

Abstract
Analyses to ensure food safety and quality are more relevant now because of rapid changes in the quantity, diversity and mobility of food. Food-contamination must be determined to maintain health and up-hold laws, as well as for ethical and cultural concerns. Real-time polymerase chain reaction (RT-PCR), a rapid and inexpensive quantitative method to detect the presence of targeted DNA-segments in samples, helps in determining both accidental and intentional adulterations of foods by biological contaminants. This review presents recent developments in theory, techniques, and applications of RT-PCR in food analyses, RT-PCR addresses the limitations of traditional food analyses in terms of sensitivity, range of analytes, multiplexing ability, cost, time, and point-of-care applications. A range of targets, including species of plants or animals which are used as food ingredients, food-borne bacteria or viruses, genetically modified organisms, and allergens, even in highly processed foods can be identified by RT-PCR, even at very low concentrations. Microfluidic RT-PCR eliminates the separate sample-processing step to create opportunities for point-of-care analyses. We also cover the challenges related to using RT-PCR for food analyses, such as the need to further improve sample handling.
Electronic supplementary material
The online version of this article (doi:10.1007/s13197-016-2205-0) contains supplementary material, which is available to authorized users.
Keywords: RT-PCR, DNA, Food analysis, GMO detection, Halal, Kosher
Introduction
Food analysis is essential to ensure the authenticity of foods, which helps avoid incidents like European horse meat scandal, and helps people avoid foods containing specific ingredients, which may pose a health risks due to a medical condition, as well as to personal taste or to religious and cultural taboos (Ahmed et al. 2010). Conventional food analysis techniques include protein-based immunoassays, enzyme-linked immunosorbent assays (ELISAs), lateral flow strip/protein strip tests and culture-based assays to detect microbial contamination. Protein immunoassays, while easy and inexpensive, are prone to cross-reactivity with non-target proteins due to their dependence on target antibodies for target tissues/species (Ahmed et al. 2014, Lim et al. 2014, Saito et al. 2008). Immunoassays cannot discriminate among phylogenetically related species in food samples. Also, tissue-specific immunoassays fail to detect species if the tested products do not contain the target tissue (López-Calleja et al. 2014). In addition, for highly processed foods, immunoassays yield false-negative results due to protein denaturation at high temperatures (López-Calleja et al. 2014). According to Shrestha et al. (2010), different tissues vary in their levels of target proteins, which restricts the quantitative determination by protein-based immunoassays. On the other hand, culture-based assays are time consuming, inaccurate, and labor intensive. Consequently, real-time polymerase chain reaction (RT-PCR) has emerged as a method of choice for food analysis due to its rapid and sensitive species identification capabilities, especially since RT-PCR can be performed when target concentrations are very low (Shrestha et al. 2010). work on different aspects of food analysis using RT-PCR increased many fold (Fig. 1), which provides a clear indication of the increasing interest in the method and its wider adoption in food analyses. Conventional PCR is a cyclic process involving denaturation, annealing, and extension steps that doubles the target sequences after each cycle. In the case of RT-PCR, which is an automated process, no post-PCR processing is required to analyze the amplification process, as it monitors the increasing copy number of amplicon in real time after each cycle (Fig. 2). The measurement is made by using the amplification plot that charts changes in fluorescence intensity against cycle number (Fig. 3) and provides the fractional cycle number. Typically presented as Cp (Crossing point), Ct (threshold cycle) or Cq (Quantification cycle), the fractional cycle number denotes the number of PCR cycles at which the amount of amplification product crosses the threshold limit and generates a detectable fluorescence signal above the noise. The fractional cycle number can be used for absolute quantification based on known amounts of targets (Fig. 4a) or for relative quantification based on the comparison with known amounts of reference targets (Fig. 4b). The chances of post-PCR contact contamination decreases in RT-PCR. Unlike conventional amplification - which requires removal of PCR products for gel-electrophoresis - we can observe and analyze RT-PCR products without removing them from the instrument. A well-validated RT-PCR can even use the RT-PCR machine as a ‘one-stop’ analyzing instrument depending on the information needed (i.e., detecting amplification, detecting SNP’s). However, it should be noted that contaminations from other sources are still possible especially during the pre-PCR preparations (e.g., contaminations from the master mix reagents and/or master mix preparation area, contaminations during extractions etc.).
Fig. 1.

Summary of the research articles published in different journals. The data was retrieved from Scopus by using the key word “Real-Time PCR for Food Analysis”
Fig. 2.

Schematic representation of DNA amplification by real time PCR
Fig. 3.
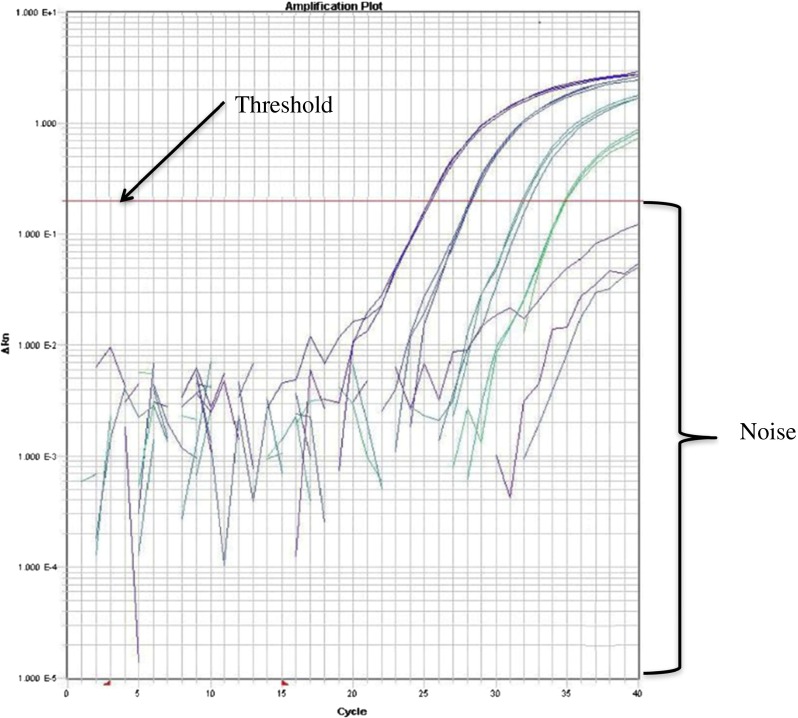
Amplification plot of tenfold serial-dilution of genetically modified maize Mon810 DNA using ABI Prism® 7900 SDS thermocycler (ABI) (adapted from Chaouachi et al., 2014b). The figure showed the threshold line which indicates the highest possible noise. The cycle number of the amplification curve at which it crosses the threshold gives the Ct/Cp/Cq value
Fig. 4.

Standard curves a single standard curve constructed from serial dilution C. jejuni (enumerated by plate counting method) used to absolutely quantify the number of cells captured by ligand-mediated capture (edited from Suh et al. 2014) b three standard curves constructed for the relative quantification of the percentages of beef and pork present in beef and pork binary mixture relative to the proportion of myostatin. (Iwobi et al. 2015)
Earlier, Shrestha et al. (2010) and Postollec et al. (2011) reviewed advancements in the RT-PCR technology in last decade. The present review, however, briefly summarizes the applications of RT-PCR in the detection of genetically modified organisms, prohibited biological ingredients, allergens, mutations, food-borne bacterial pathogen and other pathogens in various types of foods. Developments in RT-PCR platforms, instrumentation and chemistry has been addressed along with, advances in primer designing tools, the chemistry of sample preparations and data analysis, in the supplementary. The recent European horse meat scandal has been used as a case to establish the significance of food analyses and the viability of RT-PCR to combat similar challenges.
Non-specific dsDNA binding dye chemistry
A number of fluorescent dyes, with emission ranges between 487 and 560 nm, have been developed (Table 1), which show elevated fluorescence upon intercalation into double-stranded DNA (dsDNA) or by binding to the minor grooves of dsDNA compared with their unbound forms. These dyes offer an opportunity for the quantitative estimation of PCR amplicons, as the total fluorescence intensity changes in direct proportion to the amount of dsDNA in the sample. However, due to their explicit but indiscriminate intercalation or minor-groove binding into any dsDNA, such as primer-dimers in the sample, they may yield false positive results or overestimations of dsDNA, hence these dyes are not suitable for multiplexed PCR (Postollec et al. 2011). The melting curve analysis circumvents specificity issue by using the specific melting temperature (Tm) of primers (Bhagwat 2003). At the Tm, dsDNAs unwinds and release the dsDNA intercalating/minor-groove binding dyes to cause a rapid decrease in the fluorescence. Shorter non-specific products, such as primer-dimers, get denatured at lower temperatures and the target amplicons can be easily distinguished (Fig. 5a). The melting curve analysis allows for multiplexed analyses as multiple specific primer targets can be used if they have distinguishable and unique Tm (Fig. 5b). However, a quantitative melting point analysis is not possible by multiplex RT-PCR with dsDNA intercalating/minor-groove binding dyes since it cannot resolve multiple target sequences. Compared with probes, the non-specificity of dsDNA intercalating/minor-groove binding dyes allows them to be used with a wide range of primers without additional design or optimization (Postollec et al. 2011). SYBR Green I - the most widely available among the non-specific dsDNA minor-groove binding dyes - binds 100 times more easily into dsDNA than ethidium bromide. Upon binding to the minor-groove of dsDNA (Fig. 6a), its fluorescence increases more than 10,000-fold (Dragan et al. 2012). SYBR Green I’s fluorescence can be detected by most, if not all, commercial RT-PCR instruments due to its maximum emission at 520 nm (Rodríguez-Lázaro and Hernández 2013). However, at higher concentrations, the dye may inhibit PCR, thus further optimization of the amount of dye added is required to prevent inhibition (Monis et al. 2005). Commercially available dyes are also provided as part of an optimized master mix to overcome optimization challenges. However, SYBR Green dye I may shift the melting curve to a higher temperature (Ririe et al. 1997) and, at low concentrations, it may bind preferentially to long, G-C rich amplicons, making this dye unsuitable for multiplexing (Giglio et al. 2003). In addition, its occasional binding to ssDNA may lower the fluorescence level (Zipper et al. 2004). Despite these issues, SYBR Green I has remained popular, mainly due to the availability of improved proprietary formulations from different companies which address issues such as PCR inhibition, lower sensitivity, and poor fluorescence. Examples include SYBR® GreenER dye™ (Thermo Fisher Scientific, USA), BRYT Green™ Dye (Promega, USA), and the proprietary dye in the innuMIX qPCR Master Mix SyGreen (Analytik Jena, Germany). SYBR Green I alternatives have also been developed for RT-PCR applications. SYTO dyes, such as SYTO 13 and SYTO 82, and EvaGreen® (Fig. 6b) perform better, in terms of lower PCR inhibition, higher efficiencies, generally lower preferential binding temperatures, lower Tm shifts, and sharper and higher melting curve peaks (Gudnason et al. 2007; Eischeid 2011) than SYBR Green I over a larger and higher concentration ranges. However, except for EvaGreen, the use of these dyes remains limited in detection assays since they are not marketed for real-time PCR.
Table 1.
List of intercalating dyes with their absorption and emission wavelengths
| Dyes | Absorption/Emission (nm) | Company |
|---|---|---|
| SYBR Green I | 497/520 | Molecular Probes |
| SYBR® GreenER | 497/520 | Lifetechnologies |
| BRYT Green™ | 497/520 | Promega |
| SYTO 13 | 488/509 | Molecular Probes |
| SYTO 82 | 541/560 | Molecular Probes |
| EvaGreen | 500/530 | Biotium Inc. |
| LightCycler® 480 ResoLight | 450–500/487 | Roche |
Fig. 5.

a Melting curve analysis of RT-PCR of soybean oil (80 ml) (adapted from He et al. 2013) shows a lower Tm peak for the negative control which indicates the presence of primer-dimer. b Melting curve analysis of triplex RT-PCR assay for the detection of L. monocytogene, E. coli and S. typhimurium. Different Tm values for each target allows multiplex RT-PCR detection (adapted from Bhagwat 2003)
Fig. 6.
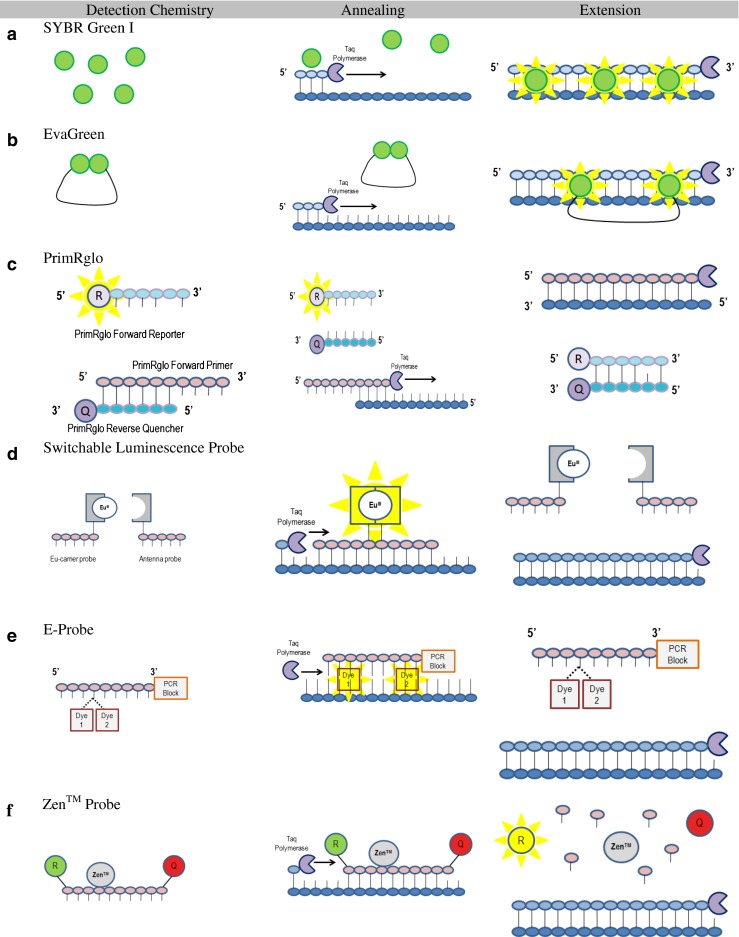
Schematics of RT-PCR fluorescence chemistries a SYBR Green I b EvaGreen c PrimRglo d Switchable luminescence probe e E-probe f Zen™ probe
Sequence-specific fluorescence detection chemistry
Hydrolysis probes, hybridization probes and fluorescent primers are examples of sequence-specific chemistries which detect the presence of the target sequence only. This specificity is usually achieved by designing additional oligonucleotide probes labeled with a fluorescence reporter and quenching system which bind to the target sequence. In the presence of target sequence, the reporter dye gets separated from the quencher, increases the fluorescence and allows the detection of target sequence. It also offers multiplexing ability by using different reporter dyes for each target (listed in Table 2) with distinguishable emissions. This approach offers unique fluorescent signal for each target compared to non-specific dsDNA binding dyes, and circumvent the necessity of melting curve analysis to differentiate the multiple targets by Tms unlike the non-specific dsDNA binding dyes chemistry as described in section 2.1. However, this type of chemistry requires users to design labeled oligonucleotides in addition to primer pairs, which can be difficult and very costly. Furthermore, multiplexing by using sequence specific chemistry is much difficult to design and optimize, compared to the unspecific dsDNA binding dye chemistry due to the necessity of multiple primers with the labeled oligonucleotides. The use of multiple oligonucleotides and primer pairs also leads to nonspecific amplifications which may affect the amplification efficiency that should remain constant for quantification purpose. This is also true for singular RT-PCR using sequence-specific chemistry. As polymerase digests oligonucleotide during PCR, reporter dye gets permanently separated from the quenching system and the fluorescence becomes indifferent to temperature changes. Therefore, melting curve analysis cannot be used for specific determination of nonspecific amplifications. It requires post-PCR gel electrophoresis which, however, increases the possibility of post-PCR cross-contaminations. In some sequence-specific chemistry, specifically hybridization probes can use melting curve analysis as the probes remained intact after the PCR cycles (Hanami et al. 2013). Several reviews have covered conventional and current sequence-specific chemistries (Buh Gašparič et al. 2010; Josefsen et al. 2012; Rodríguez-Lázaro and Hernández 2013). In this section, the prime focus will be on recent development in sequence-specific chemistries for RT-PCR.
Table 2.
List of reporter dyes with their absorption and emission wavelengths
| Reporter dyes | Absorption/Emission (nm) | Property |
|---|---|---|
| FAM™ | 494/518 | Life Technologies™ |
| HEX™ | 535/556 | Non-proprietary |
| TET™ | 522/539 | Life Technologies™ |
| VIC® | 538/554 | Life Technologies™ |
| ABY® | 568/583 | Life Technologies™ |
| JUN® | 606/618 | Life Technologies™ |
| NED™ | 546/575 | Life Technologies™ |
| Cy3™ | 550/570 | GE Healthcare |
| TEX 615 | 569/613 | N/A |
| Cy5™ | 649/670 | GE Healthcare |
| TYE™ 563 | 549/563 | IDT |
| TYE™ 665 | 645/665 | IDT |
| JOE™ | 520/548 | Life Technologies™ |
| TAMRA™ | 555/580 | Life Technologies™ |
| Texas Red® | 595/615 | Molecular Probes |
| CAL Fluor® Red 610 | 590/610 | BioSearch Technologies, Inc. |
| Alexa Fluor® 532 | 531/554 | Molecular Probes |
| Alexa Fluor® 647 | 650/668 | Molecular Probes |
| PET® | 556/595 | Life Technologies™ |
| ROX™ | 575/602 | Life Technologies™ |
Lai et al. (2012) developed a novel hybridization primers-based chemistry, the PrimRglo probe system, by employing a reverse primer, PrimRglo forward primer, PrimRglo forward reporter and PrimRglo reverse quencher (Fig. 6c). The PrimRglo forward primer and reporter are complementary sequences to the PrimRglo reverse quencher. A quencher is attached to the 3′-end of the PrimRglo forward primer and a reporter to the 5′-end, and a single-base mismatch is inserted in the PrimRglo forward reporter. As PCR starts, the PrimRglo forward primer preferentially binds to the PrimRglo reverse quencher due to the single nucleotide mismatch in the PrimRglo forward primer. This left the fluorescence from the PrimRglo forward reporter un-suppressed, thus high fluorescence level at the beginning of the PCR cycle. However, as the PCR program progresses, more copies of the PrimRglo forward primer is integrated into the amplicon and the competition for the PrimRglo reverse quencher wanes. Due to the increasing interaction between the PrimRglo forward reporter and the PrimRglo reverse quencher, the fluorescence decreases proportionally with DNA amplification. We measure fluorescence at each annealing stage. The PrimRglo system showed a comparable sensitivity to the TaqMan Probe assay with a simpler experimental design due to the omission of target-specific probes. The difficulty, however, is the length of PrimRglo forward primer (Primer-dimers) as it consists of two segments: forward primer and single-stranded sequence.
Switchable luminescence probes, developed by Lehmusvuori et al. (2012), consist of two adjacent non-fluorescent probes, one linked to lanthanide chelate (EU-carrier probe) and the other attached to the light-harvesting ligand (antenna probe) (Fig. 6d). During annealing, a highly fluorescent lanthanide chelate complex is formed as the two probes bring the lanthanide chelate and the antenna closer. During the extension stage, the chelate complex becomes destabilized. This probe offers lower detection limits with higher sensitivity in fewer amplification cycles owing to a very low background signal and highly target-specific fluorescence (Lehmusvuori et al. 2012).
A hybridization probe, named E-probe, has been developed which contains a 3′-end PCR-block and two dye moieties linked to the same nucleotide that do not fluoresce when the nucleotide is in the single-stranded form (Hanami et al. 2013) (Fig. 6e). During the annealing step, the dyes are separated from each other, intercalate into the dsDNA, and yield high levels of fluorescence. The intercalation further stabilizes the dsDNA complex and increases the Tm. As the temperature increases in preparation for the extension stage, the probe de-hybridizes, and, if it does not interfere with elongation of primers, the fluorescence signal is decreased. Consequently, measuring the amplification in real-time as well as the melting curve analysis is possible (Hanami et al. 2013).
Zen™ by Integrated DNA Technology (IDT) and Black Hole Quencher (BHQ) by BioSearch Technologies are commercialized examples of further advances in probe chemistry through the addition of internal quenchers to linear fluorescence resonance energy transfer probes. In Zen™, an additional quencher was added internally 9 base pairs from the reporter, whereas, the BHQ quencher is linked to a thymidine base placed about 10 base pairs from the fluorophore (Fig. 6f). Since BHQ is attached to a thymidine base, additional thymidine bases may also be added to the probe sequence. The addition of the internal quencher doubles the amount of quencher at work and ensures a lower background fluorescent signal due to the proximity of the quencher to the reporter, which results in a greater sensitivity and precision.
Advances in RT-PCR applications
Species identification
RT-PCR’s ability to detect and differentiate between phylogenetically related species in contrast with biochemical analyses, it has been used for species identification to check the adulteration, or the authenticity, of food products (Ng et al. 2014). In addition to the severe economic impact on the food industries, fraudulent substitutions of food products have medical, ethical, religious and cultural consequences. Recently, software applications for melting curve analysis and high resolution melting (HRM) analysis have improved automatic, multiplexed real-time PCR-based species identification protocols. HRM analysis, an improvement over the melting curve analysis, can detect mutations and polymorphisms. Vietina et al. (2013) employed software-based HRM to detect adulterants (maize oil, sunflower oil and hazelnut oil) of olive oil at a level as low as 10 % by exploiting changes in Tm due to SNPs in a 50 % conserved region of olive oil. Castiliego et al. (2015) reported the low cost simultaneous identification of seven different species of anglerfish (Lophius spp.) by using a software-based melting curve analysis with SYBR Green I dye chemistry. A SYBR GreenER™ (by Life Technologies)-based melting curve analysis traced the plant origin of edible oil DNA ‘fingerprints’ and provided a simple, cost effective approach to validate food labeling (Vietina et al. 2013).
Correct labeling helps to differentiate between adulteration due to contamination or fraud, for which, a quantification of the substitution is required at times. Drummond et al. (2013) used TaqMan MGB probe-based real-time PCR to analyze bovine adulterations in buffalo dairy and meat products. This was done by using calibration curves which were generated from a series of known bovine and buffalo mixtures, with two primer pairs and probes targeting each species instead of using eukaryotic primers to normalize the bovine DNA quantity. Another triplex TaqMan probe quantitative real-time PCR system measured the beef/pork DNA in mince meats to determine meat percentages with a 20 genome equivalent limit of detection and only 1.83 % measurement uncertainty (Iwobi et al. 2015). The primers and probes targeted beef and pork, as well as the housekeeping myostatin gene (sequence found in mammals and poultry). The quantity of the myostatin gene acted as the overall DNA content level against which the beef and pork percentages could be calculated. He et al. (2013) used nanoparticle gold colloids in real-time PCR to improve its efficiency and precision, especially for the amplification of low levels and degraded DNA, such as in the heavily processed edible oils. The addition of nanoparticles dramatically improves the efficiency of PCR and the sensitivity of the assay by taking advantage of high thermal conductivity and efficiency of nanoparticles. It significantly shortens the amplifications time by effectively homogenizing the PCR reactions temperatures (He et al. 2013). However, it should be noted that efficiency of the RT-PCR assay also depends on number of other factors such as sample preparation, optimizations of assay, presence of inhibitors etc.
Allergen detection
Exposure to dietary allergens, which pose severe health risks, can be prevented by correct food labeling. RT-PCR has proven to be useful in this area as well. A novel RT-PCR quantification method for peanut allergens co-amplified an internal competitive DNA (ICD) sequence with the peanut sequence by using separate primer pairs and probes (Holzhauser et al. 2014). The ICD sequence served as an internal amplification control (IAC) as well as an internal calibrator to normalize the tube-to-tube differences in PCR efficiency between extraction and amplification. It also replaced the standard curves for the quantification of peanuts in various food products with a single-point calibrator having known amounts/copies of peanut and ICD sequences in the mixtures. In a comparative assessment of three different real-time PCR quantification methods of sesame seeds, the ‘modified standard addition’ performed better over the other two methods (Luber et al. 2014a). The modified standard additions measured the amount of target in samples by adding varying known concentrations of targets to the individual tubes of the same sample. The sample curve plotted was dependent on the initial amount of target in sample, which could be calculated when compared to the external standard curve of target. The ‘modified standard addition’ was also successfully used in quantitative tetraplex real-time PCR of soybean, celery, white mustard and brown mustard (Luber et al. 2014b). Costa et al. (2012) successfully used RT-PCR to detect almond in a commercial food product and to distinguish almond from related species by amplifying a target gene encoding the Pru du 5 allergen in almond. Their HRM analysis detected traces of almond (0.005 % w/w) in almond/walnut mixtures. Additionally, a general fish allergen detection scheme by Herrer et al. (2014) took advantage of primers designed to target the specific 18S RNA gene sequence of fish. In comparison with commercial kits, the approach was free from cross-reactivity with other seafood products, meats and vegetables.
Identification of GMOs
The analysis of foods for screening or identifying GMOs is necessary because of legislative restrictions and public health concerns. Identification aims to distinguish and accurately quantify GMOs in food samples, while screening aims to separate GMO-containing foods from non-GMO-containing foods (Ahmed et al. 2009, Holst-Jensen, 2014, Bawa and Anilakumar 2013). Classical GMO testing requires Certified Reference Materials (CRMs) which are very difficult to obtain and produce GM tissues certified for single events (Holst-Jensen 2013; Wang et al. 2014b; Chaouachi et al. 2014b). Recently, as an easy and inexpensive method, an alternative to CRM plasmid-based reference materials has been developed, which is easy to produce or maintain (Wang et al. 2014b). Plasmid reference materials were successfully detected in GM rice lines using multiplex RT-PCR (Wang et al. 2014b) and in GM canola using duplex RT-PCR with dual plasmid calibrators (Chaouachi et al. 2014a). RT-PCR, in combination with next generation sequencing (Liang et al. 2014) or an integrated DNA walking strategy (Fraiture et al. 2014), is preferred as the first step in screening unauthorized GMOs from authorized GMOs. The combinatorial SYBR® Green real-time PCR screening (CoSYPS) strategy combined results from multiple RT-PCR reactions to detect multiple GM targets in a single plate run (Broeders et al. 2012; Barbau-Piednoir et al. 2014). CoSYPS validated six GMO targets in the laboratory and two species-specific detections with high sensitivity and specificity (Barbau-Piednoir et al. 2014).
Detection of mutations
It is imperative to detect mutations in food products from a public health perspective. Typically, allele-specific PCR is a method of choice in SNP genotyping due to its cost advantage over the conventional sequencing method (Sabui et al. 2012). This is based on the inability of polymerase (without proof-reading capability) to extend a complementary strand with a mismatch at the 3′-end of the primers. This allows the discrimination of alleles with single base difference (SNP). It is done by designing multiple primers with 3′-end specific to each allele (designed to target each alleles’ SNPs) and a single primer designed to target the conserve region of all alleles’ (Lyon et al. 2009). Typically, allele-specific PCR analyzes the target DNA in separate reactions, with each reaction containing primers specific to each allele (Lyon et al. 2009). Thus, amplification will only be observed if allele targeted by primers is present. A combination of SYBR Green and mismatch amplification mutation assay (MAMA), a variant of Allelic-specific based RT-PCR has been used to detect point mutations in the cssA and cssB genes of enterotoxigenic E. coli, which causes an allelic variation, CS6, a virulence factor (Sabui et al. 2012). In MAMA assay, forward primers have been specifically from the conserved region between cssB and cssA. whereas the 3′-end of their reverse primers have been designed at the location of mutations, with a mismatch adjacent to aforementioned 3′-end further improve specificity (Sabui et al. 2012). It presented a cost-effective and specific method of detecting allelic variants of CS6 than the conventional and more expensive DNA sequencing.
However, allele-specific RT-PCR method of detection required the design of multiple primers and the SNPs of each allele variants are detected concurrently in separate reactions. Thus, this method requires more reagents and multiple tubes to detect the allele variants. HRM analysis software available with most recent RT-PCR instruments is a simpler and more cost effective to screen SNPs of these allele variants. It is an improvement over the melting curve analysis which detects the temperature difference caused by single nucleotide variation or SNPs. HRM analysis detects allele variants in single tube with one set of primers.
Cai et al. (2013) used the HRM RT-PCR approach described above to simultaneously detect and differentially genotyped the polymorphisms between strains Cronobacter muytjensii and Cronobacter sakazaki present in powdered infant formula (Fig. 7).
Fig. 7.

Alignment of target sequences and HRM analysis of C. sazakii and C. muytjensii a the alignment of target sequences (1) C. muytjensii and (2) C. sazakii showed three bases sequence variation between the two strains b HRM analysis of C. sazakii, C. muytjensii, and 11 Cronobacter spp. isolates detected and accurately differentiated the three bases variations of the target sequence of C. sazakii and C. muytjensii
Matsuda et al. (2012) has applied HRM RT-PCR to detect and map SNPs linked to the hybrid necrosis gene, Net2, of tetraploid wheat and wild Aegilops tauschii. The presence of polymorphisms has also allowed RT-PCR to discriminate between two related species, cow and buffalo, in the milk used for making mozzarella cheese (Dalmasso et al. 2011) by targeting the SNPs between the conserved regions of buffalo and cow mitochondrial cytochrome b gene with single primer pairs and two TaqMan MGB probes.
TaqMan probe real-time PCR also detected a mutation that causes the high oleic content in peanuts, which is preferable due to its stability and health benefits (Barkley et al. 2011). This method differentiated between wild and mutant types, allowing breeders to produce high-oleic peanut butter.
Bacterial detection
An important application of RT-PCR is the quantitative and qualitative analyses of bacterial food pathogens (Singh et al. 2012, Tabit 2016). RT-PCR-based detection is a rapid alternative to the conventional culture-based methods. However, due to the pre-enrichment of samples, RT-PCR detects the amount of pathogen in an enriched sample instead of the original sample. Recent studies have circumvented the pre-enrichment phase for the detection and quantification of food pathogens. Lee and Levin (2011) treated lettuce surfaces with bentonite-coated activated charcoal to increase the recovery of E. coli O157:H7 and to remove PCR inhibitors, which allowed using the real-time PCR assay without pre-enrichment. Additionally, a pre-treatment with β-cyclodextrin and then with milk protein-coated activated carbon allowed Opet and co-workers (2014) to quantify Salmonella enterica ser. Enteritidis from ground beef sample without pre-enrichment (Opet et al., 2014).
The use of immunomagnetic separation (IMS) during sample preparation has lowered the pre-enrichment time for Alicyclobacillus acidoterrestris in fruit products to 30 min and allowed detection within 2–3 h (Wang et al. 2014a). In another development, Suh et al. (2014) replaced antibodies with nucleic acid aptamers for the magnetic capture assay to achieve lower analysis costs, analytical stability and easy manipulation. The use of aptamer-based magnetic capture real-time PCR showed better sensitivity in comparison with IMS real-time PCR assay for Campylobacter jejuni (Suh et al. 2014). Wang et al. (2014c) have combined IMS real-time PCR with sodium deoxycholate (SD) and propidium monoazide (PMA) to detect viable E. coli O15:H7 in milk with higher sensitivity than pure culture within 5 h. SD enhances the permeability of dead or injured cells, which PMA penetrates and binds to the DNA, eliminating overestimations of bacteria in samples. Thus, a pre-treatment with SD increases the efficiency of PMA (Wang et al. 2014c).
Virus detection
Multiplex real-time PCR, along with an HRM analysis, detected white spot virus, yellow-head virus and Penaeus monodon desovirus in paneid shrimp (Panichareon et al. 2011). The HRM analysis allowed these three viruses to be differentiated by their distinct Tm values, without any probe (Panichareon et al. 2011). Hematopoietic necrosis virus infects wild and farm-bred salmon populations. This virus was qualitatively detected by an RT-PCR assay that targeted the conserved region of the viral nucleocapsid gene in salmon with 100 % sensitivity and specificity (Purcell et al. 2013). Pérez et al. (2011) developed a SYBR Green reverse transcriptase RT-PCR assay for the classical swine fever virus and validated it on two different real-time PCR platforms. They also included a post-melting curve analysis to separate specific amplicons from non-specific amplifications (Pérez et al. 2011). Amer and coworkers (2011) reported a novel two-step reverse transcriptase real-time PCR assay to detect bovine coronavirus using SYBR Green I dye. An internal amplification control was also included in the assay to normalize the reaction. The bovine coronavirus and internal amplification control amplicons were differentiated from each other by using their distinct Tm in a melting curve analysis. IACs have also been developed for several food-related viruses, such as bovine polymavirus, hepatitis A and E, human and porcine adenovirus, human norovirus genogroup I and II, and also murine norovirus (Diez-Valcarce et al. 2011). IACs did not affect the limit of detection for the target viruses, and were able to detect and identify assay failures and the poor viral nucleic acid extractions. As mentioned earlier, the development of IACs is not only essential to normalize the reactions, but also to ensure that negative results are not due to reaction failures.
European horse meat scandal
Food-grade horse meat is not a health risk, rather it is a suitable alternative to beef products (Lorenzo 2013; Lorenzo et al. 2014). Yet, fraudulent labeling raises the issue of the authenticity and traceability of products, which is a huge concern among consumers (Boeri et al. 2014). Moreover, the traceability of horse meat from ‘farm to table’ is crucial since horses treated with veterinary drug phenylbutazone are not suitable for human consumption. In the beginning of 2013, the Food Safety Authority of Ireland (FSAI) published a report on the adulteration of beef burger products by horse meat and pork. The report initiated what we now call the European horse meat scandal. In FSAI tests, almost all of the samples contained very low levels of horse meat and some contained as much as 29 % horse meat. Subsequently, due to the enhanced concern, many commercial beef products were tested and most of the commercial beef products were positive for horse meat. Interestingly, some of the samples were 100 % horse meat.
The adulteration of beef products is an infringement upon consumers’ rights and can have severe economic impacts (Yamoah 2014). However, the horse meat scandal proved the importance of RT-PCR for the detection and quantitative analysis of product adulteration with other species. RT-PCR can determine if the adulteration is not accidental but fraudulent. RT-PCR assays provide both qualitative and quantitative analyses on sausage and meat samples (Köppel et al. 2011). Several methods of discriminating horse meat from beef have been developed, such as rapid Raman spectroscopy (Boyacı et al. 2014), ultra-performance liquid chromatography (Di Giuseppe et al. 2015), high performance liquid chromatography-Tandem mass spectrometry (von Bargen et al. 2014), and mid-infrared spectroscopy with multivariate analysis and soft independent modeling of class analogies (SIMCA) (Meza-Márquez et al. 2010). However, except SIMCA, all of the other methods are qualitative.
Future trends
The fast, portable, inexpensive and simple food analysis methods especially to control the ever increasing possibility of a food pathogen outbreak are in great demand. Recent research has focused on designing point-of-care RT-PCR systems for food analysis. A successful combination of microfluidics and RT-PCR into a single chip was developed by Verdoy et al. (2012) to detect Salmonella in 35 min. They injected lysed samples into the microfluidic chip and captured genomic DNAs magnetically with super paramagnetic beads. The subsequent addition of the PCR mixture into the PCR chamber amplified the sample, which was measured by probing fluorescence at the annealing step in every cycle. The platform performed similar to conventional real-time PCR. However, depending on the type of sample, adjustments during sample preparation may be required. Furutani et al. (2014) reduced the run time to 8 min for the detection of E. coli with a high sensitivity of 4 cells μL−1 using a microfluidic RT-PCR chip coated with PCR reagents. The multiplexed detection of waterborne pathogens on a microfluidic RT-PCR chip has already been achieved (Ramalingam et al. 2010).
The RAZOR EX Instrument is a commercially available, non-microfluidic portable RT-PCR bio-detection system (BioFire Defense, Salt Lake City, Utah). It detected influenza virus A in clinical samples within 90 min run time, including sample preparation (Mölsä et al. 2012). According to the Biofire Defense website (http://biofiredefense.com), the platform can be used to detect Campylobacter, L. monocytogenes and Salmonella.
There have been improvements in the sample preparation area as well. For example, immunomagnetic capture with antibodies or aptamers (Cengiz Ozalp et al. 2014; Ma et al. 2014) has been designed to reduce, or completely eliminate, the enrichment step, particularly for bacterial detection. The use of activated carbon to remove PCR inhibitors from the samples eliminated the pre-enrichment phase (Lee and Levin 2011; Opet and Levin 2014). Similarly, the inclusion of gold nanoparticles in the form of colloids in the RT-PCR assay increased efficiency, precision and sensitivity, as well as reducing the detection time (He et al. 2013).
In the discussion on future trends in food analysis digital PCR is worth mentioning. It is an end-point technique that allows absolute quantification without the construction of standard curves. Digital PCR technique involves sub-dividing the DNA sample (with master-mix) into hundreds to thousands of individual units run concurrently with each other. The individual units are then treated either as negative reactions (no DNA target present) or positive reactions (DNA target presence) (Baker 2012). The fraction of negative reactions is used for absolute quantification of initial DNA concentration of sample as it follows the Poisson distributions. It is a more precise method of DNA/RNA quantification. However, since it is still fairly new technique, further validation is required before it can become a viable replacement of RT-PCR as the gold standard of detection and quantification of DNA/RNA for food analysis.”
Conclusion
Globalization, population growth and affluence are responsible for changes in the quantity of food consumed, diversity of foods and the mobility of foods on an unforeseen scale. Therefore, the need to analyze foods qualitatively and quantitatively is becoming increasingly important from the perspectives of food standardization, authentication and certification. Additionally, because of medical, religious and cultural reasons - not all foods are suitable for consumption by all people. This necessitates the confirmation of the species composition of food ingredients and their respective amounts. Moreover, food borne pathogens pose health risks due to the improper handling and storage of foods, which demonstrates a need for pathogen testing. Traditional food testing methods are slow, costly, less sensitive, inaccurate and not amenable to multiplexing. The need to pre-treat or enrich samples before analysis, lower detection limits, non-reproducible results and the lack of portability for point-of-care applications are among the drawbacks of many existing platforms (Safavieh et al. 2014, 2015). There are emerging solutions to all of these limitations. Real time-PCR, with continued improvements in instrumentation and chemistry, is becoming a ubiquitous method in the analysis of food, to detect pathogens, such as viruses or bacteria, to identify allergens in food, and to detect what species of plants or animals are in a food, and in what proportion, with high specificity. A range of fluorescent probe chemistries are now available and nanoparticles are opening up new opportunities in RT-PCR, owing to their higher sensitivity and short detection times. However, there are many challenges yet to be addressed. Recent progress in RT-PCR analyses, such as microfluidic integrations, presents a positive outlook for gene-based point-of-care food analysis at a much lower cost in the near future.
Electronic supplementary material
(DOCX 58 kb)
References
- Ahmed MU, Hasan Q, Hossain M, Masato S, Tamiya E. Meat species identification based on the loop mediated isothermal amplification and electrochemical DNA sensor. Food Control. 2010;21:599–605. doi: 10.1016/j.foodcont.2009.09.001. [DOI] [Google Scholar]
- Ahmed MU, Saaem I, Wu PC, Brown AS. Personalized diagnostics and biosensors: a review of the biology and technology needed for personalized medicine. Crit Rev Biotechnol. 2014;34:180–196. doi: 10.3109/07388551.2013.778228. [DOI] [PubMed] [Google Scholar]
- Ahmed MU, Saito M, Hossain M, Rao M, Furui S, Hino A, Takamura Y, Takagi M, Tamiya E. Electrochemical genosensor for rapid detection of GMO using loop-mediated isothermal amplification. Analyst. 2009;134:966–972. doi: 10.1039/b812569d. [DOI] [PubMed] [Google Scholar]
- Amer HM, Almajhdi FN. Development of SYBR green I based real-time RT-PCR assay for detection and quantification of bovine coronavirus. Mol Cell Probes. 2011;25:101–107. doi: 10.1016/j.mcp.2011.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker M. Digital PCR hits its stride. Nat Methods. 2012;9:541–544. doi: 10.1038/nmeth.2027. [DOI] [Google Scholar]
- Barbau-Piednoir E, Stragier P, Koosens N, Mazzara M, Savini C, Van den Eede G, Van den Bulcke M. Inter-laboratory testing of GMO detection by combinatory SYBR® green PCR Screening (CoSYPS) Food Anal Methods. 2014;7:1719–1728. [Google Scholar]
- Barkley, N., Wang, M., and Pittman, R. (2011). A real-time PCR genotyping assay to detect FAD2A SNPs in peanuts (Arachis hypogaea L.). Electron J Biotechnol.14(1).
- Bawa AS, Anilakumar KR. Genetically modified foods: safety, risks and public concerns—a review. J Food Sci Technol. 2013;50(6):1035–1046. doi: 10.1007/s13197-012-0899-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhagwat AA. Simultaneous detection of escheria coli O157:H7, Listeria Monocytogene and Salmonella Strains by Real-Time PCR. Int J Food Microbiol. 2003;84:217–224. doi: 10.1016/S0168-1605(02)00481-6. [DOI] [PubMed] [Google Scholar]
- Boeri, M., Brown, H., and Longo, A. (2014). The implications across Europe of the ‘horse meat scandal’ on the monetary value of meat authenticity and food safety in ready to heat lasagne: evidence from six countries. Paper presented at the EAAE 2014 Congress ‘agri-food and rural innovations for healthier societies’, 26–29 august, Ljubljana, Slovenia.
- Boyacı IH, Temiz HT, Uysal RS, Velioglu HM, Yadegari RJ, Rishkan MM. A novel method for discrimination of beef and horsemeat using Raman spectroscopy. Food Chem. 2014;148:37–41. doi: 10.1016/j.foodchem.2013.10.006. [DOI] [PubMed] [Google Scholar]
- Broeders SRM, De Keersmaecker SCJ, Roosen NH (2012) How to deal with the upcoming challenges in GMO detection in food and feed. J Biomed Biotechnol 2012. doi:10.1155/2012/402418 [DOI] [PMC free article] [PubMed]
- Buh Gašparič M, Tengs T, La Paz JL, Holst-Jensen A, Pla M, Esteve T, Žel J, Gruden K. Comparison of nine different real-time PCR chemistries for qualitative and quantitative applications in GMO detection. Anal Bioanal Chem. 2010;369:2023–2029. doi: 10.1007/s00216-009-3418-0. [DOI] [PubMed] [Google Scholar]
- Cai X, Yu H, Ruan Z, Yang L, Bai J, Qiu D, Jian Z, Xiao Y, Yang J, Le TH, Zhu X (2013) Rapid detection and simultaneous genotyping of Cronobacter spp. (formerly Enterobacter sakazakii) in powdered infant formula using real-time PCR and high resolution melting (HRM) analysis. PLoS One 8(6) e67082. [DOI] [PMC free article] [PubMed]
- Castiliego L, Armani A, Tinacci L, Gianfaldoni D, Guidi A. Two alternative multiplex PCRs for identification of the seven species of anglerfish (lophius spp.) using an end-point or melting curve analysis real-time protocol. Food Chem. 2015;166:1–9. doi: 10.1016/j.foodchem.2014.06.014. [DOI] [PubMed] [Google Scholar]
- Cengiz Ozalp V, Bayramoglu G, Kavruk M, Keskin BB, Oktem HA, Yakup Arica M. Pathogen detection by core-shell type aptamer-magnetic preconcentration coupled to real-time PCR. Anal Biochem. 2014;447:119–125. doi: 10.1016/j.ab.2013.11.022. [DOI] [PubMed] [Google Scholar]
- Chaouachi M, Hafsa AB, Nabi N, Zellama MS, Said K. A new dual plasmid calibrator for the quantification of the construct specific GM canola oxy-235 with duplex real-time PCR. Food Chem. 2014;145:49–56. doi: 10.1016/j.foodchem.2013.08.026. [DOI] [PubMed] [Google Scholar]
- Chaouachi M, Zellama MS, Nabi N, Hafsa AB, Saïd K. Molecular identification of four genetically modified maize (Bt11, Bt176, Mon 810 and T25) by Duplex Quantitative Real-time PCR. Food Anal Methods. 2014;7(1):224–233. doi: 10.1007/s12161-013-9667-8. [DOI] [Google Scholar]
- Costa J, Mafra I, Beatriz M, Oliviera PP. High resolution melting analysis as new approach to detect almond DNA encoding for pru du 5 allergen in food. Food Chem. 2012;133:1062–1069. doi: 10.1016/j.foodchem.2012.01.077. [DOI] [Google Scholar]
- Dalmasso A, Civera T, La Neve F, Boterro MT. Simultaneous detection of cow and buffalo milk in mozzarella cheese by real-time PCR assay. Food Chem. 2011;124:362–366. doi: 10.1016/j.foodchem.2010.06.017. [DOI] [Google Scholar]
- Diez-Valcarce M, Kovač K, Cook N, Rodríguez-Lázaro D, Hernández M. Construction and analytical application of internal amplification controls (IAC) for detection of food supply chain-relevant viruses by real-time PCR-based assay. Food Anal Methods. 2011;4:437–445. doi: 10.1007/s12161-011-9224-2. [DOI] [Google Scholar]
- Di Giuseppe AMA, Giarretta N, Lippert M, Severino V, Di Maro A. An improved UPLC method for the detection of undeclared horse meat addition by using myoglobin as molecular marker. Food Chem. 2015;169:241–245. doi: 10.1016/j.foodchem.2014.07.126. [DOI] [PubMed] [Google Scholar]
- Dragan AI, Pavlovic R, McGivney JB, Casas-Finet JR, Bishop ES, Strouse RJ, Scenerman MA, Geddes CD. SYBR green I: fluorescence properties and interaction with DNA. J Fluoresc. 2012;22:1189–1199. doi: 10.1007/s10895-012-1059-8. [DOI] [PubMed] [Google Scholar]
- Drummond MG, Brasil BSAF, Dalsecco LS, Brasil KSAF, Teixeira LV, Oliveira DAA. A versatile real-time PCR method to quantify bovine contamination in buffalo products. Food Control. 2013;29:131–137. doi: 10.1016/j.foodcont.2012.05.051. [DOI] [Google Scholar]
- Eischeid AC. SYTO dyes and EvaGreen outperform SYBR green in real-time PCR. BMC Res Notes. 2011;4:263. doi: 10.1186/1756-0500-4-263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fraiture M, Herman P, Taverniers I, De Loose M, Deforce D, Roosens NH. An innovative and intergrated approach based on DNA waling to identify unauthorized GMOs. Food Chem. 2014;147:60–69. doi: 10.1016/j.foodchem.2013.09.112. [DOI] [PubMed] [Google Scholar]
- Furutani S, Naruishi N, Saito MA, Tamiya E, Fuchiwaka Y, Nagai H. Rapid and highly sensitive detection by real-time polymerase chain reaction using a chip coated with its reagents. Anal Sci. 2014;30:569–574. doi: 10.2116/analsci.30.569. [DOI] [PubMed] [Google Scholar]
- Giglio S, Monis PT, Saint CP. Demonstration of preferential binding of SYBR green I to specific DNA fragments in real-time multiplex PCR. Nucleic Acids Res. 2003;31(22) doi: 10.1093/nar/gng135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gudnason H, Dufva M, Bang DD, Wolff A. Comparison of multiple DNA dyes for real-time PCR: effects of dye concentration and sequence composition on DNA amplification and melting temperature. Nucleic Acids Res. 2007;35(19) doi: 10.1093/nar/gkm671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanami T, Delobel D, Kanamori H, Tanaka Y, Kimura Y, Nakasone A, Soma T, Hayashizaki Y, Usui K, Harbers M. Eprobe mediated real-time PCR monitoring and melting curve analysis. PLoS One. 2013;8(8) doi: 10.1371/journal.pone.0070942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He J, Xu W, Shang P, Mei X, Tian W. Development and optimization of an efficient method to detect the authenticity of edible oils. Food Control. 2013;31:71–79. doi: 10.1016/j.foodcont.2012.07.001. [DOI] [Google Scholar]
- Herrer B, Vietes JM, Montserrat E. Development of an in-house fast real-time PCR method for detection of fish allergen in foods and comparison with a commercial kit. Food Chem. 2014;151:415–420. doi: 10.1016/j.foodchem.2013.11.042. [DOI] [PubMed] [Google Scholar]
- Holst-Jensen A. Real-time PCR analysis of genetically modified organisms. Eds.: Caister Academic Press, Norfolk; 2013. [Google Scholar]
- Holzhauser T, Kornelia K, Janise A, Röder M. Matrix-normalised quantification of species by threshold-calibrated competitive real-time PCR: allergenic peanut in food as one example. Food Chem. 2014;163:68–76. doi: 10.1016/j.foodchem.2014.04.081. [DOI] [PubMed] [Google Scholar]
- Iwobi A, Sebah D, Kraemer I, Losher C, Fischer G, Busch U, Huber I. A multiplex real-time PCR method for the quantification of beef & pork fractions in minced meat. Food Chem. 2015;169:305–313. doi: 10.1016/j.foodchem.2014.07.139. [DOI] [PubMed] [Google Scholar]
- Josefsen MH, Löfström C, Hansen T, Reynisson E, Hoorfar J (2012) Instrumentation and fluorescent chemistries used in quantitative polymerase chain reaction. In: quantitative real-time PCR in applied microbiology, pp. 27–52. fillion, M. edn. Caister Academic Press, Norfolk
- Köppel R, Ruf J, Rentsch J. Multiplex real-time PCR for the detection and quantification of DNA from beef, pork, horse and sheep. Eur Food Res Technol. 2011;232(1):151–155. doi: 10.1007/s00217-010-1371-y. [DOI] [Google Scholar]
- Lee J, Levin RE. Detection of 5 CFU/g of Escherichia coli O157:H7 on lettuce using activated charcoal and real-time PCR without enrichment. Food Microbiol. 2011;28:562–567. doi: 10.1016/j.fm.2010.11.007. [DOI] [PubMed] [Google Scholar]
- Lai R, Liang F, Pearson D, Barnett G, Whiley D, Sloots T, Barnard RT, Corrie SR. PrimGlo: a multiplex quantitative real-time polymerase chain reaction system for nucleic acid detection. Anal Biochem. 2012;422:89–95. doi: 10.1016/j.ab.2011.12.038. [DOI] [PubMed] [Google Scholar]
- Lehmusvuori A, Karhunen U, Tapio A, Lamminmäki U, Soukka T. High-performance closed-tube PCR based on switchable luminescence probes. Anal Chim Acta. 2012;731:88–92. doi: 10.1016/j.aca.2012.04.027. [DOI] [PubMed] [Google Scholar]
- Liang C, van Dijk JP, Scholtens IM, Staats M, Prins TW, Voorhuikzen MM, da Silva AM, Arisi ACM, den Dunnene JT, Kok EJ. Detecting authorized and unauthorized genetically modified organisms containing vip3A by real-time PCR and next generation sequencing. Anal Bioanal Chem. 2014;406(11):2603–2611. doi: 10.1007/s00216-014-7667-1. [DOI] [PubMed] [Google Scholar]
- Lim SA, Yoshikawa H, Tamiya E, Yasin HM, Ahmed MU. A highly sensitive gold nanoparticle bioprobe based electrochemical immunosensor using screen printed graphene biochip. RSC Adv. 2014;4(102):58460–58466. doi: 10.1039/C4RA11066H. [DOI] [Google Scholar]
- López-Calleja IM, de la Cruz S, Pegels N, González I, Martín R, García T. Sensitive and specific detection of almond (Prunus dulcis) in commercial food products by real-time PCR. LWT Food Sci Technol. 2014;56:31–39. doi: 10.1016/j.lwt.2013.10.039. [DOI] [Google Scholar]
- Lorenzo JM. Horsemeat as a source of valuable fatty acids. Eur J Lipid Sci Technol. 2013;115:473–474. doi: 10.1002/ejlt.201300137. [DOI] [Google Scholar]
- Lorenzo JM, Sarriés MV, Tateo A, Polidori P, Franco D, Lanza M. Carcass characteristics, meat quality and nutritional value of horsemeat: A review. Meat Sci. 2014;96:1478–1488. doi: 10.1016/j.meatsci.2013.12.006. [DOI] [PubMed] [Google Scholar]
- Luber F, Demmel A, Herbet D, Husken A, Hupfer C, Huber I, Busch U, Karl-Heinz E. Comparative assessment of DNA-base approaches for quantification of food allergens. Food Chem. 2014;160:104–111. doi: 10.1016/j.foodchem.2014.03.077. [DOI] [PubMed] [Google Scholar]
- Luber F, Demmel A, Katrin P, Ulrich B, Karl-Heinz E. Simultaneously quantification of the food allergens soybean, celery, white mustard, and brown mustard via combination of tetraplex real-time PCR and standard addition. Food Control. 2014;47:246–253. doi: 10.1016/j.foodcont.2014.06.047. [DOI] [Google Scholar]
- Lyon E, Mao R, Swensen J. Mutation detection by real-time PCR. In: Logan JMJ, Edwards KJ, Saunders NA, editors. Real-time PCR: current technology and applications. Norfolk: Caister Academic Press; 2009. pp. 156–157. [Google Scholar]
- Ma K, Deng Y, Bai Y, Xu D, Chen E, Wu H, Li B. Rapid and simultaneous detection of salmonella, shigella, and Staphylococcus aureus in fresh pork using a multiplex real-time PCR assay based on immunomagnetic separation. Food Control. 2014;42:87–93. doi: 10.1016/j.foodcont.2014.01.042. [DOI] [Google Scholar]
- Matsuda R, Iehisa JCM, Takumi S. Application of real-time PCR0based detection for mapping of Net2, a causal D-genome gene for hybrid necrosis in interspecies crosses between tetraploid wheat and aehilops tauschii. Genes Genet Syst. 2012;87:137–143. doi: 10.1266/ggs.87.137. [DOI] [PubMed] [Google Scholar]
- Meza-Márquez OG, Gallardo-Velázquez T, Osorio-Revilla G. Application of mid-infrared spectroscopy with multivariate analysis and soft independent modeling of class analogies (SIMCA) for the detection of adulterants in minced beef. Meat Sci. 2010;86:511–519. doi: 10.1016/j.meatsci.2010.05.044. [DOI] [PubMed] [Google Scholar]
- Mölsä M, Koskela KA, Rönkkö E, Ikonen N, Ziegler T, Nikkari S. Detection of influenza A viruses with a portable real-time PCR instrument. J Virol Methods. 2012;181:188–191. doi: 10.1016/j.jviromet.2012.02.001. [DOI] [PubMed] [Google Scholar]
- Monis PT, Giglio S, Saint CP. Comparison of SYTO9 and SYBR green I for real-time polymerase chain reaction and investigation of the effect of dye concentration on amplification and DNA melting curve analysis. Anal Biochem. 2005;340:24–34. doi: 10.1016/j.ab.2005.01.046. [DOI] [PubMed] [Google Scholar]
- Ng J, Satkoski J, Premasuthan A, Kanthaswamy S. A nuclear DNA-based species determination and DNA quantification assay for common poultry species. J Food Sci Technol. 2014;51(12):4060–4065. doi: 10.1007/s13197-012-0893-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Opet NJ, Levin RE (2014) Use of β-cyclodextrin and activated carbon for quantification of salmonella enterica ser. Enteritidis from Ground Beef by Conventionl PCR without Enrichment Food Microbiol 38:75–79 [DOI] [PubMed]
- Panichareon B, Khawsak P, Deesukon W, Sukhumsirichart W. Multiplex real-time PCR and high resolution melting analysis for detection of white spot syndrome virus, yellow-head virus and Penaeus monodon densovirus in penaeid shrimp. J Virol Methods. 2011;178:16–21. doi: 10.1016/j.jviromet.2011.07.010. [DOI] [PubMed] [Google Scholar]
- Pérez LJ, de Arce HD, Tarradas J, Rosell R, Perera CL, Muñoz M, Frías MT, Nuñez JI, Ganges L. Development and validation of a novel SYBR green real-time RT-PCR assay for the detection of classical swine fever virus evaluated on different real-time PCR platforms. J Virol Methods. 2011;174:53–59. doi: 10.1016/j.jviromet.2011.03.022. [DOI] [PubMed] [Google Scholar]
- Postollec F, Falentin H, Pavan S, Combrisson J, Sohier D. Recent advances in quantitative PCR (qPCR) applications in food microbiology. Food Microbiol. 2011;28:848–861. doi: 10.1016/j.fm.2011.02.008. [DOI] [PubMed] [Google Scholar]
- Purcell MK, Thompson RL, Garver KA, Hawley LM, Batts WN, Sprague L, Sampson C, Winton JR. Universal reverse-transcriptase real-time PCR for infectious hematopoietic necrosis virus (IHNV) Dis Aquat Org. 2013;106:103–115. doi: 10.3354/dao02644. [DOI] [PubMed] [Google Scholar]
- Ramalingam N, Rui Z, Liu H, Dai C, Kaushik R, Ranaharika B, Gong H. Real-time PCR-based microfluidic array chip for simultaneous detection of multiple waterborne pathogens. Sensors Actuators B. 2010;145:543–552. doi: 10.1016/j.snb.2009.11.025. [DOI] [Google Scholar]
- Ririe KM, Rasmussen RP, Wittwer CT. Product differentiation by analysis of DNA melting curves during polymerase chain reaction. Anal Biochem. 1997;245:154–160. doi: 10.1006/abio.1996.9916. [DOI] [PubMed] [Google Scholar]
- Rodríguez-Lázaro D, Hernández M (2013) Introduction to the real-time polymerase chain reaction. In: real-time PCR in food science, Ch. 1. rodriguez-Lázaro, D. edn. Caister Academic Press, Norfolk
- Sabui S, Dutta S, Debnath A, Ghosh A, Hamabata T, Rajendran K, Ramamurthy T, Nataro JP, Sur D, Levine MM, Chatterjee NS. Real-time PCR-based mismatch amplification mutation assay for specific detection of CS6-expressing allelic variants of enterotoxigenic Escherichia coli and its application in assessing diarrheal cases and asymptomatic controls. J Clin Microbiol. 2012;50(4):1308–1312. doi: 10.1128/JCM.05424-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Safavieh M, Ahmed MU, Ng A, Zourob M. High throughput real time electrochemical monitoring of LAMP using a redox for pathogenic bacteria detection. Biosens Bioelectron. 2014;15(58):101–106. doi: 10.1016/j.bios.2014.02.002. [DOI] [PubMed] [Google Scholar]
- Safavieh, M., Nahar S., Zourob, M., and Ahmed, M. U. (2015). Microfluidic biosensors for high throughput screening of pathogens in food. In: High Throughput Screening for Food Safety Assessment, Biosensor Technologies, Hyperspectral Imaging and Practical Applications, pp. 327–357, Bhunia, A.K., Kim, M.S., and Taitt, C.R., Woodhead Publishing Series in Food Science, Technology and Nutrition, Cambridge. doi:10.1016/B978-0-85709-801-6.00015-0.
- Saito M, Kitsunai M, Ahmed MU, Sugiyama S, Tamiya E. Label-free electrochemical detection for food allergen using screen printed carbon electrode. Electrochem. 2008;76(8):606–609. doi: 10.5796/electrochemistry.76.606. [DOI] [Google Scholar]
- Shrestha HK, Hwu K, Chang M. Advances in detection of geneticallyengineered crops by multiplex polymerase chain reaction methods. Trends Food SciTechnol. 2010;21:442–452. doi: 10.1016/j.tifs.2010.06.004. [DOI] [Google Scholar]
- Singh J, Batish VK, Grover S. Simultaneous detection of listeria monocytogenes and salmonella spp. in dairy products using real time PCR-Melt Curve Analysis. J Food Sci Technol. 2012;49(2):234–239. doi: 10.1007/s13197-011-0278-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suh SH, Dwivedi HP, Jaykus L. Development and evaluation of aptamer magnetic capture assay in conjunction with real-time PCR for detection of campylobacter jejuni. LWT Food Sci Technol. 2014;56:256–260. doi: 10.1016/j.lwt.2013.12.012. [DOI] [Google Scholar]
- Tabit FT. Advantages and limitations of potential methods for the analysis of bacteria in milk: a review, ournal of. Food Sci Technol. 2016;53(1):42–49. doi: 10.1007/s13197-015-1993-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verdoy D, Barrenetxea Z, Beganzo J, Agirregabiria M, Ruano-López JM, Marimón JM, Olabarría G. A novel real time micro PCR based Point-Of-Care device for salmonella detection in huma clinical samples. Biosens Bioelectron. 2012;32:259–265. doi: 10.1016/j.bios.2011.12.032. [DOI] [PubMed] [Google Scholar]
- Vietina M, Agrimonti L, Marmiroli N. Detection of plant oil DNA using high resolution melting (HRM) post PCR analysis: A tool for disclosure of olive oil adulteration. Food Chem. 2013;141:3820–3826. doi: 10.1016/j.foodchem.2013.06.075. [DOI] [PubMed] [Google Scholar]
- von Bargen C, Brockmeyer J, Humpf H. Meat authentication: A new HPLC − MS/MS based method for the fast and sensitive detection of horse and pork in highly processed food. J Agric Food Chem. 2014;62(39):9428–9435. doi: 10.1021/jf503468t. [DOI] [PubMed] [Google Scholar]
- Wang Z, Cai R, Yuan Y, Niu C, Hu Z, Yue T. An immunomagnetic sepration-real-timePCR system for detection of alicyclobacillus acidoterrestris in fruit products. Int J Food Microbiol. 2014;175:30–35. doi: 10.1016/j.ijfoodmicro.2014.01.015. [DOI] [PubMed] [Google Scholar]
- Wang X, Chen X, Xu J, Wang P, Shen W. Multiplex event-specific qualitative polymerase chain reaction for detecting three transgenic rice lines and application of a standard plasmid as a quantitative reference molecule. Anal Biochem. 2014;464:1–8. doi: 10.1016/j.ab.2014.07.004. [DOI] [PubMed] [Google Scholar]
- Wang L, Li P, Zhang Z, Chen Q, Aguilar ZP, Xu H, Yang L, Xu F, Lai W, Xiong Y, Wei H. Rapid and accurate detection of viable escheria coli O157:H7 in milk using a combined IMS, sodium deoxycholate, PMA and Ral-Time Quantitative PCR Process. Food Control. 2014;36:119–125. doi: 10.1016/j.foodcont.2013.08.011. [DOI] [Google Scholar]
- Wilson IG. Inhibition and facilitation of nucleic acid amplification. Appl Environ Microbiol. 1997;63(10):3741–3751. doi: 10.1128/aem.63.10.3741-3751.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamoah FA. Assessing supermarket food shopper reaction to horsemeat scandal in UK. Int RevManag Market. 2014;4(2):98–107. [Google Scholar]
- Zipper H, Brunner H, Bernhagen J, Vitzthum F. Investigations on DNA intercalation and surface binding by SYBR green I, its structure determination and methodological implications. Nucleic Acids Res. 2004;32(12) doi: 10.1093/nar/gnh101. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
(DOCX 58 kb)