

Abstract
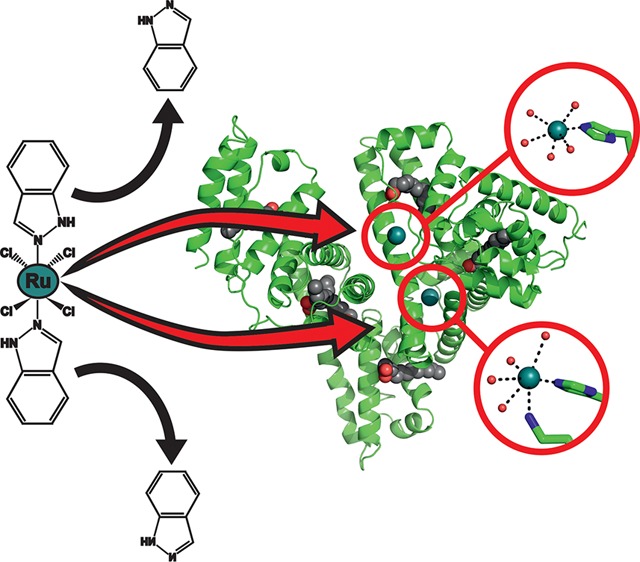
Ruthenium(III) complexes are promising candidates for anticancer drugs, especially the clinically studied indazolium trans-[tetrachlorobis(1H-indazole)ruthenate(III)] (KP1019) and its analogue sodium trans-[tetrachlorobis(1H-indazole)ruthenate(III)] (NKP-1339). Several studies have emphasized the likely role of human serum proteins in the transportation and accumulation of ruthenium(III) complexes in tumors. Therefore, the interaction between KP1019 and human serum albumin was investigated by means of X-ray crystallography and inductively coupled plasma mass spectrometry (ICP-MS). The structural data unambiguously reveal the binding of two ruthenium atoms to histidine residues 146 and 242, which are both located within well-known hydrophobic binding pockets of albumin. The ruthenium centers are octahedrally coordinated by solvent molecules revealing the dissociation of both indazole ligands from the ruthenium-based drug. However, a binding mechanism is proposed indicating the importance of the indazole ligands for binding site recognition and thus their indispensable role for the binding of KP1019.
Introduction
Metal complexes have a broad range of medical applications and are extensively used in the treatment of cancer. The pioneering and most prominent metal-based anticancer drug is platinum-containing cisplatin, which together with its later generation analogues carboplatin and oxaliplatin is still one of the most widely used anticancer agents nowadays.1,2 Despite their high efficacy in the treatment of several tumor types, there are also tumors that show primary resistance against platinum compounds. In some tumors, platinum therapy also shows strong side effects and the development of resistance. Nevertheless, there are various possibilities to develop new anticancer active drugs coming from the field of inorganic chemistry that should expand the spectrum of sensitive tumors that are better tolerated than classical antitumor agents.3,4 In this regard, ruthenium-containing compounds are very promising and are therefore considered as next generation metal-based anticancer agents due to their significant antineoplastic and antimetastatic properties causing less side effects and drug resistance.5−8 The Ru(II) and Ru(III) oxidation states are the most stable under physiological conditions with the latter being relatively inert and thus less or not at all active, which led to the not entirely undisputed “activation by reduction” theory as a possible explanation for the high selectivity of anticancer Ru(III) complexes.9,10 Complexes of both oxidation states, especially those forming octahedral species, have not only been shown to exhibit significant antiproliferative activity but also other medicinally important properties like their usage as antihypertensive, antiparasitic, and antibacterial agents.11−14 The in vivo and in vitro properties of Ru complexes can be fine-tuned through variations in the selection of the ligands, e.g., aromatic heterocycles, pyridine, arene, nitrosyl, and many more.8 There are abundant reports about Ru complexes with remarkable antiproliferative properties, which enrich the field of drug development; however, the most prominent ones are the Ru(III)-based anticancer drugs indazolium trans-[tetrachlorobis(1H-indazole)ruthenate(III)] (1, KP1019),15 its sodium salt analogue sodium trans-[tetrachlorobis(1H-indazole)ruthenate(III)] (2, NKP-1339)16 (Figure 1), and the new antimetastasis inhibitor imidazolium trans-[tetrachlorobis(1H-imidazole)(S-dimethyl sulfoxide)ruthenate(III)] (NAMI-A)17 (Figure S1), though the clinical development of the last one seems to have come to a halt.100 Compounds 1, 2, and NAMI-A have proceeded to the clinical stage of drug development and thereby successfully passed a phase 1 trial.15−19 Compounds 1 and 2 are active against a variety of solid tumors, whereas NAMI-A targets the metastatic process without affecting primary tumors; however, the mode of action of Ru-containing drugs is complex and in general less understood. Different mechanisms for different anticancer Ru complexes have been proposed, mostly describing direct DNA targeting and/or induction of oxidative/cellular stress.7,20−23
Figure 1.

Chemical structure of indazolium trans-[tetrachlorobis(1H-indazole)ruthenate(III)] (1, KP1019) and its sodium salt analogue sodium trans-[tetrachlorobis(1H-indazole)ruthenate(III)] (2, NKP-1339).
Compounds 1 and 2 exhibit outstanding efficacy in experimental models of colorectal cancer and were reported to induce a high percentage of disease stabilization (as well as a long-lasting partial remission in the case of 2) in clinical studies in patients with advanced solid tumors.16,19,24 It is believed that the cytotoxic activity of 1 and 2 is based on endoplasmic reticulum (ER) stress-related effects rather than DNA damage.7,25 Compounds 1 and 2 have been demonstrated to cause oxidative stress and perturbation of ER functions in cancer cells; in addition, the 78 kDa glucose-regulated protein (GRP78), which is a rescue factor for the tumor, is reduced to a normal level.7,26,27 In this way, apoptosis is finally induced by the intrinsic mitochondrial pathway.27 Compounds 1 and 2 are suggested to be transported and accumulated into tumor cells by serum proteins, and thus, their interaction with human serum transferrin (Tf) and albumin (HSA) has gained attention in this research field.28,29 Both serum proteins are able to bind both compounds 1 and 2, and thus, two modes of transportation have been suggested in the past to explain the reported minor side effects and tumor selectivity of these drugs, (1) Tf- and (2) HSA-mediated pathways. (1) Ruthenium exhibits chemical similarity to iron, and thus, it is able to reversibly bind to the iron transporter Tf. After metal binding, the Tf–metal complex is delivered into the cell by endocytosis, where the transport protein releases the bound metal before being recycled and transferred back to the extracellular space. Malignant tumor cells frequently express a higher level of Tf receptors than normal cells due to their higher iron demand, and therefore, it has been suggested that ruthenium compounds are able to exploit Tf as a “vehicle” to selectively reach the tumor cells in iron’s stead.29,30 (2) The HSA-mediated pathway is based on the enhanced permeability and retention (EPR) effect of tumor tissues.31 The EPR effect describes the phenomenon that macromolecules of a certain size (>40 kDa) are able to selectively accumulate in tumor tissue due to the production of blood vessels with a defective architecture. These defects in tumor blood vessels (e.g., gaps between the endothelial cells) allow macromolecules to transit from the blood vessel into the tumor tissue. Inefficient lymph drainage increases this effect. It is suggested that, after the ruthenium compounds bind to highly abundant HSA, the resulting HSA–drug complex is able to cross the blood vessels via the EPR effect and accumulates in tumor cells.32
Thus, the interaction between metallodrugs and proteins represents one major focus in drug development because it might be crucial for the biodistribution, metabolism, bioavailability, toxicity, and mode of action of some anticancer drugs. Compound 1 binds strongly to HSA, and a number of studies investigating the HSA–1 interaction have already been published.32−37 HSA is the most abundant serum protein in human plasma (∼600 μM) and serves as a transporter for hormones, fatty acids (FAs), bilirubin, and various pharmaceuticals. HSA consists of a single chain of ∼66.5 kDa, which is divided into three domains (domains I, II and III, see Figure 2), each of which is further subdivided into two helical subdomains (subdomains A and B, see Figure 2).39 The structural architecture of HSA harbors a variety of ligand binding sites, which are located within hydrophobic pockets.39,40 Despite the multitude of relevant publications about the HSA–1 interaction, the exact binding event, sites, and especially the composition of the drug during and after the binding still remain elusive.
Figure 2.
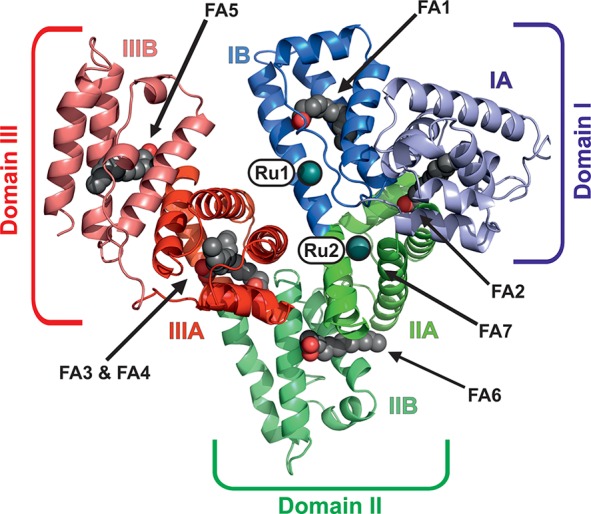
Overall structure of HSA–Myr–1 (PDB ID: 5IFO). The structure is shown as a cartoon with every domain/subdomain being differently colored and labeled (domain I, blue; domain II, green; domain III, red). The bound metal centers are represented as deep teal spheres and labeled as Ru1 and Ru2, respectively. The seven FAs bound to HSA are labeled as FA1–7 with bound FAs being displayed as sphere chains (aliphatic chain, gray spheres; carboxylate oxygens, red spheres).
Herein, we report on the X-ray crystal structure analysis of the complex of HSA with myristate (Myr) and 1. Myr is a common FA ligand of HSA, which is known to facilitate HSA crystal formation by inducing favorable conformational changes and was thus used for crystallization trails.41 The structural data provide information on the location of at least two binding sites of 1 on HSA and about the composition of the bound drug. Furthermore, the influence of FA-induced conformational changes within HSA on 1 binding and the crucial role of the indazole ligands during drug binding are discussed. The existence and number of ruthenium bound to HSA were independently proven and quantified by inductively coupled plasma mass spectrometry (ICP-MS).
Results and Discussion
Overall Structure of HSA–Myr–1
Single crystals of HSA–Myr–1 were obtained by soaking of preformed HSA–Myr crystals in 1-containing solutions, and the crystal with the highest ruthenium content diffracted to 3.2 Å resolution. The crystal belonged to the C 1 2 1 space group with one HSA molecule per asymmetric unit. The final model of HSA–Myr–1 was refined to an R-factor of 24.5% (R-free = 26.2%) with good stereochemistry (see Table S1 in the Supporting Information for further details). The single HSA chain has two ruthenium ions bound to the hydrophobic cavities of subdomains IB and IIA, respectively, and Myrs bound at six of the seven known FA binding sites on the protein (Figure 2). The overall conformation of the HSA–Myr complex is not significantly affected by binding of 1, as evidenced by the absence of major changes in the orientation of the three domains when compared to other HSA–Myr drug structures in the Protein Data Bank (PDB, www.pdb.org). For instance, the Cα rms deviation of the structure reported herein from the structure of HSA–Myr with bound 3′-azido-3′-deoxythymidine and salicylic acid (PDB ID: 3B9M(42)) is only 0.47 Å (3689 atoms included).
Details of Ru Binding Sites I and II
Analysis of the difference Fourier electron density maps clearly indicates two strong positive peaks revealing the presence of two Ru binding sites. In Ru binding site I (Figure 3A), the metal ion is bound to the imidazole nitrogen of histidine 146 (His146), which is located at the lower entrance to subdomain IB. This subdomain consists mainly of three α-helices forming a hydrophobic groove. His146 is the only proteinogenic ligand coordinated to the Ru center. The Ru–N(His146) distance is 2.1 Å and thus agrees with the average value of 2.1 Å for Ru–N distances found in the literature.43−45 The electron density around the Ru center clearly indicates the absence of bulky indazole ligands; however, despite the relatively low resolution, the existence of coordinating small ligands is still obvious (Figure 3B). Therefore, five water molecules were inserted into the density and included in the refinement. The Ru–OH2 distances range from 1.8 to 2.8 Å. Given the electron density, the Ru adopts a distorted octahedral geometry, which is the most common geometry of protein bound Ru(III).44,46
Figure 3.
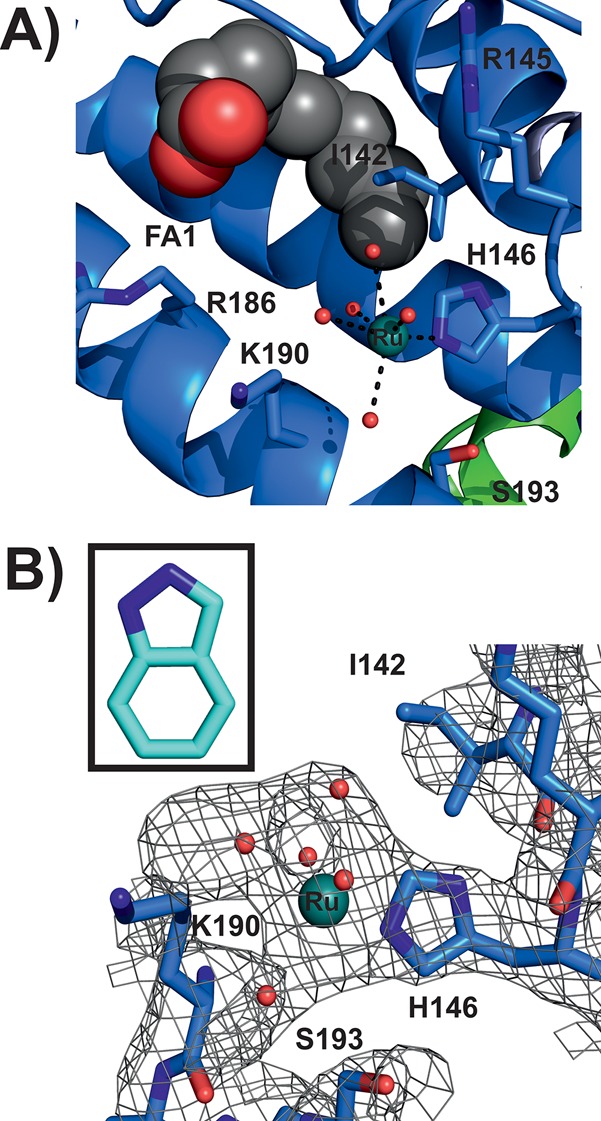
Ru binding site I (PDB ID: 5IFO). (A) HSA structure shown as a blue cartoon; side chains located within 6 Å from the Ru center are represented as sticks (color code: carbon, blue; nitrogen, dark blue; oxygen, red). The Ru center is displayed as a deep teal sphere with the coordinating water molecules shown as small red spheres. One FA is located within the binding site IB, which is shown as a chain of spheres (aliphatic chain, gray spheres; carboxylate oxygens, red spheres). (B) The electron density map (2Fo – Fc) of the binding site is shown as a gray mesh contoured at 1.0 σ. Protein side chains are shown as sticks with the remaining binding site being represented as in (A). The inset shows an indazole ligand in the corresponding size to visually indicate the absence of indazole-based electron density.
In Ru binding site II (Figure 4A), which is adjacent to Ru site I (Figure 2), the Ru atom is also bound to a histidyl nitrogen provided from His242, which is located within the hydrophobic core of HSA subdomain IIA. The Ru–N(His242) distance is 2.1 Å and thus again in perfect agreement with the literature values.43−45 The electron density of Ru binding site II is more pronounced than in Ru site I; however, again no density could be assigned to an indazole ligand (Figure 4B). In contrast to Ru site I, the Ru center at site II is additionally coordinated by a further proteinogenic ligand, lysine (Lys199), with a Ru–N(Lys199) distance of 2.7 Å. Furthermore, the electron density of the Ru species is not only merged with the density of the coordinating His and Lys ligands but also with those of residues tryptophan (Trp214) and arginine (Arg218), indicating a more sophisticated coordination sphere at Ru binding site II in comparison to that at site I. Lys199 and Arg218 are positioned at the polar entrance with His242 at the inside and Trp214 in the lower part of the hydrophobic pocket. Because no direct coordination between the Ru center and Trp214 is expected (giving rise to a merged density), a Ru-coordinating water molecule was placed between them. Another water molecule was inserted between the Ru atom and Arg218 because the Ru–Arg218 distance of 4.4 Å is rather large for a direct interaction. Thus, the incorporated water coordinates to the Ru center and is stabilized by Arg218. The distorted octahedral coordination of the Ru center is completed by two further water molecules leading to Ru–OH2 distances of 2.5–2.8 Å. Refinements resulted in an occupancy of ∼0.5 for both Ru centers and thus indicate similar Ru affinity of the two binding sites and a satisfactory degree of protein metalation.
Figure 4.
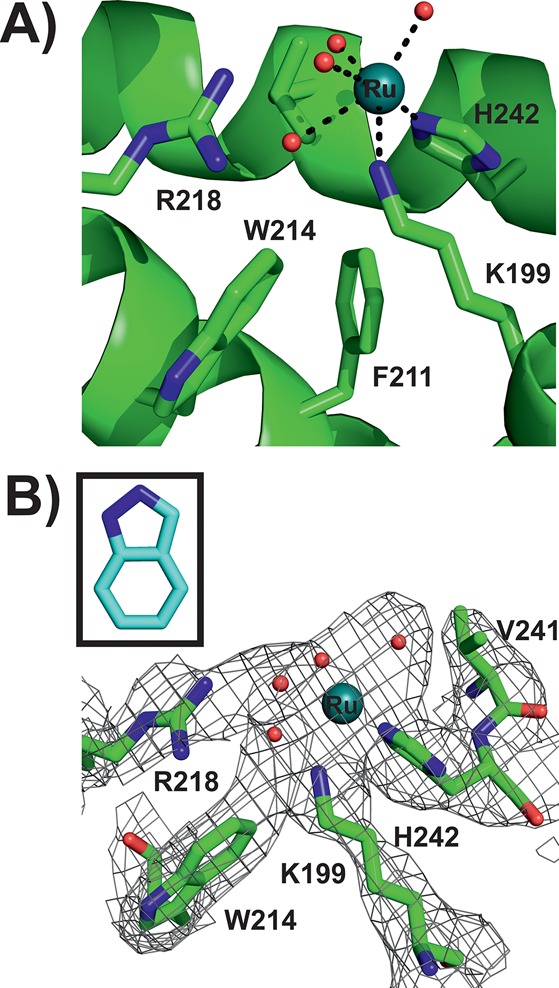
Ru binding site II (PDB ID: 5IFO). (A) HSA structure is shown as a green cartoon; side chains located within 6 Å from the Ru center are represented as sticks (color code: carbon, green; nitrogen, dark blue; oxygen, red). The Ru center is displayed as a deep teal sphere with the coordinating water molecules shown as small red spheres. (B) The electron density map (2Fo – Fc) of the binding site is shown as gray mesh contoured at 1.0 σ. Protein side chains are shown as sticks with the remaining binding site being represented as in (A). The inset shows an indazole ligand in the corresponding size to visually indicate the absence of indazole-based electron density.
Both metal centers are bound to known drug binding sites of HSA and are thus expected to be transported to their pharmacological targets. The two most prominent drug sites on HSA are Sudlow sites I and II, which are located within subdomains IIA and IIIA, respectively.47 However, Ru was only found in Sudlow site I (Ru binding site II), which is known to preferentially bind bulky heterocyclic drugs with a centrally located negative charge.48−50 These characteristics apply to 1 with its two indazole ligands being attached but not to the naked Ru atom. In contrast, Sudlow site II is known to preferentially bind aromatic carboxylates with a peripheral negative charge, which could be one reason for the lack of bound Ru moieties there.48 Thus, it seems that the indazole ligands play a crucial role in 1 binding despite their absence in the structure. In addition, the region around Sudlow site II is also a strong binding site for FAs and thus might be blocked and not accessible for 1. Another reason for the lack of Ru could be the absence of residues exhibiting strong affinity toward Ru(III) like His, aspartic acid (Asp), methionine (Met), or cysteine (Cys) in the Sudlow site II pocket.51 Therefore, the other Ru species is located within subdomain IB (Ru binding site I), which contains a strong metal binding His residue and, in addition, was reported to bind a wide range of drugs, considering it as the third major drug binding site besides the two Sudlow sites I and II.42,50,52 Subdomain IB is conformationally very flexible and provides enough spatial scope for drugs like 1.53,54 Both binding sites have been confirmed by competition studies, where 1 was shown to be able to replace the HSA site markers warfarin from Sudlow site I and bilirubin from subdomain IB.32
The previously reported structure of 1 bound to human lactoferrin showed that although 1 binds to a His residue (His253) as shown here, at least one of the heterocyclic indazole ligands remains bound to the Ru center.55 The different observations regarding the composition of the bound 1 moiety could be explained by the considerably higher amount of salt (NaCl or KCl), which was used in this study (see Experimental Section) in comparison to the crystallization experiment of 1 with human lactoferrin. Higher concentrations of NaCl or KCl in the crystallization solution could stabilize the equatorially coordinated chloride ligands by suppressing their exchange and thus giving rise to the dissociation of the axial indazole ligands, especially when considering the long incubation time (∼24 h of soaking, see Experimental Section) at this high salt concentration. However, the results here are in agreement with previously reported structures of other similar potential antiproliferative Ru(III) complexes bound to macromolecules. The potential metastasis inhibitor NAMI-A (Figure S1) was also found to bind to the imidazole ring of His64 of human carbonic anhydrase lacking all of its original ligands.56 Later, a NAMI-A analogue sodium trans-[tetrachlorobis(1H-pyridine)(S-dimethyl sulfoxide)ruthenate(III)] (AziRu) (Figure S1), which contains a pyridine instead of the imidazole ring, was crystallized with hen-egg white lysozyme, and the resulting structure showed almost the same picture, the loss of all ligands under the applied crystallization conditions.57 One Ru was coordinated by His15 and Asp87, exchanging all of its ligands. These structural results show that all crystallographically investigated anticancer Ru(III) complexes behave similarly under crystallization conditions; that is, all compounds predominantly bind to proteinogenic N-donor ligands (His) of biomacromolecules under dissociation of their own N-donor ligands (1, indazole; NAMI-A, imidazole; AziRu, pyridine). However, it is believed that both NAMI-A and AziRu readily lose their ligands in aqueous solutions, and thus, the Ru atom interacts “ligand-free” with the protein. This is not true for 1 because it is suggested that the complex is more stable and its indazole ligands remain attached for at least 8–24 h in solution, and thus, the ligands are able to interact hydrophobically with the binding site pockets of HSA during the early stages of HSA binding.58−60 Therefore, it is believed that 1 loses its indazole ligands only after a certain amount of time (∼24 h in the crystal soaking experiment). However, it has to be taken into account that under physiological conditions it might also be possible that the indazole ligands remain in the structure, and additionally, the complex could reach the tumor cell before the indazole ligands are released. Furthermore, it has to be noted that X-ray crystallography is a technique that is not able to directly examine the behavior of molecules in solution, and additionally, the study of molecular motion is also not possible. Thus, the results herein are the observations of one single conformation, and therefore, it cannot be excluded that the indazole ligands stay attached to the complex in solution.
Although the absence of the indazole ligands in the structure is obvious, it cannot be excluded that one or more Ru-coordinating water molecules are misinterpreted chloride ligands retained from the original 1 molecule. There is some evidence that at least one Ru ligand at binding site II is a chloride ion. The electron density at this site (Figure 4B) that connects the Ru atom to Trp214 could be explained by a bridging chloride ligand, which on the one side is coordinated to the Ru center and on the other side is stabilized by Trp214 via a distorted and edgewise anion-π (or quadrupole) interaction (high NaCl or KCl concentrations could suppress chloride exchange, see Experimental Section).61−63 The same could also apply to other Ru coordinating waters in the structure, especially those that are equatorially coordinated to the Ru center. The electron density of the equatorially coordinated ligands is significantly stronger than that of the axial ligands (Figures 3B and 4B). This could be explained by the existence of equatorially positioned heavy chloride ions that more likely give rise to electron density at this resolution than (axially positioned) light water molecules. However, because of the lack of clear experimental evidence for the presence of chloride ions, water molecules were incorporated into the coordination sphere of both metal centers.
The Influence of Myr Binding on the HSA Structure and Binding of 1
HSA possesses a total of seven well-characterized FA binding sites.64 Because the protein was incubated with a 10-fold molar excess of Myr prior to crystallization, the structure was searched for FAs (see Experimental Section for further details). Six Myr molecules were detected (see Figure 2), and all sites were confirmed by comparing them with FA positions of the HSA–Myr–indomethacin structure (PDB ID: 2BXM(40)), which has all known HSA FA binding sites (FA 1–7 in Figure 2) occupied. One FA site in the structure reported here is located in the hydrophobic cavity of subdomain IB, and thus, a Myr molecule was found in close proximity to Ru binding site I, however, without disturbing the coordination sphere of the bound Ru. Potential FA binding site 7 (FA7 in Figure 2), which is located within subdomain IIA, is occupied by Ru (Ru site II) herein, and thus, no Myr was found there. The known drug binding site at subdomain IIIA (Sudlow site II) has two FA chains bound and is thus fully occupied by Myr.
For the structural impact of Myr on both the protein conformation and 1 binding to be investigated, the HSA–Myr–1 structure was superimposed with FA lacking HSA–halothane structure (PDB ID:1E7B65) (Figure 5). The detected Cα rms deviation (including 3136 atoms) of 3.2 Å is the result of rigid-body rotations of domains I and III relative to domain II, leading to differences in the binding sites and a rearrangement of the hydrogen network between the polar residues within the hydrophobic cavities. The Ru atom bound at subdomain IB clashes with the His146 harboring α-helix of the FA-free HSA–halothane structure (Figure 5). This helix is shifted in the HSA–Myr–1 structure, indicating an opening of the lower part of the binding pocket and thus providing more space, which is used and occupied by a Myr molecule (FA1 in Figure 2). However, this additional space is not necessary for Ru binding to this site because the coordination sphere of the metal would not be restricted in the Myr-free conformation. In subdomain IIA, only minor changes are observed (Figure 5). Thus, it seems that these Myr-induced conformational changes have no significant impact on 1 binding at these sites although such structural alterations have been reported to have tremendous effects on the binding of some therapeutics.42
Figure 5.

Structure comparison between the FA-loaded HSA–Myr–1 complex (PDB ID: 5IFO) and the FA-free HSA–halothane complex (PDB ID: 1E7B). Both structures are shown as cartoons, where the HSA–Myr–1 complex is colored green and the HSA–halothane complex red with 30% transparency. In addition, both metal atoms are displayed as deep teal spheres. FAs are omitted for clarity in the overall structure comparison. The insets show zoomed views of the three major drug binding cavities on HSA (domains IB, IIA, and IIIA). Note that the structures in the insets are rotated in comparison to the overall structure to provide the best view of the inside of each binding pocket. Subdomains IB and IIIA harbor FAs, which are displayed as chains of spheres (aliphatic chain, gray spheres; carboxylate oxygens, red spheres). The Ru binding His residues of subdomains IB and IIA are shown as sticks (color code: carbon, green/red; nitrogen, blue) with the asterisk indicating the His residue coming from the HSA–halothane structure.
It is known that, in the presence of FAs, drug binding to subdomains IB and IIA is enhanced because the other major drug binding site at subdomain IIIA (Sudlow site II) also serves as a high affinity site for FAs and thus tends to be occupied by those lipids. Therefore, drugs switch preferentially to subdomains IB and IIA, where they can either replace weakly bound FAs or coexist with them.64,66−68 This is in accordance with the structural results represented here, where subdomain IIIA is occupied by two Myr chains (Figure 5); however, this seems not to be the main reason for the lack of Ru there as already discussed above. Rather, the absence of metal-coordinating residues like His, Asp, Cys, or Met results in no 1 binding at this site. Thus, 1 binding to Myr-free HSA (and thus freely accessible subdomain IIIA) would most likely reveal the same binding sites as shown here. However, under physiological conditions (in plasma), HSA can be loaded with up to two moles of FAs, and under certain disease states (e.g., cancer), the FA concentration is even increased.65,69,70 Furthermore, a comparison study between a series of FA-free HSA drug structures with the corresponding FA-loaded HSA drug structures revealed that, in the majority of cases, the drug binding position was not at all or only slightly changed by FA binding, and in addition, HSA exhibits so-called “breathing motions”, that is, HSA is switching between the FA-free and FA-loaded conformations in solution.40,71 All this confirms the assumption that FA binding has no significant influence on 1 binding and that, at least under extreme (tumor) physiological conditions, 1 will preferentially bind to the sites reported here (and will most likely avoid subdomain IIIA), reinforcing the potential physiological relevance of the HSA–Myr–1 structure. For the influence of FAs on 1 binding to be ruled out experimentally, ICP-MS measurements were performed applying no and different concentrations of Myr.
Identification and Quantification of the Ru Content in HSA
For unambiguously proving the presence of Ru within the crystal with an independent experiment, ICP-MS was performed with the HSA–Myr–1 crystal after X-ray data collection. For the detection of unbound Ru to be avoided, the crystal was excessively washed prior to crystal harvesting (see Experimental Section for further details). The result of the ICP-MS experiment showed 3.9 ng of Ru within the single crystal, clearly proving the presence of Ru in the structure.
For roughly estimating the expected number of bound Ru molecules per HSA molecule and confirming the structural data, the Ru content in solutions containing the preformed HSA–Myr–1 complexes were determined by a second ICP-MS experiment. Complexes consisting of different HSA/Myr/1 ratios were prepared, where the amounts of HSA and 1 were kept constant (HSA/1 ratio of 1:10) but the Myr amounts were varied (from no Myr to a 10-fold excess over HSA) to investigate possible competition reactions between 1 and Myr (see Experimental Section for more details). In addition, the complexes were prepared by incubating HSA with 1 and Myr applying different incubation times (1, 3, 6, 12, and 24 h) to examine the possible influence of Myr on the HSA–1 complex formation time. Table 1 shows the results of the second ICP-MS experiment. The number of bound moles of Ru per one mole of HSA ranges from 1 to 3 with an average value of 1.7 mol Ru per mole HSA. This is in good agreement with the crystallographic data. The ICP-MS data reveal no significant influence of Myr binding on both the number of bound Ru and the HSA–1 complex formation time. The HSA–1 complex is already formed after 1 h, which is indicated by an average of 2 bound moles of Ru per mole of HSA. However, ICP-MS does not provide information about the composition of the bound Ru moiety, and thus, it is not clear if at this time point the indazole ligands are still attached to the metal center. Because of the relatively unchanged Ru amount with increasing Myr concentration and even in the presence of equimolar Myr (see Table 1, HSA/Myr/1 = 1:10:10), it can be concluded that 1 is able to replace Myr from the binding site as already implied in the structure, where no Myr was found in Ru binding site II, although this site is also known as a potential FA binding site. Furthermore, the results indicate that Myr binding (including the structural changes) has no significant impact on 1 binding as the Ru amount is similar between the Myr-free and Myr-containing complex solutions. The structural and ICP-MS data presented here are also in good agreement with the latest report on HSA–1 interactions by Dömötör et al., who also revealed a 1/HSA ratio of 2:1 (without Myr) by ultrafiltration-UV/vis measurements.32 However, the possibility cannot be excluded that more than two Ru moieties are bound to one single HSA molecule in solution because two measurements of the ICP-MS experiment indicated the binding of three Ru molecules, and in addition, it is almost impossible to directly translate results of in vitro experiments into in vivo systems. Nevertheless, it can be concluded that a Myr-free crystal would result in binding of 1 to the same sites as reported here.
Table 1. Determination of the Bound Ru Amount by ICP-MS.
| complex ratio [HSA/Myr/1] | incubation time [h] | Ru/HSA ratioa |
|---|---|---|
| 1:0:10 | 1 | 1.48 |
| 1:0.5:10 | 1 | 2.98 |
| 1:1:10 | 1 | 2.10 |
| 1:10:10 | 1 | 1.45 |
| 1:0:10 | 3 | 1.57 |
| 1:0.5:10 | 3 | 1.45 |
| 1:1:10 | 3 | 1.66 |
| 1:10:10 | 3 | 1.52 |
| 1:0:10 | 6 | 2.72 |
| 1:0.5:10 | 6 | 1.57 |
| 1:1:10 | 6 | 1.70 |
| 1:5:10 | 6 | 1.78 |
| 1:10:10 | 12 | 1.36 |
| 1:0.5:10 | 12 | 1.24 |
| 1:1:10 | 12 | 1.45 |
| 1:10:10 | 12 | 1.63 |
| 1:0: 10 | 24 | 1.58 |
| 1:0.5:10 | 24 | 1.54 |
| 1:1: 10 | 24 | 1.71 |
| 1:10:10 | 24 | 1.75 |
Ru to HSA ratio given as n(Ru)/n(HSA).
Proposed Mechanism for Binding of 1
All structural features of Ru binding sites I and II represent ideal prerequisites for the binding of the entire 1 complex with its bulky indazole ligands being attached to the metal center. HSA contains in total 16 His residues with 10 located at the protein’s surface. Cisplatin was shown to bind to solvent-exposed His and Met (methionine) residues at the surface of HSA.72 Therefore, it is very striking that no Ru was found at the protein’s surface, which would be a more accessible binding site for Ru atoms or other Ru species lacking the bulky ligands than hydrophobically buried His residues. Thus, it is very likely that the axial indazole ligands are involved in 1 binding, which means that they remain attached to the Ru center until the complex reaches its final destination within HSA. The indazole ligands are responsible for binding site recognition via hydrophobic interactions. According to this, 1 approaches the protein in its original composition and exhibits first interactions with the hydrophobic regions of HSA and moves slowly toward the final hydrophobically buried binding destination. After the binding site is reached, the heterocyclic ligands are dissociated after a certain amount of time (∼24 h in the soaking experiment) so that the Ru(III) center is able to coordinate to the protein under ligand-exchange with protein-derived His ligands. This is possible because the imidazole groups of the His represent stronger donors toward Ru than the indazole ligands, leading to a more stable Ru–N bond.58 Cetinbas et al.37 suggested a similar two-step reaction pathway before, describing an initially rapid noncovalent binding of 1 to HSA followed by slow covalent binding. This mechanism was further confirmed by the studies of Webb et al.,60,73 emphasizing the importance of the hydrophobicity of the indazole ligands and thus the specific noncoordinate hydrophobic interactions with HSA, which are associated with lower side effects of 1. The experiments indicated that the hydrophobically coordinated species persists even after 24 h of incubation. This explains the final destination of both Ru centers and the need for large and hydrophobic binding pockets for enhanced bioavailability and cytotoxicity.
Conclusions
The structure of the HSA–Myr–1 complex reported here reveals the binding of two Ru moieties, which is independently confirmed by ICP-MS measurements. Both metal centers are bound to the imidazole nitrogen of histidines (His146 and His242), which are located within well-known drug binding sites, namely, on subdomain IB (Ru binding site 1) and IIA (Ru binding site 2) and exhibit an octahedral geometry. Structure analysis and comparison with an HSA structure lacking FAs revealed that Myr induces conformational changes within HSA, which, however, do not have a significant impact on binding of 1 at the binding sites reported here as additionally shown by ICP-MS. A two-step binding mechanism is suggested, where the indazole ligands are needed for rapid binding site recognition via hydrophobic interaction and may dissociate from the complex afterwards to enable Ru center coordination to the protein. This proposed indazole-mediated binding mechanism demonstrates the importance of the indazole ligands as binding site recognizing moieties, which promote metal binding to pharmacologically important binding sites on HSA. This inhibits unspecific metal coordination and binding to other interfering proteins in the serum, which otherwise could lead to reduced selectivity, bioavailability, and cytotoxicity. This could represent one important reason for the pharmacologically different behavior between 1 and cisplatin, which was found to bind His residues located at the surface of HSA. Furthermore, the results shown here indicate that metal binding His residues represent a major factor for Ru binding by HSA. Thus, it is suggested that 1 binds also to His sites within other proteins like Tf, which harbors two His residues in its iron binding cleft. However, HSA should represent the main vehicle for 1 on its way to the cancer cells due to its high abundance in human plasma and its innate preference for binding hydrophobic compounds. The cytotoxicity of 1 could be improved even more by attaching more hydrophobic ligands to the Ru(III) center, which might inhibit the covalent binding of the metal to the protein and instead promote noncoordinative drug binding, leading to enhanced bioavailability by facilitated metal release in the tumor cell. However, the knowledge of 1 binding sites within HSA and the proposed binding mode could provide a framework for future cancer drug development approaches.
Experimental Section
1. General Methods
All reagents and solvents were reagent grade and used as purchased from commercial sources. Compound 1 was synthesized at the Institute of Inorganic Chemistry, University of Vienna. The purity of the used batch of 1 was checked at the Microanalytical Laboratory of the University of Vienna by both elemental analysis applying a PerkinElmer 2400 CHN elemental analyzer and mass spectroscopy applying a Bruker Esquire 3000 Plus Ion Trap mass spectrometer. The results revealed a deviation of ±0.9 and ±0.1%, respectively, from the calculated value, thus confirming ≥95% purity for 1.
Protein purification was performed by size-exclusion chromatography (SEC) using the ÄKTA explorer system from GE Healthcare. The purity of the protein samples was checked by SDS-PAGE using a 15% acrylamide-containing gel and the Mini-PROTEAN Tetra Cell from Bio-Rad. For highly concentrated protein samples (75–120 mg/mL) to be obtained, ultrafiltration was performed using Vivaspin concentrators with a 30 kDa cutoff membrane from Sartorius. For the determination of protein concentrations, the protein’s absorbance at 280 nm was measured by UV–vis spectroscopy (UV-1800 from Shimadzu), and the concentrations were subsequently calculated applying the Beer–Lambert Law using the molar extinction coefficient (at 280 nm) of 36500 M–1 cm–1 for HSA.74
2. Protein Sample Preparation
FA- and globulin-free HSA (Sigma, A3782) was dissolved in 20 mM potassium phosphate buffer (pH 7.4) containing 150 mM NaCl and purified by SEC using the same buffer to remove HSA dimers and multimers. Fractions containing the monomer of the protein were pooled and incubated with a 10-fold molar excess of sodium Myr (Sigma-Aldrich) for 3 h at 37 °C. Excessive undissolved Myr was removed by centrifugation at 3200g for 15 min. The resulting HSA–Myr complexes were concentrated to 80–120 mg/mL and centrifuged at 20800g for 15 min prior to crystallization experiments.
Samples of the ternary complex HSA–Myr–1 for ICP-MS measurements were prepared as described above with the difference that HSA was incubated with both Myr and 1 at different molar ratios and for different incubation times (1, 3, 6, 12, and 24 h) at 37 °C. Complexes with the following HSA/Myr/1 ratios were prepared: 1:0:10, 1:0.5:10, 1:1:10, and 1:10:10. Excessive undissolved Myr was again removed by centrifugation followed by excessive ultrafiltration to wash off unbound 1. Concentration determination of the HSA–Myr–1 sample was performed by the Bradford method75 because the protein bound 1 species shows a highly intense band in the UV–vis spectra overlapping with the 280 nm band of the protein and thus making it impractical to determine the protein’s absorbance at this wavelength. The Bradford assay from Bio-Rad was utilized with bovine serum albumin (BSA) as standard using an Infinite M200 microplate reader (TECAN) for absorbance measurements at 595 nm.
3. ICP-MS Measurements
Milli-Q water (18.2 MΩ cm, Milli-Q Advantage) and nitric acid (≥69%, p.a., TraceSelectFluka) was used for all dilutions for ICP-MS measurements. Elemental standard solutions for ICP-MS measurements were derived from CPI International (Amsterdam, The Netherlands).
Samples were either directly diluted with 3% nitric acid or a digestion of samples was performed with 2 mL of 3% nitric acid using a microwave system Discover SP-D (CEM Microwave Technology). The following microwave parameters were applied: temperature, 200 °C; ramp time, 4 min; hold time, 6 min; and maximal power, 300 W. Digested samples were diluted with Milli-Q water resulting in nitric acid concentrations lower than 3% and Ru concentrations lower than 15 ng/g. ICP-MS measurements were performed with an ICP-quadrupole MS instrument Agilent 7500ce (Agilent Technologies) equipped with a CETAC ASX-520 autosampler and a MicroMist nebulizer at a sample uptake rate of approximately 0.25 mL/min. The instrument was tuned on a daily basis, and indium served as the internal standard for Ru. The ICP-MS was equipped with nickel cones and operated at a radio frequency (RF) power of 1550 W. The dwell time was set to 0.3 s, and the measurement was performed with 10 replicates. The Agilent MassHunter software package (Workstation Software, Version B.01.01, 2012) was used for data processing.
4. Crystallization Experiments
Crystals of the HSA–Myr–1 complex were obtained by soaking of preformed HSA–Myr crystals with 1. Crystallization of the HSA–Myr complex for the soaking experiment was performed at 20 °C by the hanging-drop vapor-diffusion method. The crystallization drops consisted of 1–2 μL of the HSA–Myr solution (75–120 mg/mL in 20 mM potassium phosphate buffer, pH 7.4) and 0.5–1 μL of reservoir solution (50 mM potassium phosphate buffer, pH 7.5–8.0, 25–30% (w/v) PEG 4000, 150 mM KCl, and 5 mM NaN3) and were equilibrated against 500 μL of reservoir solution. After 1–2 weeks, large but extremely twinned crystals grew, which were used for streak seeding to obtain high quality single crystals. For that reason, new crystallization drops were set up with 85% of the precipitant composition of the original reservoir solution that produced the first crystals. After 7 days of equilibration, streak seeding was performed, and large single crystals appeared. These crystals were then soaked in a solution containing 1–2 mM 1 in 50 mM potassium phosphate buffer, pH 7.4, 35% (w/v) PEG 4000, 150 mM KCl, and 3% DMSO for 24 h at 20 °C (DMSO was necessary to keep 1 in solution, however, most of the crystals cracked or disappeared). After the soaking procedure, the crystals were intensely washed in a series of washing solutions (50 mM potassium phosphate buffer, pH 7.4, and 150 mM KCl) containing different amounts of precipitant (30, 32, and 35% (w/v) PEG 4000) for upcoming ICP-MS measurements. The crystals were then flash frozen in liquid nitrogen after a brief soak in a cryo-protection solution containing 50 mM potassium phosphate buffer, pH 7.4, 35% (w/v) PEG 4000, 150 mM KCl, and 5 mM NaN3. The best ruthenium-containing crystal diffracted to a maximum resolution of 3.2 Å. Details of the data collection and refinement can be found in the Supporting Information.
Acknowledgments
The X-ray diffraction experiment was performed at beamline ID23-1 within the scope of proposal MX1616 at the European Synchrotron Radiation Facility (ESRF), Grenoble, France. The authors are grateful to the ESRF staff, especially to Dr. Stéphanie Monaco and Dr. Ulrich Zander, for providing assistance during the beamtime. Furthermore, the authors are grateful to Dipl.-Ing. Matthias Pretzler for valuable discussions and critically reading this article. The authors also thank Dr. Michael Jakupec for valuable discussion about the anticancer compound.
Glossary
Abbreviations Used
- Arg/R
arginine
- Asp
aspartic acid
- AziRu
sodium trans-[tetrachlorobis(1H-pyridine)(S-dimethyl sulfoxide)ruthenate(III)] (ruthenium complex and NAMI-A analogue)
- Cys
cysteine
- Cα rmsd
root-mean-square deviation of the coordinates of Cα backbone atoms
- ER
endoplasmic reticulum
- F
phenylalanine
- FA
fatty acid
- FA1–7
fatty acids binding to corresponding fatty acid binding sites 1–7
- His/H
histidine
- HSA
human serum albumin
- I
isoleucine
- ICP-MS
inductively coupled plasma mass spectrometry
- L
leucine
- Lys/K
lysine
- Met
methionine
- Myr
myristate
- NAMI-A
new antitumor metastasis inhibitor A
- PDB
protein data bank
- PEG
polyethylene glycol
- Ru
ruthenium
- Ru(III)
ruthenium ion 3+
- Ru–N(His146)/Ru–N(His242)
ruthenium bound to the imidazole nitrogen of histidine 146 or 242, respectively, of human serum albumin
- S
serine
- Tf
human serum transferrin
- Trp/W
tryptophan
- V
valine
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.jmedchem.6b00600.
Accession Codes
Atomic Coordinates and experimental data (PDB ID: 5IFO).
The research was funded by the Austrian Science Fund (FWF) P23711.
The authors declare no competing financial interest.
Supplementary Material
References
- Dabrowiak J. C.Metals in Medicine; John Wiley & Sons: Hoboken, NJ, 2013; pp 151–161. [Google Scholar]
- Yao X.; Panichpisal K.; Kurtzman N.; Nugent K. Cisplatin Nephrotoxicity: A Review. Am. J. Med. Sci. 2007, 334, 115–124. 10.1097/MAJ.0b013e31812dfe1e. [DOI] [PubMed] [Google Scholar]
- Jakupec M. A.; Galanski M.; Keppler B. K.. Tumour-Inhibiting Platinum Complexes - State of the Art and Future Perspectives. In Reviews of Physiology, Biochemistry and Pharmacology; Springer: Heidelberg, 2003; pp 1–53. [DOI] [PubMed] [Google Scholar]
- Balcerzak M. Quantification of Noble Metals in Biological and Environmental Samples. In Handbook of Trace Analysis; Baranowska I., Ed.; Springer: Berlin, 2016; pp 371–402. [Google Scholar]
- Heffeter P.; Pongratz M.; Steiner E.; Chiba P.; Jakupec M. A.; Elbling L.; Marian B.; Körner W.; Sevelda F.; Micksche M.; Keppler B. K.; Berger W. Intrinsic and Acquired Forms of Resistance against the Anticancer Ruthenium Compound KP1019 [indazolium Trans-[tetrachlorobis(1H-Indazole)ruthenate (III)] (FFC14A). J. Pharmacol. Exp. Ther. 2005, 312, 281–289. 10.1124/jpet.104.073395. [DOI] [PubMed] [Google Scholar]
- Jungwirth U.; Kowol C. R.; Keppler B. K.; Hartinger C. G.; Berger W.; Heffeter P. Anticancer Activity of Metal Complexes: Involvement of Redox Processes. Antioxid. Redox Signaling 2011, 15, 1085–1127. 10.1089/ars.2010.3663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trondl R.; Heffeter P.; Kowol C. R.; Jakupec M. A.; Berger W.; Keppler B. K. NKP-1339, the First Ruthenium-Based Anticancer Drug on the Edge to Clinical Application. Chem. Sci. 2014, 5, 2925–2932. 10.1039/C3SC53243G. [DOI] [Google Scholar]
- Dragutan I.; Dragutan V.; Demonceau A. Editorial of Special Issue Ruthenium Complex: The Expanding Chemistry of the Ruthenium Complexes. Molecules 2015, 20, 17244–17274. 10.3390/molecules200917244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bergamo A.; Sava G. Ruthenium Anticancer Compounds: Myths and Realities of the Emerging Metal-Based Drugs. Dalton Trans. 2011, 40, 7817–7823. 10.1039/c0dt01816c. [DOI] [PubMed] [Google Scholar]
- Medici S.; Peana M.; Nurchi V. M.; Lachowicz J. I.; Crisponi G.; Zoroddu M. A. Noble Metals in Medicine: Latest Advances. Coord. Chem. Rev. 2015, 284, 329–350. 10.1016/j.ccr.2014.08.002. [DOI] [Google Scholar]
- de Lima R. G.; Silva B. R.; da Silva R. S.; Bendhack L. M. Ruthenium Complexes as NO Donors for Vascular Relaxation Induction. Molecules 2014, 19, 9628–9654. 10.3390/molecules19079628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tahghighi A. Importance of Metal Complexes for Development of Potential Leishmanicidal Agents. J. Organomet. Chem. 2014, 770, 51–60. 10.1016/j.jorganchem.2014.08.007. [DOI] [Google Scholar]
- Sarniguet C.; Toloza J.; Cipriani M.; Lapier M.; Vieites M.; Toledano-Magaña Y.; García-Ramos J. C.; Ruiz-Azuara L.; Moreno V.; Maya J. D.; Azar C. O.; Gambino D.; Otero L. Water-Soluble Ruthenium Complexes Bearing Activity against Protozoan Parasites. Biol. Trace Elem. Res. 2014, 159, 379–392. 10.1007/s12011-014-9964-0. [DOI] [PubMed] [Google Scholar]
- Pavan F. R.; Poelhsitz G. V.; da Cunha L. V. P.; Barbosa M. I. F.; Leite S. R. A.; Batista A. A.; Cho S. H.; Franzblau S. G.; de Camargo M. S.; Resende F. A.; Varanda E. A.; Leite C. Q. F. In Vitro and In Vivo Activities of Ruthenium(II) Phosphine/Diimine/Picolinate Complexes (SCAR) against Mycobacterium Tuberculosis. PLoS One 2013, 8, e64242. 10.1371/journal.pone.0064242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartinger C. G.; Jakupec M. A.; Zorbas-Seifried S.; Groessl M.; Egger A.; Berger W.; Zorbas H.; Dyson P. J.; Keppler B. K. KP1019, A New Redox-Active Anticancer Agent – Preclinical Development and Results of a Clinical Phase I Study in Tumor Patients. Chem. Biodiversity 2008, 5, 2140–2155. 10.1002/cbdv.200890195. [DOI] [PubMed] [Google Scholar]
- Thompson D. S.; Weiss G. J.; Jones S. F.; Burris H. A.; Ramanathan R. K.; Infante J. R.; Bendell J. C.; Ogden A.; Von Hoff D. D. In NKP-1339: Maximum Tolerated Dose Defined for First-in-Human GRP78 Targeted Agent. Proceedings of the Annual Meeting of the American Society of Clinical Oncology; Chicago, IL. Jun 1, 2012; J. Clin. Oncol. 2012, Abstract no. 3033.22778319 [Google Scholar]
- Bergamo A.; Sava G. Linking the Future of Anticancer Metal-Complexes to the Therapy of Tumour Metastases. Chem. Soc. Rev. 2015, 44, 8818–8835. 10.1039/C5CS00134J. [DOI] [PubMed] [Google Scholar]
- Alessio E. In NAMI-A: end of the story?, Proceedings of the 17th International Conference on Biological Inorganic Chemistry; Beijing, China. Jul 20, 2015; RSC 2015, p. 93.
- Costich T. L.; Sethuraman J.; Crouse R.; Bakewell S. In IT-139 Holds Potential for Combination Therapy. Proceedings of the 107th Annual Meeting of the American Association for Cancer Research; New Orleans, LA. Apr 17, 2016; AACR 2016; Abstract no. 284. [Google Scholar]
- Sethuraman J.; Costich T. L.; Vojkovsky T.; Crouse R.; Cogner V.; Bakewell S. In IT-139 Targets GRP78 in Stressed Cancer Cells. Proceedings of the 107th Annual Meeting of the American Association for Cancer Research; New Orleans, LA. Apr 19, 2016; AACR 2016; Abstract no. 2996. [Google Scholar]
- Sharma A. R.; Gangrade D. M.; Bakshi S. D.; John J. S. Ruthenium Complexes: Potential Candidate for Anti-Tumour Activity. Int. J. ChemTech Res. 2014, 6, 828–837. [Google Scholar]
- Nazarov A. A.; Gardini D.; Baquié M.; Juillerat-Jeanneret L.; Serkova T. P.; Shevtsova E. P.; Scopelliti R.; Dyson P. J. Organometallic Anticancer Agents That Interfere with Cellular Energy Processes: A Subtle Approach to Inducing Cancer Cell Death. Dalton Trans. 2013, 42, 2347–2350. 10.1039/C2DT31936E. [DOI] [PubMed] [Google Scholar]
- Adhireksan Z.; Davey G. E.; Campomanes P.; Groessl M.; Clavel C. M.; Yu H.; Nazarov A. A.; Yeo C. H. F.; Ang W. H.; Dröge P.; Rothlisberger U.; Dyson P. J.; Davey C. A. Ligand Substitutions between Ruthenium–Cymene Compounds can Control Protein versus DNA Targeting and Anticancer Activity. Nat. Commun. 2014, 5, 3462. 10.1038/ncomms4462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Novak M. S.; Büchel G. E.; Keppler B. K.; Jakupec M. A. Biological Properties of Novel Ruthenium- and Osmium-Nitrosyl Complexes with Azole Heterocycles. JBIC, J. Biol. Inorg. Chem. 2016, 21, 347–356. 10.1007/s00775-016-1345-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lentz F.; Drescher A.; Lindauer A.; Henke M.; Hilger R. A.; Hartinger C. G.; Scheulen M. E.; Dittrich C.; Keppler B. K.; Jaehde U. Pharmacokinetics of a Novel Anticancer Ruthenium Complex (KP1019, FFC14A) in a Phase I Dose-Escalation Study. Anti-Cancer Drugs 2009, 20, 97–103. 10.1097/CAD.0b013e328322fbc5. [DOI] [PubMed] [Google Scholar]
- Bierle L. A.; Reich K. L.; Taylor B. E.; Blatt E. B.; Middleton S. M.; Burke S. D.; Stultz L. K.; Hanson P. K.; Partridge J. F.; Miller M. E. DNA Damage Response Checkpoint Activation Drives KP1019 Dependent Pre-Anaphase Cell Cycle Delay in S. Cerevisiae. PLoS One 2015, 10, e0138085. 10.1371/journal.pone.0138085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kapitza S.; Jakupec M. A.; Uhl M.; Keppler B. K. Marian, B. The Heterocyclic Ruthenium(III) Complex KP1019 (FFC14A) Causes DNA Damage and Oxidative Stress in Colorectal Tumor Cells. Cancer Lett. 2005, 226, 115–121. 10.1016/j.canlet.2005.01.002. [DOI] [PubMed] [Google Scholar]
- Flocke L. S.; Trondl R.; Jakupec M. A.; Keppler B. K. Molecular Mode of Action of NKP-1339 – a Clinically Investigated Ruthenium-Based Drug – Involves ER- and ROS-Related Effects in Colon Carcinoma Cell Lines. Invest. New Drugs 2016, 34, 261–268. 10.1007/s10637-016-0337-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pongratz M.; Schluga P.; Jakupec M. A.; Arion V. B.; Hartinger C. G.; Allmaier G.; Keppler B. K. Transferrin Binding and Transferrin-Mediated Cellular Uptake of the Ruthenium Coordination Compound KP1019, Studied by Means of AAS, ESI-MS and CD Spectroscopy. J. Anal. At. Spectrom. 2004, 19, 46–51. 10.1039/b309160k. [DOI] [Google Scholar]
- Spreckelmeyer S.; Orvig C.; Casini A. Cellular Transport Mechanisms of Cytotoxic Metallodrugs: An Overview beyond Cisplatin. Molecules 2014, 19, 15584–15610. 10.3390/molecules191015584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kratz F.; Hartmann M.; Keppler B.; Messori L. The Binding Properties of Two Antitumor Ruthenium(III) Complexes to Apotransferrin. J. Biol. Chem. 1994, 269, 2581–2588. [PubMed] [Google Scholar]
- Yin H.; Liao L.; Fang J. Enhanced Permeability and Retention (EPR) Effect Based Tumor Targeting: The Concept, Application and Prospect. JSM Clin. Oncol. Res. 2014, 2, 1010. [Google Scholar]
- Dömötör O.; Hartinger C. G.; Bytzek A. K.; Kiss T.; Keppler B. K.; Enyedy E. A. Characterization of the Binding Sites of the Anticancer Ruthenium(III) Complexes KP1019 and KP1339 on Human Serum Albumin via Competition Studies. JBIC, J. Biol. Inorg. Chem. 2013, 18, 9–17. 10.1007/s00775-012-0944-6. [DOI] [PubMed] [Google Scholar]
- Sulyok M.; Hann S.; Hartinger C. G.; Keppler B. K.; Stingeder G.; Koellensperger G. Two Dimensional Separation Schemes for Investigation of the Interaction of an Anticancer Ruthenium(III) Compound with Plasma Proteins. J. Anal. At. Spectrom. 2005, 20, 856–863. 10.1039/b508060f. [DOI] [Google Scholar]
- Hartinger C. G.; Hann S.; Koellensperger G.; Sulyok M.; Groessl M.; Timerbaev A. R.; Rudnev A. V.; Stingeder G.; Keppler B. K. Interactions of a Novel Ruthenium-Based Anticancer Drug (KP1019 or FFC14a) with Serum Proteins - Significance for the Patient. Int. J. Clin. Pharmacol. Ther. 2005, 43, 583–585. 10.5414/CPP43583. [DOI] [PubMed] [Google Scholar]
- Polec-Pawlak K.; Abramski J. K.; Semenova O.; Hartinger C. G.; Timerbaev A. R.; Keppler B. K.; Jarosz M. Platinum Group Metallodrug-Protein Binding Studies by Capillary Electrophoresis – Inductively Coupled Plasma-Mass Spectrometry: A Further Insight into the Reactivity of a Novel Antitumor Ruthenium(III) Complex toward Human Serum Proteins. Electrophoresis 2006, 27, 1128–1135. 10.1002/elps.200500694. [DOI] [PubMed] [Google Scholar]
- Groessl M.; Hartinger C. G.; Polec-Pawlak K.; Jarosz M.; Keppler B. K. Capillary Electrophoresis Hyphenated to Inductively Coupled Plasma-Mass Spectrometry: A Novel Approach for the Analysis of Anticancer Metallodrugs in Human Serum and Plasma. Electrophoresis 2008, 29, 2224–2232. 10.1002/elps.200780790. [DOI] [PubMed] [Google Scholar]
- Cetinbas N.; Webb M. I.; Dubland J. A.; Walsby C. J. Serum-Protein Interactions with Anticancer Ru(III) Complexes KP1019 and KP418 Characterized by EPR. JBIC, J. Biol. Inorg. Chem. 2010, 15, 131–145. 10.1007/s00775-009-0578-5. [DOI] [PubMed] [Google Scholar]
- Carter D. C.; Ho J. X. Structure of Serum Albumin. Adv. Protein Chem. 1994, 45, 153–203. 10.1016/S0065-3233(08)60640-3. [DOI] [PubMed] [Google Scholar]
- Ghuman J.; Zunszain P. A.; Petitpas I.; Bhattacharya A. A.; Otagiri M.; Curry S. Structural Basis of the Drug-Binding Specificity of Human Serum Albumin. J. Mol. Biol. 2005, 353, 38–52. 10.1016/j.jmb.2005.07.075. [DOI] [PubMed] [Google Scholar]
- Yang F.; Bian C.; Zhu L.; Zhao G.; Huang Z.; Huang M. Effect of Human Serum Albumin on Drug Metabolism: Structural Evidence of Esterase Activity of Human Serum Albumin. J. Struct. Biol. 2007, 157, 348–355. 10.1016/j.jsb.2006.08.015. [DOI] [PubMed] [Google Scholar]
- Zhu L.; Yang F.; Chen L.; Meehan E. J.; Huang M. A New Drug Binding Subsite on Human Serum Albumin and Drug–Drug Interaction Studied by X-Ray Crystallography. J. Struct. Biol. 2008, 162, 40–49. 10.1016/j.jsb.2007.12.004. [DOI] [PubMed] [Google Scholar]
- Mura P.; Camalli M.; Messori L.; Piccioli F.; Zanello P.; Corsini M. Synthesis, Structural Characterization, Solution Chemistry, and Preliminary Biological Studies of the Ruthenium(III) Complexes [TzH][trans-RuCl4(Tz)2] and [TzH][trans-RuCl4(DMSO) (Tz)]·(DMSO), the Thiazole Analogues of Antitumor ICR and NAMI-A. Inorg. Chem. 2004, 43, 3863–3870. 10.1021/ic0354116. [DOI] [PubMed] [Google Scholar]
- Vergara A.; Krauss I. R.; Montesarchio D.; Paduano L.; Merlino A. Investigating the Ruthenium Metalation of Proteins: X-Ray Structure and Raman Microspectroscopy of the Complex between RNase A and AziRu. Inorg. Chem. 2013, 52, 10714–10716. 10.1021/ic401494v. [DOI] [PubMed] [Google Scholar]
- Das D.; Dutta A.; Mondal P. Interactions of the Aquated Forms of Ruthenium(III) Anticancer Drugs with Protein: A Detailed Molecular Docking and QM/MM Investigation. RSC Adv. 2014, 4, 60548–60556. 10.1039/C4RA10630J. [DOI] [Google Scholar]
- Sabo-Etienne S.; Grellier M.. Ruthenium: Inorganic & Coordination Chemistry. In Encyclopedia of Inorganic Chemistry; John Wiley & Sons: Hoboken, NJ, 2006. [Google Scholar]
- Sudlow G.; Birkett D. J.; Wade D. N. The Characterization of Two Specific Drug Binding Sites on Human Serum Albumin. Mol. Pharmacol. 1975, 11, 824–832. [PubMed] [Google Scholar]
- Ascenzi P.; Bolli A.; Gullotta F.; Fanali G.; Fasano M. Drug Binding to Sudlow’s Site I Impairs Allosterically Human Serum Heme-Albumin-Catalyzed Peroxynitrite Detoxification. IUBMB Life 2010, 62, 776–780. 10.1002/iub.381. [DOI] [PubMed] [Google Scholar]
- Bal W.; Sokołowska M.; Kurowska E.; Faller P. Binding of Transition Metal Ions to Albumin: Sites, Affinities and Rates. Biochim. Biophys. Acta, Gen. Subj. 2013, 1830, 5444–5455. 10.1016/j.bbagen.2013.06.018. [DOI] [PubMed] [Google Scholar]
- Yang F.; Zhang Y.; Liang H. Interactive Association of Drugs Binding to Human Serum Albumin. Int. J. Mol. Sci. 2014, 15, 3580–3595. 10.3390/ijms15033580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levina A.; Aitken J. B.; Gwee Y. Y.; Lim Z. J.; Liu M.; Singharay A. M.; Wong P. F.; Lay P. A. Biotransformations of Anticancer Ruthenium(III) Complexes: An X-Ray Absorption Spectroscopic Study. Chem. - Eur. J. 2013, 19, 3609–3619. 10.1002/chem.201203127. [DOI] [PubMed] [Google Scholar]
- Zsila F. Subdomain IB Is the Third Major Drug Binding Region of Human Serum Albumin: Toward the Three-Sites Model. Mol. Pharmaceutics 2013, 10, 1668–1682. 10.1021/mp400027q. [DOI] [PubMed] [Google Scholar]
- Zunszain P. A.; Ghuman J.; Komatsu T.; Tsuchida E.; Curry S. Crystal Structural Analysis of Human Serum Albumin Complexed with Hemin and Fatty Acid. BMC Struct. Biol. 2003, 3, 6. 10.1186/1472-6807-3-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zunszain P. A.; Ghuman J.; McDonagh A. F.; Curry S. Crystallographic Analysis of Human Serum Albumin Complexed with 4Z,15E-Bilirubin-IXα. J. Mol. Biol. 2008, 381, 394–406. 10.1016/j.jmb.2008.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith C. A.; Sutherland-Smith A. J.; Kratz F.; Baker E. N.; Keppler B. H. Binding of Ruthenium (III) Anti-Tumor Drugs to Human Lactoferrin Probed by High Resolution X-Ray Crystallographic Structure Analyses. JBIC, J. Biol. Inorg. Chem. 1996, 1, 424–431. 10.1007/s007750050074. [DOI] [Google Scholar]
- Casini A.; Temperini C.; Gabbiani C.; Supuran C. T.; Messori L. The X-Ray Structure of the Adduct between NAMI-A and Carbonic Anhydrase Provides Insights into the Reactivity of This Metallodrug with Proteins. ChemMedChem 2010, 5, 1989–1994. 10.1002/cmdc.201000331. [DOI] [PubMed] [Google Scholar]
- Vergara A.; D’Errico G.; Montesarchio D.; Mangiapia G.; Paduano L.; Merlino A. Interaction of Anticancer Ruthenium Compounds with Proteins: High-Resolution X-Ray Structures and Raman Microscopy Studies of the Adduct between Hen Egg White Lysozyme and AziRu. Inorg. Chem. 2013, 52, 4157–4159. 10.1021/ic4004142. [DOI] [PubMed] [Google Scholar]
- Hummer A. A.; Heffeter P.; Berger W.; Filipits M.; Batchelor D.; Büchel G. E.; Jakupec M. A.; Keppler B. K.; Rompel A. X-Ray Absorption Near Edge Structure Spectroscopy to Resolve the in Vivo Chemistry of the Redox-Active Indazolium Trans-[Tetrachlorobis(1H-indazole)ruthenate(III)] (KP1019). J. Med. Chem. 2013, 56, 1182–1196. 10.1021/jm301648f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hummer A. A.; Rompel A. The Use of X-Ray Absorption and Synchrotron Based Micro-X-Ray Fluorescence Spectroscopy to Investigate Anti-Cancer Metal Compounds in Vivo and in Vitro. Metallomics 2013, 5, 597–614. 10.1039/c3mt20261e. [DOI] [PubMed] [Google Scholar]
- Webb M. I.; Wu B.; Jang T.; Chard R. A.; Wong E. W. Y.; Wong M. Q.; Yapp D. T. T.; Walsby C. J. Increasing the Bioavailability of RuIII Anticancer Complexes through Hydrophobic Albumin Interactions. Chem. - Eur. J. 2013, 19, 17031–17042. 10.1002/chem.201302671. [DOI] [PubMed] [Google Scholar]
- Jackson M. R.; Beahm R.; Duvvuru S.; Narasimhan C.; Wu J.; Wang H.-N.; Philip V. M.; Hinde R. J.; Howell E. E. A Preference for Edgewise Interactions between Aromatic Rings and Carboxylate Anions: The Biological Relevance of Anion–Quadrupole Interactions. J. Phys. Chem. B 2007, 111, 8242–8249. 10.1021/jp0661995. [DOI] [PubMed] [Google Scholar]
- Schwans J. P.; Sunden F.; Lassila J. K.; Gonzalez A.; Tsai Y.; Herschlag D. Use of Anion–Aromatic Interactions to Position the General Base in the Ketosteroid Isomerase Active Site. Proc. Natl. Acad. Sci. U. S. A. 2013, 110, 11308–11313. 10.1073/pnas.1206710110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lucas X.; Bauzá A.; Frontera A.; Quiñonero D. A Thorough Anion−π Interaction Study in Biomolecules: On the Importance of Cooperativity Effects. Chem. Sci. 2016, 7, 1038–1050. 10.1039/C5SC01386K. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simard J. R.; Zunszain P. A.; Hamilton J. A.; Curry S. Location of High and Low Affinity Fatty Acid Binding Sites on Human Serum Albumin Revealed by NMR Drug-Competition Analysis. J. Mol. Biol. 2006, 361, 336–351. 10.1016/j.jmb.2006.06.028. [DOI] [PubMed] [Google Scholar]
- Bhattacharya A. A.; Curry S.; Franks N. P. Binding of the General Anesthetics Propofol and Halothane to Human Serum Albumin. High Resolution Crystal Structures. J. Biol. Chem. 2000, 275, 38731–38738. 10.1074/jbc.M005460200. [DOI] [PubMed] [Google Scholar]
- Simard J. R.; Zunszain P. A.; Ha C.-E.; Yang J. S.; Bhagavan N. V.; Petitpas I.; Curry S.; Hamilton J. A. Locating High-Affinity Fatty Acid-Binding Sites on Albumin by X-Ray Crystallography and NMR Spectroscopy. Proc. Natl. Acad. Sci. U. S. A. 2005, 102, 17958–17963. 10.1073/pnas.0506440102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y.; Luo Z.; Shi X.; Wang H.; Nie L.; Huang M. A Fluorescent Fatty Acid Probe, DAUDA, Selectively Displaces Two Myristates Bound in Human Serum Albumin. Protein Sci. Publ. Protein Soc. 2011, 20, 2095–2101. 10.1002/pro.749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zsila F. Circular Dichroism Spectroscopic Detection of Ligand Binding Induced Subdomain IB Specific Structural Adjustment of Human Serum Albumin. J. Phys. Chem. B 2013, 117, 10798–10806. 10.1021/jp4067108. [DOI] [PubMed] [Google Scholar]
- Zhang Z.Targeted Functional Proteomics to Study Protein Post-Translational Modifications and Protein-Protein Interaction. Ph.D. Thesis, Pittsburgh, PA, 2007. [Google Scholar]
- Ito T.; Sato K.; Maekawa H.; Sakurada M.; Orita H.; Shimada K.; Daida H.; Wada R.; Abe M.; Hino O.; Kajiyama Y. Elevated Levels of Serum Fatty Acid Synthase in Patients with Gastric Carcinoma. Oncol. Lett. 2013, 7, 616–620. 10.3892/ol.2014.1793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fasano M.; Curry S.; Terreno E.; Galliano M.; Fanali G.; Narciso P.; Notari S.; Ascenzi P. The Extraordinary Ligand Binding Properties of Human Serum Albumin. IUBMB Life 2005, 57, 787–796. 10.1080/15216540500404093. [DOI] [PubMed] [Google Scholar]
- Ferraro G.; Massai L.; Messori L.; Merlino A. Cisplatin Binding to Human Serum Albumin: A Structural Study. Chem. Commun. 2015, 51, 9436–9439. 10.1039/C5CC01751C. [DOI] [PubMed] [Google Scholar]
- Webb M. I.; Walsby C. J. EPR as a Probe of the Intracellular Speciation of Ruthenium(III) Anticancer Compounds. Metallomics 2013, 5, 1624–1633. 10.1039/c3mt00090g. [DOI] [PubMed] [Google Scholar]
- Sengupta A.; Koninti R. K.; Gavvala K.; Ballav N.; Hazra P. An Anticancer Drug to Probe Non-Specific Protein–DNA Interactions. Phys. Chem. Chem. Phys. 2014, 16, 3914–3917. 10.1039/c3cp54422b. [DOI] [PubMed] [Google Scholar]
- Bradford M. M. A Rapid and Sensitive Method for the Quantitation of Microgram Quantities of Protein Utilizing the Principle of Protein-Dye Binding. Anal. Biochem. 1976, 72, 248–254. 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.