

Abstract
Background
Cation exchange high performance liquid chromatography (HPLC) is emerging as the method of choice for initial screening and diagnosis of haemoglobinopathies. The use of alkaline and acid gel electrophoresis in the developing countries may result in incorrect diagnosis of haemoglobinopathies. The aim of the study is to assess the accuracy and precision of diagnosis of haemoglobinopathies by HPLC and its possible advantage over conventional techniques.
Methods
Over a two year period, 955 patients presenting with anaemia were evaluated by HPLC for diagnosis of haemoglobinopathies. All cases showing ‘unknown peaks’ and other rare haemoglobin variants on HPLC were further analyzed by agar gel electrophoresis at alkaline pH (8.6) and at acid pH (6.0).
Result
A total of 137 (14.3%) patients showed different abnormal haemoglobins variants. Of these 91 (66.4%) were diagnosed to have beta – heterozygous thalassaemia based on high level of HbA2 (>3.9%), five (3.7%) as beta – homozygous thalassaemia (HbF 25 – 91%), 15 (10.9%) as sickle cell trait, two (1.5%) as compound heterozygous state of sickle - β+ thalassaemia and three (2.2%) patients as homozygous sickle cell anaemia (HbSS). One (0.7%) patient had unknown peak on HPLC with retention time of 4.78 minutes, constituting 16.8% of total haemoglobin. Sickling test was negative. He was diagnosed as HbQ – India heterozygous. Thirteen (9.5%) patients were diagnosed as HbE syndrome and were further sub classified as HbE trait (five cases) and HbE disease (eight cases). Seven (5.1%) patients were diagnosed as Hb – D Punjab heterozygous.
Conclusion
The simplicity of the sample preparation, superior resolution of the method and accurate quantitation of haemoglobin concentration, combined with complete automation, makes this an ideal methodology for diagnosis of haemoglobinopathies.
Key Words: Haemoglobinopathy, High performance liquid chromatography, Thalassaemia
Introduction
The use of cation - exchange high performance liquid chromatography (CE-HPLC) to separate and quantify various normal and abnormal haemoglobin (Hb) fractions has been increasing [1, 2, 3, 4]. It is a highly sensitive, specific, quick but more expensive method for diagnosis [5]. Majority of the centres in India use conventional methods for diagnosis of haemoglobinopathies, which includes clinical and family history, red cell indices, complete blood counts (CBC), HbA2, HbF estimation, sickling test and Hb electrophoresis. The limitations of which include identification of Hb variants with same electrophoretic mobility, in diagnosis of Hb S traits where low quantity of HbS is associated with negative sickling test and diagnosing certain compound heterozygous states (HbS - beta thalassaemia, HbS - HbD disease).
The Bio - Rad Variant Haemoglobin Testing system (Bio - Rad Labs, Hercules, CA), is a totally automated CE - HPLC instrument for routine quantification of HbA2, HbF and any other abnormal Hb variant. The Bio Rad variant “Beta Thalassaemia Short” program uses cation exchange HPLC to separate and elucidate the relative percentages of haemoglobin variants in whole blood. We use HPLC as a routine method for diagnosis of haemoglobinopathies in this centre. We also evaluated if this method has advantages over the conventional techniques.
Material and Methods
The present study was carried out over a two year period in a tertiary care hospital of the Armed Forces. A total of 955 (313 males and 642 females) patients of anaemia with suspected haemoglobinopathies were investigated. The age group of patients ranged from 13 months to 51 years. CBC, red blood cell (RBC) indices and peripheral blood examination were done in all cases. Sickling test was done in all relevant cases. In all abnormal cases family studies were carried out. CE- HPLC was performed in all cases. Specimens were drawn into tubes containing dipotassium EDTA (Becton Dickinson vacutainers systems). All specimens were assessed by the Bio – Rad Variant HPLC system with the use of the Variant Beta – Thalassaemia Short Program Recorder Pack (Bio – Rad Laboratories, Hercules, CA) as described in the instrument manual for the assay. After collection, the samples were stored at 2–8°C and tested within one week of collection. The sample preparation involves dilution of 5 µl of EDTA anticoagulant blood in 1 ml of haemolysis solution provided in the kit. Thereafter, the samples are loaded in the instrument. The samples are diluted with the specific haemolyzing / wash buffer and injected into an assay – specific analytic cartridge. The variant dual pumps deliver a programmed buffer gradient of increasing ionic strength to the cartridge, where the haemoglobin fractions are separated based on their ionic interaction with the cartridge material. The separated haemoglobin fractions pass through a flow cell, where absorbance is measured at 415 nm with simultaneous use of secondary wavelength of 690 nm to reduce background noise. The software delivers a printed report showing the chromatogram, with all the haemoglobin fractions eluted. The integrated peaks are assigned to manufacturer – defined “windows” derived from specific retention time (RT). This retention time is the time that elapses from the sample injection to the apex of the elution peak, of normal haemoglobin fraction and common variants (Table 1). The “windows” are established ranges in which common variants have been observed to elute using the Variant beta – thalassaemia short program. The printed chromatogram shows all the haemoglobin fractions eluted, the retention times, the areas of the peaks and the values (%) of different haemoglobin components. If a peak elutes at a retention time that is not pre defined, it is labeled as an unknown. Each analytical cycle, from sampling to printing of results takes about 6.5 minutes.
Table 1.
Manufacturer assigned windows for Bio-Rad Variant HPLC system
| Peak Name | Window (minutes) | Retention time (minutes) |
|---|---|---|
| F window | 1.00–1.30 | 1.15 |
| P2 window* | 1.30–1.60 | 1.45 |
| P3 window* | 1.60–1.90 | 1.75 |
| A0 window | 1.90–3.30 | 2.60 |
| A2 window | 3.30–3.90 | 3.60 |
| D window | 3.90–4.30 | 4.10 |
| S window | 4.30–4.70 | 4.50 |
| C window | 4.90–5.30 | 5.10 |
P2 and P3 are minor peaks associated with haemoglobin A
The instrument calibration was done by loading the method parameter via a read – only memory (ROM) card provided with each set of reagent kit together with a matched set of calibrators and reagents. Care was taken to keep the RT of HbA2 as 3.65 ± 0.05 minutes and total area between 10,00,000 – 30,00,000 microvolt seconds [6].
All cases showing ‘unknown peaks’ and other rare haemoglobin variants on HPLC were further analyzed by agar – rose gel electrophoresis at alkaline pH (8.6) and at acid pH (6.0). Results of various tests done were correlated with clinical profile of patients and family studies. Beta heterozygous thalassaemia was diagnosed when HbA2 levels were >3.9% but <9% [6]. If the values of HbA2 were borderline and between 3.2% and 3.8%, then these patients were further evaluated to rule out any associated iron deficiency anaemia. Patients with HbA2 levels higher than 9% were diagnosed as HbE syndromes, which were further sub classified as HbE disease or HbE – beta thalassaemia (HbE/A2 levels > HbA) or HbE trait HbE/A2 levels < HbA) [5, 7]. Patients, who had peak identified as HbS, were confirmed by sickling test. They were diagnosed as sickle cell trait (HbAS) (if HbA > HbS) and sickle cell disease or sickle cell – beta thalassaemia (HbSS) (if HbS > HbA). In patients with sickle cell / β0 thalassaemia there is no haemoglobin A, whereas in sickle cell/β+ thalassaemia the values of HbA varies from almost undetectable to 30% [8].
Results
Nine hundred fifty five patients of anaemia with suspected haemoglobinopathies were analyzed along with their family members and 137 (14.3%) patients showed different abnormal haemoglobins variants (Table 2, Table 3). Of these 91 (66.4%) patients were diagnosed to have beta-heterozygous thalassaemia based on the high level of HbA2 (>3.9%) (Fig. 1). These patients presented with mild anaemia (mean Hb level of 9.3gm/dl) and all showed microcytic – hypochromic blood picture with low mean corpuscular volume (MCV) of <80fl. Five of these 91 patients of beta – heterozygous thalassaemia initially had borderline HbA2 levels (3.2–3.8%) as they had concurrent iron deficiency anaemia (IDA). Their HbA2 values were >3.9% after treatment for IDA and hence they were diagnosed as beta – heterozygous thalassaemia. Five (3.7%) of 137 abnormal cases were diagnosed as beta – homozygous thalassaemia. All these five patients had increased HbF values (25–91%). Clinically they presented with severe pallor requiring regular blood transfusion and had moderate to marked splenomegaly.
Table 2.
Total cases of anaemia studied
| Total cases | Males | Females | Haemoglobinopathies diagnosed | |
|---|---|---|---|---|
| 1st year | 341 | 140 | 201 | 45 |
| 2nd year | 424 | 121 | 303 | 61 |
| 3rd year | 190 | 52 | 138 | 31 |
| (till April' 07) | ||||
| Total | 955 | 313 | 642 | 137 |
| (32.8%) | (67.2%) | (14.3%) |
Table 3.
Spectrum of haemoglobinopathies (n= 137)
| Diagnosis | Total cases (%) |
|---|---|
| β heterozygous thalassaemia | 91 (66.4%) |
| β homozygous thalassaemia | 05 (3.7%) |
| Sickle cell trait | 15 (10.9%) |
| Sickle cell disease | 03 (2.2%) |
| Sickle–β+ thalassaemia | 02 (1.5%) |
| HbE trait | 05 (3.6%) |
| HbE disease | 08 (5.9%) |
| HbD trait | 07 (5.1%) |
| HbQ India | 01 (0.7%) |
Fig. 1.
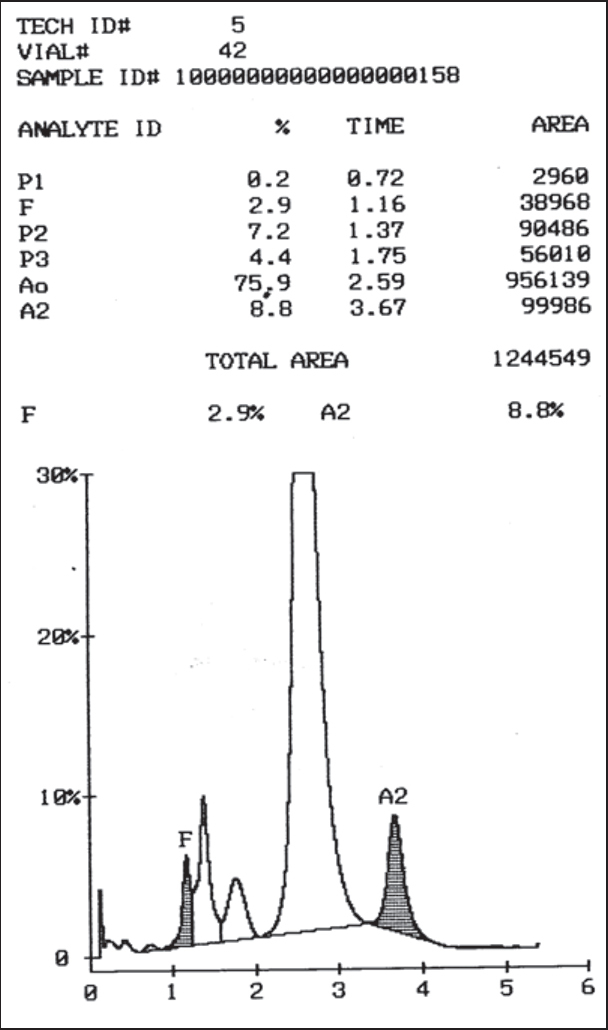
Chromatogram of β heterozygous thalassaemia
Fifteen (10.9%) patients were diagnosed as sickle cell trait, as their HbS values ranged from 25–38% and HbS was less than HbA values. Diagnosis was confirmed by positive sickling test. Five (3.7%) patients had HbS values greater than HbA. Of these, two patients had associated increase in HbA2 (>3.9%) and had HbA values between 7%-27%. They were diagnosed as compound heterozygous state of sickle - β+ thalassaemia. Three of the five patients, had increased HbS values (>70%) and normal HbA2 with increased HbF values (5–25%). On gel electrophoresis, at alkaline pH, all had abnormal band at SDG region and sickling test was positive. Clinically these three patients were symptomatic and had bony pains, jaundice and mild splenomegaly. They were diagnosed as homozygous sickle cell anaemia (HbSS) (Fig. 2). One patient (0.7%) had unknown peak on HPLC with retention time of 4.78 minutes, constituting 16.8% of total haemoglobin. Haemoglobin electrophoresis in alkaline pH revealed an abnormal band at SDG position and abnormal band between A and S positions in acid electrophoresis. Sickling test was negative. Clinically the patient was asymptomatic, had mild anaemia (Hb 9.1 gm/dl) which was found to be due to IDA. He was diagnosed as HbQ – India heterozygous (Fig. 3) [8, 9, 10].
Fig. 2.
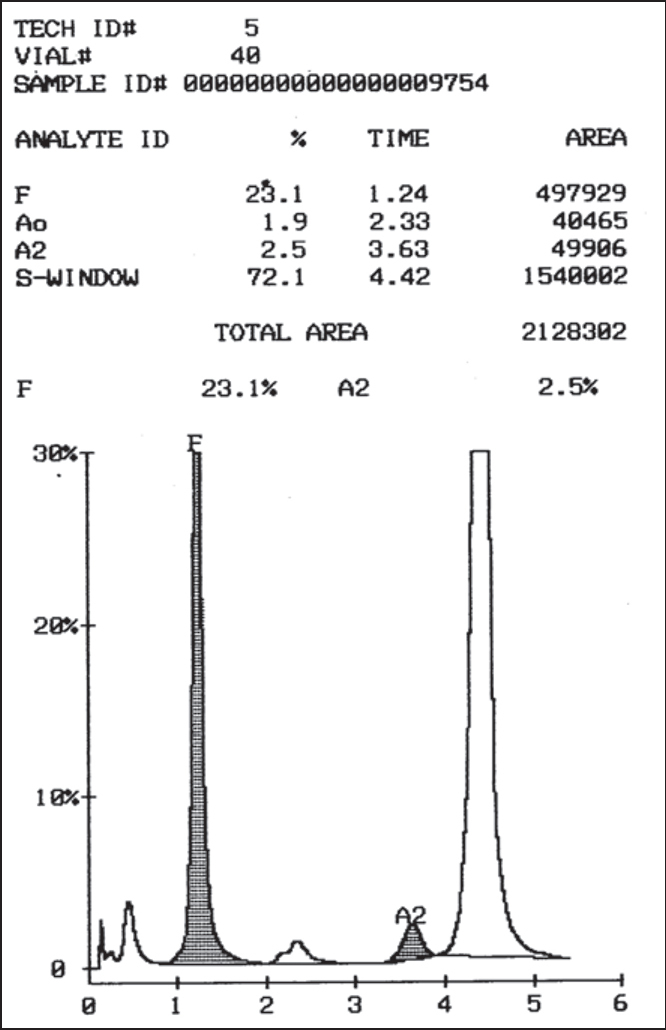
Chromatogram of sickle cell anaemia (HbSS)
Fig. 3.
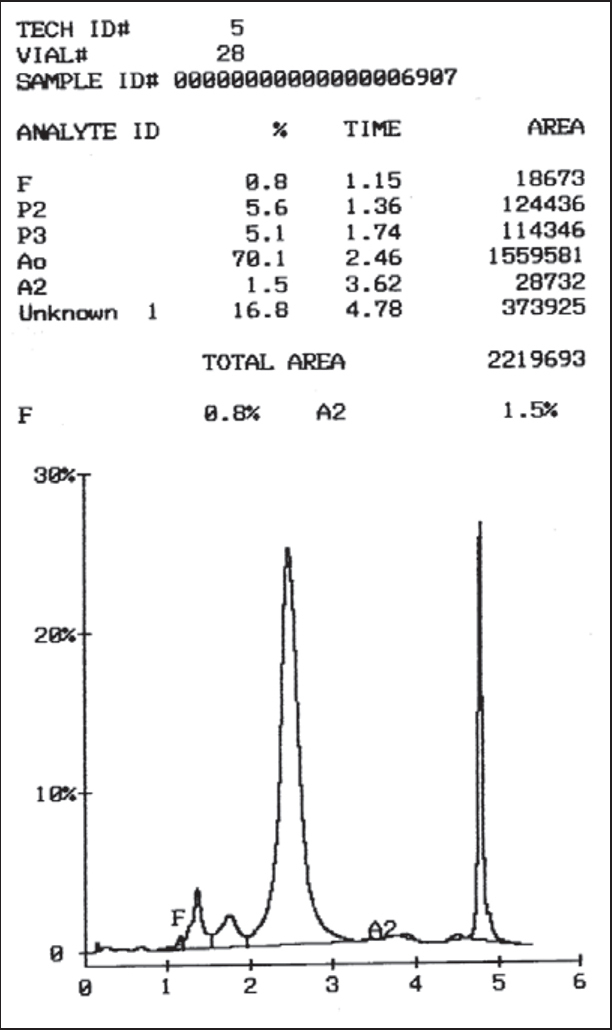
Chromatogram of HbQ India (Retention time : 4.78 minutes)
Thirteen (9.5%) patients were diagnosed as HbE syndrome on HPLC. Based on the family studies and levels of HbE / A2 and HbA, they were further sub classified as HbE trait (five cases) and HbE disease (eight cases). Clinically they were asymptomatic and were residents of North – East India / West Bengal / Orissa and one patient was from Maldives.
Seven patients (5.1%) had peak eluted at ‘D – window” with RT window of 3.90 to 4.30 minutes, this was clearly demarcated from HbS peak (RT window of 4.30 to 4.70 minutes). The abnormal haemoglobin constituted between 33–43% of total haemoglobin. On gel electrophoresis at alkaline pH, a band at HbA region and an abnormal band at SDG region were detected. In acidic electrophoresis, the abnormal band migrated with the adult haemoglobin. Sickling test was negative. Clinically these patients were normal or had mild nutritional anaemia. They were diagnosed as HbD - Punjab heterozygous.
One patient presented with anaemia (Hb 4.6gm/dl), high MCV (121fl) with normal MCH, high RDW (43%) and low RBC counts (1.1 million/cmm). Peripheral blood smear examination revealed macrocytes, polychromatic cells, macroovalocytes, and hypersegmented neutrophils. HPLC showed borderline HbA2 levels of 3.7%. The patient was treated with a trial of vitamin B12 / folic acid, subsequent to which a repeat HPLC showed normal HbA2 values.
Discussion
The laboratory diagnosis of haemoglobinopathies and thalassaemia's, both of which are common disorders in our country, is required for confirmation of sickling disorders or thalassaemia major; to find the cause of underlying haematologic abnormality (such as anaemia, microcytosis or polycythemia); for neonatal screening, to identify the abnormality in the pre symptomatic phase; to predict serious disorders of the globin – chain synthesis in the foetus and offer the option of termination of pregnancy and to permit genetic counselling of prospective parents [11].
The most common investigative tools for diagnosis of haemoglobinopathies are alkaline and acid gel electrophoresis for haemoglobin, HbA2 quantification by ion – exchange column chromatography and HbF quantification by alkaline denaturation method. The compound heterozygous disorders (HbSD – Punjab, HbSE, HbS - β thalassaemia) or unusual variants (HbQ India, HbD Punjab, HbD Iran) are all clinically significant with varying degree of severity, making precise identification important [12, 13]. None of these can be conclusively identified by a single electrophoretic technique [8]. The identification of haemoglobin variants by conventional techniques are often presumptive, based on the electrophoretic mobility of the band, quantification and / or ethnic origin of the parents.
HPLC has been shown to be rapid, sensitive, specific and reproducible alternative to conventional haemoglobin electrophoresis. HPLC offers the distinct advantage over classic haemoglobin electrophoresis as it can more accurately identify and quantitate abnormal haemoglobins. It is also very useful for paediatric group of patients, as only 5µl of blood is sufficient for analysis. The disadvantage of HPLC variant is that it requires skill in interpretation. Also, various normal and abnormal haemoglobins may have same retention time, the glycosylated variant Hb analyzer may have different retention time from non – glycosylated variant Hb analyzer. Hb E and Hb Lepore co – elute with Hb A2. Isoelectric focusing as a second line technique can resolve the issue.
In this study, beta heterozygous thalassaemia is most prevalent (66.4%), whereas, beta – homozygous thalassaemia is seen in only 3.7% of patients. This low incidence of homozygous state of the disease may be either to decrease incidence of the disease due to effective prenatal diagnosis or may be due to the affected patients group not reporting to the hospital where this study was carried out. Five patients who had associated IDA and borderline HbA2 levels had their HbA2 values >3.9% after iron therapy. However, one patient who had borderline HbA2 levels (3.7%) had associated vitamin B12/folic acid deficiency and his HbA2 levels returned to normal after vitamin B12 / folic acid therapy. So, apart from IDA, megaloblastic anaemia will also result in borderline HbA2 levels, the latter returning to normal after adequate therapy [14, 15, 16, 17, 18].
Although HbE and HbS syndromes could be diagnosed by both HPLC and gel electrophoresis, the use of HPLC helped in further sub – characterization of these syndromes based on quantification of HbE, HbS and HbA2 levels. In general, HbE syndromes and HbS trait are asymptomatic but patients with sickle cells anaemia (HbSS) are symptomatic. Use of HPLC, also helped in easier diagnosis of one case of compound heterozygous sickle - β+ thalassaemia.
Amongst the patients who had Hb band at SDG region in gel electrophoresis and negative sickling test, seven patients were detected to have HbD Punjab and one patient diagnosed as HbQ India on HPLC [19, 20]. It is thus recommended that in all cases where Hb migration occurs in SDG region on gel electrophoresis, HPLC should be definitely be performed for further sub characterization of rare Hb variants [21]. HPLC could also avoid misidentification of two haemoglobin variants having benign interaction with HbS. HbD – Punjab and HbD – Iran exhibits identical electrophoretic mobilities at alkaline pH, but their mean retention time in HPLC are unique and significantly different; HbD Punjab (RT of 4.18 min) and Hb Iran (RT of 3.49 min). These situations are clinically important as HbSD – Punjab is a significant sickling disorder, whereas HbSD – Iran is clinically benign [12, 22]. The misdiagnosis of HbD – Iran and HbD – Punjab may lead to incorrect genetic counselling in addition to undue anxiety for the family. It is also seen that relative elution time (dividing elution time of variant Hb with that of main HbA fraction) is a better method of detecting Hb variants from overlapping ones.
This is the first report in the Armed Forces, of a large prospective study on the use of HPLC for determining the presence and identities of haemoglobin variants in a clinical laboratory setting. This data demonstrates that HPLC is an excellent, powerful diagnostic tool for the direct identification of haemoglobin variants with a high degree for precision in the quantification of major and minor, normal and abnormal haemoglobin fractions. With the integration of proper algorithms involving retention time, %Hb and RBC indices, a clinical laboratory is capable of identifying about 75% of the common variants encountered without the need for confirmatory studies such as alkaline and acid electrophoresis [1]. More importantly, identification of the common haemoglobin variants (i.e. HbD – Punjab, HbE and β thalassaemia) that in combination with HbS lead to clinically significant sickling disorder which can be quickly and accurately accomplished by HPLC, without the need for confirmatory testing. In conclusion, the simplicity of sample preparation, accurate quantification of haemoglobin concentration combined with complete automation, makes HPLC an ideal methodology for the routine diagnosis of haemoglobin disorders.
Conflicts of Interest
None identified
Intellectual Contribution of Authors
Study Concept : Lt Col PK Gupta (Retd), Col H Kumar
Drafting & Manuscript Revision : Lt Col S Kumar, Lt Col PK Gupta (Retd)
Statistical Analysis : Lt Col H Kumar, Lt Col PK Gupta (Retd)
Study Supervision : Col H Kumar, Brig M Jaiprakash
References
- 1.Joutovsky A, Hadzi-Nesic J, Nardi MA. HPLC retention time as a diagnostic tool for hemoglobin variants and hemoglobinopathies: a study of 60,000 samples in a clinical diagnostic laboratory. Clin Chem. 2004;50:1736–1747. doi: 10.1373/clinchem.2004.034991. [DOI] [PubMed] [Google Scholar]
- 2.Higgins TN, Ridley B. Tentative identification of hemoglobin variants in the Bio — Rad VARIANT II Hb A1C method. Clin Biochem. 2005;38:272–277. doi: 10.1016/j.clinbiochem.2004.11.004. [DOI] [PubMed] [Google Scholar]
- 3.Riou J, Godart C, Hurtrel D, Mathis M, Bimet C. Cation — exchange HPLC evaluated for presumptive identification of hemoglobin variants. Clin Chem. 1997;43:34–39. [PubMed] [Google Scholar]
- 4.Waters HM, Howarth JE, Hyde K, Goldstone S, Cinkotai KI. An evaluation of the Bio — Rad Variant Haemoglobin Testing System for the detection of haemoglobinopathies. Clin Lab Haematol. 1998;20:31–40. doi: 10.1046/j.1365-2257.1998.00101.x. [DOI] [PubMed] [Google Scholar]
- 5.Clarke GM, Higgins TN. Laboratory investigations of haemoglobinopathies and thalassaemias. Review and update. Clin Chem. 2000;46:1284–1290. [PubMed] [Google Scholar]
- 6.Bio — Rad VARIANTIM thalassaemia short program. Instruction Manual. 2003;10 [Google Scholar]
- 7.Fucharoen S, Winichagoon P, Wisedpancihkji R. Prenatal and postnatal diagnosis of thalassemias and haemoglobinopathies by HPLC. Clin Chem. 1998;44:740–748. [PubMed] [Google Scholar]
- 8.Bain BJ. Haemoglobinopathy Diagnosis. Blackwell Science Ltd; Oxford: 2001. [Google Scholar]
- 9.Balgir RS. Burden of hemoglobinopathies in India and the challenges ahead. Current Science. 2000;79:1536–1547. [Google Scholar]
- 10.Wiwanitkit V. Secondary and tertiary structure aberration of alpha globin chain in haemoglobin Q-India disorder. Indian J Pathol Microbiol. 2006;49:491–494. [PubMed] [Google Scholar]
- 11.Working Party of the General Haematology Task Force of the British Committee for Standards in Haemotology Guideline: the laboratory diagnosis of haemoglobinopathies. Br J Haematol. 1998;101:783–792. [Google Scholar]
- 12.Steinberg MH. In: Disorders of hemoglobin: genetics, pathophysiology, and clinical management. Steinberg MH, Forget BG, Higgs DR, Nagel RL, editors. Cambridge University Press; New York: 2001. pp. 786–810. [Google Scholar]
- 13.Dash S. Hb A2 in subjects with Hb D. Clin Chem. 1998;44:2381–2382. [PubMed] [Google Scholar]
- 14.El - Aquoza I, Abu Shahla A, Sirdah M. The effects of iron deficiency anaemia on the levels of haemoglobin subtype: possible consequences for clinical diagnosis. Clin Lab Haematol. 2002;24:285–289. doi: 10.1046/j.1365-2257.2002.00464.x. [DOI] [PubMed] [Google Scholar]
- 15.Madan N, Sikka M, Sharma S, Rusia U. Haematological parameters and HbA2 levels in beta — thalassaemia trait with coincident iron deficiency. Ind J Pathol Microbiol. 1998;41:309–313. [PubMed] [Google Scholar]
- 16.Bencaiova G, Burkhardt T, Kraft A, Zimmerman R. Screening for beta — thalassaemia trait in anaemic pregnant women. Gynecol Obstet Invest. 2006;62:20–27. doi: 10.1159/000091813. [DOI] [PubMed] [Google Scholar]
- 17.Chan CW, Liu SY, Kho CS. Diagnostic clues to megaloblastic anaemia without macrocytosis. Int J Lab Hematol. 2007;29:163–171. doi: 10.1111/j.1751-553X.2007.00911.x. [DOI] [PubMed] [Google Scholar]
- 18.Das Gupta A. Abrogation of macrocytosis in pernicious anemia by beta — thalassaemia does not mask the diagnosis of vit B12 deficiency. Am J Hematol. 2002;71:61–62. doi: 10.1002/ajh.10159. [DOI] [PubMed] [Google Scholar]
- 19.Desai DV, Dhanani H, Kapoor A, Yeluri SV. HbQ — India in a Sindhi family: an uncommon hemoglobin variant. Lab Hematol. 2004;10:212–214. doi: 10.1532/lh96.04058. [DOI] [PubMed] [Google Scholar]
- 20.Abraham R, Thomas M, Britt R, Fisher C, Old J. HbQ-India: an uncommon variant diagnosed in three Punjabi patients with diabetes is identified by a novel DNA analysis test. J Clin Pathol. 2003;56:296–299. doi: 10.1136/jcp.56.4.296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tyagi S, Saxena R, Choudhry VP. HPLC — How necessary is it for haemoglobinopathy diagnosis in India? Indian J Pathol Microbiol. 2003;46:390–393. [PubMed] [Google Scholar]
- 22.Eastman JW, Lorey F, Arnopp J, Currier RJ, Sherwin J, Cunningham G. Distribution of hemoglobin F, A, S, C, E, and D quantities in 4 million newborn screening specimens. Clin Chem. 1999;45:683–685. [PubMed] [Google Scholar]