

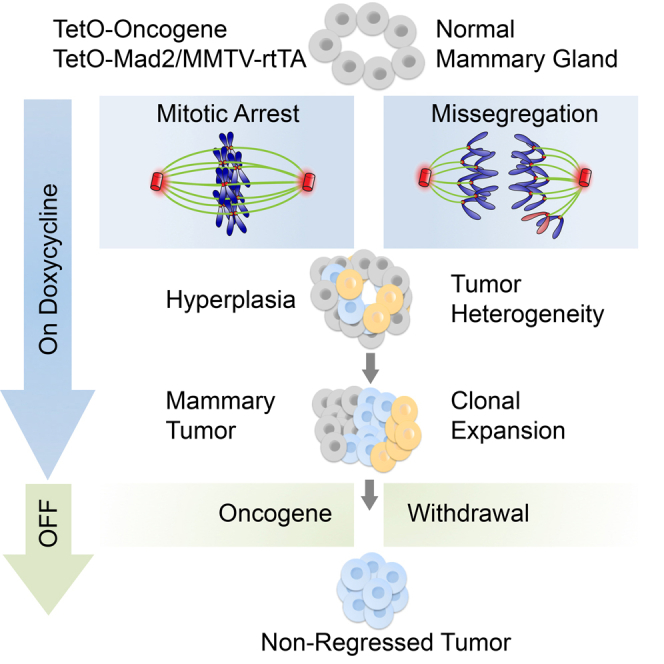
Summary
Chromosome instability (CIN) is associated with poor survival and therapeutic outcome in a number of malignancies. Despite this correlation, CIN can also lead to growth disadvantages. Here, we show that simultaneous overexpression of the mitotic checkpoint protein Mad2 with KrasG12D or Her2 in mammary glands of adult mice results in mitotic checkpoint overactivation and a delay in tumor onset. Time-lapse imaging of organotypic cultures and pathologic analysis prior to tumor establishment reveals error-prone mitosis, mitotic arrest, and cell death. Nonetheless, Mad2 expression persists and increases karyotype complexity in Kras tumors. Faced with the selective pressure of oncogene withdrawal, Mad2-positive tumors have a higher frequency of developing persistent subclones that avoid remission and continue to grow.
Graphical Abstract
Highlights
-
•
Mad2 overexpression leads to mitotic arrest, cell delamination, and cell death
-
•
High Mad2 levels delay oncogene-induced mammary tumorigenesis
-
•
Mad2 overexpression increases chromosome instability prior to and during tumor growth
-
•
Elevated Mad2 levels facilitate the development of oncogene-independent subclones
Rowald et al. report that Mad2 overexpression in the mammary gland results in mitotic arrest, chromosome missegregation, and cell depletion, causing a delay in KrasG12D-driven mammary tumorigenesis. After oncogene silencing, however, Mad2-positive tumors show a frequent occurrence of persistent tumor subclones that do not regress.
Introduction
Genomic instability is one of the main enabling features of cancer, setting the path for subsequent events of uncontrolled growth, invasion, metastasis, and drug resistance (Hanahan and Weinberg, 2011). One of the most studied mechanisms of generating genomic variability is aberrant distribution of chromosomes during cell division, known as chromosomal instability (CIN) (Boveri, 1912). Large-scale analyses of human tumors predict that 86% of solid and 72% of hematopoietic malignancies are aneuploid (Zasadil et al., 2013). CIN gene signatures have become an important prognostic tool to evaluate patient survival and therapeutic outcome (Carter et al., 2006, Habermann et al., 2009). However, deregulation of CIN genes in vitro causes a decrease in cellular fitness through p53-mediated cell death and metabolic stress because of protein imbalance (Li et al., 2010, Thompson and Compton, 2010, Torres et al., 2007). Studies in vivo show that CIN genes can have either no effect or lead to an increase in spontaneous or carcinogen-induced tumorigenesis (Schvartzman et al., 2010). In combination with loss of known tumor suppressor genes or oncogene activation, even suppressive effects on tumor induction have been reported (Janssen and Medema, 2013, Zasadil et al., 2013). To explain these ambiguities, Silk et al. (2013) proposed a model in which the level of CIN might tip the balance between tumor-promoting and -suppressive outcomes. Despite this, it is still unclear when during tumor development aneuploidy exerts its modulating effects on tumor growth.
Most researchers agree that oncogenes are the main drivers of tumorigenesis, whereas CIN contributes to genetic variation and plasticity (Chen et al., 2012, Tamborero et al., 2013). To understand how genetic instability can influence classical oncogenesis, we focused on the effect of Mad2-induced CIN during oncogene-driven tumorigenesis. Mad2 is a downstream target of two major tumor suppressors: RB and p53 (Schvartzman et al., 2011). In addition, Mad2 overexpression in a transgenic model of KrasG12D-driven lung carcinogenesis facilitated both tumor onset and relapse following oncogene silencing (Sotillo et al., 2010). Moreover, its elevated level in various cancer types (Rhodes et al., 2004) makes it an attractive target for cancer studies. Considering the detrimental effect of aneuploidy on cellular homeostasis (Torres et al., 2007) and the continued controversy with respect to both its tumor-promoting and tumor-suppressing function in vivo, it is unclear whether this synergy is a general principle of cancer.
Breast cancer is the most common malignancy in women worldwide and can be driven by a wide spectrum of oncogenes (Sanchez-Garcia et al., 2014, Stephens et al., 2012). Although Kras mutations are prevalent in 30% of all human cancers, Her2 is deregulated in 15% of human mammary tumors (Cerami et al., 2012, Gao et al., 2013). Similar to lung cancer, human breast tumors are highly aneuploid (Zasadil et al., 2013); however, animal models are less prone to CIN-induced mammary tumorigenesis (Duijf and Benezra, 2013).
We combined KrasG12D or Her2 conditional mouse models with Mad2 overexpression in the adult mammary gland to mirror the timing of tumor onset in breast cancer patients. This resulted in a strong delay in tumor formation compared with KrasG12D or Her2 expression alone. Pathologic analysis of early events after transgene induction revealed increased cell turnover caused by an initial depletion of Mad2-expressing cells and an accumulation of mitotic errors in the surviving population. Three phenotypic subgroups of the Kras/Mad2-expressing cohort were identified, all of which resulted in the generation of Mad2-overexpressing, highly aneuploid tumors. The level of aneuploidy seen in Her2 tumors was not exceeded by additional Mad2 expression, indicating that tumors self-regulate the ultimate level of CIN (Montagna et al., 2002). In both oncogenic models, Mad2-induced CIN failed to have an effect on tumor progression but led to the generation of abnormal cells that survived the strong selective pressure of oncogene withdrawal.
Results
Mad2 Overexpression Delays Tumor Onset in Kras-Driven Breast Cancer
To better understand the effect of CIN on tumor initiation we generated a mouse mammary tumor virus (MMTV)-driven, doxycycline-inducible model to overexpress hemagglutinin (HA)-tagged Mad2 (Sotillo et al., 2007) and KrasG12D (Fisher et al., 2001) in mammary glands of adult virgin mice (Gunther et al., 2002). Mice were placed on a doxycycline-enriched diet and followed weekly for tumor development. Monitored mice were divided into four cohorts depending on the presence of KrasG12D (K), Mad2 (M), both KrasG12D and Mad2 (KM), and control mice lacking transgene expression (CTRL).
CTRL and M animals developed no tumors during the aging period of 18 months, suggesting that elevated levels of Mad2 are not sufficient to induce breast cancer in this model (Figure 1A). As described previously, expression of KrasG12D in the mammary gland results in the development of breast tumors (Podsypanina et al., 2008; Figure 1A). Simultaneous overexpression of KrasG12D and Mad2 led to a significant delay in mammary tumor onset compared with KrasG12D alone. Although K mice showed palpable nodules after a median latency of 147 days (n = 107), KM mice survived 221 days (n = 75) without tumors (Figure 1A). In addition, KM mice developed a lower number of tumors per animal compared with K mice (Figure 1B). These findings show that elevated Mad2 levels are detrimental to oncogene-driven mammary tumor initiation.
Figure 1.

Mad2 Overexpression Delays Kras-Driven Tumor Initiation and Leads to Phenotypic Variance
(A) Tumor-free survival after doxycycline administration. (control, n = 33; Mad2, n = 33; KrasG12D, n = 107; KrasG12D/Mad2, n = 75), Mantel-Cox test, p < 0.0001.
(B) Tumors per animal. Mann-Whitney test, p = 0.0062.
(C) Mammary glands after 4, 30, and 100 days on doxycycline stained against HA-Mad2. Scale bar, 50 μm.
(D) Quantification of HA-positive cells per acinus after 4 and 100 days on doxycycline (M, n = 7; KM-A, n = 7; KM-B, n = 6; KM-C, n = 5).
(E) Tumor onset in K and KM subgroups (K, n = 95; KM-A, n = 14; KM-B, n = 10; KM-C, n = 7). Kruskal-Wallis test, p < 0.0001; Dunn’s multiple comparisons test. ns, not significant.
∗∗p < 0.01, ∗∗∗∗p < 0.0001. See also Figure S1.
Sequential Pathological Analysis Reveals Phenotypic Subtypes in KrasG12D/Mad2 Animals
To address the consequences of high Mad2 levels on early events during tumorigenesis, we evaluated the histological changes in mammary glands stained with an HA-specific antibody (to detect exogenous HA-tagged Mad2; Figure 1C) and Kras (Figure S1A) prior to formation of palpable tumors. Serial surgeries on different mammary glands of the same animal were performed after 4, 30, and 100 days of transgene induction. In this 3-month period, young wild-type animals showed no significant changes in tissue architecture (Figures 1C; Figure S1A) except estrous cycle-dependent fluctuations in proliferation and apoptosis (Fata et al., 2001). To control for these hormonal influences on tissue remodeling, all surgeries were performed at the pro-estrous stage.
Overexpression of KrasG12D in the mammary gland led to gradual neoplastic growth starting with a numerical expansion of acinar density 30 days post-induction. The increase in epithelial cells resulted in the formation of micro-nodules that, by 100 days, had already expanded into palpable tumors (Figure S1A).
Upon Mad2 overexpression, an initial enlargement of ducts and a predominant localization of Mad2-positive mitotic cells inside the acinar lumen were observed. The decreased number of Mad2-expressing cells and the return to normal tissue architecture at 100 days suggested epithelial cell clearance during early days of transgene induction (Figures 1C and 1D; Figures S1A–S1C). These observations are in line with the lack of carcinogenic growth seen in Mad2-overexpressing animals.
Morphological examinations of the KM cohort prompted us to partition it into three phenotypic subgroups. All animals started with a normal distribution of Mad2-expressing cells per acinus (average of 38.8% of HA-positive cells at 4 days of induction) but differed both in the Kras levels and in the expression pattern of Mad2 after 100 days on doxycycline (Figures 1C and 1D; Figures S1B–S1E). Group A continuously expressed very low amounts of Kras compared with Mad2 and, similar to the M cohort, reduced Mad2-expressing cells during the first 100 days on doxycycline. After a strong initial luminal filling of mitotic cells at 4 days, the ductal system regressed to a normal mammary architecture. Groups B and C displayed higher levels of Kras and were prone to acinar hyperplasia at 4 days. After the expansion period, mammary glands underwent a regression phase leading to an almost complete reconstitution of the normal state after 30 days on doxycycline. After 3 months, the mammary glands of Group B showed increased budding and branching from the main ducts, an augmented number of Mad2-expressing cells, and a slight increase in Kras levels. Group C showed no change in the overall percentage of Mad2-expressing cells, whereas total expression levels were reduced and Kras expression substantially augmented (Figures 1C and 1D; Figures S1B–S1E). Phenotypically, group C was reminiscent of the tumorigenic progression observed in the KrasG12D-driven model and had an equally early tumor onset at 165 days after doxycycline administration, whereas group B developed the first palpable tumors at 233 days and group A at 313 days (Figure 1E). Together, these data best fit a model in which high levels of Mad2 have a detrimental effect on mitotic progression, leading to cell depletion. Only in the presence of sufficient amounts of Kras can this phenotype be overcome. During transgene induction, the epithelial cell population can undergo different selective scenarios leading to loss of Mad2-expressing cells in the presence of low Kras levels, tolerance of high Mad2 levels in the presence of high Kras levels, or early selection of cells with lower Mad2 levels.
High Levels of Mad2 Induce Mitotic Arrest and Trigger Epithelial Cell Turnover
To understand the morphogenetic processes caused by Mad2 overexpression during early pre-malignancy, we examined epithelial cell turnover in vivo via quantification of phospho-Histone 3 (pH3) and terminal deoxynucleotidyl transferase-mediated d-UTP nick end labeling (TUNEL). Compared with controls, K mice showed an incremented number of pH3-positive cells with a punctate staining pattern at 4 days, hinting at elevated proliferation (Figures 2A and 2C). In M and KM mice, an increase was also observed, but the staining clearly exhibited morphological features of mitosis. Notably, the majority of pH3-positive cells in the M and KM cohorts were located inside the acinar lumen, suggesting delamination of mitotic cells (Figure 2A). In addition, elevated numbers of TUNEL-positive cells were found in M and KM groups compared with K and CTRL cohorts 4 days after transgene induction (Figures 2B and 2D; Figure S2A) and were clearly associated with Mad2 expression (Figure S2B). Together, these results suggest that high Mad2 levels in mammary epithelial cells in vivo may lead to mitotic arrest followed by apical extrusion and cell death. However, the presence of Mad2-positive cells in all KM subgroups at 100 days indicated that a substantial number of cells must have escaped mitotic block and apoptosis.
Figure 2.

Mad2 Overexpression Increases Epithelial Cell Turnover after 4 Days of Transgene Induction In Vivo
(A) Phospho-histone 3 staining of mammary glands after 4 days on doxycycline. Scale bar, 50 μm.
(B) Double immunofluorescence of TUNEL (red) and HA-Mad2 (green) on mammary glands after 4 days on doxycycline (DAPI, blue). Scale bar, 50 μm.
(C and D) Quantification of pH3 (C; CTRL, n = 6; M, n = 5; K, n = 6; KM, n = 13) and TUNEL-positive cells (D; CTRL, n = 6; M, n = 6; K, n = 9; KM, n = 14) per acinus at 4 days on doxycycline. Points represent single acinar measurements.
(E) Nuclear volume quantification relative to control cells at 100days on doxycycline (CTRL, n = 6; M, n = 5; K, n = 5; KM-A, n = 6; KM-B, n = 5; KM-C, n = 4). Points represent single nuclear measurements; F-test for size variability.
Data were analyzed by Kruskal-Wallis test, p < 0.0001; Dunn’s multiple comparisons test. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, ∗∗∗∗p < 0.0001. See also Figure S2.
One mechanism to escape mitotic arrest is to progress through mitosis without cytokinesis, otherwise known as mitotic slippage. To evaluate this possibility and its effect on chromosome content, we measured the nuclear size of mammary epithelial cells after 4 and 100 days of transgene expression. Numerical aberrations in the karyotype have been reported to correlate directly with nuclear size (Pérez de Castro et al., 2013). Mammary cells in KM animals acquired a higher variance of nuclear size compared with K and CTRL mice. KM group A showed no significant changes in nuclear volume. Group B reached the highest nuclear volume on day 4, together with increased cell death (Figures S2A, S2C, and S2D), returning to a less pronounced phenotype after 100 days of induction (Figure 2E). Together with the development of the first tumor-like nodules, KM group C doubled their nuclear volume at 100 days (Figure 2E). These results suggest that a fraction of cells escape the block imposed by Mad2 overexpression and accumulate errors during chromosome segregation.
Mad2-Induced Mitotic Arrest Leads to Cell Delamination, Spindle Defects, and Increased Cell Death
In vivo analysis of mammary glands following doxycycline administration indicated an increased mitotic index and a possible variance in the genomic content because of Mad2 overexpression. To better understand whether the mitotic arrest induced by high Mad2 was responsible for cell depletion, we used a 3D culture system of primary mammary epithelial cells. 3D culture offers the possibility of recapitulating the growth of mammary acini ex vivo and allows the visualization of epithelial tissues in a microscopically accessible context (Jechlinger et al., 2009).
Both control samples and KrasG12D-expressing cultures showed no evident morphological changes after 36 hr on doxycycline (Figures 3A and 3B; Figure S3A). Mad2 overexpression (in both ColA1-Mad2 and TetO-Mad2 strains; Figures S3B and S3C) led to a dramatic increase in the number of mitotic cells that mostly appeared in the acinar lumen starting 30 hr post-induction. This phenotype was exacerbated in the combination of Mad2 overexpression and mutant Kras, where the acinar structures were filled with mitotic cells and cell fragments (Figures 3A and 3B; Figure S3A). In line with previous findings, the observed phenotype is likely due to mitotic arrest as a consequence of checkpoint overactivation, given that Mad2 overexpression results in an accumulation of Cyclin B (Figures S3D and S3E) and downregulation of Aurora B (Figures S3F and S3G; Kabeche and Compton, 2012, Sotillo et al., 2007). To investigate whether the arrest was followed by apoptosis, 3D cultures were stained for cleaved Caspase 3 after 36 hr on doxycycline. K structures did not exceed the cell turnover seen in control cultures. However, M and KM spheres contained a significantly elevated number of apoptotic cells per acinus (Figures 3A and 3C). Nuclear volume counts after 4 days on doxycycline indicated that a significant number of M and KM cells became polyploid (Figure 3D). Taken together, the 3D culture system recapitulated the phenotypes seen in vivo.
Figure 3.

3D Cultures Recapitulate Increased Proliferation and Apoptosis following Mad2 Induction
(A) Representative immunofluorescence images on fixed mammary spheroid cultures after 36 hr on doxycycline. Scale bars, 25 μm.
(B and C) Quantification of mitotic cells (B; CTRL, n = 7; M, n = 4; K, n = 5; KM, n = 4) and cleaved Caspase 3-positive (C) cells per acinus after 36 hr on doxycycline (CTRL, n = 9; M, n = 6; K, n = 5; KM, n = 4).
(D) Nuclear volume quantification in spheroids relative to control cells after 4 days on doxycycline (CTRL, n = 8; M, n = 4; K, n = 3; KM, n = 4). Points represent single nuclear measurements.
Data were analyzed by Kruskal-Wallis test, p < 0.0001; Dunn’s multiple comparisons test. ColA1-HA-Mad2 animals were used: 2 in M and 2 in KM. ∗∗∗∗p < 0.0001. See also Figure S3.
Taking advantage of time-lapse imaging, we monitored the fate of single cells expressing Histone2B-GFP in acinar spheres after administration of doxycycline for 30 hr. Mitotic cell division in control cultures as well as in Kras-expressing spheroids is oriented perpendicularly to the apical and basal membrane within the epithelial layer and is completed within 1–2 hr (Figures 4A and 4C; Figure S4A; Movies S1 and S2). However, in M and KM cultures, mitosis was initiated inside the epithelial layer, but cells arrested at metaphase for over 8 hr in 100% of the cycling cells in M and 91% in KM (Figures 4A and 4C). This prolonged mitotic block led to stepwise delamination until, in some cases, contact with neighboring cells was completely lost. Afterward, cells were either degraded into apoptotic blebs (85% in M and 79% in KM; Figures 4A, center, and 4D) or completed cell division inside the epithelial rim (15% in M and 12% in KM; Figures 4A, bottom, and 4D).
Figure 4.

Time-Lapse Imaging of Organotypic Cultures Reveals Error-Prone Mitosis because of Elevated Mad2 Levels
(A) Time-lapse micrography of control and KrasG12D/Mad2 spheroids in vitro; t = 0 after 30 hr on doxycycline. The yellow circle indicates mitotic cells (H2B-GFP, green). Scale bar, 25 μm. Top: control cell entering mitosis and completing cell division inside the epithelial rim. Center: a KM cell arrests in mitosis, displays misaligned chromosomes, falls into the lumen, and dies after 11 hr. Bottom: KM cell arrests in mitosis, initiates apical extrusion, but reintegrates into the epithelial rim.
(B) Representative 3D reconstructions of a KM mitotic cell with misaligned chromosomes.
(C) Duration of mitosis in spheroid cultures (CTRL, 30 cells; K, 7 cells; M, 54 cells; KM, 42 cells); Kruskal-Wallis test, p < 0.0001; Dunn’s multiple comparisons test.
(D) Fate of mitotic cells during 20-hr time-lapse in vitro (CTRL, 30 cells; K, 7 cells; M, 54 cells; KM, 42 cells).
(E) Percentage of mitotic errors per acinus (CTRL, 29 cells; K, 7 cells; M, 45 cells; KM, 35 cells); one-way ANOVA test, p < 0.0001; Holm Sidak’s multiple comparisons test.
(F) Outcome of completed cell division after mitotic arrest in Mad2-expressing acini.
CTRL, n = 4; K, n = 3; M, n (ColA1-Mad2) = 2 and n (TetO-Mad2) = 1; KM, n (ColA1-Mad2) = 3 and n (TetO-Mad2) = 2. ∗∗∗∗p < 0.0001. See also Figure S4 and Movies S1, S2, S3, S4, and S5.
In 43% of Mad2 dividing cells and 33.7% of KM, mitotic abnormalities, including chromosome misalignments, lagging chromosomes, and micronuclei formation, were observed (Figures 4B and 4E; Figures S4B–S4D; Movies S3, S4, and S5). These aberrations are likely causes for consequent aneuploidization (Figure 4F) and/or the depletion of Mad2-expressing cells.
Mad2 Overexpression Increases Chromosome Missegregation Rates in KrasG12D-Positive Mammary Tumors
Deregulation of mitotic checkpoint genes in vivo has been shown to increase mitotic errors and is strongly associated with a CIN phenotype in human tumors (Janssen and Medema, 2013). Despite Mad2’s initial detrimental effects, analysis of tumor samples from all three KM subgroups verified the presence of both transgenes (Figures S5A and S5B), and immunohistochemistry for HA revealed Mad2-expressing cells in all tumors analyzed (Figure S5C). Nuclear size measurements proved a higher variance and a significant gain in nuclear volume in KM tumor cells compared with K (Figures S5D and S5E). In line with the finding of Kras inducing centrosome abnormalities (Zeng et al., 2010), K tumors also showed an increase in nuclear variance over control mice (Figure S5E). To determine the full extent of Mad2’s effects on karyotype composition and chromosome rearrangements, we performed low-pass, whole-genome sequencing of primary tumors (median coverage 0.11-fold), followed by somatic copy number alteration (SCNA) analysis. On average, genomes of KM tumors were more frequently affected by and had significantly more SCNAs than K tumors (mean 2.70 versus 0.85; Figures 5A and 5B). The most frequent variant types present in either cohort were whole chromosome gains and losses, followed by large partial chromosome gains and losses (Figure 5A). Although chromosomes 2 and 4 were recurrently affected in both cohorts (30% and 25% in K tumors as well as 34.7% and 52% in KM tumors, respectively), additional aneuploidies were present in nearly all other chromosomes of KM tumors (Figure S5F). No specific SCNAs were found to occur at a significantly higher frequency in KM versus K tumors, suggesting that, in this model, Mad2 serves to increase the total amount of aneuploidy in the tumor rather than selecting for specific rearrangement patterns.
Figure 5.

Mad2 Overexpression Boosts Aneuploidy and Facilitates Tumor Persistence after Oncogene Withdrawal
(A) SCNAs in 20 Kras and 23 Kras/Mad2 primary tumors. Shown are whole chromosome gain (WCG) and loss (WCL), partial chromosome gain (PCG) and loss (PCL), focal amplification (AMP), and gross chromosomal rearrangement (GCR, red). x Axis, individual tumors; y axis, SCNAs per tumor.
(B) Average SCNAs in K and KM tumors; Fisher’s exact test, p = 0.01039.
(C) Representative micrographs of KM tumor cells in vitro (H2B-GFP, green). Scale bar, 25 μm. Top: furrow regression and polyploidization. Bottom: lagging chromosome during anaphase and cytokinesis failure.
(D) Percentage of mitotic errors per tumor (K, n = 4, 74 cells; KM, n = 5, 84 cells); Mann-Whitney test, p = 0.0317.
(E) Representative examples of tumor development in K and KM tumors. The arrow indicates doxycycline withdrawal (day 0).
(F) Status of tumor regression in K and KM cohorts 2 months after doxycycline withdrawal.
∗p < 0.05. See also Figures S5 and S6.
To analyze whether Mad2 influences CIN during tumor growth, we performed time-lapse microscopy of primary tumor cells. Indeed, 30% of KM cells continued to missegregate chromosomes compared with 10% in Kras tumors (Figures 5C and 5D), suggesting that Mad2 overexpression increases CIN prior to tumorigenesis and during tumor cell expansion.
Mad2 Overexpression Facilitates Oncogene Independence
To assess the effect of Mad2-induced CIN on tumor phenotype, we analyzed tumor progression and histopathological features of primary tumors. Despite the prolonged tumor onset, K and KM cohorts displayed no differences in overall tumor growth (Figure S5G) and developed similar histopathological subtypes (Figure S5C). The tumor spectrum included mostly solid or solid-papillary carcinomas (88%) that displayed a high number of HA-Mad2-positive cells. A minor fraction of tumors was categorized as tubulo-alveolar carcinomas and epithelial to mesenchymal transition (EMT) phenotype carcinosarcomas (Rudmann et al., 2012) and contained fewer HA-Mad2-positive cells (Figure S5C). Although a low number of mice developed lung metastases, the incidence was not altered by Mad2 overexpression (data not shown). The absence of an additional Mad2 effect despite the elevated CIN in the KM tumors suggested that tumorigenesis had so far followed the strong lead of the driving oncogene.
Over the past years, it has been argued that the full potential of CIN can only be seen under selective conditions (Chen et al., 2012, Chen et al., 2015). Here, karyotype diversity can boost the adaptive potential of a cell population and enhance its fitness. We therefore set out to investigate whether the increased aneuploidy in Mad2-expressing tumors would reveal its potential when faced with strong selective pressure such as that seen with oncogene inactivation. Doxycycline was withdrawn in 48 K and 44 KM animals, each of which harbored multiple mammary tumors. In the K cohort, a total of 155 out of 166 tumors completely regressed to a non-palpable state (93.4%), demonstrating that the majority of epithelial cells remained dependent on Kras for maintenance of the transformed state. Strikingly, in the KM cohort, 23 of 108 primary tumors (21.3%) showed no signs of shrinkage, and malignant growth continued in the absence of doxycycline (Figures 5E and 5F). Activation of the same oncogenic pathway has been described as a possible mechanism of doxycycline independence (Podsypanina et al., 2008). To exclude the possibility of transgene re-expression, non-regressing tumors were analyzed for Kras and Mad2 transgene expression. Whereas 27.3% of K and 34.8% of KM tumors re-activated the transgenic system (Figure S6A), around 70% of persisting tumors remained unaccounted for. In addition, no mutations were found in the endogenous ras genes (data not shown). This finding suggests that genetic heterogeneity most likely had led to the establishment of oncogene-independent subclones, facilitating tumor persistence after transgene de-induction.
Mad2 Overexpression Delays Her2-Induced Breast Cancer
Her2 is one of the most common oncogenes overexpressed or amplified in human breast cancer (Moody et al., 2002, Sanchez-Garcia et al., 2014, Stephens et al., 2012). To test whether our findings for Kras-driven mammary tumorigenesis also apply to other breast cancer drivers, we crossed Mad2 animals into tetracycline-inducible Her2 mice (Moody et al., 2002). Her2 (H)-positive animals developed tumors with a shorter median latency (93 days, n = 60) than the ones observed with Kras (p < 0.0001). Similar to the Kras model, overexpression of Mad2 in Her2 animals (HM) led to a significant delay in tumor onset (138 days, n = 73) and a decrease in the overall tumor load per animal (Figures 6A and 6B).
Figure 6.

Elevated Levels of Mad2 Delay Her2-Driven Breast Tumorigenesis without Affecting Aneuploidy
(A) Tumor-free survival after doxycycline administration (Her2, n = 60; Her2/Mad2, n = 73); Mantel-Cox test, p < 0.0001.
(B) Number of tumors per animal; Mann-Whitney test, p = 0.0062.
(C) Representative images of phospho-Histone 3 staining of mammary glands at 4 and 100 days on doxycycline. Scale bar, 50 μm.
(D) Quantification of pH3-positive cells per acinus at 4 days on doxycycline. Points represent single acinar measurements (CTRL, n = 6; H, n = 3; HM, n = 4); Kruskal-Wallis test, p < 0.0001; Dunn’s multiple comparisons test.
(E) Average SCNAs in H and HM tumors; Fisher’s exact test, p = 1.
(F) Percentage of primary tumors of Her2 (left) and Her2/Mad2 cohort (right) containing specific structural variants per individual chromosome.
(G) Percentage of mitotic errors per tumor (H, n = 5; HM, n = 4); Mann-Whitney test, p = 0.125.
(H) Representative images showing misaligned chromosomes (arrows) in H and HM tumor cells in vitro (H2B-GFP, green). Scale bar, 20 μm.
(I) Status of tumor regression in H and HM cohort 2 months after doxycycline withdrawal.
∗∗p < 0.01, ∗∗∗∗p < 0.0001. See also Figure S6.
Histology 4 and 100 days following transgene induction revealed that Her2-driven tumorigenesis largely mimicked the continuous proliferation observed in Kras animals. Elevated Mad2 levels in this model resulted in an increase in pH3-positive cells accompanied by a predominant localization of mitotic cells to the acinar lumen (Figures 6C and 6D). The delayed tumor onset observed in the Her2/Mad2 animals might therefore also be a consequence of Mad2-induced mitotic arrest.
Unlike the KM model, in which ultimately all tumors were positive for exogenous Mad2, a minor fraction of tumors (18%) in the Her2/Mad2 cohort completely downregulated Mad2 expression (Figure S6B). The Her2 model further differed in the appearance of copy number changes. Whereas strong differences were found between K and KM, H and HM tumors showed similar numbers of SCNAs (mean 2.30 versus 2.60), with Mad2-positive tumors tending toward increased numerical aneuploidy compared with Her2 tumors (Figures 6E and 6F; Figure S6C). H and HM tumor cells also displayed similar missegregation rates in vitro (Figures 6G and 6H). The elevated number of SCNAs seen in Her2 tumors is in line with findings of centrosome abnormalities and increased aneuploidy in MMTV-Her2-induced murine breast cancer (Montagna et al., 2002). It is possible that tumorigenesis can only proceed below a certain CIN threshold (Jamal-Hanjani et al., 2015, Silk et al., 2013), restricting Mad2’s effect to increase SCNAs on Her2 mammary tumors.
These results prompted us to investigate whether tumor regression after transgene de-induction was affected in these animals. As seen previously with this model (Moody et al., 2002), of 174 Her2-positive tumors, only two tumors failed to fully regress after doxycycline withdrawal (1.15%; Figure 6I). In the HM cohort, a higher number of persistent tumors was detected, with 12 of 144 HM tumors not fully regressing upon inactivation of Her2 and Mad2 (8.33%; Figure 6I). Strikingly, 50% of HM non-regressing tumors re-expressed the oncogene (Figure S6D), suggesting that elevated Mad2 levels in the other 50% of Her2 primary tumors also offered a selective benefit to enable the escape from oncogene dependence. The presence of persistent tumors in the HM model was probably not only a result of the induced aneuploidy, given that similar levels of SCNAs were also present in the H tumors that fully regressed. It is possible that tumor cells require a certain time for the acquired CIN to generate and select fitter tumor subclones. Whereas Her2 tumors arose very early (93 days), Her2/Mad2 tumors needed a longer time to develop (138 days), allowing sufficient time for the chromosome missegregations to create oncogene-independent subclones. We speculate that the biological context established by the initiating oncogenic drivers as well as the mutational processes at work thereafter are additional determinants of the genetic adaptation.
Discussion
The consequence of chromosome instability on tumor development continues to stand at the center of a scientific debate. Depending on genetic- and tissue-specific contexts, CIN-associated gene de-regulations promote, suppress, or have no effect on spontaneous tumorigenesis (Janssen and Medema, 2013, Zasadil et al., 2013).
The overexpression of Mad2 is a frequent event in human cancers that has been modeled both in vitro and in vivo. The induction of Mad2 in adult mouse tissues leads to CIN, tumor initiation (Sotillo et al., 2007), and acceleration of KrasG12D-driven lung cancer (Sotillo et al., 2010). In this study, we extend this analysis and show that elevated levels of Mad2 in the mammary gland cause a delay in both KrasG12D- and Her2-induced breast cancer. The different outcomes upon Mad2 overexpression in the lung versus the mammary gland might be due to tissue-specific differences in cell homeostasis and signaling. The lung is one of the tissues with the highest incidence of spontaneous tumor formation in mouse models of CIN, suggesting that lung cells are specially sensitive to aneuploidy (Duijf and Benezra, 2013). Alternatively, expression levels of the reverse tetracycline-controlled transactivator (rtTA) driven by the lung-specific promoter (CCSP) could differ from the mammary-specific MMTV promoter, making it difficult to compare both models.
Murine breast cancer models allow multi-sampling of mammary glands during tumor development from the same animal. Taking advantage of this, we follow the transgenic effect in individual animals and demonstrate that early Mad2 overexpression in mammary cells in vivo leads to strong mitotic arrest and increased cell death. Moreover, time-lapse imaging of mammary spheroid cultures showed that prolonged mitotic arrest results in apical extrusion and cell degradation inside the lumen. Apical cell shedding is a common mechanism in epithelia to eliminate defective cells (Denning et al., 2012, Rosenblatt et al., 2001). Both the mitotic arrest and the higher mitotic error rate during the first rounds of division might mark Mad2-expressing cells as aberrant. It is also plausible that cells ultimately undergo anoikis because of delamination (Gilmore, 2005). The signaling pathway responsible for mitotic cell death is still unclear; however, it is possible that Myc contributes to apoptosis after prolonged checkpoint activation, as suggested recently (Topham et al., 2015).
Strikingly, not all cells expressing Mad2 die. A small percentage of cells is able to overcome the arrest and complete cell division inside the epithelial layer. In all KM and the majority of HM animals, Mad2 expression continues throughout tumorigenesis, leading to the formation of Mad2-positive tumors. The time and route to steady state, however, vary among individuals. When Mad2 expression strongly outweighs Kras (subgroup A), animals show a substantial reduction of Mad2-expressing cells over time. In subgroup B, where initial Kras and Mad2 levels are high, mammary glands can maintain or even increase the number of Mad2-expressing cells. In subgroup C, a reduction of Mad2 levels occurs, whereas Kras increases its expression. The different phenotypes depend on a tight balance between oncogenic and Mad2 signaling. Pro-survival signals downstream of Kras (Pylayeva-Gupta et al., 2011) and Her2 (Moasser, 2007) as well as supporting signals from the microenvironment (Radisky, 2012) might thereby promote cell cycle progression. Considering that Mad2 increases karyotype instability, it is possible that CIN-induced genetic changes might also take part in the detrimental effect of early Mad2 expression and contribute to overcoming checkpoint arrest or even subsequently support tumor initiation. Models combining chromosome missegregation with the process of negative selection and tolerance, as seen in the presented mouse models, are therefore good approximations to mimic human tumor growth (Valind et al., 2013).
An increased occurrence of karyotype aberrations because of Mad2 overexpression was primarily seen in KrasG12D-driven mammary tumors. Mad2-positive tumors markedly outweigh the solely KrasG12D-expressing counterparts in the genomic variance between cells as well as the number and complexity of chromosomal rearrangements. Interestingly, Mad2 did not further increase the high number of structural variants observed in Her2 mammary tumors but caused a slight trend toward whole chromosome gains and losses. Her2 has been shown to cause centrosomal defects and, thus, by itself, interferes with chromosome segregation (Montagna et al., 2002). The fact that Mad2 did not significantly contribute to additional karyotype complexity in the aneuploid Her2 tumors supports the hypothesis that a certain threshold of CIN is permissive for tumor development (Jamal-Hanjani et al., 2015, Laughney et al., 2015, Silk et al., 2013). An alternative theory suggests that the genetic setting shapes initial CIN and aneuploidy tolerance, which, after rounds of negative selection, balances into an optimized equilibrium at a specific aneuploidy index (Laughney et al., 2015, Valind et al., 2013).
Karyotypic heterogeneity can confer genetic flexibility and, in turn, increase the adaptability of a tumor cell population. Here we present in vivo evidence that Mad2-induced mitotic aberrations enhance the generation of oncogene-independent tumor subclones in breast cancer. Upon silencing of the driving oncogene, previously Mad2-expressing animals showed a higher percentage of non-regressing tumors in the K and H models. It is startling, however, that, despite the high number of structural variants in both H and HM tumors, the number of non-regressing tumors is much lower than in KM while having similar CIN levels. It may be that, given distinct initiating oncogenes, differing grades of genomic instability are necessary for an oncogene-independent subclonal population to arise. Alternatively, the stochastic appearance of fitness-improving chromosomal changes depends on the time a tumor population has to establish and expand itself. Multi-region sequencing and genomic profiling studies have verified that, although tumor growth originates from early driver mutations, fitness-contributing private alterations arise continuously during tumor development (Sottoriva et al., 2015). Therefore, both genetic instability-driving mechanisms and the time for tumor evolution might contribute to the generation of oncogene-independent cells. A previously reported MYC-induced breast cancer model supports this argument. Myc-induced mammary tumors appear at a very long latency compared with Kras and Her2 and are associated with a strong CIN phenotype (McCormack et al., 1998). Interestingly, incomplete regression following Myc inactivation is observed in 60% of tumors (Leung et al., 2012), whereas simultaneous overexpression of Mad2 in these animals results in a further increase of tumor persistence by 30% (M.M., unpublished data). The effect of transient Mad2 expression on the incidence of oncogene-independent tumors is therefore discernable in three different oncogenic settings, even if the overall adaptive capacity might depend on the initiating oncogene.
Taken together, it is becoming clear why the effect of CIN on tumor progression only becomes evident through strong selective pressures. Under such circumstances, the wide distribution of karyotypes promotes gross adaptive leaps (Chen et al., 2012, Chen et al., 2015, Endesfelder et al., 2014). Therefore, it is not surprising that CIN is found to be associated with poor prognosis and contributes to multidrug resistance (Lee et al., 2011, Zasadil et al., 2013). Their predominant role as evolutionary drivers makes CIN genes important predictive biomarkers in cancer (Carter et al., 2006, Habermann et al., 2009). Further studies aiming at understanding the contribution of individual CIN genes to oncogene independence will extend our understanding of intratumoral heterogeneity and tumor evolution. Considering the low incidence of oncogene-independent tumors in some oncogenic mouse models, the combination with CIN models might facilitate the identification of escape mechanisms from targeted therapies. The link between karyotypic and mutational changes further complicates the understanding of tumor evolution (Sheltzer et al., 2011). The role and effect of CIN-induced genetic changes has still to be determined and is likely to be an important component during tumorigenesis and acquisition of therapeutic resistance.
Experimental Procedures
Animal Husbandry
Animals were of mixed background (mainly Friend virus B [FVB]) with a heterozygous genotype: MMTV-rtTA (Gunther et al., 2002), TetO-KrasG12D (Fisher et al., 2001), TetO-rat-Her2 (Moody et al., 2002), HA-tagged-TetO-Mad2 (Sotillo et al., 2007), and H2B-GFP (Hadjantonakis and Papaioannou, 2004). ColA1-HA-Mad2 animals were included in the in vitro experiments (Supplemental Experimental Procedures). Breeding and experimentation was performed at the European Molecular Biology Laboratory (EMBL) Monterotondo, with ethical approval from the EMBL animal welfare and ethical review body and in accordance with current Italian legislation (Art. 9; January 27, 1992; no. 116) licensed by the Italian health ministry (Decree no. 233/2011-B). For animal experimentation, see the Supplemental Experimental Procedures.
Immunohistochemistry and Immunofluorescence
Immunohistochemistry was performed according to Sotillo et al. (2007). For antibody details, see the Supplemental Experimental Procedures.
3D Organotypic Assays
Mammary glands were harvested from 8- to 9-week-old female mice and prepared and stained according to published records (Jechlinger et al., 2009). The following primary antibodies were used: HA (1:,1000, Covance, MMS-101R), cleaved Caspase3 (1:200, Cell Signaling Technology, 9661S), LaminB1 (1:500, Abcam, 16048), α-Tubulin (1:500, Sigma, F2168), and Aurora B (1:150, BD Biosciences, 611082). Imaging of fixed samples was performed on a Leica TCS SP5/SP8 confocal microscope and time-lapse imaging during 20 hr on an inverted spinning disk confocal microscope (PerkinElmer Ultraview-Vox): 0.3-μm optical sectioning across a 35-μm stack, 5 frames/hr. Volocity version 6.2 (Improvision, PerkinElmer) served for image acquisition and analysis.
Tumor Cell Culture
Harvested tumors were digested with 150 U/ml Collagenase type 3 (Worthington, CLS3) and 20 mg/ml Liberase Blendzyme 2 (Roche, 11988425001) for 1 hr, washed with PBS, and dissociated with 0.25% Trypsin (Life Technologies, 25200056). Cells were cultured on 8-well chambered coverglass (Thermo Scientific, 155411) in serum-free mammary epithelial basal medium (MEBM) with supplements (Lonza, CC-3150) with 1 μg/ml doxycycline. Time-lapse imaging during 15 hr was performed on a confocal microscope (Leica TCS SP5): 2-μm optical sectioning across an 8-μm stack, 30 frames/hr. Volocity version 6.2 (Improvision, PerkinElmer) served for image analysis.
DNA Sequencing and Analysis
Genomic DNA was extracted from cells using the DNA blood mini kit (QIAGEN). Library preparation and low-coverage sequencing were pursued on an Illumina HiSeq 2500 platform (Illumina) using 50-base pair single-end reads as described previously (Mardin et al., 2015). Reads were aligned to the mm10 build of the mouse reference genome using Burrows-Wheeler Aligner (BWA; version 0.7.10). Tumor coverage files were log2-normalized to mouse genomic DNA derived from normal mammary tissue. Circular binary segmentation (CBS; R package) was applied, and somatic copy number alterations were categorized as follows. Whole chromosome gains/losses were defined as chromosome-wide shifts in the segmentation of a chromosome, whereas partial chromosome gains/losses entailed changes spanning at least one-fifth of the chromosome. Focal amplifications and deletion encompassed events smaller than this. When the number of copy number state switches on a chromosome exceeded ten, they were called as gross chromosomal rearrangements.
Statistical Analysis
Statistical analysis was carried out using Prism 6 (GraphPad). p values were as follows: ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, ∗∗∗∗p < 0.0001. Scatterplots show mean and SEM. Box-and-whisker plots show median interquartile ranges plus minimum to maximum range. The number of animals is represented with n. Control samples in vivo were animals containing the transgenes but kept on a normal diet or animals lacking MMTV-rtTA feed with doxycycline food. Non-induced cultures were considered controls in vitro.
Author Contributions
K.R. performed the experiments. M.M. collected the Her2 tumor samples. J.P. performed immunohistochemistry and the TUNEL analysis. C.B., B.R.M., and J.O.K. generated and analyzed the sequencing data. K.R. and R.S. designed the experiments, analyzed the data, and wrote the paper. M.J. and R.S. supervised the work.
Acknowledgments
We thank Giuseppe Chiapparelli and Maria Kamper for mouse husbandry, Kristina Havas-Cavalletti and Juan-Manuel Schvartzman for discussions and critical reading of the manuscript, and Krystal Timón Perez for assistance with the experimental setup. We are grateful to Vittoria Castiglioni for histopathological evaluation and to Marco Lampe and the TRLC Heidelberg for confocal microscopy assistance. We acknowledge the technical service by the EMBL Microscopy, Histology, and GeneCore Facility. Work in R.S.’s laboratory is funded by an ERC starting grant (281614), Marie Curie Grant PCIG09-GA-2011-293745, Associazione Italiana per la Ricerca sul Cancro, and the Howard Hughes Medical Institute. The M.J. laboratory is supported by Marie Curie Grant PCIG-GA-2011-294121. J.O.K.’s laboratory received funding from an ERC starting grant (336045).
Published: June 9, 2016
Footnotes
Supplemental Information includes Supplemental Experimental Procedures, six figures, and five movies and can be found with this article online at http://dx.doi.org/10.1016/j.celrep.2016.05.048.
Accession Numbers
The accession number for the sequencing data reported in this paper is ENA: PRJEB12536.
Supplemental Information
Time-lapse imaging of a cell division (red circle) in a control culture oriented perpendicular to the apical and basal membrane within the epithelial layer; H2B-GFP (green).
{kind=link}
Time-lapse imaging of cell (red circle) entering mitosis and completing cell division inside the epithelial rim in a KrasG12D-positive spheroid in vitro, t = 0 after 30 hr on doxycycline; H2B-GFP (green).
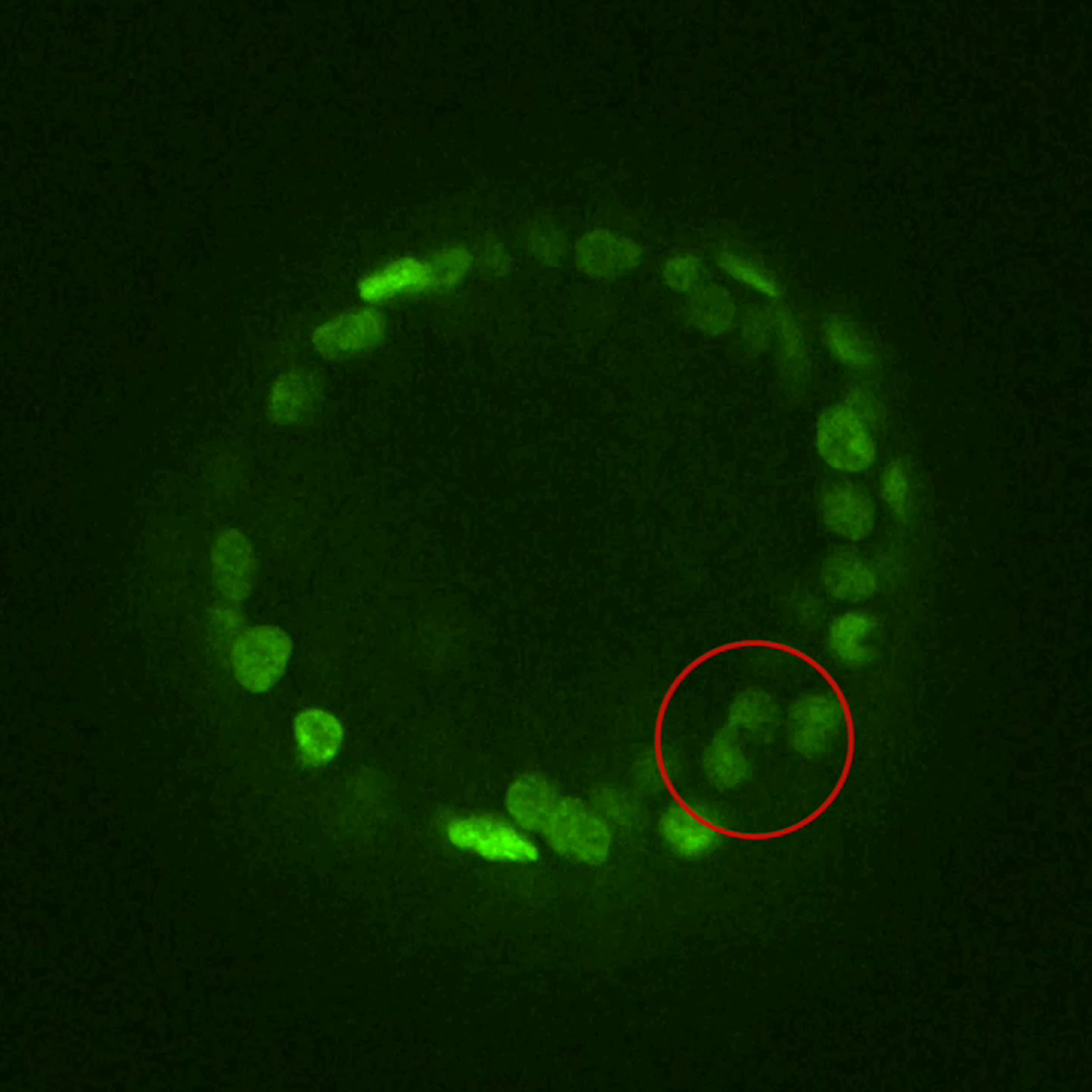{kind=link}
Time-lapse imaging of a KrasG12D/Mad2-positive acinus showing a mitotic cell (red circle) that divides and forms a micronucleus, t = 0 after 30 hr on doxycycline; H2B-GFP (green).
{kind=link}
Time-lapse imaging of KrasG12D/Mad2 positive acinus with a mitotic cell (red circle) undergoing apoptosis after severe chromosome misalignments, t = 0 after 30 hr on doxycycline; H2B-GFP (green).
{kind=link}
Time-lapse imaging of KrasG12D/Mad2-positive spheroid with a mitotic cell (red circle) failing to complete nuclear division, t = 0 after 30 hr on doxycycline; H2B-GFP (green).
{kind=link}
References
- Boveri T. ANTON DOHBN. Science. 1912;36:453–468. doi: 10.1126/science.36.928.453. [DOI] [PubMed] [Google Scholar]
- Carter S.L., Eklund A.C., Kohane I.S., Harris L.N., Szallasi Z. A signature of chromosomal instability inferred from gene expression profiles predicts clinical outcome in multiple human cancers. Nat. Genet. 2006;38:1043–1048. doi: 10.1038/ng1861. [DOI] [PubMed] [Google Scholar]
- Cerami E., Gao J., Dogrusoz U., Gross B.E., Sumer S.O., Aksoy B.A., Jacobsen A., Byrne C.J., Heuer M.L., Larsson E. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012;2:401–404. doi: 10.1158/2159-8290.CD-12-0095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen G., Rubinstein B., Li R. Whole chromosome aneuploidy: big mutations drive adaptation by phenotypic leap. BioEssays. 2012;34:893–900. doi: 10.1002/bies.201200069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen G., Mulla W.A., Kucharavy A., Tsai H.-J., Rubinstein B., Conkright J., McCroskey S., Bradford W.D., Weems L., Haug J.S. Targeting the adaptability of heterogeneous aneuploids. Cell. 2015;160:771–784. doi: 10.1016/j.cell.2015.01.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denning D.P., Hatch V., Horvitz H.R. Programmed elimination of cells by caspase-independent cell extrusion in C. elegans. Nature. 2012;488:226–230. doi: 10.1038/nature11240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duijf P.H.G., Benezra R. The cancer biology of whole-chromosome instability. Oncogene. 2013;32:4727–4736. doi: 10.1038/onc.2012.616. [DOI] [PubMed] [Google Scholar]
- Endesfelder D., Burrell R.A., Kanu N., McGranahan N., Howell M., Parker P.J., Downward J., Swanton C., Kschischo M. Chromosomal instability selects gene copy-number variants encoding core regulators of proliferation in ER+ breast cancer. Cancer Res. 2014;74:4853–4863. doi: 10.1158/0008-5472.CAN-13-2664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fata J.E., Chaudhary V., Khokha R. Cellular turnover in the mammary gland is correlated with systemic levels of progesterone and not 17beta-estradiol during the estrous cycle. Biol. Reprod. 2001;65:680–688. doi: 10.1095/biolreprod65.3.680. [DOI] [PubMed] [Google Scholar]
- Fisher G.H., Wellen S.L., Klimstra D., Lenczowski J.M., Tichelaar J.W., Lizak M.J., Whitsett J.A., Koretsky A., Varmus H.E. Induction and apoptotic regression of lung adenocarcinomas by regulation of a K-Ras transgene in the presence and absence of tumor suppressor genes. Genes Dev. 2001;15:3249–3262. doi: 10.1101/gad.947701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao J., Aksoy B.A., Dogrusoz U., Dresdner G., Gross B., Sumer S.O., Sun Y., Jacobsen A., Sinha R., Larsson E. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci Signal. 2013;6:l1. doi: 10.1126/scisignal.2004088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilmore A.P. Anoikis. Cell Death Differ. 2005;12(Suppl 2):1473–1477. doi: 10.1038/sj.cdd.4401723. [DOI] [PubMed] [Google Scholar]
- Gunther E.J., Belka G.K., Wertheim G.B.W., Wang J., Hartman J.L., Boxer R.B., Chodosh L.A. A novel doxycycline-inducible system for the transgenic analysis of mammary gland biology. FASEB J. 2002;16:283–292. doi: 10.1096/fj.01-0551com. [DOI] [PubMed] [Google Scholar]
- Habermann J.K., Doering J., Hautaniemi S., Roblick U.J., Bündgen N.K., Nicorici D., Kronenwett U., Rathnagiriswaran S., Mettu R.K.R., Ma Y. The gene expression signature of genomic instability in breast cancer is an independent predictor of clinical outcome. Int. J. Cancer. 2009;124:1552–1564. doi: 10.1002/ijc.24017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hadjantonakis A.-K., Papaioannou V.E. Dynamic in vivo imaging and cell tracking using a histone fluorescent protein fusion in mice. BMC Biotechnol. 2004;4:33. doi: 10.1186/1472-6750-4-33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanahan D., Weinberg R.A. Hallmarks of cancer: the next generation. Cell. 2011;144:646–674. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- Jamal-Hanjani M., A’Hern R., Birkbak N.J., Gorman P., Grönroos E., Ngang S., Nicola P., Rahman L., Thanopoulou E., Kelly G. Extreme chromosomal instability forecasts improved outcome in ER-negative breast cancer: a prospective validation cohort study from the TACT trial. Ann. Oncol. 2015;26:1340–1346. doi: 10.1093/annonc/mdv178. [DOI] [PubMed] [Google Scholar]
- Janssen A., Medema R.H. Genetic instability: tipping the balance. Oncogene. 2013;32:4459–4470. doi: 10.1038/onc.2012.576. [DOI] [PubMed] [Google Scholar]
- Jechlinger M., Podsypanina K., Varmus H. Regulation of transgenes in three-dimensional cultures of primary mouse mammary cells demonstrates oncogene dependence and identifies cells that survive deinduction. Genes Dev. 2009;23:1677–1688. doi: 10.1101/gad.1801809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kabeche L., Compton D.A. Checkpoint-independent stabilization of kinetochore-microtubule attachments by Mad2 in human cells. Curr. Biol. 2012;22:638–644. doi: 10.1016/j.cub.2012.02.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laughney A.M., Elizalde S., Genovese G., Bakhoum S.F. Dynamics of Tumor Heterogeneity Derived from Clonal Karyotypic Evolution. Cell Rep. 2015;12:809–820. doi: 10.1016/j.celrep.2015.06.065. [DOI] [PubMed] [Google Scholar]
- Lee A.J.X., Endesfelder D., Rowan A.J., Walther A., Birkbak N.J., Futreal P.A., Downward J., Szallasi Z., Tomlinson I.P.M., Howell M. Chromosomal instability confers intrinsic multidrug resistance. Cancer Res. 2011;71:1858–1870. doi: 10.1158/0008-5472.CAN-10-3604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leung J.Y., Andrechek E.R., Cardiff R.D., Nevins J.R. Heterogeneity in MYC-induced mammary tumors contributes to escape from oncogene dependence. Oncogene. 2012;31:2545–2554. doi: 10.1038/onc.2011.433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li M., Fang X., Baker D.J., Guo L., Gao X., Wei Z., Han S., van Deursen J.M., Zhang P. The ATM-p53 pathway suppresses aneuploidy-induced tumorigenesis. Proc. Natl. Acad. Sci. USA. 2010;107:14188–14193. doi: 10.1073/pnas.1005960107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mardin B.R., Drainas A.P., Waszak S.M., Weischenfeldt J., Isokane M., Stütz A.M., Raeder B., Efthymiopoulos T., Buccitelli C., Segura-Wang M. A cell-based model system links chromothripsis with hyperploidy. Mol. Syst. Biol. 2015;11:828. doi: 10.15252/msb.20156505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCormack S.J., Weaver Z., Deming S., Natarajan G., Torri J., Johnson M.D., Liyanage M., Ried T., Dickson R.B. Myc/p53 interactions in transgenic mouse mammary development, tumorigenesis and chromosomal instability. Oncogene. 1998;16:2755–2766. doi: 10.1038/sj.onc.1201804. [DOI] [PubMed] [Google Scholar]
- Moasser M.M. The oncogene HER2: its signaling and transforming functions and its role in human cancer pathogenesis. Oncogene. 2007;26:6469–6487. doi: 10.1038/sj.onc.1210477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montagna C., Andrechek E.R., Padilla-Nash H., Muller W.J., Ried T. Centrosome abnormalities, recurring deletions of chromosome 4, and genomic amplification of HER2/neu define mouse mammary gland adenocarcinomas induced by mutant HER2/neu. Oncogene. 2002;21:890–898. doi: 10.1038/sj.onc.1205146. [DOI] [PubMed] [Google Scholar]
- Moody S.E., Sarkisian C.J., Hahn K.T., Gunther E.J., Pickup S., Dugan K.D., Innocent N., Cardiff R.D., Schnall M.D., Chodosh L.A. Conditional activation of Neu in the mammary epithelium of transgenic mice results in reversible pulmonary metastasis. Cancer Cell. 2002;2:451–461. doi: 10.1016/s1535-6108(02)00212-x. [DOI] [PubMed] [Google Scholar]
- Pérez de Castro I., Aguirre-Portolés C., Fernández-Miranda G., Cañamero M., Cowley D.O., Van Dyke T., Malumbres M. Requirements for Aurora-A in tissue regeneration and tumor development in adult mammals. Cancer Res. 2013;73:6804–6815. doi: 10.1158/0008-5472.CAN-13-0586. [DOI] [PubMed] [Google Scholar]
- Podsypanina K., Politi K., Beverly L.J., Varmus H.E. Oncogene cooperation in tumor maintenance and tumor recurrence in mouse mammary tumors induced by Myc and mutant Kras. Proc. Natl. Acad. Sci. USA. 2008;105:5242–5247. doi: 10.1073/pnas.0801197105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pylayeva-Gupta Y., Grabocka E., Bar-Sagi D. RAS oncogenes: weaving a tumorigenic web. Nat. Rev. Cancer. 2011;11:761–774. doi: 10.1038/nrc3106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Radisky D. On the role of the microenvironment in mammary gland development and cancer. Cold Spring Harb. Perspect. Biol. 2012;4:a013458. doi: 10.1101/cshperspect.a013458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rhodes D.R., Yu J., Shanker K., Deshpande N., Varambally R., Ghosh D., Barrette T., Pandey A., Chinnaiyan A.M. ONCOMINE: a cancer microarray database and integrated data-mining platform. Neoplasia. 2004;6:1–6. doi: 10.1016/s1476-5586(04)80047-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenblatt J., Raff M.C., Cramer L.P. An epithelial cell destined for apoptosis signals its neighbors to extrude it by an actin- and myosin-dependent mechanism. Curr. Biol. 2001;11:1847–1857. doi: 10.1016/s0960-9822(01)00587-5. [DOI] [PubMed] [Google Scholar]
- Rudmann D., Cardiff R., Chouinard L., Goodman D., Küttler K., Marxfeld H., Molinolo A., Treumann S., Yoshizawa K., INHAND Mammary, Zymbal’s, Preputial, and Clitoral Gland Organ Working Group Proliferative and nonproliferative lesions of the rat and mouse mammary, Zymbal’s, preputial, and clitoral glands. Toxicol. Pathol. 2012;40(6, Suppl):7S–39S. doi: 10.1177/0192623312454242. [DOI] [PubMed] [Google Scholar]
- Sanchez-Garcia F., Villagrasa P., Matsui J., Kotliar D., Castro V., Akavia U.-D., Chen B.-J., Saucedo-Cuevas L., Rodriguez Barrueco R., Llobet-Navas D. Integration of genomic data enables selective discovery of breast cancer drivers. Cell. 2014;159:1461–1475. doi: 10.1016/j.cell.2014.10.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schvartzman J.-M., Sotillo R., Benezra R. Mitotic chromosomal instability and cancer: mouse modelling of the human disease. Nat. Rev. Cancer. 2010;10:102–115. doi: 10.1038/nrc2781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schvartzman J.-M., Duijf P.H.G., Sotillo R., Coker C., Benezra R. Mad2 is a critical mediator of the chromosome instability observed upon Rb and p53 pathway inhibition. Cancer Cell. 2011;19:701–714. doi: 10.1016/j.ccr.2011.04.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheltzer J.M., Blank H.M., Pfau S.J., Tange Y., George B.M., Humpton T.J., Brito I.L., Hiraoka Y., Niwa O., Amon A. Aneuploidy drives genomic instability in yeast. Science. 2011;333:1026–1030. doi: 10.1126/science.1206412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silk A.D., Zasadil L.M., Holland A.J., Vitre B., Cleveland D.W., Weaver B.A. Chromosome missegregation rate predicts whether aneuploidy will promote or suppress tumors. Proc. Natl. Acad. Sci. USA. 2013;110:E4134–E4141. doi: 10.1073/pnas.1317042110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sotillo R., Hernando E., Díaz-Rodríguez E., Teruya-Feldstein J., Cordón-Cardo C., Lowe S.W., Benezra R. Mad2 overexpression promotes aneuploidy and tumorigenesis in mice. Cancer Cell. 2007;11:9–23. doi: 10.1016/j.ccr.2006.10.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sotillo R., Schvartzman J.-M., Socci N.D., Benezra R. Mad2-induced chromosome instability leads to lung tumour relapse after oncogene withdrawal. Nature. 2010;464:436–440. doi: 10.1038/nature08803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sottoriva A., Kang H., Ma Z., Graham T.A., Salomon M.P., Zhao J., Marjoram P., Siegmund K., Press M.F., Shibata D., Curtis C. A Big Bang model of human colorectal tumor growth. Nat. Genet. 2015;47:209–216. doi: 10.1038/ng.3214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stephens P.J., Tarpey P.S., Davies H., Van Loo P., Greenman C., Wedge D.C., Nik-Zainal S., Martin S., Varela I., Bignell G.R., Oslo Breast Cancer Consortium (OSBREAC) The landscape of cancer genes and mutational processes in breast cancer. Nature. 2012;486:400–404. doi: 10.1038/nature11017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamborero D., Gonzalez-Perez A., Perez-Llamas C., Deu-Pons J., Kandoth C., Reimand J., Lawrence M.S., Getz G., Bader G.D., Ding L., Lopez-Bigas N. Comprehensive identification of mutational cancer driver genes across 12 tumor types. Sci. Rep. 2013;3:2650. doi: 10.1038/srep02650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson S.L., Compton D.A. Proliferation of aneuploid human cells is limited by a p53-dependent mechanism. J. Cell Biol. 2010;188:369–381. doi: 10.1083/jcb.200905057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Topham C., Tighe A., Ly P., Bennett A., Sloss O., Nelson L., Ridgway R.A., Huels D., Littler S., Schandl C. MYC Is a Major Determinant of Mitotic Cell Fate. Cancer Cell. 2015;28:129–140. doi: 10.1016/j.ccell.2015.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torres E.M., Sokolsky T., Tucker C.M., Chan L.Y., Boselli M., Dunham M.J., Amon A. Effects of aneuploidy on cellular physiology and cell division in haploid yeast. Science. 2007;317:916–924. doi: 10.1126/science.1142210. [DOI] [PubMed] [Google Scholar]
- Valind A., Jin Y., Gisselsson D. Elevated tolerance to aneuploidy in cancer cells: estimating the fitness effects of chromosome number alterations by in silico modelling of somatic genome evolution. PLoS ONE. 2013;8:e70445. doi: 10.1371/journal.pone.0070445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zasadil L.M., Britigan E.M.C., Weaver B.A. 2n or not 2n: Aneuploidy, polyploidy and chromosomal instability in primary and tumor cells. Semin. Cell Dev. Biol. 2013;24:370–379. doi: 10.1016/j.semcdb.2013.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeng X., Shaikh F.Y., Harrison M.K., Adon A.M., Trimboli A.J., Carroll K.A., Sharma N., Timmers C., Chodosh L.A., Leone G., Saavedra H.I. The Ras oncogene signals centrosome amplification in mammary epithelial cells through cyclin D1/Cdk4 and Nek2. Oncogene. 2010;29:5103–5112. doi: 10.1038/onc.2010.253. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Time-lapse imaging of a cell division (red circle) in a control culture oriented perpendicular to the apical and basal membrane within the epithelial layer; H2B-GFP (green).
Time-lapse imaging of cell (red circle) entering mitosis and completing cell division inside the epithelial rim in a KrasG12D-positive spheroid in vitro, t = 0 after 30 hr on doxycycline; H2B-GFP (green).
Time-lapse imaging of a KrasG12D/Mad2-positive acinus showing a mitotic cell (red circle) that divides and forms a micronucleus, t = 0 after 30 hr on doxycycline; H2B-GFP (green).
Time-lapse imaging of KrasG12D/Mad2 positive acinus with a mitotic cell (red circle) undergoing apoptosis after severe chromosome misalignments, t = 0 after 30 hr on doxycycline; H2B-GFP (green).
Time-lapse imaging of KrasG12D/Mad2-positive spheroid with a mitotic cell (red circle) failing to complete nuclear division, t = 0 after 30 hr on doxycycline; H2B-GFP (green).