

Abstract
Stem cell therapies for neurological disorders are rapidly moving towards use in clinical trials. Before initiation of clinical trials, extensive pre-clinical validation in appropriate animal models is essential. However, grafts of human cells into the rodent brain are rejected within weeks after transplantation and the standard methods of immune-suppression for the purpose of studying human xenografts are not always sufficient for the long-term studies needed for transplanted human neurons to maturate, integrate and provide functional benefits in the host brain. Neonatal injections in rat pups using human fetal brain cells have been shown to desensitise the host to accept human tissue grafts as adults, whilst not compromising their immune system. Here, we show that differentiated human embryonic stem cells (hESCs) can be used for desensitisation to achieve long-term graft survival of human stem cell-derived neurons in a xenograft setting, surpassing the time of conventional pharmacological immune-suppressive treatments. The use of hESCs for desensitisation opens up for a widespread use of the technique, which will be of great value when performing pre-clinical evaluation of stem cell-derived neurons in animal models.
Keywords: Transplant, Stem cell, hESC, Immune response, Desensitisation, Xenograft, Rejection, Cyclosporine
Graphical abstract
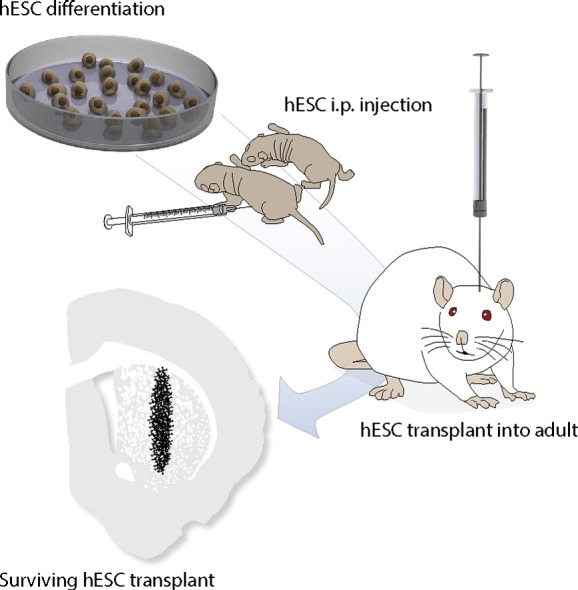
Highlights
-
•
Neonatal desensitisation prevents graft rejection in a xenograft rat model.
-
•
Successful neonatal desensitisation using hESC-derived progenitors
-
•
Grafted human neurons survive beyond the time of conventional immune suppression.
-
•
Multiple breaches of the BBB do not affect graft protection.
1. Introduction
Human embryonic stem cells (hESCs) and induced pluripotent stem cells (hiPSCs) have emerged as promising candidates to generate new neurons for repair in neurological disorders such as Parkinson's disease (Kirkeby et al., 2012, Kriks et al., 2011), Huntington's disease (Arber et al., 2015, Delli Carri et al., 2013) and stroke (Kelly et al., 2004, Thored et al., 2006). Clinical trials for diseases affecting the retina are already under way (Cyranoski, 2013, Whiting et al., 2015) and the first clinical trials for Parkinson's disease are approaching in the near future (Barker et al., 2015). In order to bring stem cell derived neurons to the clinic in a safe and effective manner, the therapeutic potential of cells must be extensively validated in preclinical rodent models of the corresponding disease. Human neurons require several months, if not years to achieve full maturation and integration after transplantation to the human (Lindvall and Bjorklund, 2004, Piccini et al., 2000) and rodent brain (Deacon et al., 1994, Pakzaban and Isacson, 1994). Therefore, long-term transplantation studies that allow for the assessment of survival, integration, and behavioral recovery are imperative. However, human to rat xenografts using naïve immune-competent hosts trigger a quick inflammation and immune response which destroys the transplants within weeks (Brundin et al., 1988, Sloan et al., 1991, Widner and Brundin, 1988). Currently, preclinical tests are therefore conducted in either pharmacologically immune-suppressed (i.e. Cyclosporine A, CsA) rats or in immune-compromised (i.e. athymic nude) rats (Brundin et al., 1985, Grealish et al., 2014, Grealish et al., 2015, Kelly et al., 2009). However, both these xenograft-models have shortcomings when it comes to neural graft assessments: CsA requires daily i.p. injections and eventually leads to declining health of the animals which limits CsA administration to a maximum of 18–20 weeks (Andoh and Bennett, 1998, Murray et al., 1985). This precludes its use for long-term testing, which is necessary to investigate maximal fibre outgrowth, graft integration, and function. For long-term studies, athymic nude rats that lack mature T-cells, can be used. However, despite being excellent animal model for long-term xenograft experiments, these animals are very prone to infections and have to be kept in specific pathogen-free conditions. Surgeries for creating lesions, cell transplantation and vector injections as well as behavioral assessments require repeated breaching of the barrier environment and thereby risking infections and early termination of the experiment. Thus, neither of these xenograft models is optimal for long-term multiplex assessment of human grafts.
In the early 1950′s it was shown that one way to circumvent the problem of graft rejection of skin grafts in two different strains of mice was to manipulate the host immune-system during its development to recognise foreign cells as self (Billingham et al., 1953, Billingham and Medawar, 1953). This concept has recently been translated to the transplantation of neural tissue (Kelly et al., 2009), and desensitising new-born rat pups at early postnatal ages with human cortical tissue obtained from elective termination of pregnancies allows for engraftment with similar cells during adulthood. This study convincingly showed that the transplants could survive in desensitised rats to a similar extent as cyclosporine treated rats without the need for additional immune-suppression (Kelly et al., 2009, Zhang et al., 2013). Several laboratories have tried to implement this approach in rats and mice with mixed success (Janowski et al., 2012, Mattis et al., 2014, Roberton et al., 2013, Zhang et al., 2013), and both the versatility and the mechanism of the desensitisation remain elusive. There are many possible reasons for the failure to desensitise rats for xenograft acceptance, but the most likely candidates are differences between host species and strains, the MHC expression of the donor cells used for inoculation as well as the state of the hosts immune system during inoculation, and the number of cells injected (Billingham and Brent, 1956, Billingham et al., 1953, Janowski et al., 2012, Kelly et al., 2009, Mattis et al., 2014, Roberton et al., 2013).
Furthermore, the widespread use of the neonatal desensitisation approach is currently dependent on the availability/access to primary fetal tissue for injection. As this tissue is harvested from elective terminations of pregnancies, it is associated with very low availability, as well as ethical and major logistical problems, making it a suboptimal cell source for desensitisation studies. From an experimental perspective, off-the-shelf cells that could be generated to the specific time of use would highly increase the usefulness of the desensitisation method for preclinical assessment of human stem cell based therapies. Here we show the ability of hESC-derived cells to be used for desensitisation with comparable efficiency to CsA and to protect against graft rejection for at least up to 24 weeks post-transplantation. The use of hESC-derived neural progenitors for desensitisation makes this method widely applicable for any centre performing pre-clinical testing of human neurons.
2. Materials and methods
All research on laboratory animals was conducted under local ethical and personal licences and approved by the local ethical committee at Lund University and the Swedish Department of Agriculture (Jordbruksverket). Time-mated dams of the Sprague-Dawley strain and adult females were purchased (Charles River, Germany) and kept under standard laboratory conditions.
2.1. Induction of desensitisation
To ensure sufficient exposure to the cells during the critical period for desensitisation we injected all pups on post-natal day 2 and 4 i.p. with 1 μl of pre-differentiated hESCs. The cells were differentiated according to previously established protocols (Kirkeby et al., 2012) and used for injections in a cell suspension of 100,000 cells/μl consisting of a mix of immature neural progenitors (day 16 of differentiation) and postmitotic neurons (day 35 of differentiation).
2.2. Preparation of hESCs for desensitisation and transplantation
All human ESC lines H9 (WA09, WiCell) and RC17 (Roslin Cells, hPSCreg #RCe021-A) were differentiated towards neural progenitors as previously described (Kirkeby et al., 2012). Human iN were derived via direct conversion of human fetal lung fibroblast as described previously (Pfisterer et al., 2011). H9-Cre cells were generated by in vitro lentiviral transduction with a Synapsin-Cre construct. For intra-striatal transplantation, 300,000 (75,000 cells/μl) cells from day 16 of differentiation were grafted as previously described (Kirkeby et al., 2012).
2.3. Transplantation
Once adult (minimum 15 weeks), all animals received lesions to the ascending dopaminergic fibre bundle as described previously (Torres et al., 2011). 4–5 weeks after the lesion the rats were engrafted using differentiated H9 and RC17, Cre-recombinase expressing H9-hESCs, or human induced neurons (hiNs). The group sizes of the respective experiments were as follows: In experiment 1 we engrafted 16 rats with H9-hESC-derived cells (CsA, n = 7; Desensitised, n = 9) and 2 rats with iNs. In experiment 2 we engrafted 28 rats using H9-hESC-derived cells (rejection, n = 7; CsA, n = 8 + 3; Desensitised, n = 8 + 2) and 12 rats using RC17 hESC (n = 8 + 4). For the assessment of breaching the blood brain barrier we engrafted 4 rats using H9-hESC-derived cells and 6 rats using Cre-recombinase expressing H9-hESC-derived cells.
All grafts were placed into the right striatum at the following coordinates: AP: + 1.2, ML: − 2.6, DV (from dura): − 4.5 and AP: + 0.5, ML: − 3.0, DV: − 4.5, with the incisor bar set at − 2.4. All cell suspensions were done in one 2 μl deposit per injection tract. Cells were infused at a speed of 1 μl per minute with 3 min for diffusion after each placement (Grealish et al., 2015).
Non-desensitised rats that were placed in the pharmacological immune-suppression group received daily i.p. injections of Cyclosporine (10 mg/kg) starting 1 day before transplantation.
2.4. Immunohistochemistry
All rats were perfused using ice-cold 4% PFA, brains were placed in 25% sucrose and cut coronal in a 1:12 series at a thickness of 35 μm. Immunohistochemistry was performed as stated in (Grealish et al., 2015). Primary antibodies against hNCAM (Santa Cruz, AB106, monoclonal, mouse, 1:1000); Ox42(CD11b) (Serotec, MCA275G, monoclonal, mouse, 1:1000), GFAP (DAKO, Z0034, polyclonal, rabbit, 1:1000), CD4 (Abcam, AB33775, monoclonal, mouse, 1:1000), CD8 (Serotec, MCA48G, monoclonal, mouse 1:1000), Ox6 (Abcam, AB23990, monoclonal, mouse, 1:500), Ox18 (Abcam, AB33752, monoclonal, mouse, 1:500), HuNu (Chemicon, MAB1281/87, monoclonal, mouse, 1:500), GFP (Abcam, AB13970, polyclonal, chicken, 1:1000), Cre-recombinase (BioLegend, PRB-106P, polyclonal, rabbit, 1:1000) were used.
2.5. Quantification of graft area and microglia response
A series of coronal sections (35 μm; 1:12) was stained for hNCAM and photographed using a Leica DMI600B microscope. The area of the graft core was quantified using ImageJ software (Version 1.50d, National Institues of Health, USA). Data are presented as the sum of all graft areas corrected for series. To estimate the microglia response we took images of three coronal sections stained for Ox42 in which the transplant location was clearly visible from the animals of the CsA-immune-suppressed, the neonatal desensitised and the rejection group (weeks 2 and 6, n = 5). Optical staining intensity was measured using ImageJ software (V1.50d, National Institues of Health, USA) calibrated with an optical density table (NIH) and Rodbard function (Heuer et al., 2012). We present the O.D. values of the Ox42 staining by subtraction of the intact striatum from the engrafted striatum.
2.6. Statistics
All data were analysed using the software package for the social science (SPSS) version 22.0 using a significance level of α = 0.05. Data were analysed using independent samples Student's t-test and one-way analysis of variance with Bonferroni corrected α for multiple comparisons, where appropriate.
3. Results
3.1. Desensitisation with pre-differentiated hESCs and subsequent engraftment of similar cells prevents graft rejection comparable to CsA
We injected new-born rat pups (n = 9) at early post-natal time-points in view of desensitising them for subsequent neural xenografts as adults. To mimic the heterogeneous composition of human fetal tissue, and to cover all cellular stages of graft maturation we used a mix of hESC-derived neural progenitors and post-mitotic neurons for injection. After a minimum of 15 weeks, the rats received unilateral 6-OHDA lesions and were transplanted 4–5 weeks later with neural progenitors prepared from the same H9 hESC line and using the same differentiation protocol as for neonatal desensitisation. In parallel, a group of naïve littermates (n = 7) rats were also lesioned and transplanted with the same batch of cells. These rats were exposed to our normal regime of daily injections of CsA (10 mg/kg) to prevent graft rejection.
When analysing the engrafted brains 18–22 weeks after transplantation, we found that 8/9 animals in the desensitised group and 7/7 animals in the CsA treated groups had surviving grafts. Staining for human specific neural cell adhesion molecule (hNCAM) confirmed that both groups contained neuron-rich grafts and revealed no difference in appearance or innervation of the grafted cells (Fig. 1, A–B; H–I; X). Estimations of graft area (Fig. 1, V) based on the hNCAM staining revealed that the grafts of CsA treated rats and neonatal desensitised rats were of similar size (t13 = − 0.873, p = n.s.). The immune response of the host after the xenografting was similar in both groups (Fig. 1, C–G and J–N), differing only on the few CD8-positive T-cells that could be found in the desensitised group (Fig. 1, E and L).
Fig. 1.

Rats desensitised with hESC-derived cells as neonates accept xenografts at a level comparable to CsA treated hosts.
(A–G) Characterisation of the survival and immune response in the brain of CsA treated rats (CsA H9; n = 7), (H–N) neonatal desensitised rats engrafted as adults with identical tissue (Des H9; n = 8) and (O–U, W) desensitised rats engrafted with hiNs (Des hfl-iN; n = 2). Graft survival (A–B, H–I, O–P) and fibre innervation (B, I) of transplants from each group were analysed using a specific marker for human NCAM. Inflammation and immune responses were assessed using the following markers respectively: microglia (Ox42: C, J, Q), T-helper cells (CD4: D, K, R), T-cells (E, L, S), MHC-class II (Ox6: F, M, T) and MHC-class I (Ox18: G, N, U). Graft sizes were not different between the CsA control group and the neonatal desensitised rats (V). Optical density analysis for Ox42 positive microglia staining revealed increased staining intensity in the rejection group (*** = p < 0.001). Representative overview of the transplant and innervation of an H9-hESC graft in a desensitised host (X). Scalebars A, H, O = 500 μM; B, I, P = 100 μM; C–G, J–N, Q–U = 250 μM; and X = 2000 μM. Data represent mean ± SEM. mean ± SEM.
Optical density (O.D.) analysis for the Ox42/cd11b microglia staining revealed an increased microglia response between the CsA immune-suppressed, the neonatal desensitised and the rejection group (F2,20, = 24.89, p < 0.001). There was a trend for slightly higher micrcroglia activation in the desensitised rats compared to CsA treated animals, but this difference failed to reach statistical significance (p = n.s.). The graft rejection group on the other hand had a significant increase in O.D. staining intensity compared to both groups (both, p < 0.001) (Fig. 1W).
This demonstrates that neonatal desensitisation using differentiated hESCs is a viable alternative to pharmacological immunosuppressive compounds. To probe the versatility of the desensitisation, animals that were desensitised using pre-differentiated H9-hESCs as new-borns, were engrafted after reaching adulthood with human induced neurons (iNs, n = 2) obtained from reprogrammed human fetal fibroblasts (Pfisterer et al., 2011). These animals displayed a full immune response against the foreign tissue (Fig. 1, P-U) and few, if any, surviving human cells/fibres could be detected 10 weeks post engraftment (Fig. 1, O–P).
3.2. Desensitisation using differentiated hESCs prevent graft rejection of cells neural cells differentiated from other hESC lines
To test the robustness of hESC-based desensitisation, and to address the specificity of the desensitisation approach further, we set up an experiment where we compared CsA treated rats (n = 8 + 2, 18 weeks and 6 weeks survival, respectively) to two groups of rats that were desensitised using H9-derived cells and subsequently engrafted them during adulthood with identically differentiated cells from either the same hESC line (H9-hESCs, n = 8 + 2) or from a different hESC line (RC17-hESCs, n = 8 + 4).
When analysing the grafts after 18–24 weeks, we found that neonatal desensitisation resulted in long-term graft survival in both desensitised groups as well as in the CsA control group (CsA H9; n = 8) to a similar extent (Fig. 2, A–C, E–F). When assessing graft sizes (Fig. 2, E) we found a significant difference between the three transplant groups (Group, F2,12 = 8.87, p < 0.01). Post-hoc assessment (Bonferroni corrected α) revealed that the difference was due to the cell line used (Des RC17 > Des H9 and Des RC17 > CsA H9, both: p < 0.05) whereas the grafts from the H9-donor cells were of similar size, irrespective of immunosuppression regime applied (Des = CsA, p = n.s.). When comparing the RC17 graft survival and appearance with other experiments where we use CsA or nude rats for immunosuppression (Supplementary Fig. 1), we did not see a major difference to the experiments in this study where we use H9 desensitised rats as hosts, and therefore we ascribe this difference in graft size to differences in the cell line, rather than differences in the host response.
Fig. 2.

H9 hESC-derived cells desensitise the host to other hESC lines.
Graft survival of (A) CsA immune-suppressed (CsA H9; n = 8 + 3), (B) identical (Des H9; n = 8 + 2, H9 to H9-desensitised) and (C) different (Des RC17; n = 8 + 4, RC17 to H9-desensitised) hESC donor – host combinations. (E) A subset of animals was sacrificed after 6 weeks. The percentage of survival was similar in all groups 18 weeks post-transplantation (E, n = 8 per group). Graft sizes were different between the three groups, with RC17 transplants being significantly larger, whilst the graft areas were similar for H9 grafts in CsA or desensitised hosts. Grafts of H9 hESC origin into naïve animals were readily rejected within 6 weeks post transplantation (D; see also Fig. 3). Scalebar = 100 μM. All data in E are presented as mean ± SEM.
In contrast, naïve rats (n = 7) that were engrafted in parallel using the same pre-differentiated H9-hESCs but received no form of immune-suppression showed a rapid rejection of the transplants within 6 weeks post engraftment (Fig. 2, D; Fig. 3 A-R). The appropriate immune reaction to the engrafted cells was accompanied by an early infiltration of microglia as well as a large amount of CD8 positive T-cells infiltrating the entire transplant. There was an abundance of MHC-class I and II positive cells and graft rejection was completed within 6 weeks post transplantation. The immune response was somewhat dampened after 18 weeks corresponding to the completed rejection of the foreign tissue. This demonstrates that the hESC-derived cells as such are immunogenic and are able to trigger an appropriate immune response, thereby leading to a rapid rejection of the transplant.
Fig. 3.

Graft rejection response.
Overview of characterisation of graft rejection of human to rat xenografts in fully immune-competent rat hosts over the time-course of 18 weeks post-engraftment. All rats were engrafted in parallel with the rats that were inoculated. Time-points for assessment of the immune and inflammation responses were 2 weeks (n = 3), 6 weeks (n = 2) and 18 weeks (n = 2) post transplantation. No graft was detected via staining for human NCAM (A–C) later than the 2 week time-point. Inflammation and immune responses were strongest at the early time-point (2 weeks) and somewhat dampened at the last time-point of assessment (18 weeks). The respective infiltration of immune cells were visualized using antibodies against microglia (Ox42, D–E), T-helper cells (G–I), T-cells (J–L), MHC-class I (M–O), and MHC-class II (P–R). Scalebar = 200 μM. The rejection seen in H9-hESC grafts in fully immune-competent hosts demonstrates the capability of these cells to elicit an immune response as well as the need and validity of the neonatal desensitisation approach.
3.3. Breaching the blood brain barrier after engraftment does not lead to subsequent rejection of the transplant
In some experimental setups it is necessary to repeatedly reopen the blood brain barrier (BBB) again after engraftment, for example when using novel tracing methods (Grealish et al., 2015, Wickersham et al., 2007) or to transduce the grafts when using optogenetic or chemogenetic techniques (Aldrin-Kirk et al., 2016, Bryson et al., 2014, Steinbeck et al., 2015, Weick et al., 2010). We tested if breaching the BBB with a second injection, leading to the unavoidable introduction of lymphocytes, would lead to spontaneous graft rejection in two different experimental paradigms. For both experiments, we used hosts that were desensitised with pre-differentiated H9-hESCs and subsequently grafted with two different batches of differentiated H9 hESCs as adults. One group (n = 4) received transplants of the unmodified hESCs, whereas a second group (n = 6) received hESCs expressing Cre-recombinase. After maturation for 20 weeks in vivo, these animals were injected with AAV vectors that either targets the host brain (hESC, n = 4) or the transplanted cells (hESC-Cre; n = 6) (Fig. 4, A). When we analysed the animals 4 weeks post-vector surgery (> 24 weeks post- transplantation), we found similar levels of survival in the AAV-GFP injected group (4/4) and in the CRE-activatable AAV-DIO-EYFP injected group (4/6), with surviving healthy transplants in both groups (Fig. 4 B and G). Selective transduction of the host (Fig. 4, F–F‴) and the graft (Fig. 4, K–L) could be demonstrated via expression of fluorescent markers. The host immune response against the graft was weak and comparable to the first two experiments (Fig. 4 C–E; H–J comp to Fig. 1, J–N), differing only in the somewhat heightening of microglial activation in the graft core of the hESC-Cre transplant (Fig. 4, H).
Fig. 4.

Characterisation of reopening the Blood Brain Barrier.
Rats that were neonatally desensitised using predifferentiated H9-hESCs and subsequently engrafted in adulthood using either H9-hESCs (Des H9 AAV; n = 4; A–F‴) or Cre-recombinase expressing H9-hESCs (Des H9-Cre AAV; n = 6; G–L) were subjected to a third opening of the blood brain barrier (after lesion and transplantation) to assess whether this would trigger an immune response causing the rejection of the transplant (A). We either used a GFP labelled AAV-vector to infect the host tissue (B–F‴) or a CRE-activatable direction inverse orientation (DIO) AAV-vector with a cassette encoding for EYFP to selectively infect the Cre-recombinase expressing donor cells (G–L) to select the cell population of interest. Transplants of both groups do survive long-term (> 24 weeks) in the rodent brain (B, G) and display limited inflammation (microglia; Ox42: C, H) and T-cell responses (CD4: D, I; CD8: E, J). Immune responses do not cause a rejection of the transplant within the four weeks after the respective AAV-transduction. Infection of the host cells was visualized using immune-fluorescent antibodies against DAPI (F) human nuclei (HuNU: F′), and GFP (F″). The composite merge shows the selective expression of the reporter in the host tissue (E‴). Selective transgene expression in the donor cells is demonstrated using fluorescent antibodies against human NCAM (J), Cre-recombinase (K′) and eYFP (K″). The composite merge (L) shows a cell that selectively expressed the EYFP reporter, clearly demonstrating the expression of nuclear Cre-recombinase and expression of EYFP. Scalebars: B–E = 500 μM; insets 50 μM; F–F‴ = 250 μM; G = 1000 μM, inset 50 μM; H–J = 1000 μM, inset 25 μM, K–K″ = 1000 μM, inset = 20 μM; L = 100 μM.
4. Discussion
In order to develop better long-term xenograft transplantations paradigms for evaluating grafts of human neurons, we sought to investigate the extent to which neonatal desensitisation using hESC-derived progenitors and neurons could prevent xenograft rejection of hESC-derived neural progenitors when engrafted into adult rats. For that purpose we set up several experiments to directly compare the neonatal desensitisation approach to conventional pharmacological immune-suppression using CsA, as well as to investigate to which extent one cell line (H9) can be used for desensitisation towards a different cell line (H9 vs RC17) or towards different types of cells (ES-derived neurons vs iNs).
First we compared the graft survival and immune reaction of rats that were transplanted with H9-hESC-derived neural progenitors into either rats that were pharmacological immune-suppressed hosts using CsA or into rats that were desensitised using identical cells at early neonatal ages. Whereas there is some discrepancy in the literature as to whether the desensitisation approach works (Janowski et al., 2012, Kelly et al., 2009), in the present setting grafts of desensitised rats survived equally well as their immune-suppressed counterparts. There was no obvious difference in the quality of the immune and inflammation response as assessed by antibody staining for T-helper cells and T-cells, as well as MHC class I and II antigens. The few CD8 + cells that could be detected in the transplant location of inoculated rats were only sparse and not enough to clear the transplant. Furthermore they are at similar levels as in reports of successful desensitisation using fetal cells from other groups (Kelly et al., 2009). We found a slight increase in the microglia activation in the desensitised rats compared to the CsA immune-suppressed animals, however this was much below the response to rejected transplants. Whether the grafts of desensitised animals are immune-activated or immunized over time (Shinoda et al., 1995, Winkler et al., 2005), could not be addressed by the current study. The long-term studies that have been conducted thus far have shown graft survival up to 40 weeks post-transplantation (Kelly et al., 2009). Important to note is that the transplanted cells are highly immunogenic as evidenced by the rapid and strong inflammation and rejection responses occurring in naïve, immune-competent rat hosts.
We noted that the desensitisation was somewhat specific to the cells used for inoculation and subsequent transplantation. Neural progenitors derived from two different hESC lines (H9 and RC17) were equally protected from graft rejection when transplanted into adult hosts that had been desensitised as neonates using H9-derived cells, whereas iN cells generated via direct neural conversion of human fetal lung fibroblast elicited a strong microglial inflammation and immune response with near complete rejection of all donor cell material.
Failures in the desensitisation that have been reported previously (Janowski et al., 2012, Mattis et al., 2014, Roberton et al., 2013), could be attributed to differences in donor cell type, strain of rats and mice, sex, age at inoculation, when compared to the initial report (Kelly et al., 2009). Similar to Kelly et al. (2009), we here used rats of the Sprague-Dawley strain which were desensitised before P5. In contrast, studies using mice (Janowski et al., 2012, Mattis et al., 2014, Roberton et al., 2013), different cells for inoculation: mix of hfNPCs and hNP1 cells, (Mattis et al., 2014), human glial-restricted progenitors or human umbilical cord blood-derived neural stem cells (Janowski et al., 2012) could not replicate the desensitisation. As such there is too much variability between the different studies to draw firm conclusion of the source of variation in successful desensitisation. However, the data in this study show that neonatal desensitisation can be successful when using this approach in rats of the Sprague-Dawley strain and using hESC-derived cells for inoculation, similar to what was reported in Kelly et al. (2009) using human fetal cells for desensitisation and grafting. One major adaptation to the protocol of Kelly et al. (2009) was the repeated injection of cells for desensitisation at post-natal days 2 and 4. The approach of using readily available hESC derived cells for neonatal desensitisation allows for repeated injections during the critical period and thereby maximizes the chances that the animals will receive a sufficient dose of cells. To expose the rats to epitopes expressed at different stages of graft maturation, we used a mixed population of progenitor cells from day 16 and mature neurons from day 35 of differentiation for the desensitisation. Whether any of these different differentiation stages individually could be used for successful desensitisation has to be determined in future studies.
We further showed that the neonatal desensitisation allowed for a second breaching of the BBB without rejection of the grafted cells opening up new avenues for the use of this model for long-term studies of human stem cell transplants using novel tracing methods (Grealish et al., 2015) or the use of optogenetic and chemogenetic approaches (Aldrin-Kirk et al., 2016, Steinbeck et al., 2015).
In summary, our data shows that long-term survival beyond the duration of conventional CsA treatment is robustly achieved when using pre-differentiated hESCs for desensitisation. Compared with human fetal tissue previously used for desensitisation, hESCs are easily accessible and can be prepared on demand, which immensely increases the utility of this system when performing pre-clinical assessment of stem cell derived neurons in xenograft models.
Author contributions
A.H., A.K. and M.P. designed the experiment. A.H, MJ, UP and A.K performed experiments, A.H. and M.P. wrote the manuscript.
The following are the supplementary data related to this article.
RC17-hESC graft in a CsA immune-suppressed host. Coronal series stained for hNCAM of a typical RC17-derived hESC graft in animals immunosuppressed with CsA (A). High power images display the level of innervation throughout the host brain (B–E). Scalebars: A = 1000 μM, B = 500 μM.
Conflict of interest
The authors declare no conflict of interest.
Acknowledgements
We would like to thank Michael Sparrenius, Bengt Mattsson, Ingar Nilsson and Jenny Johansson for technical assistance, and Dr. Anders Björklund and Dr. Shane Grealish for scientific input. This study was supported by grants from the European Community's 7th Framework Programme through NeuroStemCellRepair (nr. 602278). The Strategic Research Area at Lund University Multipark (Multidisciplinary research in Parkinson's disease), the Swedish Research Council (2015-03444, K2014-61X-20391-08-4 and K2012-99X-22324-01-5), the Swedish Parkinson Foundation (Parkinsonfonden) (772/15) and Brain Foundation (Hjärnfonden) (FO2015-0200). The research leading to these results has received funding from the European Research Council ERC Grant Agreement n. 309712.
References
- Aldrin-Kirk P., Heuer A., Mattsson B., Wang G., Lundblad M., Parmar M., Bjorklund T. Chemogenetic modulation of transplanted dopamine neurons reveals a novel serotonin dependent Parkonsonian dyskinesia mechanism mediated by the 5-HT6 receptor. Neuron. 2016;90:1–14. doi: 10.1016/j.neuron.2016.04.017. ( http://dx.doi.org/10.1016/j.neuron.2016.04.017) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andoh T.F., Bennett W.M. Chronic cyclosporine nephrotoxicity. Curr. Opin. Nephrol. Hypertens. 1998;7:265–270. doi: 10.1097/00041552-199805000-00005. [DOI] [PubMed] [Google Scholar]
- Arber C., Precious S.V., Cambray S., Risner-Janiczek J.R., Kelly C., Noakes Z., Fjodorova M., Heuer A., Ungless M.A., Rodriguez T.A., Rosser A.E., Dunnett S.B., Li M. Activin A directs striatal projection neuron differentiation of human pluripotent stem cells. Development. 2015;142:1375–1386. doi: 10.1242/dev.117093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barker R.A., Studer L., Cattaneo E., Takahashi J. G-Force PD: a global initiative in coordinating stem cell-based dopamine treatments for Parkinson's disease. Npj Parkinson's Dis. 2015;1:15017. doi: 10.1038/npjparkd.2015.17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Billingham R.E., Brent L. Acquired tolerance of foreign cells in newborn animals. Proc. R. Soc. Lond. Ser. B Biol. Sci. 1956;146:78–90. doi: 10.1098/rspb.1956.0073. [DOI] [PubMed] [Google Scholar]
- Billingham R.E., Medawar P.B. Desensitization to skin homografts by injections of donor skin extracts. Ann. Surg. 1953;137:444–449. doi: 10.1097/00000658-195304000-00002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Billingham R.E., Brent L., Medawar P.B. Actively acquired tolerance of foreign cells. Nature. 1953;172:603–606. doi: 10.1038/172603a0. [DOI] [PubMed] [Google Scholar]
- Brundin P., Nilsson O.G., Gage F.H., Bjorklund A. Cyclosporin A increases survival of cross-species intrastriatal grafts of embryonic dopamine-containing neurons. Exp. Brain Res. 1985;60:204–208. doi: 10.1007/BF00237035. [DOI] [PubMed] [Google Scholar]
- Brundin P., Strecker R.E., Widner H., Clarke D.J., Nilsson O.G., Astedt B., Lindvall O., Bjorklund A. Human-fetal dopamine neurons grafted in a rat model of Parkinson's-disease - immunological aspects, spontaneous and drug-induced behavior, and dopamine release. Exp. Brain Res. 1988;70:192–208. doi: 10.1007/BF00271860. [DOI] [PubMed] [Google Scholar]
- Bryson J.B., Machado C.B., Crossley M., Stevenson D., Bros-Facer V., Burrone J., Greensmith L., Lieberam I. Optical control of muscle function by transplantation of stem cell-derived motor neurons in mice. Science. 2014;344:94–97. doi: 10.1126/science.1248523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cyranoski D. Stem cells cruise to clinic. Nature. 2013;494:413. doi: 10.1038/494413a. [DOI] [PubMed] [Google Scholar]
- Deacon T.W., Pakzaban P., Burns L.H., Dinsmore J., Isacson O. Cytoarchitectonic development, axon-glia relationships, and long distance axon growth of porcine striatal xenografts in rats. Exp. Neurol. 1994;130:151–167. doi: 10.1006/exnr.1994.1194. [DOI] [PubMed] [Google Scholar]
- Delli Carri A., Onorati M., Lelos M.J., Castiglioni V., Faedo A., Menon R., Camnasio S., Vuono R., Spaiardi P., Talpo F., Toselli M., Martino G., Barker R.A., Dunnett S.B., Biella G., Cattaneo E. Developmentally coordinated extrinsic signals drive human pluripotent stem cell differentiation toward authentic DARPP-32 + medium-sized spiny neurons. Development. 2013;140:301–312. doi: 10.1242/dev.084608. [DOI] [PubMed] [Google Scholar]
- Grealish S., Diguet E., Kirkeby A., Mattsson B., Heuer A., Bramoulle Y., Van Camp N., Perrier A.L., Hantraye P., Bjorklund A., Parmar M. Human ESC-derived dopamine neurons show similar preclinical efficacy and potency to fetal neurons when grafted in a rat model of Parkinson's disease. Cell Stem Cell. 2014;15:653–665. doi: 10.1016/j.stem.2014.09.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grealish S., Heuer A., Cardoso T., Kirkeby A., Jonsson M., Johansson J., Bjorklund A., Jakobsson J., Parmar M. Monosynaptic tracing using modified rabies virus reveal early and extensive circuit integration of human embryonic stem cell-derived neurons in vivo. Stem Cell Rep. 2015;4:975–983. doi: 10.1016/j.stemcr.2015.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heuer A., Smith G.A., Lelos M.J., Lane E.L., Dunnett S.B. Unilateral nigrostriatal 6-hydroxydopamine lesions in mice I: motor impairments identify extent of dopamine depletion at three different lesion sites. Behav. Brain Res. 2012;228:30–43. doi: 10.1016/j.bbr.2011.11.027. [DOI] [PubMed] [Google Scholar]
- Janowski M., Jablonska A., Kozlowska H., Orukari I., Bernard S., Bulte J.W., Lukomska B., Walczak P. Neonatal desensitization does not universally prevent xenograft rejection. Nat. Methods. 2012;9:856–858. doi: 10.1038/nmeth.2146. (author reply 858) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelly S., Bliss T.M., Shah A.K., Sun G.H., Ma M., Foo W.C., Masel J., Yenari M.A., Weissman I.L., Uchida N., Palmer T., Steinberg G.K. Transplanted human fetal neural stem cells survive, migrate, and differentiate in ischemic rat cerebral cortex. Proc. Natl. Acad. Sci. U. S. A. 2004;101:11839–11844. doi: 10.1073/pnas.0404474101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelly C.M., Precious S.V., Scherf C., Penketh R., Amso N.N., Battersby A., Allen N.D., Dunnett S.B., Rosser A.E. Neonatal desensitization allows long-term survival of neural xenotransplants without immunosuppression. Nat. Methods. 2009;6:271–273. doi: 10.1038/nmeth.1308. [DOI] [PubMed] [Google Scholar]
- Kirkeby A., Grealish S., Wolf D.A., Nelander J., Wood J., Lundblad M., Lindvall O., Parmar M. Generation of regionally specified neural progenitors and functional neurons from human embryonic stem cells under defined conditions. Cell Rep. 2012;1:703–714. doi: 10.1016/j.celrep.2012.04.009. [DOI] [PubMed] [Google Scholar]
- Kriks S., Shim J.W., Piao J., Ganat Y.M., Wakeman D.R., Xie Z., Carrillo-Reid L., Auyeung G., Antonacci C., Buch A., Yang L., Beal M.F., Surmeier D.J., Kordower J.H., Tabar V., Studer L. Dopamine neurons derived from human ES cells efficiently engraft in animal models of Parkinson's disease. Nature. 2011;480:547–551. doi: 10.1038/nature10648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindvall O., Bjorklund A. Cell therapy in Parkinson's disease. NeuroRx. 2004;1:382–393. doi: 10.1602/neurorx.1.4.382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mattis V.B., Wakeman D.R., Tom C., Dodiya H.B., Yeung S.Y., Tran A.H., Bernau K., Ornelas L., Sahabian A., Reidling J., Sareen D., Thompson L.M., Kordower J.H., Svendsen C.N. Neonatal immune-tolerance in mice does not prevent xenograft rejection. Exp. Neurol. 2014;254:90–98. doi: 10.1016/j.expneurol.2014.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murray B.M., Paller M.S., Ferris T.F. Effect of cyclosporine administration on renal hemodynamics in conscious rats. Kidney Int. 1985;28:767–774. doi: 10.1038/ki.1985.196. [DOI] [PubMed] [Google Scholar]
- Pakzaban P., Isacson O. Neural xenotransplantation: reconstruction of neuronal circuitry across species barriers. Neuroscience. 1994;62:989–1001. doi: 10.1016/0306-4522(94)90338-7. [DOI] [PubMed] [Google Scholar]
- Pfisterer U., Kirkeby A., Torper O., Wood J., Nelander J., Dufour A., Bjorklund A., Lindvall O., Jakobsson J., Parmar M. Direct conversion of human fibroblasts to dopaminergic neurons. Proc. Natl. Acad. Sci. U. S. A. 2011;108:10343–10348. doi: 10.1073/pnas.1105135108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piccini P., Lindvall O., Bjorklund A., Brundin P., Hagell P., Ceravolo R., Oertel W., Quinn N., Samuel M., Rehncrona S., Widner H., Brooks D.J. Delayed recovery of movement-related cortical function in Parkinson's disease after striatal dopaminergic grafts. Ann. Neurol. 2000;48:689–695. [PubMed] [Google Scholar]
- Roberton V.H., Evans A.E., Harrison D.J., Precious S.V., Dunnett S.B., Kelly C.M., Rosser A.E. Is the adult mouse striatum a hostile host for neural transplant survival? Neuroreport. 2013;24:1010–1015. doi: 10.1097/WNR.0000000000000066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shinoda M., Hudson J.L., Stromberg I., Hoffer B.J., Moorhead J.W., Olson L. Allogeneic grafts of fetal dopamine neurons: immunological reactions following active and adoptive immunizations. Brain Res. 1995;680:180–195. doi: 10.1016/0006-8993(95)00260-w. [DOI] [PubMed] [Google Scholar]
- Sloan D.J., Wood M.J., Charlton H.M. The immune-response to intracerebral neural grafts. Trends Neurosci. 1991;14:341–346. doi: 10.1016/0166-2236(91)90159-r. [DOI] [PubMed] [Google Scholar]
- Steinbeck J.A., Choi S.J., Mrejeru A., Ganat Y., Deisseroth K., Sulzer D., Mosharov E.V., Studer L. Optogenetics enables functional analysis of human embryonic stem cell-derived grafts in a Parkinson's disease model. Nat. Biotechnol. 2015;33:204–209. doi: 10.1038/nbt.3124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thored P., Arvidsson A., Cacci E., Ahlenius H., Kallur T., Darsalia V., Ekdahl C.T., Kokaia Z., Lindvall O. Persistent production of neurons from adult brain stem cells during recovery after stroke. Stem Cells. 2006;24:739–747. doi: 10.1634/stemcells.2005-0281. [DOI] [PubMed] [Google Scholar]
- Torres E.M., Lane E.L., Heuer A., Smith G.A., Murphy E., Dunnett S.B. Increased efficacy of the 6-hydroxydopamine lesion of the median forebrain bundle in small rats, by modification of the stereotaxic coordinates. J. Neurosci. Methods. 2011;200:29–35. doi: 10.1016/j.jneumeth.2011.06.012. [DOI] [PubMed] [Google Scholar]
- Weick J.P., Johnson M.A., Skroch S.P., Williams J.C., Deisseroth K., Zhang S.C. Functional control of transplantable human ESC-derived neurons via optogenetic targeting. Stem Cells. 2010;28:2008–2016. doi: 10.1002/stem.514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whiting P., Kerby J., Coffey P., da Cruz L., McKernan R. Progressing a human embryonic stem-cell-based regenerative medicine therapy towards the clinic. Philos. Trans. R. Soc. Lond. Ser. B Biol. Sci. 2015;370 doi: 10.1098/rstb.2014.0375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wickersham I.R., Lyon D.C., Barnard R.J., Mori T., Finke S., Conzelmann K.K., Young J.A., Callaway E.M. Monosynaptic restriction of transsynaptic tracing from single, genetically targeted neurons. Neuron. 2007;53:639–647. doi: 10.1016/j.neuron.2007.01.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Widner H., Brundin P. Immunological Aspects of Grafting in the Mammalian Central Nervous-System - a Review and Speculative Synthesis. Brain Res. Rev. 1988;13:287–324. doi: 10.1016/0165-0173(88)90010-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winkler C., Kirik D., Bjorklund A. Cell transplantation in Parkinson's disease: how can we make it work? Trends Neurosci. 2005;28:86–92. doi: 10.1016/j.tins.2004.12.006. [DOI] [PubMed] [Google Scholar]
- Zhang S., Jiang Y.Z., Zhang W., Chen L., Tong T., Liu W., Mu Q., Liu H., Ji J., Ouyang H.W., Zou X. Neonatal desensitization supports long-term survival and functional integration of human embryonic stem cell-derived mesenchymal stem cells in rat joint cartilage without immunosuppression. Stem Cells Dev. 2013;22:90–101. doi: 10.1089/scd.2012.0116. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
RC17-hESC graft in a CsA immune-suppressed host. Coronal series stained for hNCAM of a typical RC17-derived hESC graft in animals immunosuppressed with CsA (A). High power images display the level of innervation throughout the host brain (B–E). Scalebars: A = 1000 μM, B = 500 μM.