

Abstract
Context
Bevacizumab (BEV) is a monoclonal antibody to vascular endothelial growth factor (VEGF) that ameliorates atheroma progression by inhibiting neovascularization.
Objective
We aimed to determine whether BEV release from echogenic liposomes (BEV-ELIP) could be enhanced by color Doppler ultrasound (US) and whether the released BEV inhibits VEGF expression by endothelial cells in vitro.
Materials and methods
BEV-ELIP samples were subjected to 6 MHz color Doppler ultrasound (MI=0.4) for 5 min. We assessed release of BEV with a direct ELISA and with fluoresceinated BEV (FITC-BEV) loaded into ELIP by the same method. Human umbilical vein endothelial cell (HUVEC) cultures were stimulated to express VEGF by 10nM phorbol-12-myristate 13-acetate (PMA). Cell-associated VEGF levels were determined using a cell-based ELISA.
Results
Overall, US caused an additional 100 μg of BEV to be released or exposed per BEV-ELIP aliquot within 60 min BEV-ELIP treated with US inhibited VEGF expression by 90% relative to non-treated controls and by 70% relative to BEV-ELIP without US. Also, US-treated BEV-ELIP inhibited HUVEC proliferation by 64% relative to untreated controls and by 45% relative to BEV-ELIP without US.
Discussion and conclusion
We have demonstrated that BEV-ELIP retains its VEGF-binding activity in a liposomal formulation and that clinical Doppler US can significantly increase that activity, both by releasing free BEV and by enhancing the surface exposure of the immunoreactive antibody.
Keywords: Controlled release/delivery, in vitro models, nanotechnology, therapeutic antibodies
Introduction
Thrombotic cardiovascular diseases, including acute myocardial infarction and ischemic stroke, continue to be the leading causes of mortality and chronic disability in the USA, as well as the rest of the developed world (Go et al., 2014). Approximately 70% of acute occlusive ischemic cardiovascular events result from rupture of “vulnerable” atherosclerotic plaques (atheroma), resulting in sudden formation of thrombi, rather than from stenotic atheroma (Casscells et al., 2003). It has been found that progression of atheroma to a vulnerable state is very dependent on adventitial neovascularization involving proliferation of the vasa vasorum (Herrmann et al., 2001; Kumamoto et al., 1995; Moulton et al., 2003; Wilson et al., 2002), which is promoted by vascular endothelial growth factor (VEGF) (Celletti et al., 2001).
Bevacizumab, a humanized monoclonal antibody to VEGF, is the first FDA-approved biological therapy designed to inhibit the formation of new blood vessels to tumors (Hurwitz et al., 2004). As such, this development represented confirmation of Judah Folkman’s seminal hypothesis that inhibition of tumor neovascularization would constitute an effective anti-cancer strategy (Folkman, 1971). Vital to testing his hypothesis was the discovery of VEGF as a major mediator of angiogenesis. Over-expression of VEGF by many transformed cells is critical to providing an adequate blood supply for growing tumors (Grunstein et al., 2000). In 2004, the FDA-approved bevacizumab for clinical treatment of metastatic colorectal cancer (Hurwitz et al., 2004). Recently, arterial implantation of bevacizumab-eluting stents has been shown to inhibit induced atheroma neovascularization and intimal hyperplasia (Stefanadis et al., 2007, 2008; Toutouzas et al., 2007).
Predictably for a therapeutic that profoundly affects a fundamental physiologic process, bevacizumab has several severe adverse effects when given systemically, including bleeding, arterial thrombosis, bowel perforation, inhibition of wound healing and hypertension (Kozloff et al., 2009). Site-specific modes of bevacizumab delivery targeted directly to the atheroma neovasculature would minimize damage to normal tissues, while enhancing the dose-dependent efficacy of the drug.
We have developed echogenic liposomes (ELIP) as a targeted ultrasound contrast agent for diagnostic applications and as an ultrasound-enhanced controlled-release vehicle for drug, gene and stem cell delivery (Herbst et al., 2010; Tiukinhoy et al., 2000, 2004). We have loaded bevacizumab into ELIP (BEV-ELIP), resulting in a formulation in which the monoclonal antibody’s VEGF-binding sites are exposed to the medium, and here report the results of investigations into BEV-ELIP echogenicity, ultrasound-triggered bevacizumab release, and ability to inhibit VEGF expression and endothelial cell growth in vitro.
Materials and methods
Preparation of BEV-ELIP
Bevacizumab-loaded ELIP were prepared by mixing the lipid components 1,2-dipalmitoyl-sn-glycero-3-phosphocholine (DPPC), 1,2-dioleyoyl-sn-glycero-3-phosphocholine (DOPC), 1,2-dipalmitoyl-sn-glycero-3-[phosphor-rac-1-glycerol] (DPPG), 1,2-dipalmitoyl-sn-glycero-3-phosphoethanolamine (DPPE) and cholesterol (CH) at a 46:23:8:8:15 molar ratio in a round-bottom flask as a chloroform solution (Huang et al., 2002). The chloroform was evaporated under argon while spinning the flask submerged in a 50 °C water bath. The consequent lipid film was placed under vacuum for 4 h at ≤100mTorr pressure for complete removal of the solvent. The dry lipid film was rehydrated with 0.2M mannitol in 12.5-fold diluted 0.02M phosphate-buffered saline, pH 7.4 (PBS), containing 0.8 mg/ml of bevacizumab (Genentech, South San Francisco, CA) to a concentration of 10 mg lipid/ml. The hydrated lipid was centrifuged at 23 000 g in a microfuge for 20 min to remove unencapsulated bevacizumab. Each aliquot (400 μg BEV in 0.5 ml) of the suspension was placed in a −80 °C freezer and lyophilized for 24–48 h. Each lyophilized 5mg dry cake was resuspended in 0.5 ml nanopure water immediately prior to use. BEV-ELIP size distribution and number were determined with a Beckman Coulter Multisizer 4 (Beckman Coulter, Inc., Brea, CA) employing a 20-μm aperture tube.
Acoustic analysis
Liposomes were imaged with a 15-MHz intravascular ultrasound catheter (Boston Scientific, Natick, MA) in a glass vial with instrumental settings for gain, zoom, compression and rejection levels constant for all samples. Relative echogenicity (apparent brightness) of the liposome formulations was objectively assessed using computer-assisted videodensitometry. This process involved image acquisition, liposome identification and gray scale quantification. Images were digitized to 640×480-pixel spatial resolution (~0.045 mm/ pixel) and 8-bit (256 level) amplitude resolution. All image processing and analyses were performed with Image-Pro Plus Software (Media Cybernetics, Rockville, MD) running on a dedicated computer. Data are reported as mean gray scale values (MGSV). Echogenicity of ELIP preparations were compared by use of an echogenicity index, E50, the MGSV of a 50-μg lipid/ml suspension.
Fractionation of BEV-ELIP samples
Reconstituted BEV-ELIP aliquots were centrifuged at 14 100 g for 20 min at room temperature. Based on optical absorbance at 440 nm, which has been established as a measure of intact liposomal concentration within the submicron size range, centrifugal pellets contained 97.4±2.1% (SD, n=6) of total ELIP (Laing et al., 2012). The phospholipid content of centrifugal fractions was also measured by the colorimetric Stewart assay (Zuidam et al., 2003), which indicated that 96.2% of total liposomal phospholipid was recovered in the pellet. Aliquots of the liposomal suspension and the resuspended pellet in 0.5% Tween-20 (total BEV and total pellet BEV), as well as the supernate and the intact resuspended pellet (pellet surface determination, in PBS) were assayed for immunoreactive (IR) BEV with a direct enzyme-linked immunosorbent assay (ELISA) as previously described (Klegerman et al., 2014). Percent encapsulation efficiency was determined as (microgram total BEV×100)/400 μg. IR BEV in intact pellet suspensions was considered to be tightly associated with ELIP, while having epitopes exposed on the liposomal surface. Binding affinities and thermodynamic association parameters were determined from ELISA data as previously described (Klegerman et al., 2007, 2014).
Negative staining transmission electron microscopy
To determine BEV-ELIP ultrastructure, a BEV-ELIP aliquot (5 mg lipid) was freshly reconstituted with nanopure water and diluted in PBS to a concentration of 1.0 mg lipid/ml. Samples were subjected to a negative staining procedure with 1% uranyl acetate on 300 mesh formvar carbon grids (EMS, Hatfield, PA). Stained samples were examined with a JEOL 1200 TEM at 60 kV. Images were captured using a Gatan BioScan 792 CCD camera.
Ultrasound-enhanced release of bevacizumab from BEV-ELIP
Bevacizumab release from BEV-ELIP, with and without application of ultrasound, was studied by measurement of IR BEV with the ELISA and by fluorescence measurement of fluorescein isothiocyanate-labeled BEV (FITC-BEV-ELIP release). For FITC labeling, 200 μg of FITC (Sigma-Aldrich Chem. Co., St. Louis, MO) in 20 μl dimethyl formamide was added to 4mg BEV in 2ml of 0.05M borate buffer, pH 8.5, and allowed to react for 1 h at room temperature in the dark. Unreacted FITC was removed with Zeba spin columns (Pierce/Thermo Scientific); the FITC-BEV product was loaded into ELIP as was unlabeled BEV. Fluorescence measurements were made in a SpectraMax M5 microplate reader (Ex=485 nm; Em=538 nm). Bevacizumab concentrations were calculated from appropriate FITC fluorescence standard curves, correcting for quench due to detergent in Tween-20 containing samples, and converting to [BEV] using specific activity values determined for each batch of FITC-BEV.
For release studies, one 5-mg BEV-ELIP or FITC-BEV-ELIP aliquot was reconstituted with 0.5 ml of nanopure water and added to 49.5 ml of PBS, human plasma (Innovative Research, Novi, MI) or porcine plasma (Innovative Research). The mixture was divided equally between two condoms. One condom was insonated in a water bath at ambient temperature or 37 °C with an HDI 5000 ultrasound system (Philips Medical Systems, Andover, MA) using an L12-5 linear array probe. Ultrasound treatment consisted of 6.0MHz duplex (color) Doppler ultrasound pulses with a mechanical index (MI) of 0.4 for 5×30 s within a 5-min period. These insonation conditions have previously been shown to cause rapid fragmentation of ELIP and optimal release of a therapeutic protein payload (Smith et al., 2010), and were used in studies of tPA-loaded ELIP efficacy (Laing et al., 2011, 2012). The other condom was carried through the same procedure without insonation. The contents of each condom were transferred to 50-ml plastic centrifuge tubes and placed on tube rockers at the same temperature as the water bath. Aliquots (0.5 ml) were removed immediately (0 min), at 30 min and at 60 min for centrifugal fractionation (as described above) and assay. Encapsulation efficiency of FITC-BEV-ELIP was determined as the sum of 0-point control supernate and total pellet BEV amounts in micrograms relative to the 400 μg total added to the lipid film during liposome production.
In vitro bevacizumab efficacy assay
Treatment materials were added to 18-h human umbilical vein endothelial cell (HUVEC; BD Biosciences, San Jose, CA) cultures in tissue culture grade 96-well plates (Falcon, BD Biosciences; 0.1 ml per well), followed by 10 nM (final concentration) phorbol 12-myristate 13-acetate (PMA; Sigma-Aldrich) to elicit VEGF expression and endothelial cell proliferation through protein kinase (PK) A and C induction, or medium alone, 4 h later and incubated for an additional 24 h. Treatments consisted of medium alone (control), bevacizumab (1 μg/ml) or BEV-ELIP (100 μg lipid/ml). After the final incubation, cells were enumerated by trypsinization and counting with a hemacytometer or by addition of 100 μl (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl-tetrazolium bromide (MTT; Sigma-Aldrich) solution per well and incubation for 4 h, after which 75 μl of medium was removed and 25 μl of dimethylsulfoxide (DMSO) was added, followed by 1 h of incubation at 37 °C. Absorbance of the wells at 540 nm was measured in the SpectraMax M5 microplate reader.
Cultured cell VEGF ELISA
Various concentrations of bevacizumab or BEV-ELIP in Medium 200 were added to PMA-treated (or untreated) HUVEC cultures in 96-well tissue culture grade microplate wells and incubated 12 h, after which cells were fixed with formalin and incubated with 5 μg/ml of bevacizumab (primary antibody) for 2 h at 37 °C. After washing, wells were incubated with 10 000-fold diluted anti-human IgG-HRP secondary antibody (Sigma-Aldrich) for 1 h at room temperature, followed by three washes. Color development with 2,2′-azino-bis(3-ethylbenzothiazoline-6-sulfonic acid) diammonium salt (ABTS; Sigma-Aldrich) substrate was carried out for 30 min at room temperature. Plate absorbance was measured at 405 nm in the SpectraMax M5 reader. For determination of ultrasound enhancement of bevacizumab in vitro cell effects, dilute BEV-ELIP/PBS suspensions (after sterilization of lyophilized BEV-ELIP aliquots by exposure to ultraviolet light for 60 min) were exposed to color Doppler ultrasound in condoms as described above.
Protein preparation and western blotting
Three groups of HUVEC grown in 24-well plates [untreated or incubated with 10nM PMA or 100nM 12-deoxyphorbol-13-O-phenylacetate (DOPPA)] were homogenized in 50mM Tris–HCl, pH 7.5, containing 0.5mM EDTA, 0.5mM EGTA and 10% glycerol (homogenation buffer) by rapid freeze-thaw cycles on dry ice, followed by centrifugation at 0 °C for 10 min at 26 000 g and retention of the supernate. To investigate the mechanism of VEGF expression mediated by PKC and PKA pathways, western blots for PKA and PKC were performed on ultracentrifugation fractions to determine translocation of the kinases from the cytosol to the endoplasmic reticulum (ER) fraction. Cell homogenates were centrifuged at 100 000 g for 1 h in a Beckman Coulter Optima L-100 XP ultracentrifuge. The supernate (containing cytosolic protein) was reserved and the pellet was resuspended in 1M sodium chloride in cold homogenization buffer, followed by centrifugation for 30 min at 100 000 g. The pellet (containing membrane protein) was then dissolved in homogenization buffer containing 2% Triton X-100. Protein concentrations were measured using the Bradford assay (Bradford, 1976) with bovine serum albumin standards.
One volume of homogenized protein was added to three volumes of Laemmli-loading buffer (Laemmli, 1970) and denatured at 90 °C for 5 min. The denatured samples were loaded onto 12% acrylamide gel lanes. Protein bands in the sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS–PAGE) gels were transferred to nitrocellulose membranes electrophoretically and then were incubated with dilutions of primary antibody (100×bevacizumab, 1 μg/ml anti-PKCα [Cell Signaling Technology, Danvers, MA] or 1 μg/ml anti-PKAα [Santa Cruz Biotechnology, Santa Cruz, CA]) at 4 °C overnight, washed and then incubated with 4000×secondary antibody (anti-human or rabbit IgG-IR Dye 800CW, Sigma-Aldrich) for 1 h at room temperature. Dry nitrocellulose membrane blots were imaged for infrared florescence with a Li-Cor Odyssey System.
Statistics
Data in comparative groups were subjected to normal variance analysis and compared for probability of population identity by Student’s t-test using Microsoft Excel 2010 software. The variance (standard error of mean, SE) of ΔH° and ΔS° of free bevacizumab binding to VEGF was calculated from standard errors of the slope and intercept, respectively, for the van’t Hoff plot linear regression, using SigmaPlot 10.0 software (Systat Software Inc., Richmond, CA). The variance of ΔG° at each temperature was calculated from these parameters using the equation of state, ΔG° =ΔH° − TΔS°.
Results
Characterization of BEV-ELIP
Initial characterization of the BEV-ELIP formulation indicated that ~30% of bevacizumab mixed with the component lipids was encapsulated in the final product, with excellent retention of ELIP echogenicity (Figure 1c and d; E50=157±34 [SD, n=7] versus 161±2 [n=4] for empty ELIP; p=0.79). The average mean number equivalent spherical diameter of BEV-ELIP>0.4 μm, measured with the Multisizer 4, was 893±105 nm (SD, n=6). Typically, >99% of ELIP are ≤100 nm in diameter (Kopechek et al., 2011), which is reflected in Figure 1(b), while Figure 1(a) shows a larger proportion of liposomes>400 nm in diameter.
Figure 1.

Negative-stained transmission electron micrographs of ELIP samples. (a) Empty ELIP (15 000×), showing a heterogeneous size distribution, including liposomes >400 nm in diameter; solid arrow denotes a vesicle ~100 nm in diameter. (b) BEV-ELIP (50 000×), demonstrating perimembranous (solid arrow) and lumenal (open arrow) protein localization, indicated by increased electron density; nearly all liposomes are ~100 nm in diameter. (c) Intravascular ultrasonogram of a BEV-ELIP suspension (200 μg lipid/ml) in a glass shell vial. (d) Dose response of BEV-ELIP echogenicity (MGSV). Points=mean of n=3–5; bars=SE.
The encapsulation efficiency of 10 lots of BEV-ELIP ranged from 22.0 to 43.9% with a mean value of 32.1±6.6 (SD) %. Based on the VEGF ELISA results, free bevacizumab was found to have an affinity for recombinant human VEGF165 of 1.1±0.3nM (n=5) at 37 °C, which is in good agreement with published values (Liang et al., 2006; Varey et al., 2008). Thermodynamic analysis indicated that the antibody-antigen interaction is slightly exothermic (ΔH° =−0.89 kcal/mole), but very hydrophobic (ΔS° = +38.2 cal/mole°; Table 1).
Table 1.
Thermodynamic parameters for free bevacizumab/VEGF 165 association.
| T (°K) | Kassoc (M−1) | ΔH° (kcal/mol) | ΔG° (kcal/mol) | ΔS° (cal/mol°) |
|---|---|---|---|---|
| 277 | 1.06×109 | −0.889±0.735a | −11.438±1.446 | +38.16±2.49 |
| 298 | 1.11×109 | −12.333±1.405 | ||
| 310 | 8.62×108 | −12.673±1.553 |
Mean±SE, based on standard errors of the van’t Hoff plot linear regression.
ELISA evaluation of IR bevacizumab in the intact ELIP centrifugal pellet relative to total pellet IR bevacizumab (detergent treated) indicated that 60.3±26.1% (n=5) of encapsulated bevacizumab was able to bind antigen with the same affinity as the free antibody, suggesting that the binding sites are exposed on the liposomal surface. This possibility was supported by negative staining TEM, which demonstrated that BEV-ELIP consists of intact liposomes with protein both integrally associated with the lipid bilayer and deposited in the vesicular lumen (Figure 1b). Protein deposition was indicated by regions of increased electron density relative to unloaded ELIP (Figure 1a).
Characterization of FITC-BEV-ELIP
Based on quantitative fluorescence measurements and protein concentrations determined from A280 (E0.1%=1.20), mean recovery of bevacizumab from the fluoresceination process was 91.9±15.0% (SD, n=9). Conjugation efficiency (specific activity) was 2.5±0.8 μg FITC/mg bevacizumab or 1.0±0.3 molecules FITC/molecule bevacizumab. Encapsulation efficiency for FITC-BEV-ELIP was 26.4±5.1% (n=6), which is not different from the value found for IR BEV-ELIP (p>0.05).
Release of IR bevacizumab from BEV-ELIP
IR bevacizumab release from BEV-ELIP in PBS, determined by ELISA analysis of centrifugal fractions, indicated that 32.3±12.3% (n=15) of encapsulated bevacizumab was spontaneously released in PBS within the first 30 min. In human plasma, color Doppler ultrasound caused an immediate average net release of 80 μg IR bevacizumab (Figure 2a) per 5mg of lipid. Expressed as percent total encapsulated antibody, the release was significant (p=0.035; Figure 2b). A reciprocal loss of IR bevacizumab from the surface (exposed) pellet fraction was observed after 30 min, but only accounted for 5.3 μg of released bevacizumab.
Figure 2.

IR bevacizumab in BEV-ELIP fractions incubated at 25 °C in human plasma. (a) Absolute quantity of bevacizumab; (b) percent 0-time control. Black bars: supernate, no US; light gray bars: supernate, US; dark gray bars: pellet surface, no US; lightest bars: pellet surface, US. Bars=SE; n=3. *p<0.05 versus no US control.
Release of FITC-BEV from FITC-BEV-ELIP
Fluorescence measurements indicated that 54.0±16.6% (n=6) of encapsulated FITC-BEV was spontaneously released in porcine plasma within 60 min at 25 °C (Figure 3). Ultrasound treatment caused 58 μg of encapsulated FITC-BEV/5 mg lipid to be released within that period (6 μg from the pellet surface). A pattern of reciprocal decrease of liposomal (pellet) FITC-BEV was evident in terms of percent total, but not absolute quantity. Ultrasound-induced release of FITC-BEV in porcine plasma was also observed at 37 °C, but the magnitude was lower (23 μg/5 mg lipid; Figure 4).
Figure 3.

Distribution of FITC-BEV in FITC-BEV-ELIP fractions incubated at 25 °C in porcine plasma, determined by fluorescence measurements. (a) Absolute quantity of FITC-BEV; (b) percent 0-time control. Black bars: supernate, no US; light gray bars: supernate, US; dark gray bars: pellet surface, no US; lightest bars: pellet surface, US. Bars=SE; n=5–6. *p<0.05 versus no US control.
Figure 4.

Distribution of FITC-BEV in FITC-BEV-ELIP fractions incubated at 37 °C in porcine plasma, determined by fluorescence measurements. (a) Absolute quantity of FITC-BEV; (b) percent 0-time control. Black bars: supernate, no US; light gray bars: supernate, US; dark gray bars: pellet surface, no US; lightest bars: pellet surface, US. Bars=SE; n=3.
Endothelial cell proliferation assay for in vitro assessment of bevacizumab efficacy
VEGF-mediated endothelial cell proliferation can be induced by PMA and 12-deoxyphorbol-13-phenylacetate-20-acetate (dPPA) through PK activation pathways that increase VEGF mRNA transcription (Bergsland & Dickler, 2004; Broughman et al., 2006; Cai et al., 2006; Xu et al., 2008). Thus, we investigated whether HUVEC proliferation secondary to VEGF expression induced by these agents could be used to demonstrate bevacizumab efficacy in vitro. Endothelial cell proliferation as an endpoint of VEGF-inducing treatments was confirmed by incubation of HUVEC cultures with various concentrations of PMA and dPPA (Figure 5). Optimal concentrations of PMA and dPPA for induction of HUVEC proliferation without noticeable toxicity were 10 and 100 nM, respectively.
Figure 5.

Effect of PMA and dPPA on HUVEC proliferation, measured by the MTT assay. (a) Effect of various concentrations of PMA on HUVEC numbers after 24 h incubation (bars=mean±SD; n=4; *p<0.01 versus control, †p<0.05 versus 50nM PMA). (b) Effect of dPPA on HUVEC proliferation (bars=mean±SD; n=4; all treatments: p<0.02 versus control; 100nM dPPA: p<0.01 versus 50nM dPPA). (c) Typical MTT standard curve. Each point=mean±SD; n=3.
Demonstration of PK-mediated VEGF expression
PMA induction of VEGF expression was demonstrated by western blot analysis of HUVEC homogenates (Figure 6). Mediation of PMA and dPPA induction of VEGF expression by a translocation of both PKAα (not shown) and PKCα from the cytosol to the ER (membrane fraction) was demonstrated by western blot analysis (Figure 7). Complete separation of the subcellular fractions was confirmed by western blots showing the ER marker ERP72 in the membrane fraction, but not the cytosolic fraction (not shown).
Figure 6.

Western blot for VEGF expression. (a) VEGF expression was induced by 10nM PMA compared to control in which no VEGF was detected. (b) Bars represent the mean ratio of detected VEGF band intensity divided by total protein band (β-actin)±SD, n=4; p<0.001 for PMA-treated versus control.
Figure 7.

Western blot for PKCα translocation. (a) PMA and dPPA induce VEGF expression by inducing PKCα translocation from the cytosol to the ER. PKCα located in the cytosol is not active. (b) Bars represent the mean ratio of detected PKC band intensity divided by the total protein band (β-actin)±SD, n=4. *p<0.01 versus corresponding control, †p<0.01 versus corresponding control.
Cultured cell VEGF ELISA as an additional in vitro assessment of bevacizumab efficacy
Quantitative ELISA measurement of VEGF expression by HUVEC after PMA or dPPA incubation paralleled the proliferation responses, indicating that 10nM PMA and 100nM dPPA were optimal concentrations of the VEGF induction agents (Figure 8). Thus, both cultured HUVEC assays were found to be suitable to determine the ability of bevacizumab and BEV-ELIP to inhibit VEGF expression and its derivative cell functions in vitro.
Figure 8.
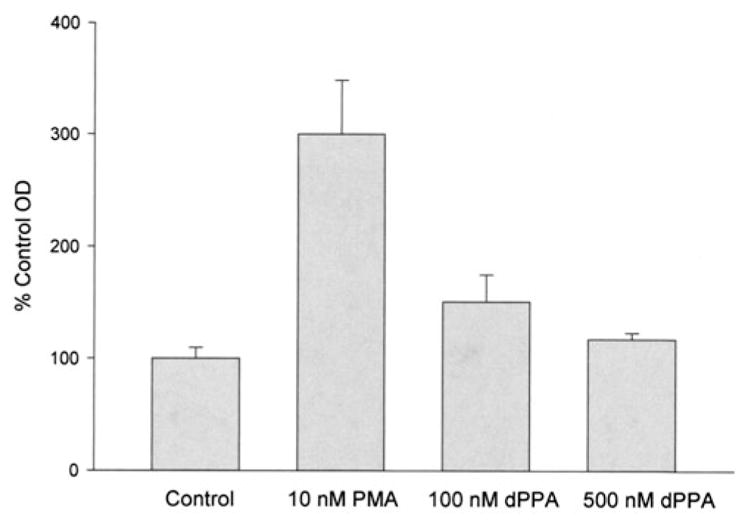
VEGF ELISA for PMA and dPPA. VEGF expression level of HUVEC treated with various concentrations of PMA and dPPA compared to control A405 (bars=mean±SD, n=4; p<0.03 for all treated HUVEC compared to controls).
Effect of bevacizumab and BEV-ELIP, with and without ultrasound, on HUVEC VEGF expression in vitro
Bevacizumab alone (10 μg/ml) inhibited VEGF expression (ELISA) by 60% relative to non-treated controls (Figure 9a). BEV-ELIP (100 μg lipid/ml; ~3 μg BEV/ml) treated with ultrasound inhibited VEGF expression by 90% relative to non-treated controls and by 70% relative to BEV-ELIP without ultrasound. Reflecting VEGF expression, bevacizumab alone inhibited HUVEC proliferation 31% relative to controls. BEV-ELIP with ultrasound inhibited HUVEC proliferation by 64% relative to untreated controls and by 45% relative to BEV-ELIP without ultrasound (Figure 9b). MTT determination of PMA induction and BEV inhibition of HUVEC proliferation was verified by hemacytometer counts to assure that increased ODs were not caused by stimulation of cell metabolism.
Figure 9.

Bevacizumab inhibition of VEGF expression. (a) VEGF ELISA showing bevacizumab (10 μg/ml) and BEV-ELIP (100 μg lipid/ml) efficacy, with optimal inhibition of VEGF expression caused by BEV-ELIP after exposure to ultrasound prior to treatment. Bars=mean±SD, n=4; *p<0.01 versus control, †p<0.01 versus BEV without ultrasound. (b) The proliferation assay shows that prior BEV-ELIP exposure to ultrasound caused the highest proliferation inhibition. Bars=mean±SD, n=4; *p<0.01 versus control, †p<0.01 versus BEV without ultrasound.
Discussion
Bevacizumab, a humanized monoclonal antibody specific for VEGF, developed and marketed by Genentech (affiliate of Hoffman LaRoche), has been approved for treatment of colorectal carcinoma by the US Food and Drug Administration (Hurwitz et al., 2004; Scott, 2010). Because of the persistent problem of the pharmacologic window, however, the antibody must be administered systemically at a dose of 1 mg/kg, resulting in severe side effects that include bowel perforation, bleeding and arterial thrombosis (Kozloff et al., 2009).
Stefanadis et al. (2006, 2007, 2008) has demonstrated that bevacizumab-eluting stents inhibit atheroma neovascularization and intimal hyperplasia, first in animals and then clinically. Ideally, however, clinical practice would dictate that anti-angiogenic therapy should be initiated prior to stenotic atheroma development requiring angioplasty, since most vulnerable atheroma causing fatal MIs and ischemic strokes are non-stenotic (Kolodgie et al., 2004; Naghavi et al., 2003). We have developed a controlled-release echogenic liposomal platform technology that features enhanced drug release induced by applied ultrasound through cavitation effects and ELIP fragmentation (Hitchcock et al., 2010; Laing et al., 2010; Smith et al., 2010). The bevacizumab-loaded form of ELIP that we constructed exhibits >20% (80 μg) encapsulation efficiency with retention of echogenicity and exposure of BEV VEGF-binding sites, obviating a need to target the formulation.
In this study, we have demonstrated that completely non-invasive conventional ultrasound under optimized conditions used in previous studies releases an appreciable amount of bevacizumab from BEV-ELIP and that the released agent is sufficient to clearly inhibit both expression of VEGF by endothelial cells and the cell proliferation that is the functional consequence of this expression, constituting an in vitro proof of probable efficacy.
Encapsulation efficiencies determined by measurement of IR bevacizumab in BEV-ELIP and by fluorescence quantitation of FITC-BEV in FITC-BEV-ELIP were in good agreement. The release data, however, indicate that the actual encapsulation efficiency may be at least 60% of total BEV added to the lipid film. Based on this figure, centrifugal fractionation of BEV-ELIP indicated that about two-thirds of ELIP-associated bevacizumab was spontaneously released within the first 30 min in 100 volumes of PBS with gentle mixing. Application of color Doppler ultrasound to diluted BEV-ELIP, using parameters that enhanced thrombolytic activity of tPA-loaded ELIP in a rabbit aorta thrombus model (Laing et al., 2011), caused release of an average of 80 μg IR BEV into porcine plasma from BEV-ELIP, in good agreement with US-induced FITC-BEV-ELIP release.
Although this represents only about one-third of ELIP-associated BEV, it produced a striking enhancement of VEGF inhibition in two cell-based assays of bevacizumab in vitro efficacy, suggesting that local release of the antibody by a targeted formulation greatly increased the therapeutic agent’s effectiveness. Thus, BEV-ELIP shows great promise for effective inhibition of VEGF-dependent neovascularization without the deleterious side effects exhibited by systemically administered bevacizumab. The bevacizumab spontaneously released from BEV-ELIP represents a dose of ~50 μg/kg, which is ≤1% of the clinical dose (Bergsland & Dickler, 2004) and therefore is unlikely to cause adverse effects. At the same time, VEGF induction in HUVEC by PKA and PKC stimulation produced two effective in vitro assays for both afferent and efferent aspects of VEGF expression.
There are a number of factors that influenced the results of these experiments. First, the discrepancy between bevacizumab encapsulation efficiencies determined from total BEV values and both BEV-ELIP and FITC-BEV-ELIP release data suggest that Tween-20 may not be as efficient in solubilizing ELIP as Triton X-100, or that detergents in general may cause underestimation of loaded BEV. Second, PMA is a relatively non-specific stimulator of VEGF expression, but we have confirmed that it acts through an activation of both PKs A and C, involving translocation of the factors from the cytoplasm to the ER. There may be some differences in BEV suppression of VEGF induced by this mechanism as opposed to the physiologically more relevant expression induced by hypoxic conditions exhibited in atheroma.
The primary challenge of demonstrating BEV-ELIP efficacy in vivo will be in optimizing an application schedule that inhibits adventitial VEGF expression at a critical stage of atheroma progression. First, an animal model featuring VEGF-dependent arteriogenesis-mediated atheroma progression to a vulnerable state approximating human atherogenic pathogenesis should be employed. The Yucatan miniswine model that we have utilized for>20 years involves histologic endpoints that may be straightforwardly assessed. The process of determining the pattern of VEGF expression that is critical to atheroma progression may require extensive study, but offers the potential for a significant advance in treatment of mid-stage atherosclerosis.
Despite these challenges, we have shown that BEV-ELIP provides a means for enhanced inhibition of VEGF expression by endothelial cells that occurs in the adventitial neovasculature of expanding atheroma. Thus, this formulation affords likelihood that atheroma progression can be minimized or stabilized by a relatively non-invasive therapeutic intervention.
Conclusions
In order to produce a therapeutic agent for inhibition of atheroma progression, we have successfully loaded the therapeutic monoclonal antibody bevacizumab into our echogenic liposomal formulation and demonstrated appreciable ultrasound-triggered release of the drug, establishing BEV-ELIP as a controlled-release delivery construct. Because a portion of the antibody, which is specific for VEGF mediating atheroma neovascularization, is associated with the lipid bilayer, antibody binding sites are exposed and, therefore, the construct is also targeted to the adventitia of progressing atheroma.
We have also demonstrated that ultrasound-treated BEV-ELIP markedly inhibits cultured endothelial cell VEGF expression and proliferation after stimulation with PMA, providing an in vitro proof-of-concept for this formulation. The basis is thus laid for demonstration of BEV-ELIP in vivo efficacy in preclinical animal studies.
Acknowledgments
This work was supported, in part, by grants from the National Institutes of Health (R01 HL074002, HL59586 and NS047603; F32 HL104916).
Footnotes
Declaration of interest
Drs Klegerman, Huang and McPherson have research-related interests in Zymo Pharmaceuticals, LLC. These interests consist of sponsored research support, as well as corporate and stock ownership.
References
- Bergsland E, Dickler MN. Maximizing the potential of bevacizumab in cancer treatment. Oncologist. 2004;9:36–42. doi: 10.1634/theoncologist.9-suppl_1-36. [DOI] [PubMed] [Google Scholar]
- Bradford MM. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem. 1976;72:248–54. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- Broughman JR, Sun L, Umar S, et al. Chronic PKC-beta activation in HT-29 Cl.19a colonocytes prevents cAMP-mediated ion secretion by inhibiting apical membrane current generation. Am J Physiol Gastrointest Liver Physiol. 2006;291:G318–30. doi: 10.1152/ajpgi.00355.2005. [DOI] [PubMed] [Google Scholar]
- Cai J, Jiang WG, Ahmed A, Boulton M. Vascular endothelial growth factor-induced endothelial cell proliferation is regulated by interaction between VEGFR-2, SH-PTP1 and eNOS. Microvasc Res. 2006;71:20–31. doi: 10.1016/j.mvr.2005.10.004. [DOI] [PubMed] [Google Scholar]
- Casscells W, Naghavi M, Willerson JT. Vulnerable atherosclerotic plaque: a multifocal disease. Circulation. 2003;107:2072–5. doi: 10.1161/01.CIR.0000069329.70061.68. [DOI] [PubMed] [Google Scholar]
- Celletti FL, Waugh JM, Amabile PG, et al. Vascular endothelial growth factor enhances atherosclerotic plaque progression. Nat Med. 2001;7:425–9. doi: 10.1038/86490. [DOI] [PubMed] [Google Scholar]
- Folkman J. Tumor angiogenesis: therapeutic implications. N Engl J Med. 1971;285:1182–6. doi: 10.1056/NEJM197111182852108. [DOI] [PubMed] [Google Scholar]
- Go AS, Mozaffarian D, Roger VL, et al. Heart disease and stroke statistics – 2014 update: a report from the american heart association. Circulation. 2014;129:e28–292. doi: 10.1161/01.cir.0000441139.02102.80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grunstein J, Masbad JJ, Hickey R, et al. Isoforms of vascular endothelial growth factor act in a coordinate fashion to recruit and expand tumor vasculature. Mol Cell Biol. 2000;20:7282–91. doi: 10.1128/mcb.20.19.7282-7291.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herbst SM, Klegerman ME, Kim H, et al. Delivery of stem cells to porcine arterial wall with echogenic liposomes conjugated to antibodies against CD34 and intercellular adhesion molecule-1. Mol Pharm. 2010;7:3–11. doi: 10.1021/mp900116r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herrmann J, Lerman LO, Rodriguez-Porcel M, et al. Coronary vasa vasorum neovascularization precedes epicardial endothelial dysfunction in experimental hypercholesterolemia. Cardiovasc Res. 2001;51:762–6. doi: 10.1016/s0008-6363(01)00347-9. [DOI] [PubMed] [Google Scholar]
- Hitchcock KE, Caudell DN, Sutton JT, et al. Ultrasound-enhanced delivery of targeted echogenic liposomes in a novel ex vivo mouse aorta model. J Control Release. 2010;144:288–95. doi: 10.1016/j.jconrel.2010.02.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang SL, Hamilton AJ, Pozharski E, et al. Physical correlates of the ultrasonic reflectivity of lipid dispersions suitable as diagnostic contrast agents. Ultrasound Med Biol. 2002;28:339–48. doi: 10.1016/s0301-5629(01)00512-9. [DOI] [PubMed] [Google Scholar]
- Hurwitz H, Fehrenbacher L, Novotny W, et al. Bevacizumab plus irinotecan, fluorouracil, and leucovorin for metastatic colorectal cancer. N Engl J Med. 2004;350:2335–42. doi: 10.1056/NEJMoa032691. [DOI] [PubMed] [Google Scholar]
- Klegerman ME, Huang S, Parikh D, et al. Lipid contribution to the affinity of antigen association with specific antibodies conjugated to liposomes. Biochim Biophys Acta. 2007;1768:1703–16. doi: 10.1016/j.bbamem.2007.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klegerman ME, Zou Y, Golunski E, et al. Use of thermodynamic coupling between antibody-antigen binding and phospholipid acyl chain phase transition energetics to predict immunoliposome targeting affinity. J Liposome Res. 2014;24:216–22. doi: 10.3109/08982104.2014.891230. [DOI] [PubMed] [Google Scholar]
- Kolodgie FD, Virmani R, Burke AP, et al. Pathologic assessment of the vulnerable human coronary plaque. Heart. 2004;90:1385–91. doi: 10.1136/hrt.2004.041798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kopechek JA, Haworth KJ, Raymond JL, et al. Acoustic characterization of echogenic liposomes: frequency-dependent attenuation and backscatter. J Acoust Soc Am. 2011;130:3472–81. doi: 10.1121/1.3626124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kozloff M, Yood MU, Berlin J, et al. Clinical outcomes associated with bevacizumab-containing treatment of metastatic colorectal cancer: the BRiTE observational cohort study. Oncologist. 2009;14:862–70. doi: 10.1634/theoncologist.2009-0071. [DOI] [PubMed] [Google Scholar]
- Kumamoto M, Nakashima Y, Sueishi K. Intimal neovascularization in human coronary atherosclerosis: its origin and pathophysiological significance. Hum Pathol. 1995;26:450–6. doi: 10.1016/0046-8177(95)90148-5. [DOI] [PubMed] [Google Scholar]
- Laemmli UK. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature. 1970;227:680–5. doi: 10.1038/227680a0. [DOI] [PubMed] [Google Scholar]
- Laing ST, Kim H, Kopechek JA, et al. Ultrasound-mediated delivery of echogenic immunoliposomes to porcine vascular smooth muscle cells in vivo. J Liposome Res. 2010;20:160–7. doi: 10.3109/08982100903218918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laing ST, Moody MR, Kim H, et al. Thrombolytic efficacy of tissue plasminogen activator-loaded echogenic liposomes in a rabbit thrombus model. Thromb Res. 2012;130:629–35. doi: 10.1016/j.thromres.2011.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laing ST, Moody M, Smulevitz B, et al. Ultrasound-enhanced thrombolytic effect of tissue plasminogen activator-loaded echogenic liposomes in an in vivo rabbit aorta thrombus model–brief report. Arterioscler Thromb Vasc Biol. 2011;31:1357–9. doi: 10.1161/ATVBAHA.111.225938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang WC, Wu X, Peale FV, et al. Cross-species vascular endothelial growth factor (VEGF)-blocking antibodies completely inhibit the growth of human tumor xenografts and measure the contribution of stromal VEGF. J Biol Chem. 2006;281:951–61. doi: 10.1074/jbc.M508199200. [DOI] [PubMed] [Google Scholar]
- Moulton KS, Vakili K, Zurakowski D, et al. Inhibition of plaque neovascularization reduces macrophage accumulation and progression of advanced atherosclerosis. Proc Natl Acad Sci USA. 2003;100:4736–41. doi: 10.1073/pnas.0730843100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naghavi M, Libby P, Falk E, et al. From vulnerable plaque to vulnerable patient: a call for new definitions and risk assessment strategies: part II. Circulation. 2003;108:1772–8. doi: 10.1161/01.CIR.0000087481.55887.C9. [DOI] [PubMed] [Google Scholar]
- Scott C. Never again. Nat Biotechnol. 2010;28:131. doi: 10.1038/nbt0210-131. [DOI] [PubMed] [Google Scholar]
- Smith DA, Vaidya SS, Kopechek JA, et al. Ultrasound-triggered release of recombinant tissue-type plasminogen activator from echogenic liposomes. Ultrasound Med Biol. 2010;36:145–57. doi: 10.1016/j.ultrasmedbio.2009.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stefanadis C, Toutouzas K, Stefanadi E, et al. First experimental application of bevacizumab-eluting PC coated stent for inhibition of vasa vasorum of atherosclerotic plaque: angiographic results in a rabbit atheromatic model. Hellenic J Cardiol. 2006;47:7–10. [PubMed] [Google Scholar]
- Stefanadis C, Toutouzas K, Stefanadi E, et al. Inhibition of plaque neovascularization and intimal hyperplasia by specific targeting vascular endothelial growth factor with bevacizumab-eluting stent: an experimental study. Atherosclerosis. 2007;195:269–76. doi: 10.1016/j.atherosclerosis.2006.12.034. [DOI] [PubMed] [Google Scholar]
- Stefanadis C, Toutouzas K, Stefanadi E, et al. Avastin-eluting stent: long-term angiographic and clinical follow up. Hellenic J Cardiol. 2008;49:188–90. [PubMed] [Google Scholar]
- Tiukinhoy SD, Khan AA, Huang S, et al. Novel echogenic drug-immunoliposomes for drug delivery. Invest Radiol. 2004;39:104–10. doi: 10.1097/01.rli.0000111207.92580.44. [DOI] [PubMed] [Google Scholar]
- Tiukinhoy SD, Mahowald ME, Shively VP, et al. Development of echogenic, plasmid-incorporated, tissue-targeted cationic liposomes that can be used for directed gene delivery. Invest Radiol. 2000;35:732–8. doi: 10.1097/00004424-200012000-00007. [DOI] [PubMed] [Google Scholar]
- Toutouzas K, Karabelas J, Vaina S, Stefanadis C. Antivascular endothelial growth factor-a treatment: new perspectives for high-risk plaque stabilization. J Am Coll Cardiol. 2007;50:186. doi: 10.1016/j.jacc.2007.05.001. [DOI] [PubMed] [Google Scholar]
- Varey AH, Rennel ES, Qiu Y, et al. VEGF 165b, an antiangiogenic VEGF-A isoform, binds and inhibits bevacizumab treatment in experimental colorectal carcinoma: balance of pro- and antiangiogenic VEGF-A isoforms has implications for therapy. Br J Cancer. 2008;98:1366–79. doi: 10.1038/sj.bjc.6604308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson SH, Herrmann J, Lerman LO, et al. Simvastatin preserves the structure of coronary adventitial vasa vasorum in experimental hypercholesterolemia independent of lipid lowering. Circulation. 2002;105:415–18. doi: 10.1161/hc0402.104119. [DOI] [PubMed] [Google Scholar]
- Xu H, Czerwinski P, Hortmann M, et al. Protein kinase C alpha promotes angiogenic activity of human endothelial cells via induction of vascular endothelial growth factor. Cardiovasc Res. 2008;78:349–55. doi: 10.1093/cvr/cvm085. [DOI] [PubMed] [Google Scholar]
- Zuidam NJ, Vrueh RD, Crommelin DJA. Characterization of liposomes. In: Torchilin VP, Weissig V, editors. Liposomes. 2. New York: Oxford University Press; 2003. pp. 31–78. [Google Scholar]