1945, G.G. Simpson published ‘The classification of mammals’ [24]. This seminal work was based on morphological comparisons and is largely reflected in the landmark phylogeny published by Novacek [25] in Nature. Novacek's [25] morphological tree portrays a basal split between an edentate group comprised of Xenarthra (e.g. armadillos, anteaters) and Pholidota (pangolins) and a larger group that includes all other placental mammal orders. The latter group includes Ungulata (all hoofed mammals), Archonta (e.g. colugos, bats, primates), Anagalida (e.g. rodents and elephant shrews), Carnivora and Insectivora (table 1). Throughout the 1990s, aspects of this topology [25] were challenged through new fossil finds and phylogenetic analyses: for example, Beard [30] suggested a closer affinity of Dermoptera (e.g. colugos) with Primates than bats, and the finding of Eocene whale fossils (Artiocetus, Rodhocetus) with diagnostic ankle bones suggested derivation of Cetacea from Artiodactyla rather than from Mesonychia [31]. Nevertheless, a recent cladistic analysis of the largest morphological dataset to date, which includes approximately 4500 characters [8], has many features in common with Novacek's [25] morphological tree and still supports numerous polyphyletic groups including an ‘insectivore’ group, a ‘spiny hedgehog’ group, an ‘ant and termite eating’ group, a ‘tree-dwelling’ group and an ‘ungulate’ group [9] (table 1). On the molecular front, features of Novacek's [25] morphological tree were both corroborated and challenged by early phylogenetic analyses of DNA sequences (reviewed by Springer et al. [14]). Morphological and molecular consensus emerged for Paenungulata (hyraxes, manatees and elephants) [24,25,32–37], and some molecular studies [35,38] agreed with morphology in supporting Glires (lagomorphs and rodents) [24,32,35,38,39]. Other morphological hypotheses including Altungulata (perissodactyls and paenungulates), Anagalida (rodents, lagomorphs, and elephant shrews), Archonta (primates, dermopterans, treeshrews and bats), Ungulata (paenungulates, perissodactyls, cetartiodactyls and aardvarks) and Volitantia (bats and colugos) were rejected by molecular data [35]. Of historical interest, early DNA studies also suggested novel or throwback hypotheses including rodent paraphyly, a basal split between rodents or hedgehogs and all other placental mammals, and a modified version of Gregory's [40] Marsupionta (monotremes and marsupials) hypothesis, all of which have now been debunked (reviewed in Novacek [25] and Springer et al. [14]). These extraordinary hypotheses potentially resulted from poor taxon sampling with its attendant long-branch misplacement problems [14]. The next wave of studies (see below) aimed to address these deficiencies with increased gene and taxon sampling.
Table 1.
Higher-level relationships of placental mammal orders based on morphology versus molecules. Orders (italics) are coloured by their superordinal membership according to molecular studies. The majority of superordinal groups based on morphology are polyphyletic and reflect ecomorphological convergence (e.g. ‘ant and termite eating group’ includes representatives from Xenarthra, Afrotheria, and Laurasiatheria).
 |
3. First wave of large-scale molecular data—comparative molecular phylogenetics
At the turn of this millennium, the first large-scale molecular phylogenetic studies were published [15,16,26] and drove the most significant revisions of Novacek's [25] phylogeny (table 1; Springer et al. [14]). These initial studies, which included representatives from all recognized mammalian orders, were based on large multigene datasets comprised of both nuclear and mitochondrial markers and provided support for four superordinal clades of placental mammals that are still recognized today: Afrotheria, Xenarthra, Euarchontoglires and Laurasiatheria [15,16,26–29]. Subsequent studies of retrotransposed elements and indels provided confirmatory support for these four superordinal clades [13,15,16,26,29,41,42]. Afrotheria contains the orders Tubulidentata (aardvarks), Afrosoricida (e.g. golden moles and tenrecs), Macroscelidea (elephant shrews), Hyracoidea (hyraxes), Sirenia (manatees, dugongs, sea cows) and Proboscidea (e.g. elephants), with the latter three orders grouping in the clade Paenungulata. Xenarthra contains Cingulata (armadillos) and Pilosa (anteaters, sloths). The remaining mammalian orders are grouped into Boreoeutheria, which is further divided into the superordinal groups Laurasiatheria and Euarchontoglires. Laurasiatheria unites phenotypically diverse orders: Chiroptera (bats), Cetartiodactyla (e.g. cetaceans, cows and pigs), Perissodactyla (e.g. rhinos and horses), Pholidota (pangolins), Carnivora (e.g. lions, dogs, seals) and Eulipotyphla (e.g. shrews and hedgehogs). Eurachontoglires contains Primates (e.g. humans and monkeys), Scandentia (treeshrews), Glires (e.g. rabbits and rodents) and Dermoptera (colugos).
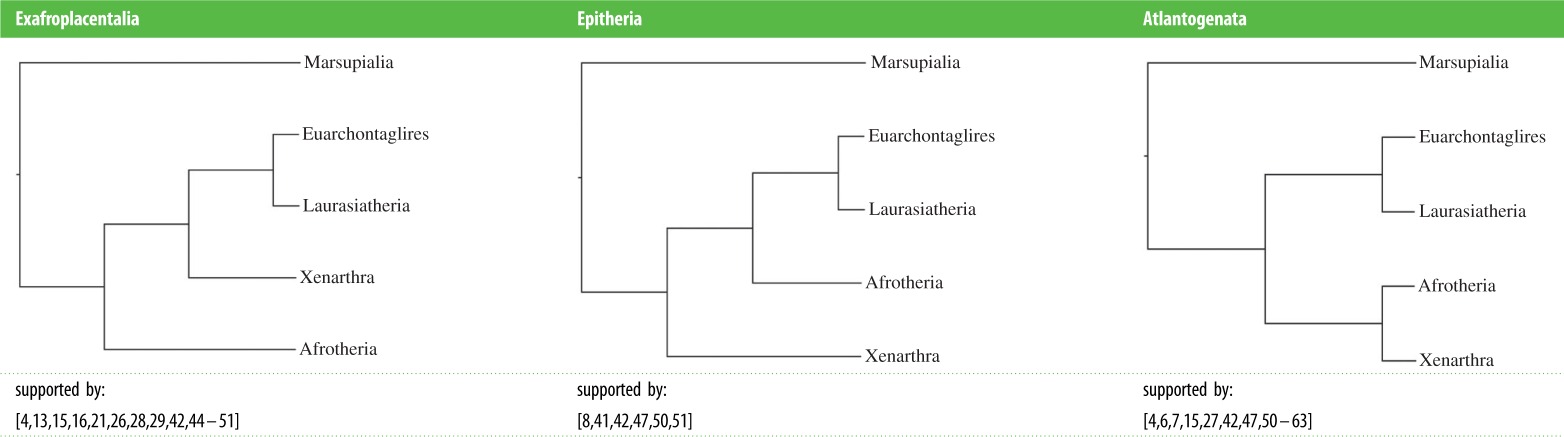
While numerous studies support the monophyly of each of the four superordinal groups, including studies with expanded gene and taxon sampling [4], their branching order at the base of Placentalia is still unresolved and remains hotly debated [43]. The three competing hypotheses for the placental root posit basal splits between (i) Exafroplacentalia and Afrotheria, (ii) Xenarthra and Epitheria, and (iii) Atlantogenata and Boreoeutheria (Teeling & Hedges [43]; table 2). Many of the early large-scale molecular phylogenetic studies favoured Afrotheria versus Exafroplacentalia [15,16,26–28,44], but Atlantogenata versus Boreoeutheria and to a lesser extent Xenarthra versus Epitheria also received support, sometimes in the same studies that provided support for Afrotheria versus Exafroplacentalia [15,16,28]. Meredith et al.'s [4] multigene dataset, which expanded gene sampling to 26 loci (totalling approx. 35.6 kb) and extended taxonomic coverage from mammalian orders to 97% of mammalian families, was insufficient to resolve the placental root and supported Afrotheria versus Exafroplacentalia (DNA analyses) or Atlantogenata versus Boreoeutheria (amino acid analyses). Analyses of coding indels and retroposons have also returned mixed results, with some studies favouring a basal split between Xenarthra and Epitheria [41] and others supporting Atlantogenata versus Boreoeutheria [52]. The most comprehensive study based on retroposon insertions provides commensurate levels of support for each of the three competing hypotheses [64].
Table 2.
Summary of the various papers supporting one of the three hypotheses for the root node of eutherian mammals: Exafroplacentalia, Epitheria or Atlantogenata.
 |
Aside from the placental root, other recalcitrant problems in higher-level placental systematics include (i) the Laurasiatheria polytomy, (ii) sister-group relationships within Paenungulata, and (iii) the position of Scandentia (treeshrews) within Euarchontoglires [14]. These difficult problems are characterized by short internal branches, which are indicative of rapid radiations, and are likely to remain difficult to resolve owing to incomplete lineage sorting (ILS) and a variety of systematic biases including long branch misplacement and model mis-specification that can hamper phylogenetic inference. Even though these nodes and branches of the tree remained unresolved, it was hoped that the promise of phylogenomic analyses would resolve these ‘tricky’ branches [65].
4. Second wave of molecular data—comparative phylogenomics
In 2001, the draft sequence of the human genome was published [66,67]. Following its initial publication, researchers aimed to discover, annotate and describe functional elements in the human genome through cross-species comparisons with other mammals [68]. In 2005, a sequencing effort began to maximize the representation of mammals from each of the four superordinal groups, which culminated in the publication of 29 mammal genomes [68]. This particular sequencing effort, along with ongoing large-scale sequencing initiatives such as Genome 10 K, which aims to sequence 10 000 vertebrate genomes, have revolutionized comparative genomics and mammalian phylogenetics [69,70]. Different approaches to analyse these computationally challenging, extremely large datasets have included standard supermatrix methods (= total evidence), with or without the incorporation of sophisticated models that accommodate tree and dataset heterogeneity [6,45,53], shortcut coalescence methods and concatalescence (= binning) approaches that combine elements of concatenation with shortcut coalescence [7]. The application of these methods to large molecular datasets has not resulted in consensus for difficult problems such as the placental root, the position of treeshrews and the Laurasiatheria polytomy. Rather, these studies often highlight incongruent results that arise from different phylogenomic analyses of the same dataset. Conflicting results of concatenation and coalescence studies, in particular, have become the subject of spirited debates [7,9–11,19,71] that hearken back to the ‘cladist/pheneticist/likelihood’ debates of the 1970s and 1980s [11].
5. Revolution in analytical methods
In the supermatrix approach, data from multiple gene fragments are concatenated to form a single matrix for subsequent phylogenetic analyses [19]. The advantage of the supermatrix approach lies in combining individual genes into a single matrix for simultaneous analysis, an approach which has the ability to decrease sampling error, offset homoplastic signals in individual genes and uncover hidden support [19,72] (table 3). Hidden support refers to increased support for a clade in combined analysis relative to the support obtained in separate analysis [19,73], and is the primary advantage of the supermatrix approach. In recent years, the supermatrix approach for species tree estimation has been criticized by champions of coalescent methods, who have correctly noted that concatenation methods do not explicitly address the problem of ILS [7,21,54,74–78]. Furthermore, it has been suggested that the supermatrix approach can overestimate nodal support when an incorrect model of sequence evolution is applied. However, coalescence methods make their own assumptions and the worthwhile goal of accounting for ILS introduces a series of potential problems that may arise if these assumptions are violated. In particular, coalescence methods assume recombination between but not within loci, gene tree heterogeneity that results exclusively from ILS, and neutral evolution [76,77].
Table 3.
Brief description and implementation of recent emerging tree reconstruction methods.
| method | coalescence | shortcut coalescence | binning | supermatrix | supertree |
|---|---|---|---|---|---|
| description | co-estimates gene trees and species tree; fully parametric | separate estimation of gene tree and species tree; partially parametric | genes are concatenated into supergenes prior to coalescence-based estimation of the species tree | estimates species tree from gene fragments concatenated into a single matrix; also referred to as concatenation | various subsets of characters are analysed to generate subtrees which are further combined to produce a species tree |
| implementation | BEST, *BEAST | STAR, STEAC, MP-EST, STEM, MDC, ASTRAL | STAR, STEAC, MP-EST, STEM, MDC, ASTRAL | wide variety of programmes that implement maximum parsimony, distance, maximum-likelihood and Bayesian methods | MRP–PAUP* |
| computation | massively computationally intensive | less computationally intensive | less computationally intensive | less computationally intensive | less computationally intensive |
| suitable for large-scale analysis | no | yesa | yes | yes | yes |
aMP-EST not suitable for datasets with large numbers of taxa [20].
Given the occurrence of unresolved nodes on the mammalian tree that are characterized by rapid divergences [65], coalescent approaches may be better suited to resolving such nodes than concatenation [21]. However, fully parametric coalescent models such as *BEAST [79] and BEST [80], which co-estimate gene trees and the species tree [81], cannot be applied to large datasets because they are massively computationally intensive [19]. As such, shortcut coalescence methods such as STEAC, STAR [76], MP-EST [54] and ASTRAL [82], which carry out separate estimation of gene trees and the species tree, have been applied to large mammalian datasets [7,21] (table 3). Criticisms aimed at recent applications of these shortcut methods to the placental tree have focused on inappropriate coalescent-gene (c-gene) size and high levels of gene tree reconstruction error [10]. One of the key assumptions of the multispecies coalescent model is that recombination occurs between c-genes but not within c-genes. To satisfy this assumption, it is necessary to use gene trees that are inferred from short stretches of DNA. Empirical evidence from primates has shown that the average length of recombination free c-genes is less than 100 bps [83,84]. This is particularly problematic for the application of coalescent approaches to large taxonomic datasets because as the number of taxa increases, c-gene size must decrease owing to the recombination rachet [19]. Recent phylogenomic studies with coalescence methods have employed widely variable mean locus lengths. McCormack et al.'s [21] mean locus length for ultraconserved elements was only 410 bp, but Song et al. [7] reported an average locus length of 3.1 kb for protein-coding sequences. However, Song et al. [7] did not consider the intervening introns and their true mean locus length, measured from start codon to stop codon, is 139.6 kb [19]. This inadvertent application of coalescence methods to ‘c-genes’ that are much too long was dubbed concatalescence by Gatesy & Springer [85], because disparately spaced exons are first concatenated into coding sequences and then subsequently analysed using shortcut coalescence approaches. A recent study using simulated data suggests that species tree estimation using coalescent methods may be more robust to recombination than previously thought [86], but these simulations were for relatively shallow divergences and only included a small number of taxa. Further, these simulations [86] did not compare the performance of coalescence methods to concatenation in their study of the effects of recombination on coalescence methods. More recently, the idea of concatalescence has been reimagined as statistical binning, in which gene trees and alignments are assigned to a bin based on ‘combinability’ before creating supergene trees for downstream coalescence analysis [87]. Accurate gene tree reconstructions are also central to the coalescent approach as each tree is given equal weight in the analysis; however, accurate reconstructions become increasingly difficult with decreasing c-gene sizes as more taxa are added to the analysis [19]. It should be noted that many of the forces impacting accurate gene tree reconstruction such as long branches, mutational saturation and weak signal are also problematic issues for supermatrix approaches [88].
At present, the relative advantages and disadvantages of coalescence versus supermatrix approaches for phylogeny reconstruction have not been fully explored. This is despite the existence of many simulation studies, which have variably supported one method over the other depending on the simulation parameters [82,87,89–93]. Future studies should simulate more realistic c-gene sizes and increase the number of taxa to better reflect empirical datasets, and examine the effects of recombination on coalescence versus concatenation methods [86].
Beyond the direct debate over coalescent and supermatrix approaches to species tree construction, alternative solutions are emerging within each of these camps. On the coalescence front, single nuclear polymorphism (SNP) methods such as SVDquartets [94] provide an alternative to fully parametric and shortcut coalescence methods that depend on gene trees. An important advantage of SNP methods is that they avoid problems with the recombination ratchet [10]. Among novel supermatrix approaches, Romiguier et al. [45] explored the effect of biased gene conversion, as measured by GC content, which is an indicator of recombination, on phylogenetic inference. Through comparative analysis of GC-rich and AT-rich datasets, they showed that GC-rich genes induced a higher amount of conflict between gene trees, whereas AT-rich datasets provided increased resolution of relationships among placental mammals [45]. Crucially, these compositionally biased datasets supported different root node hypotheses and in doing so highlight the potentially confounding role composition bias can play in phylogenetic inference if not adequately accounted for during data selection or through appropriate model selection. Previous large-scale supermatrix analyses of the placental tree have used homogeneous models of sequence evolution [4,95]. However, among placental mammals, there exists considerable heterogeneity among genes, lineage-specific substitution rates and sequence compositional bias [53]. In an analysis that accounted for heterogeneity across the phylogeny using NDRH/NDCH node-discrete rate matrix heterogeneous/node-discrete composition heterogeneous models and CAT to model heterogeneity across the data, Morgan et al. [53] showed that employing models that account for heterogeneity greatly improves phylogenetic resolution compared to homogeneous models.
6. Can comparative genomics resolve problematic nodes in the placental tree?
Despite the abundance of genomic data for placental mammals and the development of novel methods for phylogeny reconstruction, the ‘tricky’ nodes are still unresolved. The placental root node remains a matter of contention with claims of strong support emerging for one or a number of the three competing hypotheses using different data types and analytical methods: Exafroplacentalia [21,45,46], Epitheria [8] and Atlantogenata [4,7,53]. However, the most recent study of the placental tree has taken a ‘total evidence’ approach to resolve the root by analysing morphological, nucleotide and micro-RNA data using a supermatrix approach with heterogeneous modelling, coalescent approaches with unbinned gene trees and statistical binning [6]. In this study, Tarver et al. [6] recovered high support for the Atlantogenata hypothesis (originally described from coding indels by Murphy et al. [52]) from their supermatrix and concatalescence analyses of nucleotides, as well as from their micro-RNA dataset. Furthermore, to investigate previous studies that supported other hypotheses for the placental root, they re-analysed the datasets of O'Leary et al. [8], Hallström & Janke [46] and the AT-rich dataset of Romiguier et al. [45], and found that model fit for these datasets could be improved using heterogeneous modelling, and that reanalysis of the datasets of O'Leary et al. [8] and Hallström & Janke [46] supported Atlantogenata [6]. While the re-analysis of the Romiguier et al. [45] dataset only yielded minimal support for Altlantogenata (approx. 0.5 posterior probability), the dataset no longer recovered strong support for Exafroplacentalia [6]. While consensus may be tipping in favour of an Atlantogenata versus Boreoeutheria root, advocates of this and other hypotheses for the placental root have not provided compelling explanations for Nishihara et al.'s [64] retroposon study that provides commensurate levels of support for each of the three competing hypotheses, i.e. 22, 25 and 21 L1 insertions favouring Exafroplacentalia, Epitheria and Atlanatogenata, respectively. Unfortunately, despite the large datasets, the other hard nodes also remain to be resolved. At present, it is unclear if treeshrews (Order Scandentia) are more closely related to Glires (rodents and lagomorphs), Primatomorpha (primates and colugos) or Dermoptera (colugos). Also, relationships among the laurasiatherian clades Chiroptera, Ostentoria (carnivorans and pangolins), Perissodactyla and Cetartiodactyla await elucidation. There is still much research ahead for future mammal phylogeneticists.
7. Dating the mammal tree—a time bomb
The timing of the origin and diversification of placental mammals is a highly contentious topic in phylogenetics. At its core lies disagreement on the timing of interordinal and intraordinal divergences in relationship to the KPg boundary. Archibald and Deutchsmann [96] proposed three models to characterize the results of different studies. More recently, each of these models has received support from one or more large-scale studies of placental mammal phylogeny. Archibald & Deutchsmann's [96] three models are (i) the explosive model, (ii) the short fuse model and (iii) the long fuse model. The explosive model is generally favoured by palaeontologists and proposes that both interordinal and intraordinal clagodenesis within Placentalia occurred after the KPg boundary (approx. 66 Ma) in response to newly available ecospace vacated by non-avian dinosaurs after the extinction event [8]. This model rejects a crown position for all Mesozoic eutherians and is consistent with O'Leary et al.'s [8] parsimony analysis of a combined phenomic–genomic dataset for representative extant and extinct eutherians.
By contrast, most molecular timetrees posit much older interordinal divergences, wherein placental mammals originate in the Cretaceous and persisted at low diversity before eventually experiencing an explosion in diversification [96]. Where molecular diversification models differ is in the duration of the lag period between the origin of placentals and their burst of diversification. The long fuse model posits interordinal cladogenesis in the Cretaceous followed by intraordinal cladogenesis after the KPg mass extinction, and is broadly supported by recent studies that have employed large molecular datasets with different models for branch rates (i.e. autocorrelated, independent) and multiple calibrations from the fossil record [4–6,12,97]. We analysed a dataset for Meredith et al.'s 26 genes that was expanded to included 286 mammals and four outgroups (26 loci; see electronic supplementary material, table S1). We included only therian taxa that are represented by at least 16 of 26 genes. Timetree analyses were performed with mcmctree [98] using autocorrelated rates with 99 hard-bounded calibrations (see electronic supplementary material, table S2). The results are highly congruent with Meredith et al.'s [4] original timetree analyses and estimates from dos Reis et al. [5] (figure 1). An estimated eutherian origin around approximately 170 Ma is in agreement with the fossil record following the discovery of a stem eutherian fossil Juramaia sinensis [101] from the Jurassic [4,5] and these results support the long fuse model (figure 1). While limited intraordinal cladogenesis may have commenced in the Late Cretaceous for a few orders (e.g. Eulipotyphla), the vast majority of placental intraordinal diversification takes place in the Cenozoic. The long fuse model is also consistent with a major role for the KPg extinction in promoting morphological and ecological diversification in the wake of the ecological vacuum that ensued after the KPg extinction event [4,5]. The short fuse model agrees with the long fuse model in positing interordinal cladogenesis in the Cretaceous, and further suggests that many intraordinal divergences occurred well before the KPg boundary. Among recent studies, support for this model is more limited and derives mostly from Bininda-Emonds et al.'s [95] mammalian supertree.
Figure 1.

Timetree based on mcmctree supports a long fuse model of mammalian diversification [4,5]. Divergence time estimates were obtained under a GTR + Γ model of sequence evolution, 99 calibrated nodes (see electronic supplementary material table S2 for details), and autocorrelated rates of evolution. Divergence time estimates for clades of interest are shown at each node, with the 95% CI denoted by blue bars. The dataset included 26 tree of life gene fragments [4] and was expanded to include 286 taxa (see electronic supplementary material, table S1 for taxa, gene fragments and accession numbers). GenBank sequences for single genes were supplemented with available genome data that were mined using BLAST searches. The tree was estimated using RAxML [99] on the CIPRES web server [100] under a fully partitioned model where each partition had its own GTR + Γ model of sequence evolution. Paintings by Carl Buell. (Online version in colour.)
Both the short and long fuse models suggest that there should be crown placentals from the Cretaceous, but recent morphological parsimony analysis studies provide no support for this prediction and instead, position all Cretaceous taxa outside of crown Placentalia [8,102–104]. Explanations for this discrepancy are that (i) reconstructed divergence times in the Cretaceous based on molecular data are inaccurate [8], (ii) the placental fossil record is highly incomplete and it is simpler to propose ghost lineages in the Cretaceous than virus-like rates of molecular evolution in early placental mammals [9] and (iii) morphological parsimony analyses that position Cretaceous eutherians outside of crown Placentalia are inherently unreliable owing to pervasive convergence in morphological characters and clustering of the recent, which is a form of long branch attraction that can result in stemward slippage of fossil taxa. Below, we discuss each of these explanations for the perceived discrepancies between molecular and palaeontological estimates for interordinal divergence times.
O'Leary et al.'s [8] timetree for Placentalia provides the most explicit formulation of the explosive model. Importantly, O'Leary et al.'s [8] estimated nodal ages for interordinal divergences are minimum ages that are younger than true ages, because the fossil record is incomplete [12]. O'Leary et al.'s [8] enforcement of a maximum age of 66 Ma for the most recent common ancestor of Placentalia suggests that as many as 10 interordinal divergences may have occurred in the 200 000 years just after the KPg boundary. This hypothesis requires rates of molecular evolution that were accelerated more than 60× in early crown placentals relative to the stem placental ancestor [9]. Such rates would be commensurate with those of double-stranded DNA viruses. For these reasons, we reject the explosive model of placental mammal diversification and prefer the alternate explanations that ghost lineages extend into the Cretaceous and/or morphological parsimony analyses that position Cretaceous eutherians outside of Placentalia are incorrect. Phillips [105] suggested that Meredith et al.'s [4] timetree dates require more than 250 Myr of ghost lineages in Boreoeutheria, and for this reason proposed a soft explosive model wherein most interordinal diverences in Placentalia occurred after the KPg boundary. Phillip's [105] soft explosive model only allows for the emergence of the stem Afrotheria, stem Xenarthra, stem Laurasiatheria and stem Euarchontoglires lineages in the Cretaceous. Phillips [105] also suggested that Meredith et al.'s [4] interordinal divergences were inflated because of rate-transference errors, and that these errors can be mitigated by eliminating calibrations in clades that are characterized by large body sizes and long lifespans that are the source of rate transference errors. An alternate explanation is that Phillips' [105] interordinal divergence dates are too young because they are dragged forward by divergence dates in clades with large body size and long lifespan that are also too young. Indeed, Phillip's preferred timetree, which is based on calibrations at only 27 nodes (contra 82 in Meredith et al. [4]), has estimated dates at 62 of 136 internal nodes in Placentalia that are younger than minimum ages implied by the fossil record [106]. Included among these nodes are numerous interordinal divergences as well as some divergences in clades that are characterized by smaller body sizes (e.g. Eulipotyphla). By contrast, timetree analyses that exclude taxa based on large body size and/or long lifespan thresholds provide support for the long fuse model of placental diversification [29].
The final explanation for the perceived disagreement between molecular studies that support Cretaceous interordinal divergences and morphological parsimony analyses that place Cretaceous eutherians outside of crown Placentalia is that higher-level placental relationships in the latter are inaccurate and unreliable. Along these lines, Sansom & Wills [107] documented fundamental taphonomic biases that will cause stemward slippage of fossils. In addition, pseudo-extinction analyses [13] have shown that the majority of living placental orders move to a different superordinal group when molecular data are recoded as missing and that some orders are rendered polyphyletic or paraphyletic. Finally, cladistic analyses of the largest morphological datasets [8,103] exhibit massive homoplasy problems that call into question the results of analyses based on these datasets. In addition to problems with O'Leary et al.'s [8] morphological dataset that were noted above, Halliday et al.'s [103] dataset results in equally surprising relationships and their unconstrained morphological parsimony tree (their fig. 3) supports treeshrew diphyly, talpid diphyly, elephant shrew diphyly, etc., and fails to recover virtually every superordinal clade of placental mammals that is well supported by numerous molecular datasets.
Molecular scaffolds or combined analyses with molecular and morphological data can effectively rescue extant taxa, but the results of pseudo-extinction analyses suggest that molecular scaffolds/combined analyses are not effective for extinct taxa in higher-level placental phylogenetics. Finally, several authors (e.g. [108] for arctoid carnivorans) have suggested that parallel evolutionary trends can result in excessive homoplasy among living taxa that obscures relationships when diachronous terminals (i.e. extinct and extant taxa) are included in the same analyses. In view of these difficulties, Cretaceous interordinal divergences should not be rejected because they disagree with morphological parsimony trees. New approaches for molecular dating include total evidence or tip dating, but these approaches are dependent on accurate phylogenies and should be used cautiously given potential problems with morphological and palaeontological character data for placental mammals (e.g. correlated homoplasy, taphonomic biases, incompleteness) that are surpassed by the quantity, quality and unambiguity of molecular data [109–111]. Future clever and novel integrative research based on molecules, fossils and timetree estimation methods will be required to resolve the timing of origination and diversification of mammals.
8. The future
We have come so far and have greatly advanced our understanding of mammalian evolutionary history in the past twenty years. These advances have resulted from (i) new methods in molecular technologies enabling the fast and rapid generation of huge amounts of sequence data from novel taxa [69,70], (ii) the conception and implementation of novel analytical methods for accurately modelling sequence evolution and allowing independent rates and models across branches and (iii) the availability of fast and powerful computers that enable computationally intensive phylogenetic analyses. However, despite these advances, there are still three key areas that must be addressed before we can have ‘mammal tree’ resolution.
First, the unresolved ‘hard’ nodes in the mammal tree of life must be resolved. Whole genomes should provide the required data, which if aligned and analysed correctly, should aid in the resolution of these nodes. One requirement is that the sequence data must be excellent, the genomes must be at high coverage and ultimately at a chromosome level assembly [112]. New sequencing technologies, such as Dovetail and 10X Genomics, promise to aid in the difficult task of scaffolding de novo genomes and coupled with long sequence reads, such as PacBio sequencing, excellent chromosome level assemblies of de novo genomes are possible in the near future [112]. Appropriate and correct analyses of these data are required to resolve these problems. At present, the field advances on two fronts. On one front, the supermatrix approaches to solve these problems have been advanced through the application of better fitting heterogeneous models of sequence evolution, compared with analyses employing widely and in some cases inappropriately used homogeneous models [6,53]. On the other front, as outlined above, the limitations, powers and pitfalls of novel gene tree estimation methods must be elucidated to ascertain the best methods to analyse these new whole genome data; however, the refinement of statistical binning [82] shows promise to address some of the criticisms of this approach. Potentially, some of the ‘tricky’ nodes may not be resolved with phylogenomic analyses alone, but whole genome data will provide accurate insertion positions of novel transposable elements and insertion/deletion events that will provide independent synapomorphic characters, indicative of true shared ancestry. Non-molecular data can also be used to resolve these hard nodes. Haeckel [113] originally proposed that the study of morphological ontogenic development of embryos may reveal phylogenetic affinities not present in the adults [114]. New imaging technologies promise to provide novel non-lethal ontogenic evo/devo data of developing embryos [115], thus allowing for the study of protected and diverse species, never possible before. Molecular phylogenetic trees can act as a scaffold to differentiate between homoloplastic and homologous morphological characters, ultimately providing true morphological synapomorphic characters to help provide the data to resolve the difficult nodes [116]. All of these data, interpreted together should provide the information needed to resolve the mammal tree of life.
Second, to determine divergence times for this topology, we still must accurately assign fossils to the appropriate branches, whether across the tree or just at key basal nodes. This is a more difficult task as certain lineages are extensively missing fossil data (e.g. bats are missing more than 70% of fossil history; Teeling et al. [117]), and also the key homologous structures may be lost or modified during preservation. Uncovering key transitions fossils, e.g. Eocene whale fossils (Artiocetus, Rodhocetus), which contain diagnostic characters, could ultimately resolve the tricky nodes. Therefore, more fossil finds, particularly in the under-represented Southern Hemisphere are required. The development in new sequencing technologies has revolutionized the field of ancient DNA. It is now possible to sequence the whole genome of recent fossils and ultimately place them on a tree, from as little as a single bone (e.g. Denisovan [118]). This greatly advances our understanding of fossil placement in the tree. The integration of both molecular and morphological data and the contribution of each data type to phylogenetic inference must also be explored. Potentially these data cannot be analysed together, but each type of data offers unique insights into evolutionary history and therefore must be considered. Further advances in divergence time analyses are required to refine estimates, recent simulation studies, which incorporate the multispecies coalescent model into estimates of divergence time show promise with small datasets [119] and surely, the application of such an approach to large empirical datasets is just around the corner. With the new technological advances in sequencing, imaging, computation and analytical methods, coupled with future fossil finds, the next 20 years of phylogenetic research promise to be exciting and dynamic, and ultimately, should result in the resolution and dating of the mammal tree of life.
Supplementary Material
Acknowledgements
We thank Phil Donoghue and Ziheng Yang for the invitation to contribution. We also thank Mario dos Reis, Robert Asher and one anonymous reviewer for helpful comments and suggestions.
Authors' contributions
E.C.T. conceived and designed the study. M.S.S. assembled the datasets and built trees. All authors contributed to the timetree analysis, interpretation of results and writing, editing the manuscript.
Competing interests
We have no competing interests.
Funding
E.C.T and N.M.F are supported through a European Research Council Research Grant awarded to E.C.T (ERC-20120StG311000). M.S.S is supported through an NSF United States grant no. DEB-1457735
References
- 1.Wilson DE, Reeder DM. 2005. Mammal species of the world: a taxonomic and geographic reference. Baltimore, MD: Johns Hopkins University Press. [Google Scholar]
- 2.McKenna MC, Bell SK. 1997. Classification of mammals. New York, NY: Columbia University Press. [Google Scholar]
- 3.Benton MJ. 2010. The origins of modern biodiversity on land. Phil. Trans. R. Soc. B 365, 3667–3679. ( 10.1098/rstb.2010.0269) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Meredith RW, et al. 2011. Impacts of the Cretaceous Terrestrial Revolution and KPg extinction on mammal diversification. Science 334, 521–524. ( 10.1126/science.1211028) [DOI] [PubMed] [Google Scholar]
- 5.dos Reis M, Inoue J, Hasegawa M, Asher RJ, Donoghue PC, Yang Z. 2012. Phylogenomic datasets provide both precision and accuracy in estimating the timescale of placental mammal phylogeny. Proc. R. Soc. B 279, 3491–3500. ( 10.1098/rspb.2012.0683) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Tarver JE, et al. 2016. The interrelationships of placental mammals and the limits of phylogenetic inference. Genome Biol. Evol. 8, 330–344. ( 10.1093/gbe/evv261) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Song S, Liu L, Edwards SV, Wu S. 2012. Resolving conflict in eutherian mammal phylogeny using phylogenomics and the multispecies coalescent model. Proc. Natl Acad. Sci. USA 109, 14 942–14 947. ( 10.1073/pnas.1211733109) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.O'Leary MA, et al. 2013. The placental mammal ancestor and the post–K–Pg radiation of placentals. Science 339, 662–667. ( 10.1126/science.1229237) [DOI] [PubMed] [Google Scholar]
- 9.Springer MS, Meredith RW, Teeling EC, Murphy WJ. 2013. Technical comment on ‘The placental mammal ancestor and the post–K-Pg radiation of placentals’. Science 341, 613 ( 10.1126/science.1238025) [DOI] [PubMed] [Google Scholar]
- 10.Springer MS, Gatesy J. 2016. The gene tree delusion. Mol. Phylogenet. Evol. 94, 1–33. ( 10.1016/j.ympev.2015.07.018) [DOI] [PubMed] [Google Scholar]
- 11.Edwards SV, et al. 2016. Implementing and testing the multispecies coalescent model: a valuable paradigm for phylogenomics. Mol. Phylogenet. Evol. 94, 447–462. ( 10.1016/j.ympev.2015.10.027) [DOI] [PubMed] [Google Scholar]
- 12.dos Reis M, Donoghue PC, Yang Z. 2014. Neither phylogenomic nor palaeontological data support a Palaeogene origin of placental mammals. Biol. Lett. 10, 20131003 ( 10.1098/rsbl.2013.1003) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Springer MS, Burk-Herrick A, Meredith R, Eizirik E, Teeling E, O'Brien SJ, Murphy WJ. 2007. The adequacy of morphology for reconstructing the early history of placental mammals. Syst. Biol. 56, 673–684. ( 10.1080/10635150701491149) [DOI] [PubMed] [Google Scholar]
- 14.Springer MS, Stanhope MJ, Madsen O, de Jong WW. 2004. Molecules consolidate the placental mammal tree. Trends Ecol. Evol. 19, 430–438. ( 10.1016/j.tree.2004.05.006) [DOI] [PubMed] [Google Scholar]
- 15.Madsen O, et al. 2001. Parallel adaptive radiations in two major clades of placental mammals. Nature 409, 610–614. ( 10.1038/35054544) [DOI] [PubMed] [Google Scholar]
- 16.Murphy WJ, Eizirik E, Johnson WE, Zhang YP, Ryder OA, O'Brien SJ. 2001. Molecular phylogenetics and the origins of placental mammals. Nature 409, 614–618. ( 10.1038/35054550) [DOI] [PubMed] [Google Scholar]
- 17.Welker F, et al. 2015. Ancient proteins resolve the evolutionary history of Darwin's South American ungulates. Nature 522, 81–84. ( 10.1038/nature14249) [DOI] [PubMed] [Google Scholar]
- 18.Delsuc F, et al. 2016. The phylogenetic affinities of the extinct glyptodonts. Curr. Biol. 26, R155–R156. ( 10.1016/j.cub.2016.01.039) [DOI] [PubMed] [Google Scholar]
- 19.Gatesy J, Springer MS. 2014. Phylogenetic analysis at deep timescales: unreliable gene trees, bypassed hidden support, and the coalescence/concatalescence conundrum. Mol. Phylogenet. Evol. 80, 231–266. ( 10.1016/j.ympev.2014.08.013) [DOI] [PubMed] [Google Scholar]
- 20.Prum RO, Berv JS, Dornburg A, Field DJ, Townsend JP, Lemmon EM, Lemmon AR. 2015. A comprehensive phylogeny of birds (Aves) using targeted next-generation DNA sequencing. Nature 526, 569–573. ( 10.1038/nature15697) [DOI] [PubMed] [Google Scholar]
- 21.McCormack JE, Faircloth BC, Crawford NG, Gowaty PA, Brumfield RT, Glenn TC. 2012. Ultraconserved elements are novel phylogenomic markers that resolve placental mammal phylogeny when combined with species-tree analysis. Genome Res. 22, 746–754. ( 10.1101/gr.125864.111) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Springer MS. 2013. Phylogenetics: bats united, microbats divided. Curr. Biol. 23, R999–R1001. ( 10.1016/j.cub.2013.09.053) [DOI] [PubMed] [Google Scholar]
- 23.Novacek MJ. 1999. 100 million years of land vertebrate evolution: the Cretaceous–early Tertiary transition. Ann. Missouri Bot. Garden 86, 230–258. ( 10.2307/2666178) [DOI] [Google Scholar]
- 24.Simpson GG. 1945. The principles of classification and a classification of mammals. Bull. Am. Mus. Nat. Hist. 85, 1–350. [Google Scholar]
- 25.Novacek MJ. 1992. Mammalian phylogeny: shaking the tree. Nature 365, 121–125. ( 10.1038/356121a0) [DOI] [PubMed] [Google Scholar]
- 26.Murphy WJ, et al. 2001. Resolution of the early placental mammal radiation using Bayesian phylogenetics. Science 294, 2348–2351. ( 10.1126/science.1067179) [DOI] [PubMed] [Google Scholar]
- 27.Scally M, Madsen O, Douady CJ, de Jong WW, Stanhope MJ, Springer MS. 2001. Molecular evidence for the major clades of placental mammals. J. Mamm. Evol. 8, 239–277. ( 10.1023/A:1014446915393) [DOI] [PubMed] [Google Scholar]
- 28.Delsuc F, Scally M, Madsen O, Stanhope MJ, De Jong WW, Catzeflis FM, Springer MS, Douzery EJ. 2002. Molecular phylogeny of living xenarthrans and the impact of character and taxon sampling on the placental tree rooting. Mol. Biol. Evol. 19, 1656–1671. ( 10.1093/oxfordjournals.molbev.a003989) [DOI] [PubMed] [Google Scholar]
- 29.Springer MS, Murphy WJ, Eizirik E, O'Brien SJ. 2003. Placental mammal diversification and the Cretaceous–Tertiary boundary. Proc. Natl Acad. Sci. USA 100, 1056–1061. ( 10.1073/pnas.0334222100) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Beard KC. 1993. Origin and evolution of gliding in early Cenozoic Dermoptera (Mammalia, Primatomorpha). In Primates and their relatives in phylogenetic perspective (ed. MacPhee RDE.), pp. 63–90. New York, NY: Springer. [Google Scholar]
- 31.Gingerich PD, ul Haq M, Zalmout IS, Khan IH, Malkani MS. 2001. Origin of whales from early artiodactyls: hands and feet of Eocene Protocetidae from Pakistan. Science 293, 2239–2242. ( 10.1126/science.1063902) [DOI] [PubMed] [Google Scholar]
- 32.Novacek MJ, Wyss AR, McKenna MC. 1988. The major groups of eutherian mammals. Phylogeny Classif. Tetrapods 2, 31–71. [Google Scholar]
- 33.Springer MS, Kirsch JA. 1993. A molecular perspective on the phylogeny of placental mammals based on mitochondrial 12S rDNA sequences, with special reference to the problem of the Paenungulata. J. Mamm. Evol. 1, 149–166. ( 10.1007/BF01041592) [DOI] [Google Scholar]
- 34.Springer MS, Burk A, Kavanagh JR, Waddell VG, Stanhope MJ. 1997. The interphotoreceptor retinoid binding protein gene in therian mammals: implications for higher level relationships and evidence for loss of function in the marsupial mole. Proc. Natl Acad. Sci. USA 94, 13 754–13 759. ( 10.1073/pnas.94.25.13754) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Springer MS, Cleven GC, Madsen O, de Jong WW, Waddell VG, Amrine HM, Stanhope MJ. 1997. Endemic African mammals shake the phylogenetic tree. Nature 388, 61–64. ( 10.1038/40386) [DOI] [PubMed] [Google Scholar]
- 36.Porter CA, Goodman M, Stanhope MJ. 1996. Evidence on mammalian phylogeny from sequences of exon 28 of the von Willebrand factor gene. Mol. Phylogenet. Evol. 5, 89–101. ( 10.1006/mpev.1996.0008) [DOI] [PubMed] [Google Scholar]
- 37.Asher R, Seiffert E. 2010. Systematics of endemic African mammals, pp. 911–928. Berkeley, CA: University of California Press. [Google Scholar]
- 38.Stanhope MJ, Smith MR, Waddell VG, Porter CA, Shivji MS, Goodman M. 1996. Mammalian evolution and the interphotoreceptor retinoid binding protein (IRBP) gene: convincing evidence for several superordinal clades. J. Mol. Evol. 43, 83–92. ( 10.1007/BF02337352) [DOI] [PubMed] [Google Scholar]
- 39.Luckett W, Hartenberger J-L. 1993. Monophyly or polyphyly of the order Rodentia: possible conflict between morphological and molecular interpretations. J. Mamm. Evol. 1, 127–147. ( 10.1007/BF01041591) [DOI] [Google Scholar]
- 40.Gregory WK. 1947. The monotremes and the palimpsest theory. Bull. Am. Mus. Nat. Hist. 88, 1–52. [Google Scholar]
- 41.Kriegs JO, Churakov G, Kiefmann M, Jordan U, Brosius J, Schmitz J. 2006. Retroposed elements as archives for the evolutionary history of placental mammals. PLoS Biol. 4, e91 ( 10.1371/journal.pbio.0040091) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Nishihara H, Hasegawa M, Okada N. 2006. Pegasoferae, an unexpected mammalian clade revealed by tracking ancient retroposon insertions. Proc. Natl Acad. Sci. USA 103, 9929–9934. ( 10.1073/pnas.0603797103) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Teeling EC, Hedges SB. 2013. Making the impossible possible: rooting the tree of placental mammals. Mol. Biol. Evol. 30, 1999–2000. ( 10.1093/molbev/mst118) [DOI] [PubMed] [Google Scholar]
- 44.Amrine-Madsen H, Koepfli K-P, Wayne RK, Springer MS. 2003. A new phylogenetic marker, apolipoprotein B, provides compelling evidence for eutherian relationships. Mol. Phylogenet. Evol. 28, 225–240. ( 10.1016/S1055-7903(03)00118-0) [DOI] [PubMed] [Google Scholar]
- 45.Romiguier J, Ranwez V, Delsuc F, Galtier N, Douzery EJ. 2013. Less is more in mammalian phylogenomics: AT-rich genes minimize tree conflicts and unravel the root of placental mammals. Mol. Biol. Evol. 30, 2134–2144. ( 10.1093/molbev/mst116) [DOI] [PubMed] [Google Scholar]
- 46.Hallström BM, Janke A. 2010. Mammalian evolution may not be strictly bifurcating. Mol. Biol. Evol. 27, 2804–2816. ( 10.1093/molbev/msq166) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Waddell PJ, Shelley S. 2003. Evaluating placental inter-ordinal phylogenies with novel sequences including RAG1, γ-fibrinogen, ND6, and mt-tRNA, plus MCMC-driven nucleotide, amino acid, and codon models. Mol. Phylogenet. Evol. 28, 197–224. ( 10.1016/S1055-7903(03)00115-5) [DOI] [PubMed] [Google Scholar]
- 48.Gibson A, Gowri-Shankar V, Higgs PG, Rattray M. 2005. A comprehensive analysis of mammalian mitochondrial genome base composition and improved phylogenetic methods. Mol. Biol. Evol. 22, 251–264. ( 10.1093/molbev/msi012) [DOI] [PubMed] [Google Scholar]
- 49.Nikolaev S, Montoya-Burgos JI, Margulies EH, Rougemont J, Nyffeler B, Antonarakis SE, Program NCS. 2007. Early history of mammals is elucidated with the ENCODE multiple species sequencing data. PLoS Genet. 3, e2 ( 10.1371/journal.pgen.0030002) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Asher RJ. 2007. A web-database of mammalian morphology and a reanalysis of placental phylogeny. BMC Evol. Biol. 7, 1 ( 10.1186/1471-2148-7-108) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Churakov G, Kriegs JO, Baertsch R, Zemann A, Brosius J, Schmitz J. 2009. Mosaic retroposon insertion patterns in placental mammals. Genome Res. 19, 868–875. ( 10.1101/gr.090647.108) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Murphy WJ, Pringle TH, Crider TA, Springer MS, Miller W. 2007. Using genomic data to unravel the root of the placental mammal phylogeny. Genome Res. 17, 413–421. ( 10.1101/gr.5918807) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Morgan CC, Foster PG, Webb AE, Pisani D, McInerney JO, O'Connell MJ. 2013. Heterogeneous models place the root of the placental mammal phylogeny. Mol. Biol. Evol. 30, 2145–2156. ( 10.1093/molbev/mst117) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Liu L, Yu L, Edwards SV. 2010. A maximum pseudo-likelihood approach for estimating species trees under the coalescent model. BMC Evol. Biol. 10, 302 ( 10.1186/1471-2148-10-302) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Hudelot C, Gowri-Shankar V, Jow H, Rattray M, Higgs PG. 2003. RNA-based phylogenetic methods: application to mammalian mitochondrial RNA sequences. Mol. Phylogenet. Evol. 28, 241–252. ( 10.1016/S1055-7903(03)00061-7) [DOI] [PubMed] [Google Scholar]
- 56.Van Rheede T, Bastiaans T, Boone DN, Hedges SB, de Jong WW, Madsen O. 2006. The platypus is in its place: nuclear genes and indels confirm the sister group relation of monotremes and therians. Mol. Biol. Evol. 23, 587–597. ( 10.1093/molbev/msj064) [DOI] [PubMed] [Google Scholar]
- 57.Hallström BM, Kullberg M, Nilsson MA, Janke A. 2007. Phylogenomic data analyses provide evidence that Xenarthra and Afrotheria are sister groups. Mol. Biol. Evol. 24, 2059–2068. ( 10.1093/molbev/msm136) [DOI] [PubMed] [Google Scholar]
- 58.Wildman DE, et al. 2007. Genomics, biogeography, and the diversification of placental mammals. Proc. Natl Acad. Sci. USA 104, 14 395–14 400. ( 10.1073/pnas.0704342104) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Kjer KM, Honeycutt RL. 2007. Site specific rates of mitochondrial genomes and the phylogeny of eutheria. BMC Evol. Biol. 7, 8 ( 10.1186/1471-2148-7-8) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Nishihara H, Okada N, Hasegawa M. 2007. Rooting the eutherian tree: the power and pitfalls of phylogenomics. Genome Biol. 8, R199 ( 10.1186/gb-2007-8-9-r199) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Hallström BM, Janke A. 2008. Resolution among major placental mammal interordinal relationships with genome data imply that speciation influenced their earliest radiations. BMC Evol. Biol. 8, 1 ( 10.1186/1471-2148-8-162) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Prasad AB, Allard MW, Green ED. 2008. Confirming the phylogeny of mammals by use of large comparative sequence data sets. Mol. Biol. Evol. 25, 1795–1808. ( 10.1093/molbev/msn104) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Arnason U, Adegoke JA, Gullberg A, Harley EH, Janke A, Kullberg M. 2008. Mitogenomic relationships of placental mammals and molecular estimates of their divergences. Gene 421, 37–51. ( 10.1016/j.gene.2008.05.024) [DOI] [PubMed] [Google Scholar]
- 64.Nishihara H, Maruyama S, Okada N. 2009. Retroposon analysis and recent geological data suggest near-simultaneous divergence of the three superorders of mammals. Proc Natl. Acad. Sci. USA 106, 5235–5240. ( 10.1073/pnas.0809297106) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Murphy WJ, Pevzner PA, O'Brien SJ. 2004. Mammalian phylogenomics comes of age. Trends Genet. 20, 631–639. ( 10.1016/j.tig.2004.09.005) [DOI] [PubMed] [Google Scholar]
- 66.Venter JC, et al. 2001. The sequence of the human genome. Science 291, 1304–1351. ( 10.1126/science.1058040) [DOI] [PubMed] [Google Scholar]
- 67.Lander ES, et al. 2001. Initial sequencing and analysis of the human genome. Nature 409, 860–921. ( 10.1038/35057062) [DOI] [PubMed] [Google Scholar]
- 68.Lindblad-Toh K, et al. 2011. A high-resolution map of human evolutionary constraint using 29 mammals. Nature 478, 476–482. ( 10.1038/nature10530) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Haussler D, et al. 2009. Genome 10 K: a proposal to obtain whole-genome sequence for 10 000 vertebrate species. J. Hered. 100, 659–674. ( 10.1093/jhered/esp086) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Koepfli K-P, Paten B, O'Brien SJ. 2015. The genome 10 K project: a way forward. Annu. Rev. Anim. Biosci. 3, 57–111. ( 10.1146/annurev-animal-090414-014900) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Wu S, Song S, Liu L, Edwards SV. 2013. Reply to Gatesy and Springer: the multispecies coalescent model can effectively handle recombination and gene tree heterogeneity. Proc. Natl Acad. Sci USA 110, E1180 ( 10.1073/pnas.1300129110) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Gatesy J, O'Grady P, Baker RH. 1999. Corroboration among data sets in simultaneous analysis: hidden support for phylogenetic relationships among higher level artiodactyl taxa. Cladistics 15, 271–313. ( 10.1111/j.1096-0031.1999.tb00268.x) [DOI] [PubMed] [Google Scholar]
- 73.Thompson RS, Bärmann EV, Asher RJ. 2012. The interpretation of hidden support in combined data phylogenetics. J. Zool. Syst. Evol. Res. 50, 251–263. ( 10.1111/j.1439-0469.2012.00670.x) [DOI] [Google Scholar]
- 74.Edwards SV. 2009. Is a new and general theory of molecular systematics emerging? Evolution 63, 1–19. ( 10.1111/j.1558-5646.2008.00549.x) [DOI] [PubMed] [Google Scholar]
- 75.Liu L, Edwards SV. 2009. Phylogenetic analysis in the anomaly zone. Syst. Biol. 58, 452–460. ( 10.1093/sysbio/syp034) [DOI] [PubMed] [Google Scholar]
- 76.Liu L, Yu L, Pearl DK, Edwards SV. 2009. Estimating species phylogenies using coalescence times among sequences. Syst. Biol. 58, 468–477. ( 10.1093/sysbio/syp031) [DOI] [PubMed] [Google Scholar]
- 77.Liu L, Xi Z, Wu S, Davis CC, Edwards SV. 2015. Estimating phylogenetic trees from genome-scale data. Ann. NY Acad. Sci. 1360, 36–53. ( 10.1111/nyas.12747) [DOI] [PubMed] [Google Scholar]
- 78.Ren F, Tanaka H, Yang Z. 2009. A likelihood look at the supermatrix–supertree controversy. Gene 441, 119–125. ( 10.1016/j.gene.2008.04.002) [DOI] [PubMed] [Google Scholar]
- 79.Heled J, Drummond AJ. 2010. Bayesian inference of species trees from multilocus data. Mol. Biol. Evol. 27, 570–580. ( 10.1093/molbev/msp274) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Liu L. 2008. BEST: Bayesian estimation of species trees under the coalescent model. Bioinformatics 24, 2542–2543. ( 10.1093/bioinformatics/btn484) [DOI] [PubMed] [Google Scholar]
- 81.Warnow T. 2015. Concatenation analyses in the presence of incomplete lineage sorting. PLoS Curr. 7 ( 10.1371/currents.tol.8d41ac0f13d1abedf4c4a59f5d17b1f7) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Mirarab S, Reaz R, Bayzid MS, Zimmermann T, Swenson MS, Warnow T. 2014. ASTRAL: genome-scale coalescent-based species tree estimation. Bioinformatics 30, 541–i548. ( 10.1093/bioinformatics/btu462) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Hobolth A, Christensen OF, Mailund T, Schierup MH. 2007. Genomic relationships and speciation times of human, chimpanzee, and gorilla inferred from a coalescent hidden Markov model. PLoS Genet. 3, e7 ( 10.1371/journal.pgen.0030007) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Hobolth A, Dutheil JY, Hawks J, Schierup MH, Mailund T. 2011. Incomplete lineage sorting patterns among human, chimpanzee, and orangutan suggest recent orangutan speciation and widespread selection. Genome Res. 21, 349–356. ( 10.1101/gr.114751.110) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Gatesy J, Springer MS. 2013. Concatenation versus coalescence versus ‘concatalescence’. Proc. Natl Acad. Sci. USA 110, E1179 ( 10.1073/pnas.1221121110) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Lanier HC, Knowles LL. 2012. Is recombination a problem for species-tree analyses? Syst. Biol. 61, 691–701. ( 10.1093/sysbio/syr128) [DOI] [PubMed] [Google Scholar]
- 87.Mirarab S, Bayzid MS, Boussau B, Warnow T. 2014. Statistical binning enables an accurate coalescent-based estimation of the avian tree. Science 346, 1250463 ( 10.1126/science.1250463) [DOI] [PubMed] [Google Scholar]
- 88.Zhong B, Liu L, Penny D. 2014. The multispecies coalescent model and land plant origins: a reply to Springer and Gatesy. Trends Plant Sci. 19, 270–272. ( 10.1016/j.tplants.2014.02.011) [DOI] [PubMed] [Google Scholar]
- 89.Leaché AD, Rannala B. 2010. The accuracy of species tree estimation under simulation: a comparison of methods. Syst. Biol. 60, 126–137. ( 10.1093/sysbio/syq073) [DOI] [PubMed] [Google Scholar]
- 90.Bayzid MS, Warnow T. 2013. Naive binning improves phylogenomic analyses. Bioinformatics 29, 2277–2284. ( 10.1093/bioinformatics/btt394) [DOI] [PubMed] [Google Scholar]
- 91.Patel S, Kimball RT, Braun EL. 2013. Error in phylogenetic estimation for bushes in the tree of life. J. Phylogenet. Evol. Biol. 1, 110. [Google Scholar]
- 92.Mirarab S, Bayzid MS, Warnow T. 2014. Evaluating summary methods for multilocus species tree estimation in the presence of incomplete lineage sorting. Syst. Biol. 65, 366–380. ( 10.1093/sysbio/syu063) [DOI] [PubMed] [Google Scholar]
- 93.Zimmermann T, Mirarab S, Warnow T. 2014. BBCA: improving the scalability of *BEAST using random binning. BMC Genomics 15(Suppl 6), S11 ( 10.1186/1471-2164-15-S6-S11) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Chifman J, Kubatko L. 2014. Quartet inference from SNP data under the coalescent model. Bioinformatics 30, 3317–3324. ( 10.1093/bioinformatics/btu530) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Bininda-Emonds OR, et al. 2007. The delayed rise of present-day mammals. Nature 446, 507–512. ( 10.1038/nature05634) [DOI] [PubMed] [Google Scholar]
- 96.Archibald JD, Deutschman DH. 2001. Quantitative analysis of the timing of the origin and diversification of extant placental orders. J. Mamm. Evol. 8, 107–124. ( 10.1023/A:1011317930838) [DOI] [Google Scholar]
- 97.Emerling CA, Springer MS. 2015. Genomic evidence for rod monochromacy in sloths and armadillos suggests early subterranean history for Xenarthra. Pro. R. Soc. B 282, 20142192 ( 10.1098/rspb.2014.2192) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Yang Z. 2007. PAML 4: phylogenetic analysis by maximum likelihood. Mol. Biol. Evol. 24, 1586–1591 ( 10.1093/molbev/msm088) [DOI] [PubMed] [Google Scholar]
- 99.Stamatakis A, Hoover P, Rougemont J. 2008. A rapid bootstrap algorithm for the RAxML web servers. Syst. Biol. 57, 758–771. ( 10.1080/10635150802429642) [DOI] [PubMed] [Google Scholar]
- 100.Miller MA, Schwartz T, Pickett BE, He S, Klem EB, Scheuermann RH, Passarotti M, Kaufman S, O'Leary MA. 2015. A RESTful API for access to phylogenetic tools via the CIPRES science gateway. Evol. Bioinformatics Online 11, 43–48. ( 10.4137/EBO.S21501) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Luo Z-X, Yuan C-X, Meng Q-J, Ji Q. 2011. A Jurassic eutherian mammal and divergence of marsupials and placentals. Nature 476, 442–445. ( 10.1038/nature10291) [DOI] [PubMed] [Google Scholar]
- 102.Wible JR, Rougier GW, Novacek MJ, Asher RJ. 2009. The eutherian mammal Maelestes gobiensis from the Late Cretaceous of Mongolia and the phylogeny of Cretaceous Eutheria. Bull. Am. Mus. Nat. Hist. 327, 1–123. ( 10.1206/623.1) [DOI] [Google Scholar]
- 103.Halliday TJ, Upchurch P, Goswami A. 2015. Resolving the relationships of Paleocene placental mammals. Biol. Rev. ( 10.1111/brv.12242) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Zhou C-F, Wu S, Martin T, Luo Z-X. 2013. A Jurassic mammaliaform and the earliest mammalian evolutionary adaptations. Nature 500, 163–167. ( 10.1038/nature12429) [DOI] [PubMed] [Google Scholar]
- 105.Phillips MJ. 2015. Geomolecular dating and the origin of placental mammals. Syst. Biol. 65, 546–557. ( 10.1093/sysbio/syv115) [DOI] [PubMed] [Google Scholar]
- 106.Springer MS, Emerling CA, Meredith RW, Janecka JE, Eizirik E, Murphy WJ. Submitted. Waking the undead: implications of a soft explosive model for the timing of placental mammal diversification. Mol. Phylogenet. Evol. [DOI] [PubMed]
- 107.Sansom RS, Wills MA. 2013. Fossilization causes organisms to appear erroneously primitive by distorting evolutionary trees. Sci. Rep. 3, 2545 ( 10.1038/srep02545) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Wang X, McKenna MC, Dashzeveg D. 2005. Amphicticeps and Amphicynodon (Arctoidea, Carnivora) from Hsanda Gol Formation, central Mongolia and phylogeny of basal arctoids with comments on zoogeography. Am. Mus. Novit. 3438, 1–60. ( 10.1206/0003-0082(2005)483%5B0001:AAAACF%5D2.0.CO;2) [DOI] [Google Scholar]
- 109.O'Reilly JE, dos Reis M, Donoghue PC. 2015. Dating tips for divergence-time estimation. Trends Genet. 31, 637–650. ( 10.1016/j.tig.2015.08.001) [DOI] [PubMed] [Google Scholar]
- 110.Kumar S, Hedges SB. 2016. Advances in time estimation methods for molecular data. Mol. Biol. Evol. 33, 863–869. ( 10.1093/molbev/msw026) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.dos Reis M, Donoghue PC, Yang Z. 2016. Bayesian molecular clock dating of species divergences in the genomics era. Nat. Rev. Genet. 17, 71–80. ( 10.1038/nrg.2015.8) [DOI] [PubMed] [Google Scholar]
- 112.Jarvis ED. 2016. Perspectives from the avian phylogenomics project: questions that can be answered with sequencing all genomes of a vertebrate class. Annu. Rev. Anim. Biosci. 4, 45–59. ( 10.1146/annurev-animal-021815-111216) [DOI] [PubMed] [Google Scholar]
- 113.Haeckel E. 1866. Systematische Einleitung in die allgemeine Entwicklungsgeschichte. Berlin, Germany: George Reimer. [Google Scholar]
- 114.Wanninger A. 2015. Morphology is dead–long live morphology! Integrating MorphoEvoDevo into molecular EvoDevo and phylogenomics. Front. Ecol. Evol. 3, 54 ( 10.3389/fevo.2015.00054) [DOI] [Google Scholar]
- 115.Gregg CL, Recknagel AK, Butcher JT. 2015. Micro/nano-computed tomography technology for quantitative dynamic, multi-scale imaging of morphogenesis. In Tissue morphogenesis: methods and protocols, methods in molecular biology (ed. Nelson CM.), pp. 47–61. New York, NY: Springer Science and Business Media. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Springer MS, DeBry RW, Douady C, Amrine HM, Madsen O, de Jong WW, Stanhope MJ. 2001. Mitochondrial versus nuclear gene sequences in deep-level mammalian phylogeny reconstruction. Mol. Biol. Evol. 18, 132–143. ( 10.1093/oxfordjournals.molbev.a003787) [DOI] [PubMed] [Google Scholar]
- 117.Teeling EC, Springer MS, Madsen O, Bates P, O'Brien SJ, Murphy WJ. 2005. A molecular phylogeny for bats illuminates biogeography and the fossil record. Science 307, 580–584. ( 10.1126/science.1105113) [DOI] [PubMed] [Google Scholar]
- 118.Krause J, Fu Q, Good JM, Viola B, Shunkov MV, Derevianko AP, Pääbo S. 2010. The complete mitochondrial DNA genome of an unknown hominin from southern Siberia. Nature 464, 894–897. ( 10.1038/nature08976) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Angelis K, dos Reis M. 2015. The impact of ancestral population size and incomplete lineage sorting on Bayesian estimation of species divergence times. Curr. Zool. 61, 874–885. ( 10.1093/czoolo/61.5.874) [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.