

Abstract
Described herein is a catalytic asymmetric total synthesis of (−)-actinophyllic acid, with the key step being a chiral phosphine-catalyzed [3 + 2] annulation between an imine and an allenoate to form a pyrroline intermediate in 99% yield and 94% ee. The synthesis also features CuI-catalyzed coupling between a ketoester and a 2-iodoindole to shape the tetrahydroazocine ring; intramolecular alkylative lactonization; SmI2-mediated intramolecular pinacol coupling between ketone and lactone subunits to assemble the complex skeleton of (−)-actinophyllic acid; and an unprecedented regioselective dehydroxylation.
In a search for therapeutic agents for the treatment of cardiovascular disorders, Quinn, Carroll, et al. isolated (−)-actinophyllic acid (1) from the leaves of the tree Alstonia actinophylla growing in Cape York Peninsula, Far North Queensland, Australia (Scheme 1).1 (−)-Actinophyllic acid was reported to be a potent inhibitor of the zinc-dependent carboxypeptidase U (CPU), with an IC50 of 0.84 μM. CPU is an endogenous inhibitor of fibrinolysis, the breakage of fibrin clots. Consequently, inhibitors of CPU can facilitate fibrinolysis and inhibit the blood clot formation that is a cause of various cardiovascular disorders.2 There have not, however, been any subsequent biological studies reported, presumably because of the scarcity of the natural product, due to its low isolation yield (0.0072%).3 Therefore, any efficient de novo syntheses of this potent CPU inhibitor should benefit explorations of its biomedical potential.
Scheme 1.

Retrosynthesis of (−)-Actinophyllic Acid
Structurally, (−)-actinophyllic acid contains the cage-like scaffold of a 1,2,3,5,6,7,8,10a-octahydro-1,7-methanopyrrolo-[1,2-a]azocine, highlighted in red in Scheme 1, with five contiguous stereogenic centers, one of which is a quaternary carbon, bridged by a tetrahydrofuran lactol.4 This unprecedented architecture, along with great biomedical potential, has garnered widespread attention from the synthetic community. In 2008, Overman et al. accomplished an elegant total synthesis of (±)-actinophyllic acid through aza–Cope/Mannich cascade strategy (Figure 1).5a Later, the same group reported a second-generation synthesis toward (−)-actinophyllic acid based on diastereoselective coupling between a 2-indole malonate and diacetoxypiperidine.5b,6 In 2013, Martin’s group revealed an alternative synthesis of (±)-actinophyllic acid, spotlighting a remarkable cascade reaction between a seven-membered ring dienamine and a tertiary 2-indolyl acetoxylate.7 Contemporarily, the Wood, Taniguchi, Maldonado, and Coldham groups reported their synthetic studies toward this novel monoterpene indole alkaloid.8 Previous efforts, and our own experience, exposed that establishing the cis stereochemistry between the C19 ketone and the C21 indole substituents on the pyrrolidine ring (4 to 3, Scheme 1) is challenging. In both Maldonado’s and Coldham’s synthetic attempts, intramolecular Mannich reactions between an indole-3-carboxaldehyde and an azocinone resulted in the 1-azabicyclo[4.2.1]nonane scaffold with incorrect trans stereochemistry between the C19 ketone and the C21 indole (actinophyllic acid numbering), presumably due to steric congestion between the indoyl substituent and the adjacent C19 acyl chain.7c,7d Both Overman and Martin brought this challenge under control through their early stage construction of the indole-fused heptenone (in pink, Figure 1). We, on the other hand, addressed it through intramolecular lactonization (from 7 to 6, Scheme 1). Herein, we report a catalytic asymmetric synthesis of (−)-actinophyllic acid, featuring a chiral phosphinecatalyzed [3 + 2] annulation between an imine and an electron-deficient allene.
Figure 1.

Key steps in previous attempts toward actinophyllic acid.
In the retrosynthetic sense, we originally envisioned that (−)-actinophyllic acid could be obtained from the diester 2 through Overman’s decarboxylation, hydroxymethylation, and lactol formation sequence (Scheme 1). We targeted forming the C15–C16 bond in 2 through oxidative coupling between the malonate and ketone units of intermediate 3. The 1-azabicyclo[4.2.1]nonan-5-one scaffold (in green) of compound 3 was to be fashioned from pyrroline 4 via azepinone ring formation. The pyrroline 4, in turn, could be assembled through a well-established phosphine-catalyzed [3 + 2] annulation between an indole imine and an electron-deficient allene.9 While reduction of the pyrroline 4 to the 2,3-cis-substituted pyrrolidine was readily accomplished, the formation of the bridged 1-azabicycl[4.2.1]nonan-5-one system (in green) proved difficult, due to epimerization at C19, even under mild conditions.10 To circumvent these obstacles, we devised an alternative route in which the hexahydroazocine ring would be built first (from 9 to 8). Diastereoselective hydrogenation of the pyrroline should, then, bring the carbonyl groups at C15 and C18 in close proximity to form the azepane ring through pinacol coupling (from 6 to 5). The two carbonyl groups would be brought even closer, we surmised, after intramolecular alkylative lactonization (from 7 to 6). The azocinone ring in compound 8 should be accessible through coupling between the indole C2 atom and the C16 atom of the β-ketoester in 9, which should be readily preparable from intermediate 4.
Our synthetic campaign commenced with an exploration of the key [3 + 2] annulation between benzyl allenoate and the N-(o-nitrobenzenesulfonyl) (o-nosyl) imine 10, which could be synthesized according to the known procedure from indole 3-carboxaldehyde.9c Initially, when using PPh3, the desired racemic pyrroline was obtained in 99% yield after 6 h (Table 1, entry 1). Our previous studies on the enantioselective synthesis of pyrrolines foretold that the endo-phenyl Kwon [2.2.1] bicyclic phosphine A should produce the desired (S)-enantiomer 11.9c,d When we applied 20 mol % of phosphine A to the reaction, the (S)-enantiomer was indeed formed in 97% yield and 75% ee after 5 h at room temperature in CHCl3 (entry 2).11,12 The more nucleophilic phosphine B improved the enantioselectivity to 83% ee. The ee increased further, to 91%, after decreasing the reaction temperature to 0 °C (entries 3 and 4). Further lowering of the temperature did not improve the ee.13 From a mechanistic perspective, we suspected that hydrogen bonding would facilitate the proton-transfer steps14 and rigidify the transition-state assembly,15 thereby improving the enantioselectivity. Among a variety of tested hydrogen-bond donors, we found that phenol and derivatives accelerated the reaction and improved the enantioselectivity. Addition of 20 mol % phenol or biphenol decreased the reaction time to 2 h, albeit without improving the enantioselectivity (entries 5 and 6). When 20 mol % s-BINOL or r-BINOL was used as the additive, the enantioselectivity improved to 94% without decreasing the yield, with the reaction occurring within 2 h (entries 7 and 8).15
Table 1.
Phosphine-Catalyzed Pyrrolidine Synthesis

| ||||||
|---|---|---|---|---|---|---|
|
| ||||||
| entry | cat. | temp. (°C) | additive | time (h) | yield (%)a | ee (%)b |
| 1 | PPh3 | rt | 6 | 99 | ||
| 2 | A | rt | 5 | 97 | 75 | |
| 3 | B | rt | 5 | 99 | 83 | |
| 4 | B | 0 | 5 | 99 | 91 | |
| 5 | B | 0 | phenol | 2 | 99 | 91 |
| 6 | B | 0 | biphenol | 2 | 99 | 91 |
| 7 | B | 0 | s-BINOL | 2 | 99 | 94 |
| 8 | B | 0 | r-BINOL | 2 | 99 | 94 |
Isolated yield after silica gel FCC
Determined using HPLC
With the annulation product in hand, we attempted to form the hexahydroazocinone ring through oxidative coupling between the ketoester and the C2 atom of indole.16 Quick access to the ketoester 13′ was secured in 92% yield through deprotection of the o-nosyl group, performed with sodium benzenethiolate in MeCN at room temperature, followed by reaction with ethyl 3-oxopent-4-enoate (Scheme 2).17 We examined a list of oxidants, including Fe3+, Cu2+, Mn3+, Co2+, Ag+, and I2, to facilitate the oxidative coupling of the substrates 13′ and 13″, but obtained no fruitful results.
Scheme 2.

Attempted Oxidative Coupling
Instead of using an oxidative coupling approach, we anticipated that a redox-neutral coupling reaction between indole 2-iodide and ketoester subunits might give the desired cyclization product 14′.18 Following the procedure used for the synthesis of 13′, the iodoketoester 13 was obtained in 80% yield over two steps (Scheme 3). Having efficient access to the necessary iodoketoester 13, several transition-metal catalysts were probed. Pleasingly, subjecting the iodoketoester 13 to CuI in DMSO at room temperature yielded (82%) the desired cyclization product 14, which existed exclusively in its enol form. One recrystallization increased the ee to 99%.19 As far as we know, this CuI-catalyzed coupling is the first between a 2-iodoindole and a ketoester to generate a tetrahydroazocine cycle.18d To our delight, a high pressure of H2 gas over Pd/C formed the cis hydrogenation product along with deprotection of the benzyl group in one pot. Exchanging the solvent to DMF and treating the resulting carboxylic acid with chloroiodomethane and K2CO3 readily manufactured the chloromethyl ester 15 in 78% yield for the two steps. After significant experimentation, we found that 40 equiv of NaI in DMF with K2CO3 as base furnished the lactone 16 in 35–48% yield. Although modestly yielded, this alkylative lactonization framed another challenging eight-membered tetrahydrooxocine portion of (−)-actinophyllic acid and set the stage for the final azepane segment, and concomitant tetrahydrofuran part, formation through intramolecular ketone–lactone pinacol coupling. The pinacol coupling strategy departs significantly from the lactol formation approach adopted by Overman and Martin.
Scheme 3.

Synthesis of (−)-Actinophyllic Acid Hydrochloride
Continuing with the synthetic venture, we evaluated the effects of several single-electron-transfer reagents, including Ti3+, Li, Na, and Sm2+, on the pinacol coupling.20 Only SmI2 combined with 10 equiv of t-BuOH provided the desired coupling product 17, in quantitative yield; its structure was confirmed through X-ray crystallographic analysis.21 At this stage, the complete heavy atom arrangement of (−)-actinophyllic acid was in place. What remained was regioselective removal of the hindered C15 hydroxyl group from compound 17.
To this end, we turned our attention to radical dehydroxylation.22 The idea was that the thiocarbonate 18, when treated with a tributyltin radical, would undergo homolytic cleavage of the tertiary carbon–oxygen bond preferably, due to electronic differentiation (the rigid multicyclic framework of actinophyllic acid would not allow the necessary stereoelectronic alignment of the lone pair of electrons on the tetrahydrofuran oxygen with the ensuing α-radical). The dihydroxy compound 17 was reacted with thiophosgene in the presence of DMAP at −15 °C, transforming into the thionocarbonate 18 in 72% yield. The standard conditions of n-Bu3SnH and AIBN at 90 °C in toluene worked efficiently to furnish the desired lactol product. Finally, global deprotection, through the effect of aqueous HCl under microwave heating at 100 °C for 30 min, furnished (−)-actinophyllic acid hydrochloride in 90% yield over two steps . The spectral data of our synthetic sample matched those reported in the literature.5
In conclusion, we have successfully completed a catalytic asymmetric total synthesis of (−)-actinophyllic acid in 13 steps from a known aldehyde in 12.4% yield. Our synthesis exhibits several salient features: (i) chiral phosphine-catalyzed [3 + 2] annulation between an allenoate and an indole imine; (ii) CuI-catalyzed coupling between a 2-iodoindole and a ketoester to assemble a hexahydro-1H-azocino[4,3-b]indole system; (iii) intramolecular alkylative lactonization to form a tetrahydrooxocine ring; (iv) highly efficient pinacol coupling between a ketone and a lactone to form the caged scaffold of (−)-actinophyllic acid; and (v) regioselective removal of a tertiary alcohol by taking advantage of a vicinal hemiketal. Our strategy not only circumvented the difficulties typically associated with forming the correct stereochemistry around the pyrrolidine ring but also resulted in the first enantioselective total synthesis of (−)-actinophyllic acid in which the asymmetric synthesis employs the same starting materials as the racemic synthesis.6 Detailed screening of the biological activity of (−)-actinophyllic acid is ongoing.
Supplementary Material
Acknowledgments
We thank the NIH (GM071779) for financial support. This study was supported by shared instrumentation grants from the NSF (CHE-1048804) and the NIH NCRR (S10RR025631). Dedicated to Professor Stuart L. Schreiber on the occasion of his 60th Birthday.
Footnotes
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/jacs.6b00567.
Experimental details and data (PDF)
Crystallographic data for 16(CIF)
Crystallographic data for 17 (CIF)
Notes
The authors declare no competing financial interest.
References
- 1.(a) Carroll AR, Hyde E, Smith J, Quinn RJ, Guymer G, Forster PI. J Org Chem. 2005;70:1096. doi: 10.1021/jo048439n. [DOI] [PubMed] [Google Scholar]; Later, Morita et al. isolated (+)-actinophyllic methyl ester from Alstonia pneumatophore in 0.0009% yield; see:; (b) Koyama K, Hirasawa Y, Hosoya T, Hoe T, Chan K, Morita H. Bioorg Med Chem. 2010;18:4415. doi: 10.1016/j.bmc.2010.04.086. [DOI] [PubMed] [Google Scholar]; In 2005, Luo et al. isolated Alstonia scholarine E, which is an ethyl ketal of actinophyllic acid, from Alstonia scholaris in 0.000006% yield; see:; (c) Qin X, Zhao Y, Lunga P, Yang X, Song C, Cheng G, Liu L, Chen Y, Liu Y, Luo X. Tetrahedron. 2015;71:4372. [Google Scholar]
- 2.(a) Leurs J, Nerme V, Sim Y, Hendriks DJ. J Thromb Haemostasis. 2004;2:416. doi: 10.1111/j.1538-7836.2004.00605.x. [DOI] [PubMed] [Google Scholar]; (b) Polla MO, Tottie L, Nordén C, Linschoten M, Musil M, Trumpp-Kallmeyer S, Aurkurst IR, Ringom R, Holm KH, Neset SM, Sandberg M, Thurmond J, Yu P, Hategan G, Anderson H. Bioorg Med Chem. 2004;12:1151. doi: 10.1016/j.bmc.2003.12.039. [DOI] [PubMed] [Google Scholar]; (c) Leurs J, Hendriks D. Thromb Haemostasis. 2005;94:417. doi: 10.1160/TH04-07-0454. [DOI] [PubMed] [Google Scholar]; (d) Marder VJ, Aird WC, Bennett JS, Schulman S, White GC, editors. Hemostasis and Thrombosis: Basic Principles and Clinical Practice. 6th. Lippincott Williams & Wilkins; Philadelphia: 2013. [Google Scholar]
- 3.The natural actinophyllic acid was isolated as a brown gum, but synthetic (−)-actinophyllic acid hydrochloride is a white solid.
- 4.Taniguchi T, Martin CL, Monde K, Nakanishi K, Berova N, Overman LE. J Nat Prod. 2009;72:430. doi: 10.1021/np800665s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.(a) Martin CL, Overman LE, Rohde JM. J Am Chem Soc. 2008;130:7568. doi: 10.1021/ja803158y. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Martin CL, Overman LE, Rohde JM. J Am Chem Soc. 2010;132:4894. doi: 10.1021/ja100178u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Having failed to introduce chirality through catalytic asymmetric heteroarylation of piperidone with indole (a strategy employed in the earlier racemic synthesis), Overman’s key transformation was based on diastereoselective heteroarylation of piperidine diacetoxylate. The enantioriched piperidine diol derivative was synthesized through Noyori’s asymmetric hydrogenation.
- 7.(a) Granger BA, Jewett IT, Butler JD, Hua B, Knezevic CE, Parkinson EI, Hergenrother PJ, Martin SF. J Am Chem Soc. 2013;135:12984. doi: 10.1021/ja4070206. [DOI] [PubMed] [Google Scholar]; (b) Granger BA, Jewett IT, Butler JD, Martin SF. Tetrahedron. 2014;70:4094. doi: 10.1016/j.tet.2014.03.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.(a) Vaswani RG, Day JJ, Wood JL. Org Lett. 2009;11:4532. doi: 10.1021/ol901746c. [DOI] [PubMed] [Google Scholar]; (b) Zaimoku H, Taniguchi T, Ishibashi H. Org Lett. 2012;14:1656. doi: 10.1021/ol300280s. [DOI] [PubMed] [Google Scholar]; (c) Galicia IZ, Maldonado LA. Tetrahedron Lett. 2013;54:2180. [Google Scholar]; (d) Mortinmer D, Whiting M, Harrity JPA, Jones S, Coldham I. Tetrahedron Lett. 2014;55:1255. [Google Scholar]; (e) Meketa ML. PhD Thesis. Pennsylvania State University; Dec, 2008. Part One: Total Syntheses Of Ageladine A; Part Two: Studies Directed Towards a Total Synthesis of Actinophyllic Acid. [Google Scholar]
- 9.(a) Zhu XF, Henry CE, Kwon O. Tetrahedron. 2005;61:6276. [Google Scholar]; (b) Andrews IP, Kwon O. Org Synth. 2011;88:138. [Google Scholar]; (c) Andrews IP, Kwon O. Chem Sci. 2012;3:2510. doi: 10.1039/C2SC20468A. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Henry CE, Xu Q, Fan YC, Martin TJ, Belding L, Dudding T, Kwon O. J Am Chem Soc. 2014;136:11890. doi: 10.1021/ja505592h. [DOI] [PMC free article] [PubMed] [Google Scholar]; For reports on Lu’s original allene–imine [3 + 2] annulation, see:; (e) Xu Z, Lu X. Tetrahedron Lett. 1997;38:3461. [Google Scholar]; (f) Xu Z, Lu X. J Org Chem. 1998;63:5031. [Google Scholar]
- 10.A weak base (e.g., K2CO3), a weak acid (e.g., p-TSA), or neutral conditions (e.g., NaBH3CN or PhSNa) could induce the epimerization.
- 11.Both chiral phosphines are commercially available from Sigma-Aldrich. Phosphine A: catalog no. L512397; phosphine B: catalog no. L512478.
- 12.After removing the crystalline racemic pyrroline 11, we could enrich the ee of the mother liquid up to 94%.
- 13.See the Supporting Information for further details regarding optimization of the conditions.
- 14.Xia Y, Liang Y, Chen Y, Mang M, Jiao L, Huang F, Liu S, Yu Z. J Am Chem Soc. 2007;129:3470. doi: 10.1021/ja068215h. [DOI] [PubMed] [Google Scholar]
- 15.(a) Shi M, Chen LH. Chem Commun. 2003;1310 doi: 10.1039/b301863f. [DOI] [PubMed] [Google Scholar]; (b) Han X, Zhong F, Wang Y, Lu Y. Angew Chem Int Ed. 2012;51:767. doi: 10.1002/anie.201106672. [DOI] [PubMed] [Google Scholar]; (c) Yao W, Dou X, Lu Y. J Am Chem Soc. 2015;137:54. doi: 10.1021/ja5109358. [DOI] [PubMed] [Google Scholar]
- 16.(a) Paquette LA, Bzowej EI, Branan BM, Stanton KJ. J Org Chem. 1995;60:7277. [Google Scholar]; (b) Baran PS, Richter JM, Lin DW. Angew Chem Int Ed. 2005;44:609. doi: 10.1002/anie.200462048. [DOI] [PubMed] [Google Scholar]; c) Magolan J, Kerr MA. Org Lett. 2006;8:4561. doi: 10.1021/ol061698+. [DOI] [PubMed] [Google Scholar]; (d) Richter JM, Whitefield BW, Maimone TJ, Lin DW, Castroviejo MP, Baran PS. J Am Chem Soc. 2007;129:12857. doi: 10.1021/ja074392m. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Chen P, Cao L, Tian W, Wang X, Li C. Chem Commun. 2010;46:8436. doi: 10.1039/c0cc03428b. [DOI] [PubMed] [Google Scholar]; (f) Zi W, Xie W, Ma D. J Am Chem Soc. 2012;134:9126. doi: 10.1021/ja303602f. [DOI] [PubMed] [Google Scholar]; (g) Oisaki K, Abe J, Kanai M. Org Biomol Chem. 2013;11:4569. doi: 10.1039/c3ob40855h. [DOI] [PubMed] [Google Scholar]; (h) Jiang Q, Sheng W, Tian M, Tang J, Guo C. Eur J Org Chem. 2013;2013:1861. [Google Scholar]; (i) Wang H, Guo LN, Duan XH. Chem Commun. 2013;49:10370. doi: 10.1039/c3cc46114a. [DOI] [PubMed] [Google Scholar]; (j) Matsubara T, Takahashi K, Ishihara J, Hatakeyama S. Angew Chem Int Ed. 2014;53:757. doi: 10.1002/anie.201307835. [DOI] [PubMed] [Google Scholar]; For a brief review, see:; (k) Csaky AG. Plumet Chem Soc Rev. 2001;30:313. [Google Scholar]; (l) Guo F, Clift MD, Thomson RJ. Eur J Org Chem. 2012;2012:4881. doi: 10.1002/ejoc.201200665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Barcan GA, Patel A, Houk KN, Kwon O. Org Lett. 2012;14:5388. doi: 10.1021/ol302265z. in this paper PhSK was generated from KH and thiophenol, this approach worked well for our reactions. PhSNa could be obtained at lower cost by mixing thiophenol and NaOH and drying in a high-vacuum oven. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.(a) Hamann BC, Beare NA, Hartwig JF. J Am Chem Soc. 1997;119:12382. [Google Scholar]; (b) Hennessy EJ, Buchwald SL. Org Lett. 2002;4:269. doi: 10.1021/ol017038g. [DOI] [PubMed] [Google Scholar]; (c) Xie X, Cai G, Ma D. Org Lett. 2005;7:4693. doi: 10.1021/ol0518838. [DOI] [PubMed] [Google Scholar]; (d) Coste A, Toumi M, Wright K, Razafimahaleo V, Couty F, Marrot J, Evano G. Org Lett. 2008;10:3841. doi: 10.1021/ol8015513. [DOI] [PubMed] [Google Scholar]; Recent reviews on enolate coupling, see:; (e) Culkin DA, Hartwig JF. Acc Chem Res. 2003;36:234. doi: 10.1021/ar0201106. [DOI] [PubMed] [Google Scholar]; (f) Johansson CCC, Colacot TJ. Angew Chem Int Ed. 2010;49:676. doi: 10.1002/anie.200903424. [DOI] [PubMed] [Google Scholar]; (g) Bellina F, Rossi R. Chem Rev. 2010;110:1082. doi: 10.1021/cr9000836. [DOI] [PubMed] [Google Scholar]
- 19.Because we could not identify conditions for the separation of the two enantiomers of compound 14, we proceeded to compound 15 to determine the enantiomeric excess. See the SI for more details.
- 20.(a) Auer E, Gossinger E, Graupe M. Tetrahedron Lett. 1997;38:6577. [Google Scholar]; (b) Molander GA, Wolfe CN. J Org Chem. 1998;63:9031. [Google Scholar]; (c) Yamamoto Y, Matsumi D, Itoh K. Chem Commun. 1998;34:875. [Google Scholar]; (d) Liu Y, Zhang Y. Tetrahedron Lett. 2001;42:5745. [Google Scholar]; (e) Hasegawa E, Kentaro O, Tanikawa N, Nakamura M, Iwaya K, Hoshi T, Suzuki T. Tetrahedron Lett. 2006;47:7715. [Google Scholar]; (f) Ge HM, Zhang LD, Tan RX, Yao ZJ. J Am Chem Soc. 2012;134:12323. doi: 10.1021/ja305261h. [DOI] [PubMed] [Google Scholar]; (g) Frey G, Luu HT, Bichovski P, Feurer M, Streuff D. Angew Chem Int Ed. 2013;52:7131. doi: 10.1002/anie.201302460. [DOI] [PubMed] [Google Scholar]; (h) Bichovski P, Haas TM, Kratzert D, Streuff J. Chem-Eur J. 2015;21:2339. doi: 10.1002/chem.201405852. [DOI] [PubMed] [Google Scholar]; For more recent reviews on SmI2, see:; (i) Edmonds DJ, Johnston D, Procter D. J Chem Rev. 2004;104:3371. doi: 10.1021/cr030017a. [DOI] [PubMed] [Google Scholar]; (j) Nicolaou KC, Ellery SP, Chen JS. Angew Chem Int Ed. 2009;48:7140. doi: 10.1002/anie.200902151. [DOI] [PMC free article] [PubMed] [Google Scholar]
-
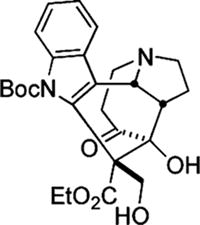
21.More than 10 equiv of t-BuOH slowed down the reaction and necessitated the use of increased amounts of SmI2, and <10 equiv of t-BuOH resulted in formation of the following rearrangement side product:
- 22.(a) Emmer G, Roth SW. Tetrahedron. 1992;48:5861. [Google Scholar]; (b) Myers AG, Tom NJ, Fraley ME, Cohen SB, Madar DJ. J Am Chem Soc. 1997;119:6072. [Google Scholar]; (c) Brüggemann M, McDonald AI, Overman LE, Rosen MD, Schwink L, Scott JP. J Am Chem Soc. 2003;125:15284. doi: 10.1021/ja0388820. [DOI] [PubMed] [Google Scholar]; (d) Nicolaou KC, Sun YP, Korman H, Sarlah D. Angew Chem Int Ed. 2010;49:5875. doi: 10.1002/anie.201003500. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.