

INTRODUCTION
Immunoproliferative small intestinal disease (IPSID), a term adopted by WHO in 1978, is a variant of extranodal marginal zone lymphoma of mucosa associated lymphoid tissue (MALT) in which defective heavy chains are secreted. Subsequently most cases are characterized by a loss of ability to synthesize light chains. It is largely common in the Mediterranean region and relatively rare in South-East Asia. Common sites of involvement are small intestine (duodenum and jejunum) and mesenteric lymph nodes.1 The serum contains free a heavy chains without associated the light chains and hence is also known as a heavy chain disease (HCD).
We present a case of IPSID with classical clinical presentation and laboratory findings.
CASE REPORT
A 43-year-old male patient presented with complaints of weight loss, small bowel type diarrhoea, features of malabsorption, and pedal oedema. Initial evaluation revealed normal renal and liver function tests, normal haemogram and a negative Mantoux test. Hypoproteinemia was consistently noted. All tumour markers were within the normal range. Serum antibodies to tissue transglutaminase (TTG) were not raised and HIV, HBsAg and HCV were negative. CT scan abdomen showed diffuse mural thickening of the small intestine more in the jejunum together with mesenteric and retroperitoneal lymphadenopathy. PET scan showed increased FDG uptake along the bowel loops.
Jejunal biopsy revealed severe villous blunting and an intense lymphoplasmacytic infiltrate filling and expanding the lamina propria with well-preserved surface epithelium (Figure 1). Broad villi and shortened crypts were prominent and lympho-epithelial lesions were also noted. Immunohistochemistry (IHC) was done and showed CD20 to be diffusely and strongly positive in tumour cells (Figure 2). CD5, CD10 and Cyclin D1 were negative (ruling out mantle cell and follicular lymphoma). No kappa or lambda light chain restriction was present. IgA was focally expressed (Figure 3) and positive staining for CD138 was noted in the plasma cells (Figure 4).
Figure 1.
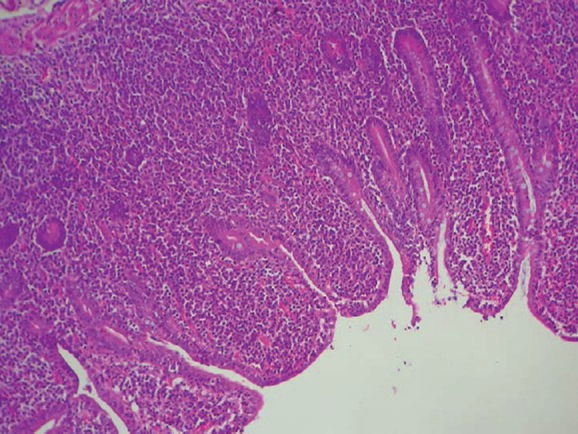
Jejunal biopsy (100×): severe villous blunting and dense lymphoplasmacytic infiltrate in lamina.
Figure 2.
Immunohistochemistry: strong CD20 staining of tumour cells.
Figure 3.
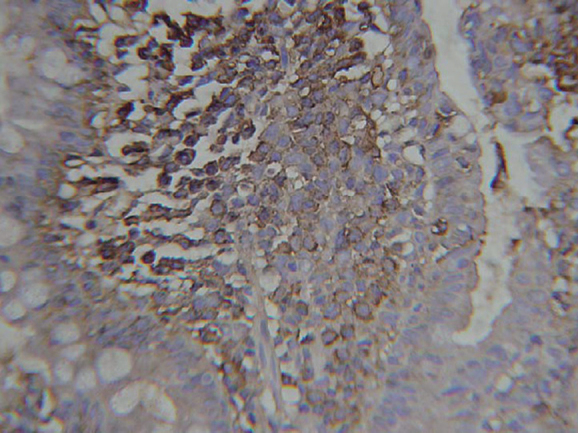
Immunohistochemistry: IgA positivity in tumour cells.
Figure 4.
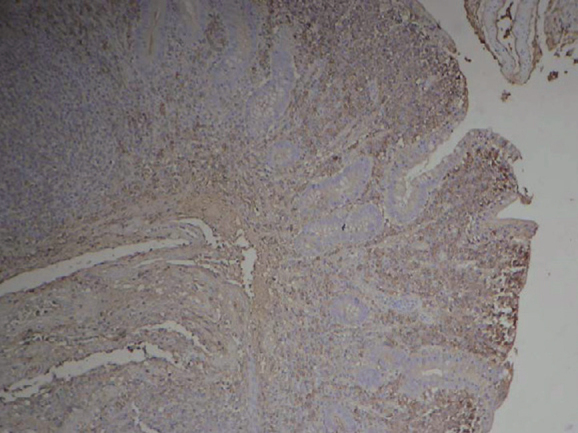
Immunohistochemistry: CD138 staining plasma cells.
Serum protein electrophoresis (SPE) with immunofixation was subsequently carried out, which showed a distinct band in the IgA region but without corresponding light chains (Figure 5).
Figure 5.
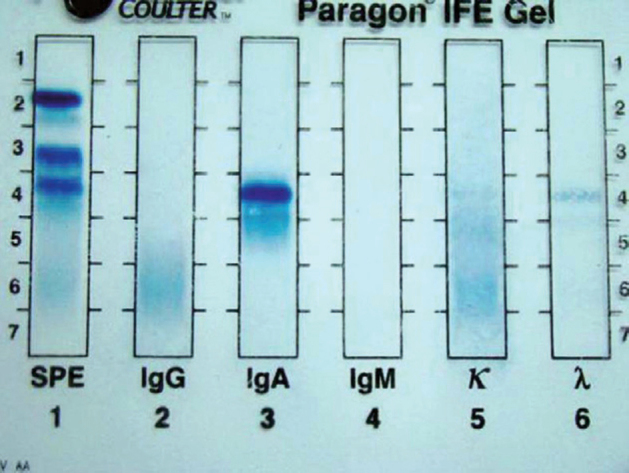
Serum electrophoresis with immunofixation: A distinct band identified in the IgA region anodal to point of origin. No corresponding light chain identified.
The M-component typed as IgA (denoted by the band seen in the IgA lane), but without a corresponding light chain (only faint polyclonal pattern was seen in kappa and lambda lanes). Histomorphology, IHC profile, and SPE with immunofixation were consistent with a diagnosis of IPSID. Patient was treated with cap doxycycline and showed significant improvement. He is on regular follow-up and subsequent jejunal biopsies have not shown any evidence of transformation to diffuse large B cell lymphoma.
DISCUSSION
IPSID also known as Mediterranean lymphoma is now considered to be a distinct subtype of extranodal marginal zone B-Cell lymphoma.1 IPSID tends to be associated with people of the lower socio-economic status. Patients present with abdominal pain, malabsorption, diarrhoea, and weight loss of months to years duration.2, 3, 4 Obstruction, bleeding, and perforation are uncommon in contrast to patients with the other types of small intestinal lymphomas.2 At laparotomy, patients may have one or more recognizable intestinal masses, diffuse mural thickening, and/or luminal dilatation or they may have normal appearing bowel. Abnormalities involve the proximal small intestine, the entire small intestine, or rarely just the ileum or the small intestine in combination with either stomach or colon. Mesenteric lymph nodes are often enlarged.3, 4 Our case also presented with diarrhoea and weight loss, and CT scan abdomen showed extensive small bowel thickening, more in the jejunum. Mesenteric lymphadenopathy was also present.
Molecular genetic analysis has shown clonal rearrangement of heavy and light chains, even in early cases responsive to antibiotic therapy. In the past, early phase of IPSID was thought to be inflammatory, but evidence of clonality indicates the neoplastic nature of this process, despite response to antibiotics.5, 6, 7 Characteristic laboratory abnormality noted in IPSID cases is that of serum containing free heavy chains without associated light chains. Internal deletions in the alpha heavy chain gene are present that result in the expression of a defective heavy chain protein that cannot bind light chains to form a complete immunoglobulin molecule. The deletion involves the VH region (variable region of immunoglobulin heavy chain gene) and CHl region (first segment, constant region of immunoglobulin heavy chain gene).4, 8 Secreted heavy chains are more likely to be found in early stages of IPSID.
It has been suggested that IPSID occurs in patients with recurrent or persistent intestinal infection leading to chronic antigenic stimulation of IgA-secreting lymphoid tissue in this site with a resultant clonal population that acquires mutations leading to the production of a heavy chain with the internal deletion described previously. Subsequently most cases are characterized by a loss of ability to synthesize light chains.4 Serum protein electrophoresis with immunofixation was carried out for our case and the M component was typed as IgA without any corresponding light chains.
Immunohistochemically cells express monoclonal cytoplasmic alpha chains without light chains. Cells express CD20 and are CD5 and CD10 negative. Plasma cells are CD20 negative and CD138 positive.6 Strong and diffuse CD20 expression by tumour cells and plasma cell CD138 positivity was seen in our case. No light chain restriction was seen on IHC.
Cytogenetic abnormalities have been reported in rare single cases. Translocation t(11;18) associated with gastric and pulmonary MALT lymphomas has not been described.9 One reported case had t(9;14) (p11;q32).9
Both IPSID and celiac disease are characterized by lymphoplasmacytic intestinal mucosal infiltrate and villous atrophy. Histological features in favour of celiac disease include total villous atrophy, hyperplastic elongated crypts, increased intraepithelial lymphocytes, and surface epithelial damage.4 Serum protein electrophoresis (SPE) with immunofixation and serum antibodies to tissue transglutaminase (TTG) are important diagnostic tests for differentiation of the two entities. Patient demographic data and response to gluten-free diet also provide a clue. High-grade lymphoma may be found in association with either disease, but lymphoma that complicates celiac disease is a T-cell lymphoma prone to cause perforations.
Distinguishing nonspecific chronic inflammation from early-stage IPSID may be difficult, particularly on a small biopsy. In favour of IPSID is a dense, predominantly plasmacytic infiltrate that distorts the mucosal architecture in a patient with clinical features compatible with IPSID. Immunohistochemical stains that show plasma cells expressing only a heavy chains confirm the diagnosis.
IPSID is a rare disorder in the Indian subcontinent. Early phase of IPSID can completely remit with antibiotic therapy. Patient needs follow-up in view of the fact that transformation to diffuse large B cell lymphoma (DLBCL) can occur.
REFERENCES
- 1.Harris NL, Isaacson PG, Grogan TM, Jaffe ES. In: Who Classification of Tumours of Haematopoietic and Lymphoid Tissue 4th ed. IARC 2008:198-199.
- 2.Salem P, El-Hashimi L, Anassie E. Primary Small Intestinal lymphoma in adults. A comparative study of IPSID versus non-IPSID in the Middle East. Cancer. 1987;59:1670–1676. doi: 10.1002/1097-0142(19870501)59:9<1670::aid-cncr2820590925>3.0.co;2-d. [DOI] [PubMed] [Google Scholar]
- 3.Tabbane F, Mourali N, Commoun M. Results of laparotomy in immunoproliferative small intestine disease. Cancer. 1988;61:1699–1706. doi: 10.1002/1097-0142(19880415)61:8<1699::aid-cncr2820610831>3.0.co;2-e. [DOI] [PubMed] [Google Scholar]
- 4.Fine K, Stone M. a-heavy chain disease, Mediterranean lymphoma and immunoproliferative disease. Am J Gastroenterol. 1999;94:1139–1152. doi: 10.1111/j.1572-0241.1999.01057.x. [DOI] [PubMed] [Google Scholar]
- 5.Smoth W, Price S, Isaacson PG. Immunoglobulin gene rearrangement in immunoproliferative small intestinal disease (IPSID) J Clin Pathol. 1987;40:1291–1297. doi: 10.1136/jcp.40.11.1291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Isaacson PG, Dogan A, Price S. Immunoproliferative small intestinal disease. An immunohistiochemical study. Am J Surg Pathol. 1989;13:1023–1033. doi: 10.1097/00000478-198912000-00004. [DOI] [PubMed] [Google Scholar]
- 7.Isaacson PG. Gastrointestinal lymphoma. Hum Pathol. 1994;25:1020–1029. doi: 10.1016/0046-8177(94)90060-4. [DOI] [PubMed] [Google Scholar]
- 8.Fermand JP, Bronet JC. Heavy chain disease. Hematol Oncol Clin North Am. 1999;13:1281–1294. doi: 10.1016/s0889-8588(05)70127-1. [DOI] [PubMed] [Google Scholar]
- 9.Pellet P, Berger R, Beinheim A, Bronet JC, Tsapis A. Molecular analysis of t(9;14) (p11:q32) translocation occurring in a case of human alpha heavy chain disease. Oncogene. 1989;4:653–657. [PubMed] [Google Scholar]