

Abstract
In recent years, electron microscopy (EM) and single particle analysis have emerged as essential tools for investigating the architecture of large biological complexes. When high resolution is achievable, crystal structure docking and de-novo modeling allows for precise assignment of individual protein domain sequences. However, the achievable resolution may limit the ability to do so, especially when small or flexible complexes are under study. In such cases, protein labeling has emerged as an important complementary tool to characterize domain architecture and elucidate functional mechanistic details. All labeling strategies proposed to date are either focused on the identification of the position of protein termini or require multi-step labeling strategies, potentially interfering with the final labeling efficiency. Here we describe a strategy for determining the position of internal protein domains within EM maps using a recombinant one-step labeling approach named Efficient Mapping by Internal Labeling (EMIL). EMIL takes advantage of the close spatial proximity of the GFP’s N- and C-termini to generate protein chimeras containing an internal GFP at desired locations along the main protein chain. We apply this method to characterize the subunit domain localization of the human Polycomb Repressive Complex 2.
Keywords: Protein labeling, GFP, Electron microscopy, Structure, Domain mapping
During the past several years, electron microscopy (EM) and single particle analysis have described the architecture and function of several macromolecular machineries (Nogales and Scheres, 2015). When high resolution is achievable, docking of available atomic coordinates or de-novo modeling of protein structures allow for precise assignment of individual components and localization of protein domains (Wiedenheft et al., 2011; He et al., 2013; Chang et al., 2015; Baskaran et al., 2014). When high resolution is not achievable or atomic coordinates are unavailable, additional structural information is required to describe both architectural and mechanistic details. This is particularly true for small or flexible macromolecular complexes. Different approaches have been developed to reconstitute and visualize protein complexes containing individual subunits labeled at specific sites. The majority of labeling studies utilize fusion protein tags, expressed in-frame at either the N- or the C-terminal region of the protein of interest. Successful applications of this technology include Maltose Binding Protein (MBP) (Lander et al., 2012; Ciferri et al., 2012; Baskaran et al., 2014), Green Fluorescence Protein (GFP) (Choy et al., 2009; Ciferri et al., 2012), Dynein Interacting Domain (DID) (Flemming et al., 2010) and actin polymer (Stroupe et al., 2009). While these approaches offer the advantage of reconstituting homogeneously labeled protein complexes, they are also best suited for the labeling of small subunits, where the localization of the N or C-termini matches reasonably well the position of the entire protein (Lander et al., 2012). In contrast, this subunit localization can be ambiguous if the N- and C-termini of the labeled protein are several nanometer away from each other or distant from important functional domains of interest. To overcome this limitation, other additional strategies have been adopted thus far. The first one makes use of monoclonal antibodies raised against specific protein domains (Hutchins et al., 2010; Chittuluru et al., 2011). While this technology has the potential of being very efficient, generating a complete set of monoclonal antibodies for each individual domain is often difficult and, when possible, low labeling efficiency or high-flexibility of the bound antibody could make the detection of the labeling challenging. A second strategy, termed DOLORS, utilizes monovalent streptavidin added post-translationally to an avi-tag sequence positioned within the main chain of the protein of interest (Lau et al., 2012). This method has the great advantage of specifically labeling any desired domain within a protein complex, without using costly and labor-intensive antibody production. However, a potential pitfall is represented by the multi-step process utilized for the labeling, which could diminish the overall labeling efficiency and tag-occupancy of the EM images.
In this manuscript, we present a strategy named Efficient Mapping by Internal Labeling (EMIL) to identify and localize internal domains within a multi subunit complex by electron microscopy. This method takes advantage of the close spatial proximity of the N- and C-termini of GFP (Supplementary Fig. 1A) and combines the advantages of fusion protein-based tags with the spatial resolution of the internal labeling. Previous work, utilizing similar concepts, has been used to characterize functional fusion proteins (Kratz et al., 1999; Roberts et al., 2009; Cockrell et al., 2011; Sun et al., 2014).
We designed vectors for Escherichia coli, insect cell and mammalian cell expression systems for the production of protein chimeras containing an internal GFP, connected through a short loop, to desired locations along the main protein chain (Supplementary Fig. 1B and C). GFP is a compact 27 kDa protein that can be easily visualized by electron microscopy when attached to the surface of a larger protein complex at defined location (Choy et al., 2009). For this reason, GFP can be inserted inside a polypeptide and serve as a marker for the identification of a specific domain within a protein complex. We use this method to characterize the domain organization of the Polycomb Repressive Complex 2 (PRC2) bound to AEBP2 (Ciferri et al., 2012). The results are presented here with a particular focus on the vector design and cloning strategy used to reconstitute different complexes carrying the internal labeling (Fig. 1A and Supplementary Fig. 1C). Complementing other labeling systems and universally applicable to any protein complex and expression system, this method can provide unique information not achievable with other techniques.
Fig. 1.
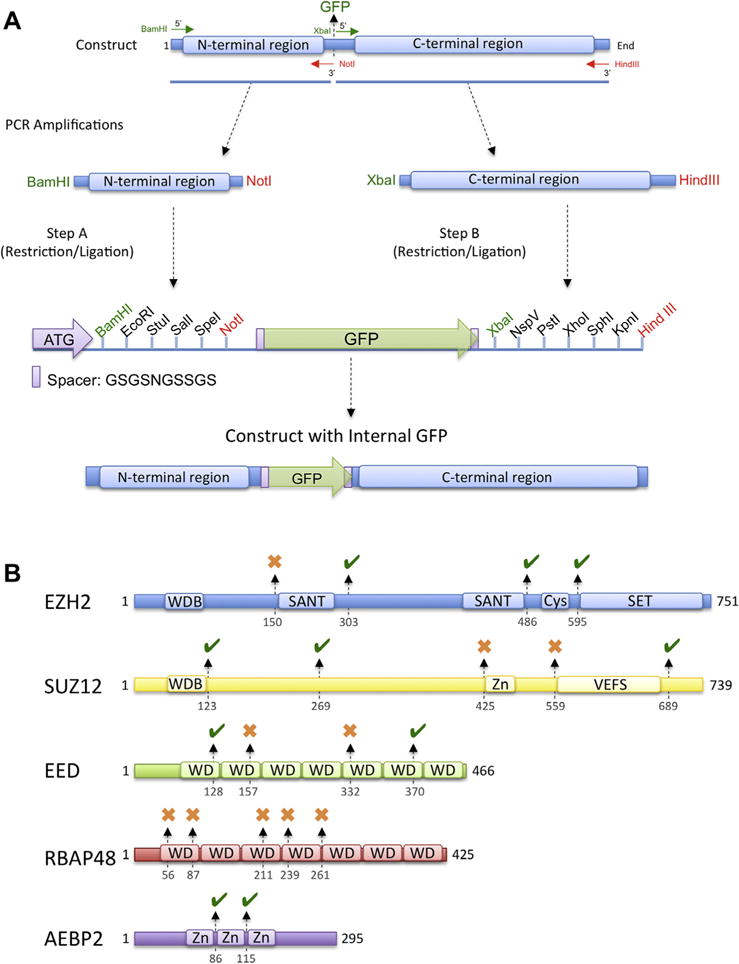
EMIL tagging strategy and its utilization in the characterization of the PRC2 complex. (A) EMIL tagging cloning strategy. Protein chimeras, carrying GFP at desired locations, can be generated with a two-step cloning procedure. In the first step, the DNA preceding the desired point of GFP insertion is cloned into the first MCS. Successively, the remaining DNA sequence, following the site of GFP insertion, is cloned into the second MCS of a vector already containing the first insert. This procedure generates a chimera composed of the N-terminal portion of the protein of interest fused to GFP through two ten-residue antiparallel spacers, followed by its C-terminal region. (B) EMIL tagging applied to the domain characterization of the PRC2–AEBP2 complex. Black arrows and numbers indicate the position of the GFP insertions into the main chains. A green mark indicates insertions with successful expression used for domain localization. Red marks indicate chimeras that proved to be not amenable for structural studies.
1. pEMIL vector design
We designed a set of vectors for protein expression in E. coli, insect cells and mammalian cells to generate chimeras containing an internal GFP at desired sites along the main protein chain (Supplementary Fig. 1C). Each of these plasmids was designed to have the GFP sequence flanked by a DNA sequence encoding for the 10-amino acid spacer GSGSNGSSGS and two multi cloning sites (MCS), each with unique restriction enzyme sequences (Supplementary Fig. 1C).
Protein chimeras, containing internal GFP at desired locations, can be generated with a two-step cloning procedure indicated in Fig. 1A. In the first step, the DNA coding for the protein sequence preceding the desired point of GFP insertion, is cloned into the first MCS. Successively, the DNA coding for the remaining sequence following the site of GFP insertion, is cloned into the second MCS of a vector already containing the first insert. The order of cloning can be swapped based on the presence of specific restriction sites within the two halves of the protein of interest. Using this procedure, it is possible to obtain a protein chimera composed of the N-terminal portion of the protein of interest fused to GFP through two ten-residue antiparallel spacers, followed by its C-terminal portion (Fig. 1A). This method has several advantages. First, it is applicable to all expression systems, allowing production of labeled proteins even when post-translational modifications are needed. Second, it is possible to simultaneously clone different protein boundaries into the MCS, enabling generation of tag insertion to multiple subunits at different desired positions. Third, the entire cassette carrying the protein chimeras can be quickly moved from one vector for a specific expression system to another. Fourth, the usage of GFP as a labeling system allows for fast assessment of tag expression and incorporation into larger complexes using UV light. Finally, covalent incorporation of the tag during protein production ensures homogeneously labeled sample preparation reflecting in maximum occupancy during single-particle EM analysis.
2. Preparation of labeled complexes and EM analysis
Several factors were taken into consideration when designing the specific position at which to incorporate the GFP tag. To localize protein domains with a known crystal structure, we introduced GFP into non-conserved exposed loops, not likely involved in protein–protein interaction, projecting towards the outside. Analysis of the protein surface, obtained using common structural biology software applications, could be very informative in indicating hydrophobic regions or charged pockets, likely mediating protein interaction. We generally designed our GFP chimeras in regions other than these since it is expected that GFP insertion in proximity of these regions could potentially disrupt complex formation. When high-resolution structures are not available, a larger number of constructs might be required, using a trial-error approach. In these cases, we found it effective to insert the GFP tag into non-conserved loops localized inside, or in proximity of, the domain of interest.
We used EMIL tagging to define the spatial organization of each subunit and determine the localization of all PRC2 functional domains. PRC2 is composed of the subunits EZH2, Suz12, EED and RBAP48 (reviewed in Margueron and Reinberg (2011)) and its activity is stimulated by the cofactor AEBP2. The structure of the PRC2–AEBP2 complex was solved by negative stain electron microscopy at a resolution of 19 Å (Ciferri et al., 2012), which does not provide enough details to allow unambiguous docking of the available EED, RBAP48 and EZH2 atomic coordinates. PRC2 architecture consists of four large lobes: A–D, interconnected by two narrower arms, Arm 1 at the top, and Arm 2 in the center (Ciferri et al., 2012). We tested a total of 20 different PRC2–AEBP2 complexes, each incorporating the GFP tag internally to one of the subunits at a specific location (Fig. 1B). In the absence of the high-resolution structure of the entire PRC2 complex, it is expected that some GFP incorporation could interfere with proper folding or complex formation. GFP insertions that assembled into functional complexes are indicated in Fig. 1B with a green mark, while those that did not are indicated by a red cross (Fig. 1B).
All complexes containing GFP insertions were purified and prepared for electron microscopy analysis as described in Ciferri et al., 2012. Samples were analyzed by negative staining EM and imaged using a CCD camera. We used reference-free 2D classification to sort particles positioned in different orientations, and cross correlation to measure similarity between GFP-labeled and unlabeled complexes oriented in the same view. We concentrated the analysis on two orthogonal views where the structural features of the PRC2 complex are clearly identifiable (Fig. 2A).
Fig. 2.
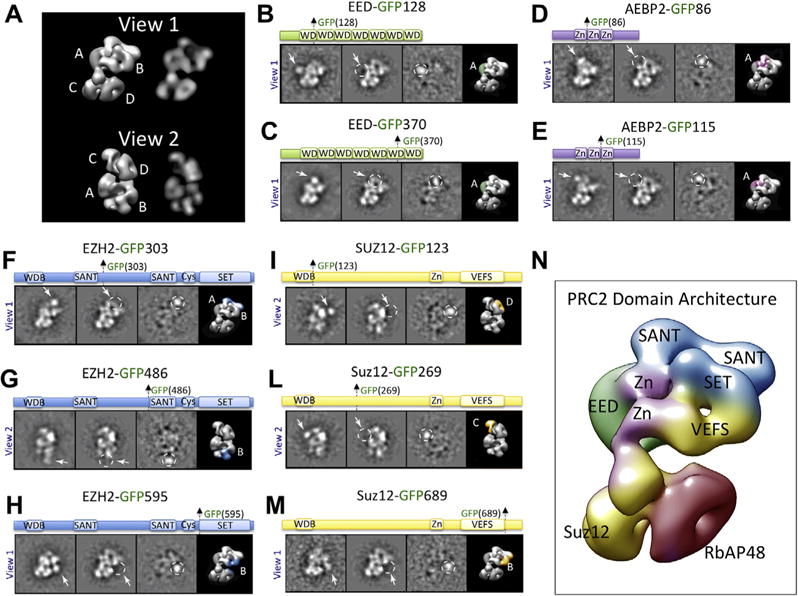
Domain organization of the PRC2 complex. (A) Architecture of the PRC2 complex in two orthogonal views (View 1 and View 2), shown as a 3D reconstruction and as 2D forward projection images. Lobes (A)–(D) are indicated. (B–M) Reference-free 2D classes of the labeled and unlabeled sample, the difference map between them, and the 3D view of the complex with the assigned localization for different PRC2 subunit domains color-coded. (N) Summarized domain architecture of the PRC2–AEBP2 complex. Individual domains are color-coded based on their original protein sequence as indicated in panels (B)–(M).
GFP labels, visible as protruding additional rounded densities with a diameter of 40 Å, were observed for all the constructs that assembled into stable complexes (Figs. 1 and 2). This analysis allowed us to identify the position of EED and of all the domains of EZH2, Suz12 and AEBP2 (Fig. 2A–H, L, M, Ciferri et al., 2012).
When GFP incorporation in a specific domain disrupts complex formation, labeling of protein regions interacting with this specific domain could be used to obtain similar results.
In the case of PRC2, none of the GFP insertions into RbAP48 subunit assembled into a stable complex amenable to EM analysis, suggesting that the GFP incorporation interferes with complex formation (data not shown). To localize RbAP48, we inserted GFP immediately after the WDB region of Suz12 (Suz12-GFP123), shown to interact with RbAP48 (Nowak et al., 2011; Schmitges et al., 2011). EMIL tagging localized Suz12 WDB domain (Suz12-GFP123), and consequently RbAP48, within lobe D (Fig. 2I). A summary of the complete domain architecture of the PRC2–AEBP2 complex is summarized in Fig. 2N.
In conclusion, we have developed a technology to reconstitute protein complexes carrying an internal label at a desired location for structural characterization. We found that placing the GFP tag into non-structured protein loops inside, or immediately adjacent to, the specific protein domain of interest is particularly effective in identifying subunits and regions of interest. Designing GFP fusion chimeras is especially straightforward when crystal structures are available, but it is successful only if the GFP insertion does not interfere with subunit incorporation and complex formation.
We were able to use the EMIL tagging to quickly characterize the domain architecture of the PRC2–AEBP2 complex (Fig. 2A–N). This technique can be successfully used to generate 3D reconstructions of labeled complexes if enough particles and different views are available (Supplementary Fig. 2). EMIL tagging, complementing other biochemical methods and being applicable to any expression system, can inform on domain localization and complex architecture, even in cases where only low or moderate resolution is available.
Supplementary Material
Acknowledgments
We are grateful to Emily M. Ciferri for contributing to the original design strategy, and Alberto Estevez and Sara Sun for technical support. This project was funded in part by an NIGMS grant to E.N. (GM63072). E.N. is a Howard Hughes Medical Institute investigator. C.C. is a recipient of the American Italian Cancer Foundation Fellowship. G.C.L. is a recipient of the Damon Runyon Cancer Research Foundation Fellowship.
Appendix A. Supplementary data
Supplementary data associated with this article can be found, in the online version, at http://dx.doi.org/10.1016/j.jsb.2015.09.016.
References
- Baskaran S, Carlson LA, Stjepanovic G, Young LN, Kim do J, Grob P, Stanley RE, Nogales E, Nogales E, Hurley JH. Architecture and dynamics of the autophagic phosphatidylinositol 3-kinase complex. Elife. 2014;3 doi: 10.7554/eLife.05115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang L, Zhang Z, Yang J, McLaughlin SH, Barford D. Atomic structure of the APC/C and its mechanism of protein ubiquitination. Nature. 2015;522:450–454. doi: 10.1038/nature14471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choy RM, Kollman JM, Zelter A, Davis TN, Agard DA. Localization and orientation of the gamma-tubulin small complex components using protein tags as labels for single particle EM. J Struct Biol. 2009;168:571–574. doi: 10.1016/j.jsb.2009.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciferri C, Lander GC, Maiolica A, Herzog F, Aebersold R, Nogales E. Molecular architecture of human polycomb repressive complex 2. Elife. 2012;1:e00005. doi: 10.7554/eLife.00005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chittuluru JR, Chaban Y, Monnet-Saksouk J, Carrozza MJ, Sapountzi V, Selleck W, Huang J, Utley RT, Cramet M, Allard S, Cai G, Workman JL, Fried MG, Tan S, Cote J, Asturias FJ. Structure and nucleosome interaction of the yeast NuA4 and Piccolo-NuA4 histone acetyltransferase complexes. Nat Struct Mol Biol. 2011;18:1196–1203. doi: 10.1038/nsmb.2128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cockrell SK, Huffman JB, Toropova K, Conway JF, Homa FL. Residues of the UL25 protein of herpes simplex virus that are required for its stable interaction with capsids. J Virol. 2011;85(10):4875–4887. doi: 10.1128/JVI.00242-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flemming D, Thierbach K, Stelter P, Bottcher B, Hurt E. Precise mapping of subunits in multiprotein complexes by a versatile electron microscopy label. Nat Struct Mol Biol. 2010;17:775–778. doi: 10.1038/nsmb.1811. [DOI] [PubMed] [Google Scholar]
- He Y, Fang J, Taatjes DJ, Nogales E. Structural visualization of key steps in human transcription initiation. Nature. 2013;495:481–486. doi: 10.1038/nature11991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hutchins JR, Toyoda Y, Hegemann B, Poser I, Heriche JK, Sykora MM, Augsburg M, Hudecz O, Buschhorn BA, Bulkescher J, Conrad C, Comartin D, Schleiffer A, Sarov M, Pozniakovsky A, Slabicki MM, Schloissnig S, Steinmacher I, Leuschner M, Ssykor A, Lawo S, Pelletier L, Stark H, Nasmyth K, Ellenberg J, Durbin R, Buchholz F, Mechtler K, Hyman AA, Peters JM. Systematic analysis of human protein complexes identifies chromosome segregation proteins. Science. 2010;328:593–599. doi: 10.1126/science.1181348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kratz PA, Bottcher B, Nassal M. Native display of complete foreign protein domains on the surface of hepatitis B virus capsids. Proc Natl Acad Sci USA. 1999;96:1915–1920. doi: 10.1073/pnas.96.5.1915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lander GC, Estrin E, Matyskiela ME, Bashore C, Nogales E, Martin A. Complete subunit architecture of the proteasome regulatory particle. Nature. 2012;482:186–191. doi: 10.1038/nature10774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lau PW, Potter CS, Carragher B, MacRae IJ. DOLORS: versatile strategy for internal labeling and domain localization in electron microscopy. Structure. 2012;20:1995–2002. doi: 10.1016/j.str.2012.10.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Margueron R, Reinberg D. The polycomb complex PRC2 and its mark in life. Nature. 2011;469:343–349. doi: 10.1038/nature09784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nogales E, Scheres SH. Cryo-EM: a unique tool for the visualization of macromolecular complexity. Mol Cell. 2015;58:677–689. doi: 10.1016/j.molcel.2015.02.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nowak AJ, Alfieri C, Stirnimann CU, Rybin V, Baudin F, Ly-Hartig N, Lindner D, Muller CW. Chromatin-modifying complex component Nurf55/p55 associates with histones H3 and H4 and polycomb repressive complex 2 subunit Su(z)12 through partially overlapping binding sites. J Biol Chem. 2011;286:23388–23396. doi: 10.1074/jbc.M110.207407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts AJ, Numata N, Walker ML, Kato YS, Malkova B, Kon T, Ohkura R, Arisaka F, Knight PJ, Sutoh K, Burgess SA. AAA+ Ring and linker swing mechanism in the dynein motor. Cell. 2009;136(3):485–495. doi: 10.1016/j.cell.2008.11.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmitges FW, Prusty AB, Faty M, Stutzer A, Lingaraju GM, Aiwazian J, Sack R, Hess D, Li L, Zhou S, Bunker RD, Wirth U, Bouwmeester T, Bauer A, Ly-Hartig N, Zhao K, Chan H, Gu J, Gut H, Fischle W, Muller J, Thoma NH. Histone methylation by PRC2 is inhibited by active chromatin marks. Mol Cell. 2011;42:330–341. doi: 10.1016/j.molcel.2011.03.025. [DOI] [PubMed] [Google Scholar]
- Stroupe ME, Xu C, Goode BL, Grigorieff N. Actin filament labels for localizing protein components in large complexes viewed by electron microscopy. RNA. 2009;15:244–248. doi: 10.1261/rna.1313609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun J, Fernandez A, Riera A, Tognetti S, Yuan Z, Stillman B, Speck C, Li H. Structural and mechanistic insights into Mcm2–7 double-hexamer assembly and function. Genes Dev. 2014;28(20):2291–2303. doi: 10.1101/gad.242313.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiedenheft B, Lander GC, Zhou K, Jore MM, Brouns SJ, van der Oost J, Doudna JA, Nogales E. Structures of the RNA-guided surveillance complex from a bacterial immune system. Nature. 2011;477:486–489. doi: 10.1038/nature10402. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.