

Abstract
The 8p11‐p12 amplicon occurs in approximately 15% of breast cancers in aggressive luminal B‐type tumors. Previously, we identified WHSC1L1 as a driving oncogene from this region. Here, we demonstrate that over‐expression of WHSC1L1 is linked to over‐expression of ERα in SUM‐44 breast cancer cells and in primary human breast cancers. Knock‐down of WHSC1L1, particularly WHSC1L1‐short, had a dramatic effect on ESR1 mRNA and ERα protein levels. SUM‐44 cells do not require exogenous estrogen for growth in vitro; however, they are dependent on ERα expression, as ESR1 knock‐down or exposure to the selective estrogen receptor degrader fulvestrant resulted in growth inhibition. ChIP‐Seq experiments utilizing ERα antibodies demonstrated extensive ERα binding to chromatin in SUM‐44 cells under estrogen‐free conditions. ERα bound to ERE and FOXA1 motifs under estrogen‐free conditions and regulated expression of estrogen‐responsive genes. Short‐term treatment with estradiol enhanced binding of ERα to chromatin and influenced expression of many of the same genes to which ERα was bound under estrogen‐free conditions. Finally, knock‐down of WHSC1L1 in SUM‐44 cells resulted in loss of ERα binding to chromatin under estrogen‐free conditions, which was restored upon exposure to estradiol. These results indicate the SUM‐44 cells are a good model of a subset of luminal B breast cancers that have the 8p11‐p12 amplicon, over‐express WHSC1L1, and over‐express ERα, but are independent of estrogen for binding to chromatin and regulation of gene expression. Breast cancers such as these, that are dependent on ERα activity but independent of estradiol, are a major cause of breast cancer mortality.
Keywords: Breast cancer, Oncogenes, Epigenomics, Estrogen receptor, Estrogen‐independence
Highlights
SUM44 is a model cell line for ERα positive breast cancer with the 8p11 amplicon.
WHSC1L1 is a driving oncogene from the 8p11 amplicon in SUM44 cells.
SUM44 breast cancer cells have high ERα expression, regulated by WHSC1L1 knockdown.
ERα is required for growth and survival of SUM44 cells but is estrogen‐independent.
WHSC1L1 knock‐down re‐sensitizes ERα to estradiol for binding to essential genes.
1. Introduction
Gene amplification is an important mechanism of oncogene activation, particularly in solid human malignancies. It is now understood that oncogenes are typically amplified in groups, referred to as amplicons, that contain many amplified genes, some of which are over‐expressed and transforming (Ciriello et al., 2013). Thus, an important challenge that remains is determining, within complex amplicons, which genes are true driving oncogenes and which are passengers. The 8p11‐p12 amplicon has been studied for many years and is most often associated with breast cancer, but more recently, this amplicon has been shown to occur commonly in other cancer types, including squamous cell lung cancers and bladder cancers (Chen et al., 2014; Zack et al., 2013). The 8p11‐p12 amplicon spans approximately 60 kB and contains approximately 55 genes. Moreover, the amplicon has been shown to consist of four sub‐regions that can be amplified independently (Gelsi‐Boyer et al., 2005). This finding alone is strong evidence for the existence of multiple driving oncogenes in the 8p11 amplicon, and indeed several laboratories, including our own, have provided compelling evidence of roles for a number of genes, including WHSC1L1, ZNF703, FGFR1, RAB11FIP1, IKBKB, LSM1, BAG4, TC1 and others (Adelaide et al., 1998, 2008, 2011, 2005, 2009, 2004, 2001, 2007, 2014, 2012, 2004, 2010, 2006, 2009). WHSC1L1 was first identified as a possible breast cancer oncogene by Chambon and co‐workers, who identified it as a third member of the nuclear set domain family (NSD3), which connected it to other NSD family members known to play a role in Wolf–Hirschhorn syndrome (Angrand et al., 2001). Since then, we and others have provided direct evidence that WHSC1L1 is a potent transforming gene and a likely driving oncogene in breast cancer (He et al., 2013; Stec et al., 2001; Tonon et al., 2005; Yang et al., 2010; Zhou et al., 2010). As a result of the TCGA project, interest in WHSC1L1 has increased as evidence continues to mount implicating this oncogene as a major driver in the 8p11 region. Ciriello et al. (2013) identified the 8p11 region as one of key importance in several cancer types, including breast cancer. Zack et al. (2013) performed GISTIC analysis of 4934 cancer specimens, including 880 breast cancers, and identified 70 recurrently amplified regions, each with a peak region most likely to contain driving oncogenes. In this analysis, the 8p11 amplicon was the 10th most significant peak and WHSC1L1 was considered the most likely candidate gene from the region. In a separate analysis of TCGA data, Chen et al. (2014) combined GISTIC data with gene expression data to identify the most likely driving oncogenes that are also druggable. WHSC1L1 was the top ranked gene among 42 driver genes identified, and they validated WHSC1L1 as a driving oncogene in several cell lines, confirming earlier reports from our laboratory. Thus, agnostic analyses of robust genomic data sets have consistently identified WHSC1L1 as an important driver oncogene in breast and other cancer types.
In this report, we provide further evidence for an important role for WHSC1L1 in luminal breast cancer and demonstrate that WHSC1L1 plays a role in driving over‐expression of the ESR1 gene and ERα protein, resulting in ERα that is transcriptionally active in an estrogen‐independent manner. These findings are consistent with data showing that the 8p11 amplicon most often occurs in luminal B breast cancers, which are typically ER‐positive and associated with a poor outcome.
2. Results
2.1. Role of WHSC1L1 in cell proliferation and gene expression
In previous work, we demonstrated that WHSC1L1 is amplified and over‐expressed in SUM‐44 cells, and that knock‐down of this gene has a dramatic effect on their proliferative capacity (Yang et al., 2010). Since WHSC1L1 codes for a histone methyltransferase (HMT) that modifies the epigenome, we investigated the effect of WHSC1L1 knock‐down on gene expression in SUM‐44 cells. To carry out these experiments, SUM44 cells were transduced with two different pGIPZ lentiviral vectors and selected using puromycin resistance. mRNA extracted from shLacZ control and shWH SUM‐44 cells was hybridized to Agilent arrays and the expression profiles for the two conditions were compared. The experiment was performed using two biological replicates, with dye‐swap groups used as technical replicates. This experiment identified several genes of importance in breast cancer that were significantly reduced in expression following knock‐down of WHSC1L1, including ESR1, ERBB3, ERBB4, CD44, CD24, MYB, and others (Table 1).
Table 1.
Changes in expression of select genes following knock‐down of WHSC1L1.
| Gene symbol | Absolute change | Fold‐change | p‐Value (corrected) |
|---|---|---|---|
| MYCN | −7766.5165 | −7.4258156 | 0.02 |
| ESR1 | −47,330.30575 | −7.3267975 | 0.004 |
| CXCR4 | −4656.73475 | −4.8752847 | 0.007 |
| ID2 | −17,146.6466 | −4.296558 | 0.004 |
| RET | −4582.921 | −3.7028813 | 0.008 |
| MYB | −17,224.83965 | −2.9244578 | 0.016 |
| CD24 | −1,57,749.3623 | −2.6231372 | 0.010 |
| ALDH2 | −45,419.4645 | −2.549547 | 0.002 |
| ERBB3 | −5371.384175 | −2.5006907 | 0.009 |
| ERBB4 | −3791.247525 | −2.1241639 | 0.002 |
We next performed experiments to validate the observation that WHSC1L1 is involved in expression of ESR1. Figure 1A and B show that SUM‐44 cells express significantly higher levels of ERα than MCF‐7 cells, and that knock‐down of WHSC1L1 reduced levels of ERα protein (the ESR1 gene is neither amplified nor point‐mutated in SUM‐44 cells). We have also observed repeatedly that the short isoform of WHSC1L1 is expressed at higher levels than the long isoform in SUM‐44 cells as well as other cell lines with the 8p11 amplicon (Figure 1C). TCGA data confirm that WH‐short is expressed at higher levels than WH‐long in primary breast cancers as well (Figure 1D). The importance of the short isoform of WHSC1L1 is further indicated by the observation that in some breast cancers with the 8p11 amplicon, only the exons that code for the short isoform of WHSC1L1 are copy number‐amplified, resulting in over‐expression of the short isoform and not the long isoform (data not shown). To determine if high‐level expression of the short isoform of WHSC1L1 influences ESR1 expression, we used a lentiviral shRNA construct that targets a 3′‐UTR sequence specific to the short isoform (shWH‐short). We found that knock‐down of WHSC1L1‐short alone had an even more dramatic effect on ESR1 expression than knock‐down of both the long and short isoforms of WHSC1L1 (Figure 1E). Knock‐down of WHSC1L1‐short also had a more dramatic effect on cell proliferation than knock‐down of both isoforms of WHSC1L1 (Figure 1F). These results suggest that WHSC1L1, particularly the short isoform, plays a role in the over‐expression of ERα observed in SUM‐44 cells.
Figure 1.
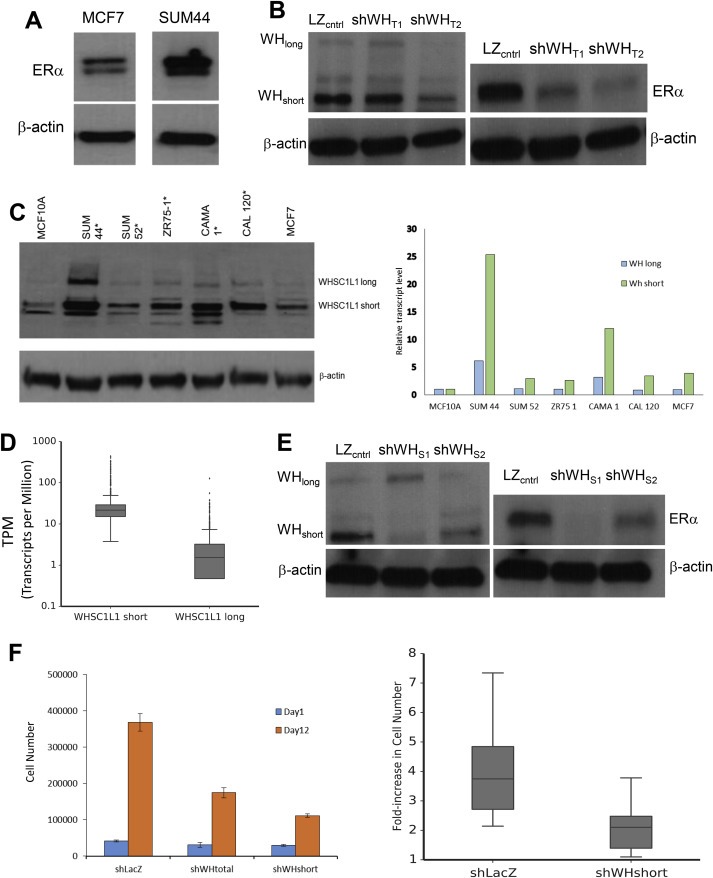
A. Immunoblot for ERα in MCF‐7 and SUM‐44 cells. β‐actin was used to confirm equal loading. B. Immunoblot showing WHSC1L1 (left) and ERα (right) protein levels in whole cell lysates from SUM‐44 cells lentivirally transduced with shRNA against LacZ or two shRNAs against both isoforms of WHSC1L1. β‐actin was used to confirm equal loading. C. (Left) Immunoblot showing WHSC1L1‐long and WHSC1L1‐short isoform levels in several breast cancer cell lines and MCF10A cells. Cell lines with increased WHSC1L1 copy number are denoted by an asterisk (*). (Right) Real‐time PCR results showing transcript levels for WHSC1L1‐long or WHSC1L1‐short in several breast cancer cell lines relative to levels in normal breast epithelial cells, represented here by MCF10A cells. D. Transcript expression (RNAseqV2) of WHSC1L1‐long and WHSC1L1‐short isoforms from Cancer Genome Atlas (TCGA) breast cancer samples (N = 964). E. Immunoblot showing WHSC1L1 (left) and ERα (right) protein levels in whole cell lysates from SUM‐44 cells lentivirally‐transduced with shRNAs against LacZ or two shRNAs against WHSC1L1‐short. β‐actin was used to confirm equal loading. F. (Left) Proliferation assay showing cell number after 12 days of culture in SFIH media for SUM‐44 WH‐total and WHSC1L1‐short shRNA knock‐downs compared to shLacZ control cells. The error bars represent 1.96 multiplied by the standard error of the mean (1.96*SEM). (Right) Boxplot showing the results of five replicates of the proliferation assay in SUM‐44 shWHshort_1 vs. shLacZ control cells.
To explore this observation further, we examined ESR1 expression levels in a panel of primary breast tumors available in our lab. In previous work, we studied 100 primary breast cancers and identified those that were positive for the 8p11‐p12 amplicon. Based on the correlation of copy number increase and gene amplification, we then identified WHSC1L1 as a top candidate oncogene (14). This group of 8p11 amplicon‐bearing breast cancers included six breast cancers with amplification and over‐expression of WHSC1L1, and in all six of these tumor specimens, ESR1 mRNA levels were high and comparable to the level expressed by SUM‐44 cells (Figure 2A). We also observed that some breast cancers over‐express ESR1/ERα in the absence of WHSC1L1 amplification, consistent with published data on other mechanisms of ESR1 over‐expression in breast cancer (26–28). However, in contrast to the SUM‐44 cells, none of the other breast cancer cell lines that have evidence for the presence of the 8p11 amplicon over‐express either WHSC1L1 or ESR1/ERα (Figure 2A,B). These results indicate that the SUM‐44 cell line is the only line that can be used as a model for the subset of breast cancers with WHSC1L1 and ESR1 over‐expression. TCGA data provides further support for the relationship between amplification and over‐expression of WHSC1L1 and ESR1. Figure 2C shows that ESR1 mRNA levels are significantly higher (p < 0.005) in breast cancers that over‐express WHSC1L1 compared to breast cancers without amplification and over‐expression of this oncogene.
Figure 2.
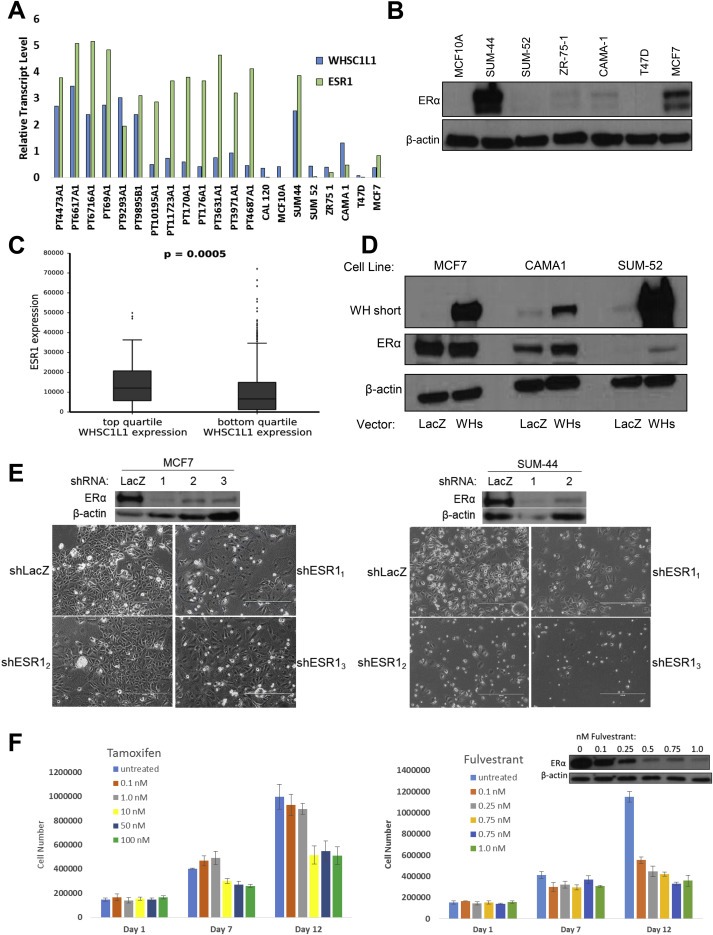
A. WHSC1L1 and ESR1 transcript levels in breast cancer tissue samples and breast cancer cell lines with and without the 8p11‐p12 amplicon. B. Immunoblot showing ERα protein levels in a panel of breast cancer cell lines. C. Boxplot of RNAseqV2 transcript expression levels for TCGA breast cancer samples grouped by high or low WHSC1L1 expression. Samples were ranked based on WHSC1L1 expression and grouped by quartile. The top and bottom quartiles for WHSC1L1 gene‐level expression were used to plot ESR1 transcript levels as shown. Student's t‐test was performed to obtain a p‐value of 0.0005. D. Immunoblot showing expression of WHSC1L1‐short and ERα in three breast cancer cell lines following transduction with a lentiviral expression vector for the short isoform of WHSC1L1. E. MCF‐7 (left) or SUM‐44 (right) cells were transfected with either LacZ or three different ESR1 shRNAs and grown for 9 days in culture before being photographed. The western blots above each panel show the expression level of ERα at day 9 after infection (except for shESR13 in SUM‐44 cells, for which an insufficient amount of protein was obtained.) F. SUM‐44 cells were seeded in triplicate wells in 6‐well plates and treated with the indicated concentrations of tamoxifen (left) or fulvestrant (right) daily for 12 days. Additional wells were seeded and treated with the indicated concentrations of fulvestrant daily for 12 days prior to being harvested for western blot analysis of ERα expression (right, inset).
Since WHSC1L1 is not over‐expressed in other cell lines with the amplicon, we infected several of these lines with a lentiviral expression vector for the short isoform of WHSC1L1. Figure 2D shows that over‐expression of WHSC1L1‐short in CAMA1 cells and SUM‐52 cells, but not MCF‐7 cells, resulted in increased expression of ERα. Furthermore, over‐expression of WHSC1L1‐short in CAMA1 cells not only increased expression of ERα protein, but also expression of some ERα target genes, including FOXA1 and IER3 (Supplementary Figure 1). These results help to confirm and extend the link between over‐expression of WHSC1L1 and ERα expression in breast cancer cells bearing the 8p11 amplicon.
We next determined if estrogen is required for the growth of SUM‐44 cells in vitro. Because SUM‐44 cells are cultured in serum‐free Ham's F12 medium, which has 10‐fold less phenol red (a weak estrogen) than conventional culture media; these cells have always been maintained under low‐estrogen conditions. Therefore, we compared the growth of SUM‐44 cells under normal conditions to their growth under phenol red‐free conditions. Omission of phenol red from the medium had no effect on the proliferation of SUM‐44 cells (Supplementary Figure 2A). Next, we cultured cells in phenol red‐free medium and examined the influence of exogenous 17‐β‐estradiol on growth of these cells. Like phenol red, 17‐β‐estradiol did not stimulate proliferation of SUM‐44 cells at any concentration tested (Supplementary Figure 2B). To examine the importance of the estrogen receptor itself in the growth and viability of SUM‐44 cells, we knocked‐down ESR1 using three different shRNAs that target the gene in both SUM‐44 and MCF‐7 cells. Figure 2E shows that knock‐down of ESR1 had a dramatic effect on the viability of SUM‐44 cells, and reduced, but did not completely block, the growth of MCF‐7 cells. We then examined the influence of the selective estrogen receptor modulator tamoxifen and the selective estrogen receptor degrader fulvestrant on the proliferation of SUM‐44 cells. Figure 2F shows that tamoxifen had a small inhibitory effect on SUM‐44 cell proliferation which was greatest at 1 nM. However, SUM‐44 cells still proliferated at this concentration, and increasing concentrations of tamoxifen, even to 100 nM, did not have further inhibitory effects on cell proliferation. By contrast, fulvestrant had a more dramatic, dose‐dependent effect on cell proliferation and ERα expression, which peaked at 1 nM, consistent with the effects of ESR1 knock‐down. These results indicate that amplification of WHSC1L1 in SUM‐44 cells plays a role in over‐expression of the ESR1 gene and ERα protein, and that SUM‐44 cells require ERα for growth and survival.
To extend our understanding of the relationship between WHSC1L1 over‐expression and ESR1/ERα over‐expression, we performed Illumina bead array expression profiling in SUM‐44 cells following knock‐down of WHSC1L1‐short, WHSC1L1‐total, or ESR1. Since WHSC1L1 regulates the expression of ESR1/ERα, which in turn regulates the expression of a specific set of genes, we predicted a significant overlap in the genes regulated by both genes, as well as a set of genes regulated by WHSC1L1 but not ESR1/ERα. Figure 3 shows that indeed, both WHSC1L1 and ESR1 regulated a common set of genes. Bioinformatic analysis of this gene set demonstrated highly significant enrichment for genes related to cell cycle progression. Similar gene expression profiles of SUM‐44 cells following estrogen treatment have recently been published by Sikora et al. (2014). By contrast, the gene set regulated by WHSC1L1 and not by ERα was enriched for genes associated with ER to Golgi vesicle‐mediated transport as well as a number of metabolic processes.
Figure 3.
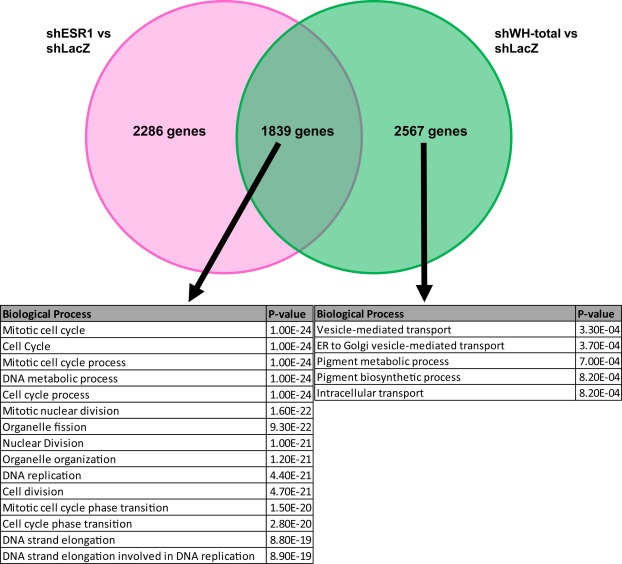
Venn diagram showing the number of differentially expressed genes identified in SUM‐44 cells following lentivirally mediated knock‐down of WHSC1L1 or ESR1 comparted to LacZ. Table below shows the Biological Processes enriched by the overlapping gene sets or by the gene set influenced by WHSC1L1 alone.
2.2. ERα binds chromatin in the absence of estrogen in SUM‐44 cells
To explore the activity of ERα in SUM‐44 cells, we measured ERα binding to chromatin and examined the relationship between ERα binding and gene expression. SUM‐44 cells were cultured in phenol red‐free media for 72 h and ChIP‐Seq was performed using an ERα antibody cocktail. Both biological and technical replicates of each sample were prepared and sequenced. After alignment and peak calling, we identified a consensus peak set containing binding regions found to be present in at least two of the three replicates for each condition.
In these experiments, we identified 32,994 ERα binding sites in SUM‐44 cells under estrogen‐free conditions (Figure 4A). We then annotated this peak set to identify genes associated with these binding sites and used RNA‐Seq to determine which genes were transcriptionally active. This analysis demonstrated that under estrogen‐free conditions, ERα was bound within 25 KB of the transcriptional start site of 5795 genes, and the majority of these genes (3501) were transcriptionally active (Supplementary Table 1). Motif enrichment analysis of the peak set revealed enrichment of several known sequence motifs. Table 2a lists the top ten motifs by enrichment score for cells grown in estrogen‐free conditions and shows that the ERE motif was the most significantly enriched, followed by the FOXA1 motif. While enrichment for ERE was stronger than for FOXA1, a total of 70.26% of ERα peaks in minus‐estrogen cells were within 200 bases of a FOXA1 motif, while 17.29% of peaks were on or near ERE motifs. The higher score for ERE indicates that, despite fewer peaks on or near ERE motifs, the mean binding intensity of ERα was higher on or near ERE motifs, and ERE motifs were more often closer than FOXA1 motifs to the center of the ERα peak. Bioinformatic analysis of the transcriptionally active genes to which ERα was bound identified several biological processes important in breast cancer, including Regulation of Ras Protein Signal Transduction, Cell Death/Programmed Cell Death/Apoptosis, and Intercellular Signaling (Table 3). Key canonical pathways enriched by this gene set include Insulin Signaling, the Phosphatidylinositol Signaling System, mTOR Signaling, and MAPK Signaling. These results suggest that in SUM‐44 cells, ERα is active under estrogen‐free conditions, because it is present at binding sites/motifs associated with well‐known ERα binding motifs and target genes that are transcriptionally active, such as CCND1, XBP1, ESR1, FOXA1, TSKU and others, which are associated with biological processes and canonical pathways of known importance in breast cancer. To validate further the role of ERα and FOXA1 in the expression of a subset of these genes, we examined their expression by RT‐PCR following siRNA‐mediated knock‐down of either ESR1 or FOXA1. The results shown in Figure 4B demonstrate that knock‐down of either ESR1 or FOXA1 had a significant effect on expression of these genes, consistent with a role for ERα binding in their transcriptional activation. Together, these results indicate that in SUM‐44 cells under estrogen‐free conditions, ERα binds to and regulates the expression of a set of genes associated with ERα activity and breast cancer biology.
Figure 4.
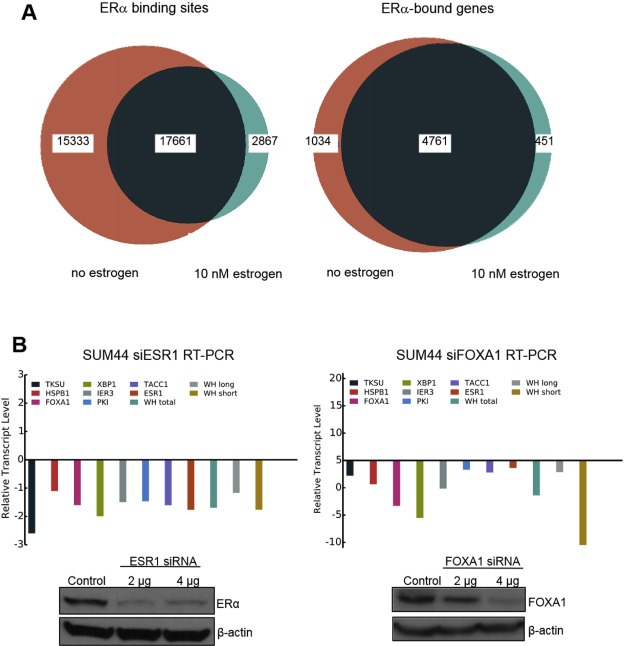
A. Venn diagrams showing ERα binding sites (left) or ERα‐bound genes (right) in SUM‐44 cells under estrogen‐free conditions or following administration of 10 nM estrogen. B. RT‐PCR analysis of changes in the transcript levels of several ER‐bound genes upon siRNA‐mediated knock‐down of ESR1 (left) or FOXA1 (right). Western blots showing the protein expression levels of ESR1 and FOXA1 following siRNA transfection are shown below each graph.
Table 2a.
Motif enrichment analysis of ERα peaks observed under estrogen free conditions.
| Motif name | Consensus | %Peaks | q‐Value (FDR) |
|---|---|---|---|
| ERE | VAGGTCACNSTGACC | 17.29 | 1e‐2268 |
| FOXA1 | WAAGTAAACA | 70.26 | 1e‐2186 |
| Foxa2 | CYTGTTTACWYW | 28.17 | 1e‐2131 |
| Fox:Ebox | NNNVCTGWGYAAACASN | 28.52 | 1e‐1764 |
| AP‐2γ | SCCTSAGGSCAW | 25.58 | 1e‐914 |
| AP‐2α | ATGCCCTGAGGC | 21.38 | 1e‐841 |
| FOXP1 | NYYTGTTTACHN | 12.64 | 1e‐718 |
| GRHL2 | AAACYKGTTWDACMRGTTTB | 10.08 | 1e‐545 |
| BATF | DATGASTCAT | 14.17 | 1e‐537 |
| Atf3 | DATGASTCATHN | 14.07 | 1e‐519 |
Table 3.
Selected GO Biological Processes associated with transcriptionally active genes to which ERα was bound within 25 kB of the transcriptional start site.
| Category | ID | Name | q‐Value Bonferroni | Hit count in query list | Hit count in genome |
|---|---|---|---|---|---|
| GO: Biological Process | GO:0007264 | Small GTPase mediated signal transduction | 3.07E‐11 | 219 | 739 |
| GO: Biological Process | GO:1902531 | Regulation of intracellular signal transduction | 6.57E‐11 | 390 | 1518 |
| GO: Biological Process | GO:0007265 | Ras protein signal transduction | 3.13E‐10 | 165 | 524 |
| GO: Biological Process | GO:0051056 | Regulation of small GTPase mediated signal transduction | 3.29E‐10 | 155 | 483 |
| GO: Biological Process | GO:0007169 | Transmembrane receptor protein tyrosine kinase signaling pathway | 7.51E‐08 | 204 | 726 |
| GO: Biological Process | GO:0033554 | Cellular response to stress | 1.22E‐07 | 368 | 1489 |
| GO: Biological Process | GO:0030036 | Actin cytoskeleton organization | 3.11E‐07 | 152 | 509 |
| GO: Biological Process | GO:0007266 | Rho protein signal transduction | 5.32E‐07 | 85 | 238 |
| GO: Biological Process | GO:0007010 | Cytoskeleton organization | 9.50E‐07 | 257 | 987 |
| GO: Biological Process | GO:0006915 | Apoptotic process | 1.25E‐06 | 428 | 1806 |
We recently completed a genome‐scale shRNA screen of the entire SUM line panel, including SUM‐44 cells, and identified the genes essential for the growth and survival of these cells. Among the 3596 genes to which ERα was bound, 168 were hits in this screen, (p‐value for the number of hits occurring within the set of 3596 ER‐bound genes is 0.0005142) and therefore functionally important to SUM‐44 cells, including CDK6, FOXA1, and BCL2 (Supplementary Table 2). These results are consistent with an important role for ERα in regulating the expression of important genes in SUM‐44 cells under estrogen‐free conditions.
Next, we measured the influence of short‐term estrogen treatment on binding of ERα to chromatin and gene expression. SUM‐44 cells were cultured in serum‐free, phenol red‐free medium for 72 h and treated with 10 nM 17‐beta estradiol for 45 min before harvesting for chromatin isolation and ChIP‐Seq analysis. As shown in Figure 4A, ChIP‐Seq identified approximately 20,528 peaks in triplicate experiments, and 17,661 of these peaks overlapped with peaks identified in cells growing under estrogen‐free conditions. Figure 5A shows that short‐term estrogen treatment sometimes resulted in the appearance of new peaks within genes that already exhibited ERα binding and influenced the intensity of some peaks previously identified under estrogen‐free conditions. Table 2b shows the results of motif enrichment analysis of SUM‐44 cells following exposure to estrogen. The motifs enriched in the estrogen‐treated samples were similar to those observed under estrogen‐free conditions. ERα was bound within 25 kb of the transcriptional start site of 5212 genes, 4761 of which were common between the minus‐ and plus‐estrogen conditions (Figure 4A, Supplementary Table 3). Some of the genes common to both estrogen‐free and estrogen‐treated conditions were known estrogen target genes, such as CCND1, XBP1, CTSD, and TFF1. To determine the influence of estrogen treatment on expression of these genes, we performed RNA‐Seq profiling of SUM‐44 cells at 3, 6, and 12 h after administration of 10 nM 17‐β‐estradiol. The heat map in Figure 6 summarizes the influence of estrogen treatment on the expression of ERα‐bound genes that map to the highly enriched biological processes and canonical pathways shown in Table 3. These results demonstrate that in SUM‐44 cells, ERα remains capable of interacting with the hormone in a manner that influences chromatin binding and gene expression.
Figure 5.
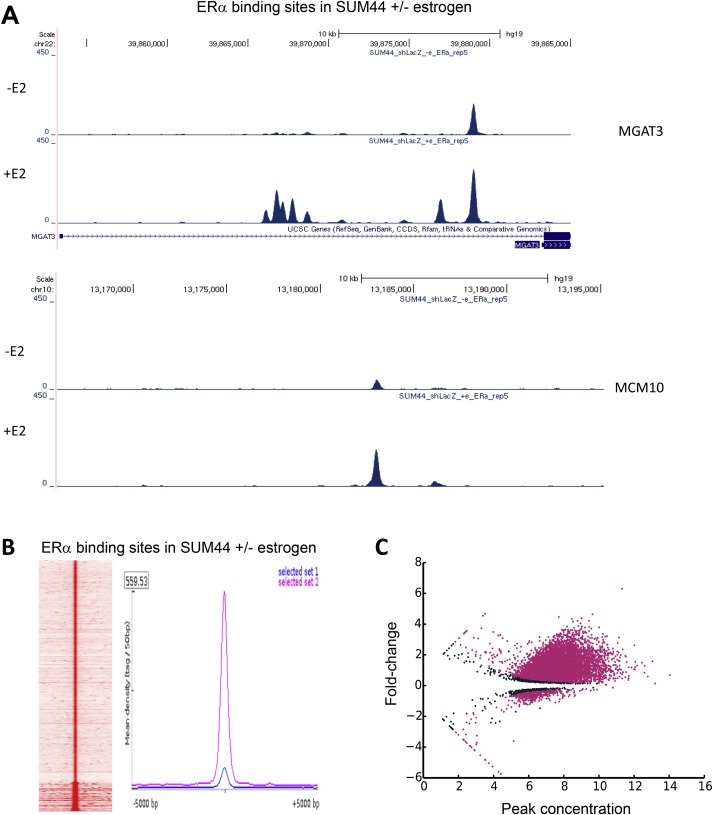
A. Genome traces showing the changes in several ERα binding regions in MGAT3 and MCM10 genes following treatment of SUM‐44 cells with 10 nM estrogen. B. Heatmap showing ERα peaks in SUM‐44 cells under estrogen‐free conditions and a mean‐profile plot of ERα peaks in SUM‐44 cells under 10 nM estrogen (magenta) and estrogen‐free (blue) conditions (left panel). C. MA‐plot showing results of ChIP‐Seq analysis of original peak intensity and fold‐change for significantly differentially‐bound ERα binding sites observed in the absence versus presence of estrogen in SUM‐44 cells. The y‐axis shows log2‐fold‐change in ERα binding intensity, and the x‐axis represents the initial peak intensity in the estrogen‐free control sample. Pink points represent significantly differentially‐bound peaks.
Table 2b.
Motif enrichment analysis of ERα peaks observed following administration of 10 nM estradiol.
| Motif name | Consensus | %Peaks | q‐Value (FDR) |
|---|---|---|---|
| ERE | VAGGTCACNSTGACC | 21.68 | 1e‐4750 |
| FOXA1 | WAAGTAAACA | 61.3 | 1e‐2326 |
| Foxa2 | CYTGTTTACWYW | 24.23 | 1e‐2236 |
| Fox:Ebox | NNNVCTGWGYAAACASN | 24.55 | 1e‐1670 |
| AP‐2γ | SCCTSAGGSCAW | 22.88 | 1e‐1013 |
| AP‐2α | ATGCCCTGAGGC | 19.25 | 1e‐955 |
| FOXP1 | NYYTGTTTACHN | 11.03 | 1e‐782 |
| BATF | DATGASTCAT | 13.63 | 1e‐677 |
| Atf3 | DATGASTCATHN | 13.41 | 1e‐628 |
| GRHL2 | AAACYKGTTWDACMRGTTTB | 8.48 | 1e‐479 |
Figure 6.
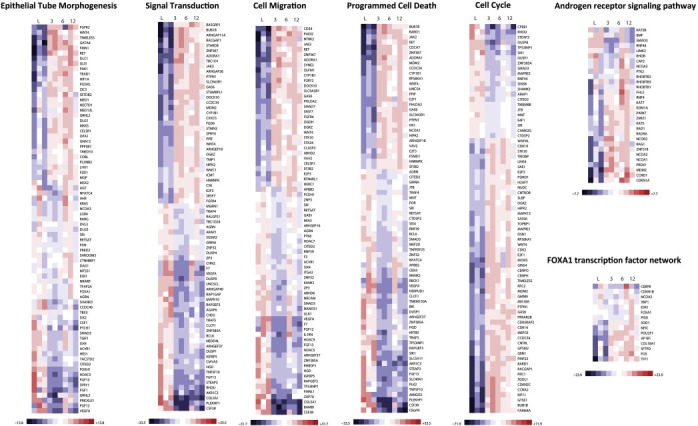
Heat maps showing the effect of estrogen treatment on the expression of ERα‐bound genes in SUM‐44 cells that map to selected biological processes and canonical pathways.
Finally, we attempted to perform ERα ChIP‐Seq in SUM‐44 cells following knock‐down of WHSC1L1‐short, both in the absence and presence of estrogen. In repeated attempts, we failed to identify specific ERα ChIP‐Seq peaks in WH‐short knock‐down SUM‐44 cells cultured under estrogen‐free conditions (data not shown). This is consistent with the effect of WHSC1L1 knock‐down on levels of ERα protein, as shown in Figure 1. However, treatment of WH‐short knock‐down SUM‐44 cells with 10 nM estrogen resulted in the identification of 11,212 peaks (Figure 7) within 25 kb of the transcriptional start site of 3064 genes (Supplementary Table 4). Bioinformatic analysis of this gene set showed enrichment of a number of biological processes and canonical pathways relevant to breast cancer cells (Table 4). Critical genes such as ESR1, FOXA1, CCND1 and MYB are included in this gene set, indicating that under conditions in which ERα levels have been reduced as a result of WH‐short knock‐down, ERα reverts to estrogen‐dependency for binding to genes important for cell proliferation and other cancer cell phenotypes.
Figure 7.
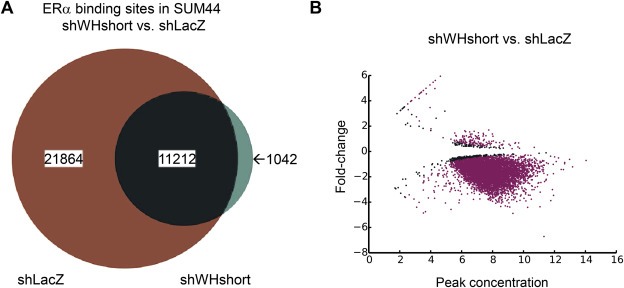
A. ERα binding sites in SUM‐44 shWHshort vs. shLacZ control cells following treatment with 10 nM estradiol as determined by ChIP‐Seq analysis. In this experiment, no peaks were detected in WHSC1L1‐short knock‐down cells in the absence of estrogen. B. MA‐plot showing original peak intensity and fold‐change for significantly differentially‐bound ERα binding sites observed in the presence of estrogen in SUM‐44 shWHshort vs shLacZ cells as determined by ChIP‐Seq analysis. The y‐axis shows the log2‐fold‐change in ERα binding intensity, and the x‐axis represents the initial peak intensity in the estrogen‐free control sample. Pink points represent significantly differentially‐bound peaks.
Table 4.
Selected GO Biological Processes associated with transcriptionally active genes to which ERα was bound within 25 kB of the transcriptional start site following estrogen treatment of WHSC1L1 knock‐down SUM‐44 cells.
| Category | ID | Name | q‐Value Bonferroni | Hit count in query list | Hit count in genome |
|---|---|---|---|---|---|
| GO: Biological Process | GO:0009888 | Tissue development | 1.27E‐08 | 389 | 1794 |
| GO: Biological Process | GO:1902531 | Regulation of intracellular signal transduction | 3.81E‐08 | 336 | 1518 |
| GO: Biological Process | GO:0060429 | Epithelium development | 3.36E‐05 | 246 | 1110 |
| GO: Biological Process | GO:0012501 | Programmed cell death | 4.54E‐05 | 376 | 1832 |
| GO: Biological Process | GO:0008202 | Steroid metabolic process | 1.40E‐04 | 86 | 304 |
| GO: Biological Process | GO:0051056 | Regulation of small GTPase mediated signal transduction | 2.67E‐04 | 122 | 483 |
| GO: Biological Process | GO:0030855 | Epithelial cell differentiation | 4.68E‐04 | 142 | 589 |
| GO: Biological Process | GO:0048545 | Response to steroid hormone | 6.45E‐04 | 100 | 381 |
| GO: Biological Process | GO:0009725 | Response to hormone | 6.98E‐04 | 195 | 871 |
| GO: Biological Process | GO:0007264 | Small GTPase mediated signal transduction | 7.41E‐04 | 170 | 739 |
| GO: Biological Process | GO:0007265 | Ras protein signal transduction | 9.73E‐04 | 128 | 524 |
In summary, these experiments demonstrate that the SUM‐44 cell line is a good model for an important subset of breast cancers characterized by the presence of the 8p11 amplicon, amplification and over‐expression of the WHSC1L1 oncogene, and high‐level expression of the ESR1 gene and ERα protein. Further, these results suggest that in such breast cancer cells, over‐expression of WHSC1L1 and ERα results in estrogen‐independent transcriptional regulation of a large number of genes that play important roles in the malignant phenotype of these cells.
3. Discussion
Estrogen receptor‐positive breast cancer is the most prevalent form of the disease. Currently, there are several drugs that target the estrogen receptor. These include SERMs, such as tamoxifen, pure anti‐estrogens that are also selective estrogen receptor degraders (SERDs), such as fulvestrant, and aromatase inhibitors, such as anastrozole (Charehbili et al., 2014). All of these drugs are effective at blocking estrogen‐mediated receptor activity. Nevertheless, thousands of patients die every year from ERα‐positive breast cancers, because the disease progresses to a stage at which these drugs are no longer effective. Since many of these patients succumb to disease in which ERα is still highly expressed and functional, estrogen‐independent activity of the receptor is now understood to be an important mechanism for the acquisition of resistance to hormonally‐based therapies.
An important limitation of fulvestrant is its relatively low bioavailability, which limits the biologically effective concentrations that can be achieved in patients. Indeed, our results show that SUM‐44 cells lose viability when ESR1 is knocked down, or when ERα protein is degraded following treatment with fulvestrant. However, the concentrations of fulvestrant required to achieve sufficient ERα degradation for significant levels of cell death are not always achievable in patients. This result suggests that the poor bioavailability of fulvestrant is an important reason for the lack of efficacy observed in patients that express high levels of ERα. This result also predicts that the next generation of SERDs, which are being designed to have improved bioavailability, will be more effective in this setting. Indeed, the presence of the 8p11 amplicon may be a good biomarker to predict the need or efficacy of these new SERD drugs.
There are several mechanisms by which breast cancer cells can become independent of estrogen but remain dependent on the estrogen receptor for viability and growth. Several years ago, Fuqua and co‐workers identified mutations in the ESR1 gene that result in estrogen‐independent ERα activity (Fuqua et al., 1993, 1991). It was subsequently found that such mutations are rare in primary breast cancers. However, more recent studies have shown that ESR1 mutations are relatively common in metastatic breast cancer cells (Jeselsohn et al., 2014). There is also emerging evidence that the ESR1 gene can be amplified and over‐expressed in metastatic breast cancer (Brown et al., 2008; Nielsen et al., 2011; Nikolsky et al., 2008). Thus, two molecular mechanisms have emerged that help explain how some breast cancer patients continue to express high levels of ERα but become unresponsive to hormonally‐based therapies. In this study, we have identified another mechanism for this phenomenon; the amplification of genes in the 8p11 amplicon, and over‐expression of the WHSC1L1 oncogene, which occurs predominantly in aggressive luminal B‐type breast cancers. The data reported here indicate a role for over‐expression of WHSC1L1 in the high level expression of ESR1 message and ERα protein in SUM‐44 cells. We originally identified ESR1 as a gene that was dramatically down‐regulated in SUM‐44 cells following transduction of an shRNA targeting WHSC1L1. We have now validated this finding using a different set of shRNAs and demonstrated down‐regulation of ESR1 at the message level and ERα at the protein level following knock‐down of WHSC1L1. Down‐regulation of ESR1 was also confirmed by the results of a second gene expression profiling study, which also showed that knock‐down of WHSC1L1 and ESR1 regulate the expression of an overlapping set of genes that map to several Biological Processes associated with cell cycle progression and DNA replication. This result demonstrates that the consistently observed effects of WHSC1L1 knock‐down on cell proliferation and viability are mediated via its effects on ESR1/ERα. In addition, the expression profiling results showed that WHSC1L1 regulates other gene sets that are independent of ESR1. Thus, as an amplified and over‐expressed breast cancer oncogene, targeting WHSC1L1 directly is likely to have profound effects on cell viability via its effects on ERα and its downstream targets, and via its effects on regulating expression of genes associated with endosome trafficking and metabolism. Recently, Shen et al. (2015) demonstrated that the short isoform of WHSC1L1 also plays a critical role in regulating expression of key genes in acute myeloid leukemia by acting as an adapter protein that bridges BRD4 with the chromatin remodeling protein CHD8. In these cells, as in breast cancer cells, WHSC1L1‐short is expressed to much higher levels than the long‐isoform, thus providing the first evidence for an important epigenomic role for WHSC1L1‐short despite its lack of a catalytic SET domain.
Data obtained from primary human breast cancers by our laboratory as well as data from the TCGA substantiate the connection between amplification and over‐expression of WHSC1L1 and high levels of ESR1 expression (Cerami et al., 2012; Gao et al., 2013). These results are consistent with results reported recently by Feng et al. (2014), who demonstrated a role for the related gene, WHSC1, in regulating expression of ESR1 in MCF‐7 cells. As we and others have reported previously, when the 8p11 amplicon is present in breast cancer cells, many genes are coordinately over‐expressed (Ray et al., 2004; Yang et al., 2006). Among these genes are the oncogenes ASH2L and KAT6A. Like WHSC1L1, ASH2L is a histone methyltransferase and is almost always coordinately over‐expressed with WHSC1L1. In addition, Qi et al. (2014) reported that ASH2L can influence expression of the ESR1 gene in luminal breast cancer cells. Recently, we published evidence that KAT6A, a histone acetyl transferase, is a transforming oncogene from the 8p11 amplicon and demonstrated that it also influences expression of ESR1 (Turner‐Ivey et al., 2014). SUM‐44 cells, like the majority of breast cancers with the 8p11 amplicon, coordinately over‐express WHSC1L1, ASH2L, and KAT6A. Together, these reports link the presence of the 8p11 amplicon, and WHSC1L1 over‐expression in particular, with high‐level expression of ESR1/ERα. We hypothesize that these three 8p11 amplicon oncogenes interact to drive high level expression of ESR1 in breast cancer cells. In addition, our data suggest that this results in estrogen‐independent ERα activity and subsequently, estrogen‐independent expression of ERα target genes.
In a recent landmark paper, Carroll and co‐workers identified a gene set associated with ERα binding in breast cancer patients who had experienced a poor outcome that was distinct from the ERα‐associated gene set found in patients with a good outcome (Ross‐Innes et al., 2012). Interestingly, WHSC1L1, as well as other genes from the 8p11 amplicon, were included in the poor outcome gene set. The results presented here suggest that the presence of the 8p11 amplicon, especially WHSC1L1, in the poor outcome gene set is due to its role in driving estrogen‐independent ERα activity, and hence resistance to hormonally‐based drugs.
One finding that resulted from our experiments is that in a panel of known ER‐positive human breast cancer cell lines, SUM‐44 cells express the highest levels of ESR1 mRNA and ERα protein. Even other ER‐positive breast cancer cell lines that have the 8p11 amplicon, such as ZR‐75‐1 and CAMA‐1, do not express high levels of ESR1. The reasons for this are unclear and in stark contrast to data obtained from primary breast cancers in our own panel as well as the TCGA database (Figure 2), which demonstrates a positive association between over‐expression of WHSC1L1 and ESR1. Thus, the SUM‐44 cell line is the only cell line that is representative of this important class of ER‐positive breast cancers, and exhibits estrogen‐independent ERα transcriptional activity. Indeed, SUM‐44 cells, despite their lack of dependence on exogenous estrogen, are highly dependent on ERα for proliferation and survival, as evidenced by their response to ESR1 knock‐down and fulvestrant treatment, which resulted in ERα protein degradation. These results bode well for the development of new, highly bioavailable SERDs, many of which are currently being tested in clinical trials (Lai et al., 2015). Furthermore, the presence of the 8p11 amplicon, and particularly the amplification and over‐expression of WHSC1L1, could serve as a biomarker to identify cancers that will be responsive to treatment with these drugs rather than aromatase inhibitors, which are ineffective in cells that depend on ERα but no longer require estrogen.
In summary, the results reported here, coupled with previous publications from our laboratory and others, point to WHSC1L1 as an important oncogene in aggressive luminal B breast cancers, particularly those that have become unresponsive to SERMs or aromatase inhibitors, and provide a direct link between amplification and over‐expression of WHSC1L1, ESR1/ERα over‐expression, and estrogen‐independent ERα activity.
4. Methods
4.1. shRNA vectors
Lentiviral shRNA expression vectors from the pLKO shRNA catalog were purchased from Sigma. The vectors used were the WHSC1L1‐short vector TRCN0000415241, and the WHSC1L1‐total vectors, TRCN0000425711, TRCN0000015615, and TRCN0000015616.
4.2. Lentiviral infection protocol
Lentivirus for lentiviral transduction of shRNAs into SUM‐44 cells was prepared using the Sigma Mission lentiviral packaging system (Sigma, shp‐001) in 293FT packaging cells following the manufacturer's protocol. Briefly, each construct was co‐transfected into 293FT cells with Sigma pLKO shRNA vectors and virus was harvested according to the manufacturer's instructions. Target cells were transduced with packaged virus in growth media supplemented with 5 μg/ml polybrene. Cells were cultured for three days to allow for expression of the resistance marker. Non‐transduced cells were eliminated from the culture by addition of the selection agent puromycin (3 μg/ml).
4.3. Antibodies
The WHSC1L1 antibody was purchased from ProteinTech Group Inc. (Cat. No. 11345‐1‐AP). The estrogen receptor alpha antibody was purchase from Bethyl Labs (Cat. No. A300‐498A). ChIP‐grade anti‐estrogen receptor alpha antibodies were purchased from Santa Cruz (sc‐543x) and Thermo Scientific (MA5‐13065). ChIP‐grade histone antibodies were used for both ChIP and immunoblotting, and were purchased from Abcam and Millipore: total H3 (Abcam ab1791), H3K36me3 (Abcam ab9050), H3K36me2 (Abcam ab9049), and H3K4me3 (Millipore 17‐678).
4.4. Cell culture
SUM‐44PE cells were cultured in serum‐free Ham's F‐12 with supplements (Ham's F‐12 with 1 μg/ml hydrocortisone, 1 mg/ml bovine serum albumin, 10 mM HEPES, 5 mM ethanolamine, 5 μg/ml transferrin, 10 nM triiodothyronine, 50 nm sodium selenite, 25 μg/ml gentamicin, 2.5 μg/ml fungizone, and 5 μg/ml insulin), as described previously (Ethier et al., 1993). For estrogen‐free culture conditions, phenol red‐free Ham's F‐12 media was substituted for normal Ham's F‐12; all supplements were as in normal media. Cells transduced with lentiviral shRNAs were selected and maintained in 3 μg/mL puromycin.
4.5. Proliferation assays
Proliferation assays were performed in 6‐well culture plates seeded with 5 × 105 cells per well. At 24 h and 10 days after seeding, cells were washed three times with PBS and agitated on a rocker table with 0.5 ml of a HEPES/MgCl2 buffer (0.01 M HEPES and 0.015 M MgCl2) for 5 min. Cells were then lysed for 10 min with a solution of ethyl hexadecyldimethylammonium, and the nuclei were counted using a Z1 Coulter Counter (Beckman Coulter, Brea, CA, USA).
4.6. Two‐color gene expression microarrays
The quality of mRNA was measured using an Agilent Bioanalyzer. All samples had a minimum RNA integrity (RIN) score of 8. Sample labeling was performed using 500 ng of total RNA along with the Target 1‐Round Aminoallyl‐aRNA Amplification Kit 101 (Epicentre, Madison, WI) and Agilent spike‐in controls to produce aminoallyl‐aRNA according to the vendor's protocol. Five μg of each aminoallyl‐aRNA sample and Alexa Fluor 555 or Alexa Fluor 647 (Molecular probes/Life Technologies, Foster City, CA) were used for labeling. Samples were incubated with the dye for 30 min at room temperature. After incubation, samples were run through RNeasy Mini Elute Cleanup columns (Qiagen, Valencia, CA) to remove unincorporated dye. Sample concentration and dye incorporation were then checked on a NanoDrop. Labeled samples were prepared for hybridization following Agilent's “Two‐Color Microarray‐Based GE Analysis” protocol. For microarray hybridization, 825 ng of Alexa Fluor 555 labeled aminoallyl‐aRNA and 825 ng of Alexa Fluor 647 labeled aminoallyl‐aRNA were mixed and allowed to co‐hybridize on the Agilent 60‐mer oligo array (Human Gene Expression V.2, 4X44K) for 17 h at 65°C at 10 rpm in a hybridization oven. Slides were washed with Agilent GE Wash Buffers following Agilent's protocol.
Slides were immediately scanned with the Agilent dual laser scanner with SureScan High Resolution Technology. Tiff images were analyzed using Agilent feature extraction software version 10.7.1.1 and protocol GE2_107_Sep09 to obtain fluorescence intensities for each spot on the array. Data were normalized using the linear and lowess method. Normalized data were imported into Genespring for analysis. Statistical significance of differentially expressed genes was determined using four replicate measurements for each probe (gene). A t‐test against zero was performed with normalized log ratios reflecting the change in gene expression (44/NS) The Benjamini and Hochberg multiple test correction was used to determine the false discovery rate.
4.7. Chromatin immunoprecipitation
For ChIP‐Seq analysis of ERα binding sites was performed following the procedures originally developed by Carroll, Brown and co‐workers (Carroll et al., 2005, 2006) and personal communications). Cells were cultured in 150 mm plates to ∼90% confluence (approximately 1 × 108 cells). Cells were treated with 2 mM disuccinyl‐glutamate in PBS for 30 min at room temperature and then treated with 1% fresh formaldehyde for 2.5 min before quenching with 125 mM glycine for 5 min. Cells were washed with cold PBS, and chromatin was prepared using the Covaris High‐Cell SDS chromatin shearing kit with SDS buffer (Cat. #520076) according to the manufacturer's recommended protocol. Chromatin was then sheared with a Covaris S220 sonicator using standard settings for 8 min. Chromatin preparation was optimized for SUM‐44 cells. ChIP was performed using the Magna ChIP A/G ChIP Kit (17‐10085) with an antibody cocktail containing equal concentrations of the ERα antibodies from Thermo Scientific and Santa Cruz. Sequencing libraries from ERα ChIP DNA were prepared with Rubicon ThruPLEX library preparation kits and sequenced as 35 bp single‐end reads on an Illumina Hi‐Seq 2500. Libraries for three biological replicates were prepared and sequenced for each sample type. Read data for each sample was aligned with Bowtie2, and peaks were called with both the MACS version 2 and HOMER FindPeaks peak callers. To generate consensus peak sets for downstream analysis, the called peak sets from each peak caller for each sample were input into DiffBind, which created consensus peak sets consisting of peaks present in at least two replicates for each sample type. These consensus peak sets were used for downstream analysis, including differential occupancy and differential binding.
4.8. Western blotting
Cells were plated and grown to 90% confluency. Where indicated, the cells were treated with 500 nM gefitinib for the indicated times. Cells were then lysed in buffer containing 20 mM Tris–HCl (pH 8.0), 137 mM NaCl, 1% NP40, 10% glycerol, 1 mM Na3VO4, and 1x Protease Inhibitor cocktail (Calbiochem, 539131). Protein concentrations were measured by Bradford assay (Bio‐Rad). Laemmli sample buffer was added to the lysates and the samples were boiled for 5 min before being separated by electrophoresis on SDS‐polyacrylamide gels (Bio‐Rad). After transferring the proteins to polyvinylidene difluoride (PVDF) membranes, blots were probed overnight at 4°C with the indicated antibodies.
4.9. RNA expression profiling
Total RNA was prepared using a Qiagen RNeasy Plus Mini Kit and processed by the MUSC Genomics core for 2 × 101 cycles, paired‐end RNA sequencing on an Illumina HiScanSQ. RNA integrity was verified on an Agilent 2200 TapeStation (Agilent Technologies, Palo Alto, CA). A total of 100–200 ng of total RNA was used to prepare RNA‐Seq libraries using the TruSeq RNA Sample Prep kit following the protocol described by the manufacturer (Illumina, San Diego, CA). Sequencing was performed on an Illumina HiScanSQ. Samples were demultiplexed using CASAVA (Illumina, San Diego, CA). Fastq files were used to map reads to the human genome (hg19, UCSC) utilizing Tophat2 with default settings. A total of 100–200 ng of total RNA was used to prepare RNA‐Seq libraries using the TruSeq RNA Sample Prep kit following the protocol described by the manufacturer (Illumina, San Diego, CA).
4.10. Bioinformatics and statistical analysis
Paired end sequencing was performed on RNA samples using an Illumina HiSeq2500 with each sample sequenced to a depth of ∼10 million reads. Data was subjected to Illumina quality control (QC) procedures (>80% of the data yielded a Phred score of 30), and preprocessing using Trimmomatic, which removed adapter sequences and filtered low quality reads (Bolger et al., 2014). Further data QC was performed using FastQC prior to aligning the reads to the human genome HG19 using Tophat2 (Kim et al., 2013). The resulting SAM files were inputted into the Python package HTSeq and quantitative readouts for each sample obtained in the form of count data. In order to infer differential signal within the data sets with robust statistical power, we utilize DEseq2, a method which tests for differential expression based on a model using negative binomial distribution (Kozak et al., 2013). Transcript count data from DESeq2 analysis was ranked according to q‐value, which is the smallest false discovery rate (FDR) at which a transcript is called significant. Statistical analysis of Pathways and Gene Ontology terms was carried out using this sorted transcript list (Subramanian et al., 2005) which was subjected to Gene Set Enrichment Analysis (GSEA) using the ToppGene Suite (Chen et al., 2009).
Each ChIP‐Seq experiment was repeated three times. Fastq files were aligned to the UCSC hg19 human reference genome assembly using Bowtie2. Aligned reads were converted from SAM to BAM format, sorted and indexed. BED format files were obtained from BAM files. Quality control tools, including FastQC and HOMER makeTagDirectory, were employed to verify read quality and the strength of the ChIP signal. After QC, aligned reads for ChIP and input samples were analyzed using MACS version 2 to call peaks. Peaks were also called with Homer's findPeaks and MACS version 1.4. Called peak sets were utilized for downstream analyses, including motif enrichment analysis via HOMER's findMotifsGenome and gene annotation with HOMER's annotatePeaks. Bedgraph files output by MACS v2 were converted to bigWig format for peak visualization on the UCSC Genome Browser. CEAS and HOMER's annotatePeaks were used to determine which genomic features were enriched by called peaks. Other tools were used for visualization and analysis of ChIP‐Seq peak set data, including DiffBind to measure ERα peak occupancy and differential binding intensity SeqMiner was used to perform unsupervised K‐means clustering of peaks. To generate a consensus peak set for a specific experimental condition, the DiffBind package was used to generate a peak set consisting only of peaks called in two or more replicates. Consensus peak sets were then used for differential occupancy and differential binding analyses using DiffBind. SeqMiner was used to perform unsupervised K‐means clustering of peaks.
Supporting information
The following are the supplementary data related to this article:
Supplementary data
Supplementary data
Supplementary data
Supplementary data
Supplementary Figure 1 A. Immunoblot showing over‐expression of WHSC1L1‐short in CAMA1 cells and increased expression of ERα in CAMA1 cells. B. Transcript levels of a panel of ERα target genes in CAMA1 cells following over‐expression of WHSC1L1‐short.
Supplementary Figure 2 A. Proliferation assay showing the proliferation of SUM‐44 cells in serum‐free Ham's F12 medium with or without phenol red. B. Proliferation assay showing the response of SUM‐44 shWHshort and shLacZ control cells to varying concentrations of 17‐β‐estradiol. Error bars represent 1.96*SEM of three experiments.
Acknowledgments
This work was supported by grant number RO1 CA100724 and by the Cell Evaluation & and Genomics Shared Resource Facilities, Hollings Cancer Center, Medical University of South Carolina (P30 CA138313). We gratefully acknowledge the expert technical assistance of Jennifer Schulte and the technical and scientific advice of Drs. Myles Brown and Tom Westerling.
Supplementary data 1.
1.1.
Supplementary data related to this article can be found at http://dx.doi.org/10.1016/j.molonc.2016.02.003.
Irish Jonathan C., Mills Jamie N., Turner-Ivey Brittany, Wilson Robert C., Guest Stephen T., Rutkovsky Alexandria, Dombkowski Alan, Kappler Christiana S., Hardiman Gary, Ethier Stephen P., (2016), Amplification of WHSC1L1 regulates expression and estrogen‐independent activation of ERα in SUM‐44 breast cancer cells and is associated with ERα over‐expression in breast cancer, Molecular Oncology, 10, doi: 10.1016/j.molonc.2016.02.003.
Contributor Information
Jonathan C. Irish, Email: jirish9@gmail.com
Jamie N. Mills, Email: millsjn@musc.edu
Brittany Turner-Ivey, Email: turnerbp@musc.edu.
Robert C. Wilson, Email: wilsorc@musc.edu
Stephen T. Guest, Email: guests@musc.edu
Alexandria Rutkovsky, Email: rutkovsk@musc.edu.
Alan Dombkowski, Email: domski@wayne.edu.
Christiana S. Kappler, Email: kapplerc@musc.edu
Gary Hardiman, Email: hardiman@musc.edu.
Stephen P. Ethier, Email: ethier@musc.edu
References
- Adelaide, J. , Chaffanet, M. , Imbert, A. , Allione, F. , Geneix, J. , Popovici, C. , van Alewijk, D. , Trapman, J. , Zeillinger, R. , Borresen-Dale, A.L. , Lidereau, R. , Birnbaum, D. , Pebusque, M.J. , 1998. Chromosome region 8p11-p21: refined mapping and molecular alterations in breast cancer. Genes Chromosomes Cancer. 22, 186–199. [DOI] [PubMed] [Google Scholar]
- Angrand, P.O. , Apiou, F. , Stewart, A.F. , Dutrillaux, B. , Losson, R. , Chambon, P. , 2001. NSD3, a new SET domain-containing gene, maps to 8p12 and is amplified in human breast cancer cell lines. Genomics. 74, 79–88. [DOI] [PubMed] [Google Scholar]
- Bernard-Pierrot, I. , Gruel, N. , Stransky, N. , Vincent-Salomon, A. , Reyal, F. , Raynal, V. , Vallot, C. , Pierron, G. , Radvanyi, F. , Delattre, O. , 2008. Characterization of the recurrent 8p11-12 amplicon identifies PPAPDC1B, a phosphatase protein, as a new therapeutic target in breast cancer. Cancer Res. 68, 7165–7175. [DOI] [PubMed] [Google Scholar]
- Bolger, A.M. , Lohse, M. , Usadel, B. , 2014. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics. 30, 2114–2120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown, L.A. , Hoog, J. , Chin, S.-F. , Tao, Y. , Zayed, A.A. , Chin, K. , Teschendorff, A.E. , Quackenbush, J.F. , Marioni, J.C. , Leung, S. , 2008. ESR1 gene amplification in breast cancer: a common phenomenon?. Nat. Genet. 40, 806–807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carroll, J.S. , Liu, X.S. , Brodsky, A.S. , Li, W. , Meyer, C.A. , Szary, A.J. , Eeckhoute, J. , Shao, W. , Hestermann, E.V. , Geistlinger, T.R. , Fox, E.A. , Silver, P.A. , Brown, M. , 2005. Chromosome-wide mapping of estrogen receptor binding reveals long-range regulation requiring the forkhead protein FoxA1. Cell. 122, 33–43. [DOI] [PubMed] [Google Scholar]
- Carroll, J.S. , Meyer, C.A. , Song, J. , Li, W. , Geistlinger, T.R. , Eeckhoute, J. , Brodsky, A.S. , Keeton, E.K. , Fertuck, K.C. , Hall, G.F. , Wang, Q. , Bekiranov, S. , Sementchenko, V. , Fox, E.A. , Silver, P.A. , Gingeras, T.R. , Liu, X.S. , Brown, M. , 2006. Genome-wide analysis of estrogen receptor binding sites. Nat. Genet. 38, 1289–1297. [DOI] [PubMed] [Google Scholar]
- Cerami, E. , Gao, J. , Dogrusoz, U. , Gross, B.E. , Sumer, S.O. , Aksoy, B.A. , Jacobsen, A. , Byrne, C.J. , Heuer, M.L. , Larsson, E. , Antipin, Y. , Reva, B. , Goldberg, A.P. , Sander, C. , Schultz, N. , 2012. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2, 401–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charehbili, A. , Fontein, D.B. , Kroep, J.R. , Liefers, G.J. , Mieog, J.S. , Nortier, J.W. , van de Velde, C.J. , 2014. Neoadjuvant hormonal therapy for endocrine sensitive breast cancer: a systematic review. Cancer Treat. Rev. 40, 86–92. [DOI] [PubMed] [Google Scholar]
- Chen, J. , Bardes, E.E. , Aronow, B.J. , Jegga, A.G. , 2009. ToppGene Suite for gene list enrichment analysis and candidate gene prioritization. Nucleic Acids Res. 37, W305–W311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen, Y. , McGee, J. , Chen, X. , Doman, T.N. , Gong, X. , Zhang, Y. , Hamm, N. , Ma, X. , Higgs, R.E. , Bhagwat, S.V. , Buchanan, S. , Peng, S.B. , Staschke, K.A. , Yadav, V. , Yue, Y. , Kouros-Mehr, H. , 2014. Identification of druggable cancer driver genes amplified across TCGA datasets. PloS One. 9, e98293 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciriello, G. , Miller, M.L. , Aksoy, B.A. , Senbabaoglu, Y. , Schultz, N. , Sander, C. , 2013. Emerging landscape of oncogenic signatures across human cancers. Nat. Genet. 45, 1127–1133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dutt, A. , Ramos, A.H. , Hammerman, P.S. , Mermel, C. , Cho, J. , Sharifnia, T. , Chande, A. , Tanaka, K.E. , Stransky, N. , Greulich, H. , Gray, N.S. , Meyerson, M. , 2011. Inhibitor-sensitive FGFR1 amplification in human non-small cell lung cancer. PloS One. 6, e20351 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ethier, S.P. , Mahacek, M.L. , Gullick, W.J. , Frank, T.J. , Weber, B.L. , 1993. Differential isolation of normal luminal mammary epithelial cells and breast cancer cells from primary and metastatic sites using selective media. Cancer Res. 53, 627–635. [PubMed] [Google Scholar]
- Feng, Q. , Zhang, Z. , Shea, M.J. , Creighton, C.J. , Coarfa, C. , Hilsenbeck, S.G. , Lanz, R. , He, B. , Wang, L. , Fu, X. , Nardone, A. , Song, Y. , Bradner, J. , Mitsiades, N. , Mitsiades, C.S. , Osborne, C.K. , Schiff, R. , O'Malley, B.W. , 2014. An epigenomic approach to therapy for tamoxifen-resistant breast cancer. Cell Res. 24, 809–819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuqua, S.A.W. , Chamness, G.C. , Mcguire, W.L. , 1993. Estrogen receptor mutations in breast cancer. J. Cell Biochem. 51, 135–139. [DOI] [PubMed] [Google Scholar]
- Fuqua, S.A.W. , Fitzgerald, S.D. , Chamness, G.C. , Tandon, A.K. , McDonnell, D.P. , Nawaz, Z. , O'Malley, B.W. , Mcguire, W.L. , 1991. Variant human breast tumor estrogen receptor with constitutive transcriptional activity. Cancer Res. 51, 105–109. [PubMed] [Google Scholar]
- Gao, J. , Aksoy, B.A. , Dogrusoz, U. , Dresdner, G. , Gross, B. , Sumer, S.O. , Sun, Y. , Jacobsen, A. , Sinha, R. , Larsson, E. , Cerami, E. , Sander, C. , Schultz, N. , 2013. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci. Signaling. 6, l1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gelsi-Boyer, V. , Orsetti, B. , Cervera, N. , Finetti, P. , Sircoulomb, F. , Rouge, C. , Lasorsa, L. , Letessier, A. , Ginestier, C. , Monville, F. , Esteyries, S. , Adelaide, J. , Esterni, B. , Henry, C. , Ethier, S.P. , Bibeau, F. , Mozziconacci, M.J. , Charafe-Jauffret, E. , Jacquemier, J. , Bertucci, F. , Birnbaum, D. , Theillet, C. , Chaffanet, M. , 2005. Comprehensive profiling of 8p11-12 amplification in breast cancer. Mol. Cancer Res. 3, 655–667. [DOI] [PubMed] [Google Scholar]
- He, C. , Li, F. , Zhang, J. , Wu, J. , Shi, Y. , 2013. The methyltransferase NSD3 has chromatin-binding motifs, PHD5-C5HCH, that are distinct from other NSD (nuclear receptor SET domain) family members in their histone H3 recognition. J. Biol. Chem. 288, 4692–4703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeselsohn, R. , Yelensky, R. , Buchwalter, G. , Frampton, G. , Meric-Bernstam, F. , Gonzalez-Angulo, A.M. , Ferrer-Lozano, J. , Perez-Fidalgo, J.A. , Cristofanilli, M. , Gomez, H. , Arteaga, C.L. , Giltnane, J. , Balko, J.M. , Cronin, M.T. , Jarosz, M. , Sun, J. , Hawryluk, M. , Lipson, D. , Otto, G. , Ross, J.S. , Dvir, A. , Soussan-Gutman, L. , Wolf, I. , Rubinek, T. , Gilmore, L. , Schnitt, S. , Come, S.E. , Pusztai, L. , Stephens, P. , Brown, M. , Miller, V.A. , 2014. Emergence of constitutively active estrogen receptor-alpha mutations in pretreated advanced estrogen receptor-positive breast cancer. Clin. Cancer Res.: Official J. Am. Assoc. Cancer Res. 20, 1757–1767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim, D. , Pertea, G. , Trapnell, C. , Pimentel, H. , Kelley, R. , Salzberg, S.L. , 2013. TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol. 14, R36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kozak, I. , Sasik, R. , Freeman, W.R. , Sprague, L.J. , Gomez, M.L. , Cheng, L. , El-Emam, S. , Mojana, F. , Bartsch, D.U. , Bosten, J. , Ayyagari, R. , Hardiman, G. , 2013. A degenerative retinal process in HIV-associated non-infectious retinopathy. PloS One. 8, e74712 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwek, S.S. , Roy, R. , Zhou, H. , Climent, J. , Martinez-Climent, J.A. , Fridlyand, J. , Albertson, D.G. , 2009. Co-amplified genes at 8p12 and 11q13 in breast tumors cooperate with two major pathways in oncogenesis. Oncogene. 28, 1892–1903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai, A. , Kahraman, M. , Govek, S. , Nagasawa, J. , Bonnefous, C. , Julien, J. , Douglas, K. , Sensintaffar, J. , Lu, N. , Lee, K.J. , Aparicio, A. , Kaufman, J. , Qian, J. , Shao, G. , Prudente, R. , Moon, M.J. , Joseph, J.D. , Darimont, B. , Brigham, D. , Grillot, K. , Heyman, R. , Rix, P.J. , Hager, J.H. , Smith, N.D. , 2015. Identification of GDC-0810 (ARN-810), an orally bioavailable Selective Estrogen Receptor Degrader (SERD) that demonstrates robust activity in tamoxifen-resistant breast cancer xenografts. J. Med. Chem. 58, 4888–4904. [DOI] [PubMed] [Google Scholar]
- Nielsen, K.V. , Ejlertsen, B. , Müller, S. , Møller, S. , Rasmussen, B.B. , Balslev, E. , Lænkholm, A.-V. , Christiansen, P. , Mouridsen, H.T. , 2011. Amplification of ESR1 may predict resistance to adjuvant tamoxifen in postmenopausal patients with hormone receptor positive breast cancer. Breast Cancer Res. Treat. 127, 345–355. [DOI] [PubMed] [Google Scholar]
- Nikolsky, Y. , Sviridov, E. , Yao, J. , Dosymbekov, D. , Ustyansky, V. , Kaznacheev, V. , Dezso, Z. , Mulvey, L. , Macconaill, L.E. , Winckler, W. , Serebryiskaya, T. , Nikolskaya, T. , Polyak, K. , 2008. Genome-wide functional synergy between amplified and mutated genes in human breast cancer. Cancer Res. 68, 9532–9540. [DOI] [PubMed] [Google Scholar]
- Qi, J. , Huo, L. , Zhu, Y.T. , Zhu, Y.J. , 2014. Absent, small or homeotic 2 like protein (ASH2L) enhances the transcription of estrogen receptor alpha gene through GATA binding protein 3 (Gata3). J. Biol. Chem. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ray, M.E. , Yang, Z.Q. , Albertson, D. , Kleer, C.G. , Washburn, J.G. , Macoska, J.A. , Ethier, S.P. , 2004. Genomic and expression analysis of the 8p11–12 amplicon in human breast Cancer cell lines. Cancer Res. 64, 40–47. [DOI] [PubMed] [Google Scholar]
- Ross-Innes, C.S. , Stark, R. , Teschendorff, A.E. , Holmes, K.A. , Ali, H.R. , Dunning, M.J. , Brown, G.D. , Gojis, O. , Ellis, I.O. , Green, A.R. , Ali, S. , Chin, S.F. , Palmieri, C. , Caldas, C. , Carroll, J.S. , 2012. Differential oestrogen receptor binding is associated with clinical outcome in breast cancer. Nature. 481, 389–393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen, C. , Ipsaro, J.J. , Shi, J. , Milazzo, J.P. , Wang, E. , Roe, J.S. , Suzuki, Y. , Pappin, D.J. , Joshua-Tor, L. , Vakoc, C.R. , 2015. NSD3-Short is an adaptor protein that couples BRD4 to the CHD8 chromatin remodeler. Mol. Cell. 60, 847–859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sikora, M.J. , Cooper, K.L. , Bahreini, A. , Luthra, S. , Wang, G. , Chandran, U.R. , Davidson, N.E. , Dabbs, D.J. , Welm, A.L. , Oesterreich, S. , 2014. Invasive lobular carcinoma cell lines are characterized by unique estrogen-mediated gene expression patterns and altered tamoxifen response. Cancer Res. 74, 1463–1474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stec, I. , v.O., G.J. , den Dunnen, J.T. , 2001. WHSC1L1, on human chromosome 8p11.2, closely resembles WHSC1 and maps to a duplicated region shared with 4p16.3. Genomics. 76, 5–8. [DOI] [PubMed] [Google Scholar]
- Streicher, K.L. , Yang, Z.Q. , Draghici, S. , Ethier, S.P. , 2007. Transforming function of the LSM1 oncogene in human breast cancers with the 8p11-12 amplicon. Oncogene. 26, 2104–2114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subramanian, A. , Tamayo, P. , Mootha, V.K. , Mukherjee, S. , Ebert, B.L. , Gillette, M.A. , Paulovich, A. , Pomeroy, S.L. , Golub, T.R. , Lander, E.S. , Mesirov, J.P. , 2005. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. U.S.A. 102, 15545–15550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tonon, G. , Wong, K.K. , Maulik, G. , Brennan, C. , Feng, B. , Zhang, Y. , Khatry, D.B. , Protopopov, A. , You, M.J. , Aguirre, A.J. , Martin, E.S. , Yang, Z. , Ji, H. , Chin, L. , Depinho, R.A. , 2005. High-resolution genomic profiles of human lung cancer. Proc. Natl. Acad. Sci. U.S.A. 102, 9625–9630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turner-Ivey, B. , Guest, S.T. , Irish, J.C. , Kappler, C.S. , Garrett-Mayer, E. , Wilson, R.C. , Ethier, S.P. , 2014. KAT6A, a chromatin modifier from the 8p11-p12 amplicon is a candidate oncogene in luminal breast cancer. Neoplasia. 16, 644–655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, G. , Liu, G. , Wang, X. , Sethi, S. , Ali-Fehmi, R. , Abrams, J. , Zheng, Z. , Zhang, K. , Ethier, S. , Yang, Z.Q. , 2012. ERLIN2 promotes breast cancer cell survival by modulating endoplasmic reticulum stress pathways. BMC Cancer. 12, 225 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang, Z.Q. , Albertson, D. , Ethier, S.P. , 2004. Genomic organization of the 8p11-p12 amplicon in three breast cancer cell lines. Cancer Genet. Cytogenet. 155, 57–62. [DOI] [PubMed] [Google Scholar]
- Yang, Z.Q. , Liu, G. , Bollig-Fischer, A. , Giroux, C.N. , Ethier, S.P. , 2010. Transforming properties of 8p11-12 amplified genes in human breast cancer. Cancer Res. 70, 8487–8497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang, Z.Q. , Streicher, K.L. , Ray, M.E. , Abrams, J. , Ethier, S.P. , 2006. Multiple interacting oncogenes on the 8p11-p12 amplicon in human breast cancer. Cancer Res. 66, 11632–11643. [DOI] [PubMed] [Google Scholar]
- Zack, T.I. , Schumacher, S.E. , Carter, S.L. , Cherniack, A.D. , Saksena, G. , Tabak, B. , Lawrence, M.S. , Zhang, C.Z. , Wala, J. , Mermel, C.H. , Sougnez, C. , Gabriel, S.B. , Hernandez, B. , Shen, H. , Laird, P.W. , Getz, G. , Meyerson, M. , Beroukhim, R. , 2013. Pan-cancer patterns of somatic copy number alteration. Nat. Genet. 45, 1134–1140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, J. , Liu, X. , Datta, A. , Govindarajan, K. , Tam, W.L. , Han, J. , George, J. , Wong, C. , Ramnarayanan, K. , Phua, T.Y. , Leong, W.Y. , Chan, Y.S. , Palanisamy, N. , Liu, E.T. , Karuturi, K.M. , Lim, B. , Miller, L.D. , 2009. RCP is a human breast cancer-promoting gene with Ras-activating function. J. Clin. Invest. 119, 2171–2183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou, Z. , Thomsen, R. , Kahns, S. , Nielsen, A.L. , 2010. The NSD3L histone methyltransferase regulates cell cycle and cell invasion in breast cancer cells. Biochem. Biophys. Res. Commun. 398, 565–570. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
The following are the supplementary data related to this article:
Supplementary data
Supplementary data
Supplementary data
Supplementary data
Supplementary Figure 1 A. Immunoblot showing over‐expression of WHSC1L1‐short in CAMA1 cells and increased expression of ERα in CAMA1 cells. B. Transcript levels of a panel of ERα target genes in CAMA1 cells following over‐expression of WHSC1L1‐short.
Supplementary Figure 2 A. Proliferation assay showing the proliferation of SUM‐44 cells in serum‐free Ham's F12 medium with or without phenol red. B. Proliferation assay showing the response of SUM‐44 shWHshort and shLacZ control cells to varying concentrations of 17‐β‐estradiol. Error bars represent 1.96*SEM of three experiments.