

Abstract
Cornelia de Lange Syndrome (CdLS) is characterized by a wide variety of structural and functional abnormalities in almost every organ system of the body. CdLS is now known to be caused by mutations that disrupt the function of the cohesin complex or its regulators, and studies of animal models and cell lines tell us that the effect of these mutations is to produce subtle yet pervasive dysregulation of gene expression. With many hundreds of mostly small gene expression changes occurring in every cell type and tissue, identifying the etiology of any particular birth defect is very challenging. Here we focus on limb abnormalities, which are commonly seen in CdLS. In the limb buds of the Nipbl-haploinsufficient mouse (Nipbl+/− mouse), a model for the most common form of CdLS, modest gene expression changes are observed in several candidate pathways whose disruption is known to cause limb abnormalities, yet the limbs of Nipbl+/− mice develop relatively normally. We hypothesized that further impairment of candidate pathways might produce limb defects similar to those seen in CdLS, and performed genetic experiments to test this. Focusing on Sonic hedgehog (Shh), Bone morphogenetic protein (Bmp), and Hox gene pathways, we show that decreasing Bmp or Hox function (but not Shh function) enhances polydactyly in Nipbl+/− mice, and in some cases produces novel skeletal phenotypes. However, frank limb reductions, as are seen in a subset of individuals with CdLS, do not occur, suggesting that additional signaling and/or gene regulatory pathways are involved in producing such dramatic changes.
Keywords: Nipped-B-like (NIPBL) gene, polydactyly, limb development, Hox genes, Shh gene, bone morphogenetic proteins (BMPs)
INTRODUCTION
The most frequent cause of Cornelia de Lange Syndrome (CdLS) is haploinsufficiency for NIPBL, which encodes a highly-conserved protein that is involved in loading cohesin onto chromosomes [Gillis et al., 2004; Krantz et al., 2004; Tonkin et al., 2004; Dorsett and Krantz, 2009; Liu and Krantz, 2009]. Studies in mice, zebrafish, fruit flies and human cell lines all point to altered gene expression as being the ultimate cause of the developmental and physiological abnormalities in CdLS [Kawauchi et al., 2009; Liu et al., 2009; Schaaf et al., 2009; Muto et al., 2011, 2014], a view that is consistent with an emerging understanding that cohesin plays key roles in gene regulation [Dorsett, 2011; Dorsett and Merkenschlager, 2013]. In cells and tissues examined to date, the gene expression changes caused by NIPBL haploinsufficiency usually number in the many hundreds, differ from one cell type to another, and are almost always modest (increases or decreases of rarely more than 1.5-fold) [Kawauchi et al., 2009; Liu et al., 2009; Muto et al., 2011, 2014]. Among dysregulated genes are often ones known to be controlled by long-range enhancers [Chien et al., 2011], consistent with a proposed role for NIPBL and cohesin in DNA-looping [Kagey et al., 2010; Chien et al., 2011; Dorsett, 2011; Guo et al., 2012; Remeseiro et al., 2013].
In the mouse model of Nipbl haploinsufficiency [Kawauchi et al., 2009], hereinafter referred to as the Nipbl+/− mouse, many of the structural abnormalities of CdLS are faithfully replicated, including small body size, craniofacial anomalies and heart defects. In contrast, abnormalities of limb development have not been noted in Nipbl+/− mice, whereas they are relatively common in CdLS, with an overall incidence of 20–30% [Jackson et al., 1993; Gillis et al., 2004; Oliver et al., 2010], which rises to ~40% among cases with documented frame-shift, splice site or nonsense mutations in NIPBL [Gillis et al., 2004]. The severity of limb alterations in CdLS differs greatly among individuals, varying from reductions in the sizes of hands and feet, to digit abnormalities (clinodactyly of fifth fingers, oligodactyly, proximally placed thumbs, syndactyly of the second and third toes), to frank reductions of the upper limbs.
Recent work suggests that the gene expression changes that give rise to these structural abnormalities might occur, at least to some degree, in Nipbl+/− mice, despite the lack of overt phenotype. Specifically, in a study of nipbl-deficient zebrafish [Muto et al., 2014], in which reduction defects of the pectoral fins (homologues of mammalian forelimbs) were linked to expression changes in key growth and patterning genes, we found that many of the same genes are similarly misregulated in Nipbl+/− mouse limb buds [Muto et al., 2014]. Genes affected in Nipbl+/− limb buds included Sonic hedgehog (Shh) genes, Hox genes, and genes encoding molecules involved in fibroblast growth factor (FGF), bone morphogenetic protein (BMP), and Wnt signaling pathways.
In most cases, gene expression changes in Nipbl+/− mouse limb buds were smaller than what was observed for the equivalent genes in nipbl-deficient fin buds, suggesting that the relative lack of phenotypic effect in mice may be due to a failure to reach a threshold of functional impairment. If this view is correct, we reasoned that Nipbl+/− mice might provide a “sensitized background” in which to test other genes or signaling pathways that, when reduced in mice that were already Nipbl-deficient, could cause that threshold to be crossed. Such experiments would identify pathways that are likely to be directly involved in limb abnormalities in CdLS, as well as help explain the wide variability in severity of limb defects observed in individuals with CdLS. Here, we use this strategy to specifically implicate Hoxd and Bmp gene expression in the etiology of CdLS limb abnormalities.
MATERIALS AND METHODS
Mice
All animal experiments were approved by the University of California, Irvine, Institutional Animal Care and Use Committee. Nipbl+/− mice [Kawauchi et al., 2009] were maintained on a CD1 (Charles River) background, since their survival on inbred backgrounds is poor. ShhFlox/Flox mice (Shhtm2Amc/J) were obtained from Jackson Laboratories, and were maintained on a C57Bl6/J (Jackson Labs) background. Nanog-Cre mice were a kind gift from A. Economides [Economides et al., 2013], and Shh heterozygous mutant mice (Shh+/−) were generated by crossing Shhflox/flox mice with Nanog-Cre mice. HoxdΔ11-13 mice [Zakany and Duboule, 1996] were a kind gift from S. Mackem and were maintained on a FVB/N background; these mice have a simultaneous deletion of Hoxd11, Hoxd12, and Hoxd13. Mice harboring a targeted, inactivating insertion of a lacZ-neo fusion into the Bmp4 locus were a kind gift from B. Hogan [Lawson et al., 1999] and were maintained on both CD1 and C57Bl6/J backgrounds; they are referred to here as Bmp4+/−. Noon on the day of vaginal plug detection was designated as gestational day E0.5.
Genotyping
Nipbl+/−; Bmp4+/− embryos and pups were genotyped with primers that detect the Nipbl null allele (Forward 5′-tcccgaaaaccaaagaagaa and Reverse 5′-gtgctgcaaggcgattaagt) and the Bmp4 null allele (Forward 5′-tacatcttgaccgtcggttg and Reverse 5′-gtgctgcaaggcgattaagt). Shh+/− mice were genotyped with the following primers: Forward 5′-ggacacc attctatgcaggg; Reverse 5′-gaagagatcaag gcaagctctggc. HoxdΔ11-13/+ mice were genotyped with the following primers: Forward 5′-ccaccctgctaaataaacgctg; Reverse 5′-ggttgcctcttttcctctgtctc).
Bone Staining
Bone and cartilage staining was perfomed using Alician Blue and Alazarin Red S as described [Ovchinnikov, 2009], with minor modifications. Briefly, embryos or pups were euthanized and tissue fixed in 4% paraformaldehyde or 10% formalin until used. Carcasses were rinsed in several changes of water over 3–4 days. Skin was removed, and carcasses were dehydrated in 95% ethanol for 1–3 days, followed by staining in Alcian Blue solution for 2 days, rehydration, and incubation in 1% KOH for 2 days. Final staining was in Alizarin Red S for 2 days, after which excess tissue was removed, skeletons were cleared in glycerol, and photographs taken.
Whole Mount In Situ Hybridization
Embryos were collected at E13.5 and placed in 4% paraformaldehyde overnight. Whole mount in situ hybridization was performed as published [Kawauchi et al., 1999]. The 463 bp Sox9 probe was generated by RT-PCR using the following primers (Forward 5′-accaatacttgccacccaac; Reverse 5′-taggagccggagttctgatg).
RESULTS
Postaxial Polydactyly in Nipbl+/− Mice
In our initial study of Nipbl+/− mice [Kawauchi et al., 2009], only mild skeletal changes were reported. The long bones of mutant mice were found to be ~10% shorter than those of wildtype littermates (consistent with overall smaller body size), and the process of ossification was somewhat delayed. We also noted some abnormalities in the shape of the olecranon process Kawauchi et al., 2009]. More recently, by carefully examining a large number of adult animals, we found that between one-fourth and one-third of Nipbl+/− mice possess a very small, supernumerary digit on the postaxial side of either forelimb (Fig. 1A). Bone staining (Fig. 1B) in such animals typically reveals a single phalangeal bone. No other structural abnormalities were observed in the fore- or hindlimbs of Nipbl+/− mice. Interestingly, polydactyly (postaxial and insertional) has also been reported in CdLS, at a frequency of ~6% [Kline et al., 2007]. Thus, although mice fail to reproduce the more severe limb alterations that are seen in CdLS, they do replicate at least one structural abnormality.
Figure 1.

Postaxial polydactyly in Nipbl+/− mice. (A) Forelimb of a postnatal P24 Nipbl+/− mouse shows a postaxial digit proximal to digit 5 (white arrowhead). (B) Alizarin red staining of the same forelimb in (A) indicates the presences of a rudimentary phalanx (arrowhead, inset).
Reducing Shh Function Fails to Elicit New Limb Abnormalities
As described previously [Muto et al., 2014], the limb buds of Nipbl+/− mice at E10.5—an early stage in growth and patterning—exhibit small changes in the expression of genes in a variety of signaling pathways. Some of the largest and highest-confidence changes are observed in Shh (Sonic hedgehog; decreased ~50%), 5′-Hox genes (e.g., Hoxd12, Hoxd13; reduced ~45–50%), and Bmp genes (Bmp2, Bmp4; reduced 10–20%). Shh plays a key role in directing both the growth and patterning of limbs, and limb-specific deletion of Shh in mice is known to produce dramatic distal truncation phenotypes [Galli et al., 2010], which bear strong similarity to the reduction defects observed in individuals with CdLS (e.g., ulnar hypoplasia terminating with a single digit).
In view of these observations, we wondered whether limb truncations could be elicited in Nipbl+/− mice by further reducing Shh function. This was accomplished by crossing Nipbl+/− mice onto backgrounds haploinsufficient for Shh (Shh+/− mice); haploinsufficient for Smoothened, which encodes the cell surface molecule that transduces Shh signaling (Smo+/− mice); or haploinsufficient for both genes together (N = 16 for Nipbl+/−; Shh+/−; N =15 for Nipbl+/−; Smo+/−; and N = 5 for Nipbl+/−; Shh+/−; Smo+/−). The resulting compound heterozygous mice were scored for limb abnormalities at E16.5–E17.5. In no cases were limb truncations observed. Furthermore, the frequency of polydactyly in such mice was not significantly different than that observed in Nipbl+/−mice alone (data not shown). These results imply that, even though Shh expression is reduced in developing Nipbl+/−limbs (see Fig. 4 in Kawauchi et al. [2016]), mice can tolerate even greater reduction in Shh function without a disruption in limb development. These findings suggest that genes other than those involved in the Sonic hedgehog pathway likely play more important roles in the etiology of limb defects in Nipbl-deficient models, and in CdLS.
Bmp4 Haploinsufficiency Enhances Limb Abnormalities
The genes encoding at least two BMPs (Bmp2 and Bmp4) display reduced expression in Nipbl+/− limb buds [Muto et al., 2014]. To test whether further reducing Bmp expression could enhance Nipbl-deficiency phenotypes, Nipbl+/− and Bmp4+/− mice were intercrossed, and progeny examined at E17.5. The results are shown in Figure 2. Like Nipbl+/− mice, Bmp4+/− mice are known to display partially-penetrant postaxial polydactyly [Selever et al., 2004; Goldman et al., 2006]. In Nipbl+/−; Bmp4+/− compound mutants, however, the incidence of this phenotype increases substantially; moreover, rather than being predominantly confined to forelimbs, both fore- and hindlimbs are frequently involved (Fig. 2A). Overall, the total proportion of polydactylous limbs (calculated by adding the number of unilateral cases to twice the number of bilateral cases) increased from 11% in Nipbl+/− mice and 27% in Bmp4+/− mice, to 60% in the compound mutants, implying a synergistic interaction between the two genotypes. Importantly, in forelimbs, the incidence of polydactyly in compound mutants was 96%, and was mostly bilateral; whereas in the single mutants, unilateral and bilateral polydactyly were about equally represented (Fig. 2A). Despite this dramatic increase in polydactyly, neither truncations nor other severe limb abnormalities were observed, even in the compound mutants. Phenotypes were similar whether the parental Bmp4+/− strain was maintained on outbred (CD1) or inbred (C57Bl6/J) backgrounds.
Figure 2.

Deficiency of Bmp4 enhances the penetrance of forelimb and hindlimb polydactyly in Nipbl+/− mice. (A) Polydactyly observed in E17.5 embryos. Numbers in bar graph are the total number of mice of a given genotype that have no (white), unilateral (yellow) or bilateral (blue) polydactyly. R, right. L, left. (B) Forelimbs and hindlimbs from Nipbl+/− and Nipbl+/−; Bmp4+/− E17.5 mice. 27% of Nipbl+/− mice, and nearly all Nipbl+/−; Bmp4+/− mice display postaxial polydactyly of the forelimb (arrowheads). Postaxial polydactyly and preaxial polydactyly (white asterisk) of the hindlimbs is also observed in some Nipbl+/−; Bmp4+/− mice. (C) Whole-mount in situ hybridization for Sox9 reveals emergence of an extra postaxial digit in Nipbl+/−; Bmp4+/− forelimbs (arrowhead) as early as E13.5. 1–5 mark the corresponding digits.
Interestingly, the polydactyly detected in Nipbl+/− mice, Bmp4+/− mice, and compound mutants was disproportionately observed on the right side of the body (Fig. 2A; see breakdown in yellow bars). Overall, 77% of unilateral forelimb polydactyly involved the right forelimb and 89% of unilateral hindlimb polydactyly involved the right hindlimb. A similar right-sided prevalence has been noted for the limb abnormalities that occur in CdLS, including the severe reduction defects [Mehta and Krantz, 2016].
The forelimb polydactyly we observe is, in all cases, postaxial, and the extra digit is never complete, although in compound (Nipbl+/−; Bmp4+/−) mutants the extra digit often appears larger and more fully-formed (Fig. 2B), especially when the Bmp4+/− allele has been introduced from a CD1 background (the same background on which Nipbl+/− mice are maintained). Using whole mount in situ hybridization for Sox9, a pre-chondrogenic marker, it is possible to visualize the formation of extra digits in Nipbl+/−; Bmp4+/− forelimbs as early as E13.5 (Fig. 2C). In the hindlimbs of Bmp4+/− and Nipbl+/−; Bmp4+/− mice, preaxial polydactyly is also sometimes observed (Fig. 2B). Unilateral preaxial polydactyly of the hindlimbs has previously been noted to occur in Bmp4+/− mice [Dunn et al., 1997; Goldman et al., 2006].
To better analyze the skeletal changes in these animals, Alizarin Red and Alcian Blue staining of E17.5 embryos and neonatal pups was carried out to reveal bone and cartilage, respectively (Fig. 3). Because penetration of Alcian Blue requires removal of skin, the smallest incomplete extra digits could not be stained by this dye, as it was impossible to remove the skin over them without disrupting or destroying them. Nevertheless, the absence of Alizarin Red staining in such structures (Alizarin Red penetrates skin well) suggested that they were primarily cartilaginous (Fig. 3A, top row, arrowheads). In larger supernumerary digits, which tolerated skin removal, Alcian Blue confirmed the presence of cartilage (Fig. 3A, lower row, arrows).
Figure 3.

Forelimb and hindlimb skeletal abnormalities in Nipbl+/−; Bmp4+/− mice. (A) Top row: Bilateral postaxial polydactyly of left (white arrowheads) and right (black arrowheads) forepaws in postnatal day 2 Nipbl+/−; Bmp4+/− mice. Cartilage and bone staining of these forepaws is shown in right two panels. Bottom row: Preaxial polydactyly in a hindlimb from a postnatal day 2 Nipbl+/−; Bmp4+/− mouse. Cartilage and bone staining indicate the presence of a triphalangeal digit 1* (middle panel), which larger magnification image shows a shared metatarsal (M) with digit1. Joints between phalanges are indicated by P. (B) Cartilage and bone staining of forepaws of wildtype (left two panels) and Nipbl+/−; Bmp4+/− (right two panel) littermates at E17.5. Nipbl+/−; Bmp4+/− mice on a CD1 background all (6/6) displayed abnormally-shaped falciform bones that extended towards the distal phalanx of digit 1 or was fused to a bifid distal phalanx of digit 1. Falciform bones indicated by arrowheads; higher magnification image of each forepaw, slightly tilted to view the falciform, is shown at right. Reduced red staining in forepaws of Nipbl+/−; Bmp4+/− mice, relative to wildtype, indicates reduced or delayed ossification, a consistent finding. (C) Abnormalities of the deltoid tuberosity were seen in 92% of Nipbl+/−; Bmp4+/− mice. The deltoid tuberosity is missing from the left humerus (asterisk), but is present on the right humerus (arrowhead) of a postnatal day 2 Nipbl+/−; Bmp4+/− mouse. Insets show higher magnification views of the affected areas.
In those cases in which preaxial polydactyly occurred in the hindlimbs of Nipbl+/−; Bmp4+/−mice, Alizarin Red/Alcian Blue staining showed a wide range of degrees of digit duplication. For example, in one case examined at postnatal day 2, we observed a triphalangeal digit 1 that shared a metatarsal with the adjoining digit 1 (Fig. 3A, lower row, 1*). In another case, complete duplication of digit 1 was observed on a right hindlimb (not shown). These abnormalities are similar to what Dunn et al., [1997] observed in Bmp4+/−mice.
Abnormalities were also observed in the carpal (wrist) bones of Nipbl+/−; Bmp4+/− forelimbs. Particularly striking were malformations of the falciform bone, which displayed a boomerang-like shape in both forelimbs in 6 of 13 Nipbl+/−; Bmp4+/− embryos (Fig. 3B, arrowheads); such malformations were not observed in wildtype or Nipbl+/− mice. Falciform bone malformation was only seen in animals in which the Bmp4+/− allele was introduced from a CD1 background, and was occasionally also present, unilaterally and in milder form, in Bmp4+/− mice on this background (one of four examined). As described previously for Nipbl+/− mice [Kawauchi et al., 2009], staining also revealed delayed bone ossification in Nipbl+/−; Bmp4+/− mice (compare wildtype with compound mutant in Fig. 3B).
Although duplications or reductions involving the long bones of the limb were not observed in any of the animals investigated, we did find that Nipbl+/−; Bmp4+/− mice frequently exhibited a small, misshapen, or completely absent deltoid tuberosity (asterisk, Fig. 3C) on one or both forelimbs (92%, N = 13). This phenotype was never observed in wildtype or Nipbl+/− mice, and was seen in only 1 of 10 Bmp4+/− mice.
Axial skeletal phenotypes in Nipbl+/−; Bmp4+/− Mice
In Nipbl+/−; Bmp4+/−mice, defects were frequently observed in the axial skeleton as well as in the digits (Fig. 4). Rib abnormalities were seen in 85% (N = 13) of compound mutants. Severe abnormalities included thoracic rib fusions (Fig. 4A, asterisks) and misalignment of ribs at the sternum (Fig 4B). Less drastic anomalies, including blunting/squaring of the ends of free ribs (Fig. 4C) and lateral protrusions on the ribs (Fig. 4D, arrowhead) were also observed. Rare phenotypes included absent sacral and caudal vertebrae as well as occasional fused caudal vertebrae (not shown). None of these phenotypes was observed in wildtype mice or mice that were Nipbl+/− or Bmp4+/− alone. Interestingly, we did not observe the misalignment of ossification centers in the sternum that Smith et al. [2014] noted in a different strain of Nipbl+/− mice, although we did observe a further decrease in ossification of sternabrae in Nipbl+/−; Bmp4+/− embryos, compared to Nipbl+/− mice (not shown).
Figure 4.

Axial skeletal phenotypes of Nipbl+/−; Bmp4+/− mice. (A) Examples of rib fusion (asterisks), involving ribs 3 and 4 (on right and left) and ribs 5 and 6 (on the right side). (B) Misalignment of ribs at the sternum. (C) Minor anomalies such as blunting/squaring of rib ends and floating ribs (arrowheads and inset). Fusion of ribs 6 and 7 (on left side of sternum) is also observed in this case. (D) Lateral protrusions (arrowhead, dashed line), suggestive of costal rib bridging, were also observed. (E) Nipbl+/− and Nipbl+/−; Bmp4+/− mice on a hybrid CD1/C57Bl6 background often had an extra (14th) pair of stunted ribs (black arrows). (F) Bifurcated neural arches of the C2 cervical vertebra (yellow arrowhead) were observed in 4 of 6 Nipbl+/−; Bmp4+/− mice on CD1 background.
A phenotype of high penetrance emerged in Nipbl+/− mice when they were on a hybrid CD1;C57Bl6/J background: the development of an extra (14th) pair of ribs (92%, N = 12; Fig. 4E). On this hybrid background, wildtype and Bmp4+/− mice (N = 8) have 13 pairs of ribs, but both Nipbl+/− and Nipbl+/−; Bmp4+/− mice display 14 pairs of ribs at a frequency greater than 90%. We noted that in 8 of 11 mice with supernumerary ribs, these ribs had a stunted appearance; stunted extra thoracic ribs were also noted by Smith et al. [2014] in their study of a different line of Nipbl+/− mice, and are, importantly, a common finding in CdLS [Braddock et al., 1993; Bhuiyan et al., 2006]. Interestingly, supernumerary ribs were never seen when mice were maintained on a CD1 background, in which 14 ribs is the normal wildtype number.
Apart from the abnormalities listed above, the only consistent skeletal change we detected was bifurcation of the neural arch of the C2 cervical vertebra, which occurred in 4 out of 6 Nipbl+/−; Bmp4+/− mice on the CD1 background (arrowhead, Fig. 4F).
Enhancement of Polydactyly in Nipbl+/−; HoxdΔ11-13/− Mice
The effect of Nipbl haploinsufficiency on Hox gene expression in developing mouse limbs mirrors what is seen in nipbl-deficient zebrafish fin buds, with expression of genes at the 5′ ends of Hox clusters being reduced [Muto et al., 2014]. In E10.5 mouse limb buds, the most significantly affected genes are those at the extreme 5′ end of the Hoxd cluster, namely Hoxd11, Hoxd12, and Hoxd13 [Muto et al., 2014]. The availability of a targeted mutant allele in which all three of these genes have been deleted [Zakany and Duboule, 1996] made it possible to test whether simultaneous haploinsufficiency for all three Hox genes might enhance Nipbl+/− limb phenotypes.
Nipbl+/− mice were thus crossed with HoxdΔ11-13/+ mice and progeny examined at E17.5. The data are shown in Figure 5. HoxdΔ11-13/+ mice exhibited some polydactyly on their own (25%), which involved the forelimbs, and was always unilateral. This phenotype has not been reported before in HoxdΔ11-13/+ heterozyogotes, although it is seen in homozygotes [Zakany and Duboule, 1996; Gonzalez-Martin et al., 2014]. It is possible that the genetic background of these crosses (CD1 × FVB/N hybrid) has some influence on the expressivity of these traits.
Figure 5.
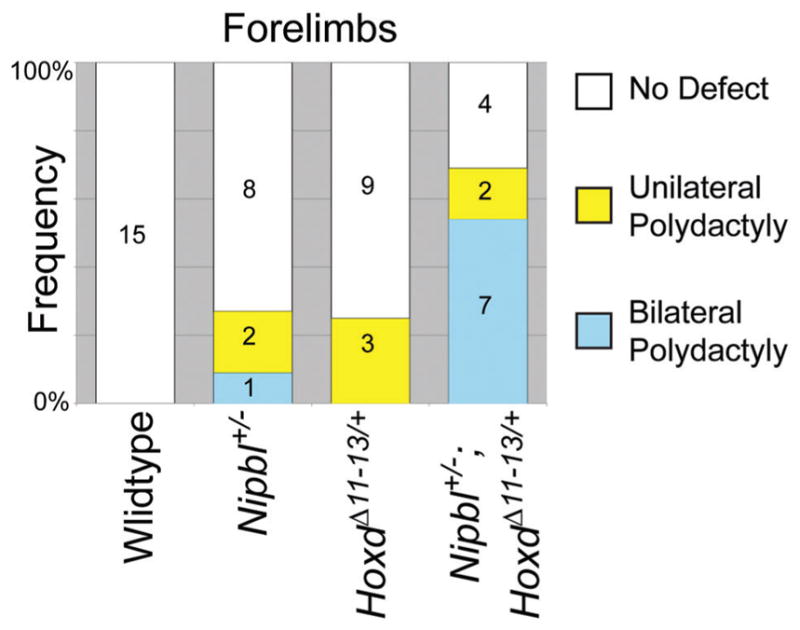
Deficiency of Hoxd11, Hoxd12, and Hoxd13 enhances the penetrance of forelimb polydactyly in Nipbl+/− mice. E17.5 embryos of the indicated genotypes were examined for the presence of unilateral (yellow) or bilateral (blue) polydactyly. Numbers in bar graph are the total number of mice in each category.
In Nipbl+/−; HoxdΔ11-13/+ mice, the incidence of forelimb polydactyly was markedly increased over the levels observed in either Nipbl+/− or HoxdΔ11-13/+ single mutants, with the total number of affected limbs rising to 62% and a marked shift toward bilateral presentation (Fig. 5). Overall the supernumerary digits in these animals were smaller than those observed in Nipbl+/−; Bmp4+/− mice (not shown). Hindlimb polydactyly was not seen in any of these animals, nor were defects such as limb truncations. These results suggest that reduced 5′-Hoxd function plays a role in the etiology of a subset of the limb defects produced by Nipbl deficiency.
DISCUSSION
Mouse, zebrafish and fly models of Nipbl-deficiency have provided considerable insight into the gene expression changes that must underlie the structural and functional abnormalities of CdLS [Kawauchi et al., 2009, 2016; Schaaf et al., 2009; Muto et al., 2011, 2014]. Given the modest sizes of most gene expression changes in Nipbl-deficient tissues/cells, it is likely that most abnormalities are the result of collective effects at multiple genes [Muto et al., 2011, 2014]. Identifying which combinations explain any particular defect is inherently challenging, given the very large number of gene expression changes that one must consider. Here we took advantage of observations suggesting that the genes dysregulated in Nipbl+/− mouse limbs are likely to include those that cause limb defects in CdLS, yet collectively seem to fall below a threshold level of impairment needed to bring about the same phenotypes. This enabled us to use the Nipbl+/− mouse as a sensitized background in which to ask whether further reduction in function of Shh, Bmp or Hox gene pathways could elicit severe limb abnormalities.
With Shh, a variety of mutant backgrounds that reduce Shh expression or function failed to show any significant genetic interaction with Nipbl haploinsufficiency, even though Shh expression is markedly reduced in Nipbl+/− limb buds, and complete inactivation of Shh in limbs is known to cause truncating anomalies [Galli et al., 2010]. In contrast, synergistic interactions were observed between Nipbl haploinsufficiency and haploinsufficiency for Bmp4, as well as haploinsufficiency for the three 5′-most genes of the Hoxd cluster. In both cases, compound mutants exhibited enhanced penetrance of polydactyly, a known—albeit relatively uncommon—limb defect observed in individuals with CdLS [Kline et al., 2007]. Several aspects of the polydactyly that was enhanced by Bmp4 and Hox mutant backgrounds bear strong resemblance to what is seen in CdLS: These include predominantly post-axial positioning, and a much greater prevalence on fore- versus hind-limbs. In addition, polydactyly was strongly right-side biased, just as is observed with more severe reduction defects in CdLS [Mehta and Krantz, 2016, this issue].
Nipbl+/−; Bmp4+/− mice also displayed additional, intriguing skeletal abnormalities (Fig. 4), several of which have been reported to occur in CdLS. In no cases, however, did we observe overt limb truncations.
Overall, our findings suggest that dysregulation of Bmp4 and Hox gene expression may play an important role in the etiology of some limb defects in CdLS, but we will probably need to look elsewhere to find the causes of the most severe abnormalities. In this regard, it is noteworthy that the limb buds of E10.5 Nipbl+/− mice also exhibit alterations in the expression of genes involved in Wnt and Fibroblast growth factor signaling [Muto et al., 2014]. It is also possible that there are additional gene expression changes that occur only at earlier or later developmental stages, and play a causal role in the etiology of limb defects in CdLS; such stage-specific influences on limb growth in CdLS remain to be investigated.
Acknowledgments
These studies were supported by grants from the National Institutes of Health (P01-HD052860 and P50-GM076516) and the Cornelia de Lange Syndrome Foundation.
Biographies
Martha E. Lopez-Burks, M.S., is a Staff Research Associate who specializes in molecular biology. During the past decade, her research has focused on molecular analysis of Nipbl-deficient mice.
Rosaysela Santos, Ph.D., is a Postdoctoral Scholar who has been a lead researcher in developing and analyzing mouse models of CdLS since she was a graduate student in the Calof lab. Dr. Santos’s research interests lie in using animal models to understand the origins of birth defects associated with developmental and genetic disorders in man.
Shimako Kawauchi, Ph.D., is an Associate Project Scientist who has been a primary researcher in developing and analyzing mouse models of CdLS for over a decade. Dr. Kawauchi’s research interests are in the regulation of gene expression and pattern formation during mammalian development.
Anne L. Calof, Ph.D., is Professor of Anatomy and Neurobiology at the University of California, Irvine. She serves as Co-Director of UCI’s Center of Excellence in Research on Cornelia de Lange Syndrome and as a Theme Leader in the Center for Complex Biological Systems. Dr. Calof’s research is focused on understanding how cell differentiation, tissue morphogenesis, and tissue size are regulated, both during normal development and when pathological conditions result in birth defects.
Arthur D. Lander, M.D., Ph.D., is Donald Bren Professor of Developmental and Cell Biology, Director of the Center for Complex Biological Systems, and Co-Director the Center of Excellence in Research on Cornelia de Lange Syndrome at the University of California, Irvine. Dr. Lander’s research uses experimental, mathematical and computational approaches to elucidate the engineering principles that underlie human development, with a focus on patterning, growth control, and the origins of complex traits.
References
- Bhuiyan ZA, Zilfalil BA, Hennekam RC. A Malay boy with the Cornelia de Lange syndrome: Clinical and molecular findings. Singapore Med J. 2006;47:724–727. [PubMed] [Google Scholar]
- Braddock SR, Lachman RS, Stoppenhagen CC, Carey JC, Ireland M, Moeschler JB, Cunniff C, Graham JM., Jr Radiological features in Brachmann-de Lange syndrome. Am J Med Genet. 1993;47:1006–1013. doi: 10.1002/ajmg.1320470714. [DOI] [PubMed] [Google Scholar]
- Chien R, Zeng W, Kawauchi S, Bender MA, Santos R, Gregson HC, Schmiesing JA, Newkirk DA, Kong X, Ball AR, Jr, Calof AL, Lander AD, Groudine MT, Yokomori K. Cohesin mediates chromatin interactions that regulate mammalian beta-globin expression. J Biol Chem. 2011;286:17870–17878. doi: 10.1074/jbc.M110.207365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dorsett D. Cohesin: Genomic insights into controlling gene transcription and development. Curr Opin Genet Dev. 2011;21:199–206. doi: 10.1016/j.gde.2011.01.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dorsett D, Krantz ID. On the molecular etiology of Cornelia de Lange syndrome. Ann N Y Acad Sci. 2009;1151:22–37. doi: 10.1111/j.1749-6632.2008.03450.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dorsett D, Merkenschlager M. Cohesin at active genes: A unifying theme for cohesin and gene expression from model organisms to humans. Curr Opin Cell Biol. 2013;25:327–333. doi: 10.1016/j.ceb.2013.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunn NR, Winnier GE, Hargett LK, Schrick JJ, Fogo AB, Hogan BL. Haploinsufficient phenotypes in Bmp4 heterozygous null mice and modification by mutations in Gli3 and Alx4. Dev Biol. 1997;188:235–247. doi: 10.1006/dbio.1997.8664. [DOI] [PubMed] [Google Scholar]
- Economides AN, Frendewey D, Yang P, Dominguez MG, Dore AT, Lobov IB, Persaud T, Rojas J, McClain J, Lengyel P, Droguett G, Chernomorsky R, Stevens S, Auerbach W, Dechiara TM, Pouyemirou W, Cruz JM, Jr, Feeley K, Mellis IA, Yasenchack J, Hatsell SJ, Xie L, Latres E, Huang L, Zhang Y, Pefanis E, Skokos D, Deckelbaum RA, Croll SD, Davis S, Valenzuela DM, Gale NW, Murphy AJ, Yancopoulos GD. Conditionals by inversion provide a universal method for the generation of conditional alleles. Proc Natl Acad Sci USA. 2013;110:E3179–E3188. doi: 10.1073/pnas.1217812110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galli A, Robay D, Osterwalder M, Bao X, Benazet JD, Tariq M, Paro R, Mackem S, Zeller R. Distinct roles of Hand2 in initiating polarity and posterior Shh expression during the onset of mouse limb bud development. PLoS Genet. 2010;6:e1000901. doi: 10.1371/journal.pgen.1000901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gillis LA, McCallum J, Kaur M, DeScipio C, Yaeger D, Mariani A, Kline AD, Li HH, Devoto M, Jackson LG, Krantz ID. NIPBL mutational analysis in 120 individuals with Cornelia de Lange syndrome and evaluation of genotype-phenotype correlations. Am J Hum Genet. 2004;75:610–623. doi: 10.1086/424698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldman DC, Hackenmiller R, Nakayama T, Sopory S, Wong C, Kulessa H, Christian JL. Mutation of an upstream cleavage site in the BMP4 prodomain leads to tissue-specific loss of activity. Development. 2006;133:1933–1942. doi: 10.1242/dev.02368. [DOI] [PubMed] [Google Scholar]
- Gonzalez-Martin MC, Mallo M, Ros MA. Long bone development requires a threshold of Hox function. Dev Biol. 2014;392:454–465. doi: 10.1016/j.ydbio.2014.06.004. [DOI] [PubMed] [Google Scholar]
- Guo Y, Monahan K, Wu H, Gertz J, Varley KE, Li W, Myers RM, Maniatis T, Wu Q. CTCF/cohesin-mediated DNA looping is required for protocadherin alpha promoter choice. Proc Natl Acad Sci USA. 2012;109:21081–21086. doi: 10.1073/pnas.1219280110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson L, Kline AD, Barr MA, Koch S. De Lange syndrome: A clinical review of 310 individuals. Am J Med Genet. 1993;47:940–946. doi: 10.1002/ajmg.1320470703. [DOI] [PubMed] [Google Scholar]
- Kagey MH, Newman JJ, Bilodeau S, Zhan Y, Orlando DA, van Berkum NL, Ebmeier CC, Goossens J, Rahl PB, Levine SS, Taatjes DJ, Dekker J, Young RA. Mediator and cohesin connect gene expression and chromatin architecture. Nature. 2010;467:430–435. doi: 10.1038/nature09380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawauchi S, Calof AL, Santos R, Lopez-Burks ME, Young CM, Hoang MP, Chua A, Lao T, Lechner MS, Daniel JA, Nussenzweig A, Kitzes L, Yokomori K, Hallgrimsson B, Lander AD. Multiple organ system defects and transcriptional dysregulation in the Nipbl(+/−) mouse, a model of Cornelia de Lange Syndrome. PLoS Genet. 2009;5:e1000650. doi: 10.1371/journal.pgen.1000650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawauchi S, Santos R, Muto A, Lopez-Burks ME, Schilling TF, Lander AD, Calof AL. Using mouse and zebrafish models to understand the etiology of developmental defects in Cornelia de Lange syndrome. Am J Med Genet Part C Semin Med Genet. 2016 doi: 10.1002/ajmg.c.31484. (in this issue) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawauchi S, Takahashi S, Nakajima O, Ogino H, Morita M, Nishizawa M, Yasuda K, Yamamoto M. Regulation of lens fiber cell differentiation by transcription factor c-Maf. J Biol Chem. 1999;274:19254–19260. doi: 10.1074/jbc.274.27.19254. [DOI] [PubMed] [Google Scholar]
- Kline AD, Grados M, Sponseller P, Levy HP, Blagowidow N, Schoedel C, Rampolla J, Clemens DK, Krantz I, Kimball A, Pichard C, Tuchman D. Natural history of aging in Cornelia de Lange syndrome. Am J Med Genet C Semin Med Genet. 2007;145C:248–260. doi: 10.1002/ajmg.c.30137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krantz ID, McCallum J, DeScipio C, Kaur M, Gillis LA, Yaeger D, Jukofsky L, Wasserman N, Bottani A, Morris CA, Nowaczyk MJ, Toriello H, Bamshad MJ, Carey JC, Rappaport E, Kawauchi S, Lander AD, Calof AL, Li HH, Devoto M, Jackson LG. Cornelia de Lange syndrome is caused by mutations in NIPBL, the human homolog of Drosophila melanogaster Nipped-B. Nat Genet. 2004;36:631–635. doi: 10.1038/ng1364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawson KA, Dunn NR, Roelen BA, Zeinstra LM, Davis AM, Wright CV, Korving JP, Hogan BL. Bmp4 is required for the generation of primordial germ cells in the mouse embryo. Genes Dev. 1999;13:424–436. doi: 10.1101/gad.13.4.424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J, Krantz ID. Cornelia de Lange syndrome, cohesin, and beyond. Clin Genet. 2009;76:303–314. doi: 10.1111/j.1399-0004.2009.01271.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J, Zhang Z, Bando M, Itoh T, Deardorff MA, Clark D, Kaur M, Tandy S, Kondoh T, Rappaport E, Spinner NB, Vega H, Jackson LG, Shirahige K, Krantz ID. Transcriptional dysregulation in NIPBL and cohesin mutant human cells. PLoS Biol. 2009;7:e1000119. doi: 10.1371/journal.pbio.1000119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mehta D, Krantz I. Characterization of limb differences in Cornelia de Lange Syndrome. Am J Med Genet. 2016 doi: 10.1002/ajmg.c.31498. [DOI] [PubMed] [Google Scholar]
- Muto A, Calof AL, Lander AD, Schilling TF. Multifactorial origins of heart and gut defects in nipbl-Deficient zebrafish, a model of Cornelia de Lange Syndrome. PLoS Biol. 2011;9:e1001181. doi: 10.1371/journal.pbio.1001181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muto A, Ikeda S, Lopez-Burks ME, Kikuchi Y, Calof AL, Lander AD, Schilling TF. Nipbl and mediator cooperatively regulate gene expression to control limb development. PLoS Genet. 2014;10:e1004671. doi: 10.1371/journal.pgen.1004671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oliver C, Bedeschi MF, Blagowidow N, Carrico CS, Cereda A, Fitzpatrick DR, Gervasini C, Griffith GM, Kline AD, Marchisio P, Moss J, Ramos FJ, Selicorni A, Tunnicliffe P, Wierzba J, Hennekam RC. Cornelia de Lange syndrome: Extending the physical and psychological phenotype. Am J Med Genet Part A. 2010;152A:1127–1135. doi: 10.1002/ajmg.a.33363. [DOI] [PubMed] [Google Scholar]
- Ovchinnikov D. Alcian blue/alizarin red staining of cartilage and bone in mouse. Cold Spring Harb Protoc. 2009;2009 doi: 10.1101/pdb.prot5170. pdb.prot5170. [DOI] [PubMed] [Google Scholar]
- Remeseiro S, Cuadrado A, Losada A. Cohesin in development and disease. Development. 2013;140:3715–3718. doi: 10.1242/dev.090605. [DOI] [PubMed] [Google Scholar]
- Schaaf CA, Misulovin Z, Sahota G, Siddiqui AM, Schwartz YB, Kahn TG, Pirrotta V, Gause M, Dorsett D. Regulation of the Drosophila Enhancer of split and invected-engrailed gene complexes by sister chromatid cohesion proteins. PLoS ONE. 2009;4:e6202. doi: 10.1371/journal.pone.0006202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selever J, Liu W, Lu MF, Behringer RR, Martin JF. Bmp4 in limb bud mesoderm regulates digit pattern by controlling AER development. Dev Biol. 2004;276:268–279. doi: 10.1016/j.ydbio.2004.08.024. [DOI] [PubMed] [Google Scholar]
- Smith TG, Laval S, Chen F, Rock MJ, Strachan T, Peters H. Neural crest cell-specific inactivation of Nipbl or Mau2 during mouse development results in a late onset of craniofacial defects. Genesis. 2014;52:687–694. doi: 10.1002/dvg.22780. [DOI] [PubMed] [Google Scholar]
- Tonkin ET, Wang TJ, Lisgo S, Bamshad MJ, Strachan T. NIPBL, encoding a homolog of fungal Scc2-type sister chromatid cohesion proteins and fly Nipped-B, is mutated in Cornelia de Lange syndrome. Nat Genet. 2004;36:636–641. doi: 10.1038/ng1363. [DOI] [PubMed] [Google Scholar]
- Zakany J, Duboule D. Synpolydactyly in mice with a targeted deficiency in the HoxD complex. Nature. 1996;384:69–71. doi: 10.1038/384069a0. [DOI] [PubMed] [Google Scholar]